Transition-metal-catalyzed asymmetric denitrogenative transannulation
Wen-Ge Guo
1,2
,
Ren-Rong Liu
1,*

*Correspondence to:
Ren-Rong Liu, College of Pharmaceutical Sciences, Guizhou University, Guiyang 550025, Guizhou, China.
E-mail: rrliu@gzu.edu.cn
Chiral Chem. 2025;1:202504. 10.70401/cc.2025.0001
Received: September 17, 2025Accepted: November 03, 2025Published: November 06, 2025
Abstract
Over the past few decades, denitrogenation has proven to be an effective method for synthesizing high-value chiral heterocyclic compounds. These compounds find widespread applications in pharmaceutical chemistry, drug development, and natural product synthesis. Denitrogenation demonstrates high activity and can engage in cyclization reactions with olefins, alkynes, carbon-heterocycles, aldehydes, and other reagents. This one-step operation enables the rapid construction of chiral heterocycles such as pyrroles and indoles, significantly shortening complex synthetic pathways. Innovations in chiral ligands, optimization of catalytic systems, and detailed studies on mechanisms have significantly enhanced the enantioselectivity and substrate applicability of denitrogenation reactions. This review highlights recent advancements in the synthesis of chiral heterocycles via denitrogenation reactions and systematically examines the reaction characteristics of various metal catalytic systems.
Graphical Abstract
Keywords
Denitrogenation, transition-metal-catalyzed, heterocycles, cyclization, enantioselective
1. Introduction
Heterocyclic compounds are organic compounds that contain at least one heteroatom within their cyclic structure. Serving as fundamental frameworks for many natural and synthetic functional molecules, heterocyclic compounds have extensively impacted crucial domains such as pharmaceutical research and development[1-4], pesticide creation[5,6], dye synthesis[7,8], catalytic technology[9,10], and polymer materials[11]. Among them, chiral heterocyclic compounds are not only important members of the heterocyclic compound family but also widely exist as core skeletons in natural products, bioactive molecules, and clinical drugs. Data indicate that approximately 59% of small-molecule drugs approved by the U.S. Food and Drug Administration feature chiral nitrogen-containing heterocyclic segments in their molecular structures, highlighting their core status in the field of medicinal chemistry[12,13].
Within organisms, distinct pharmacological activities and varying toxic side effects are frequently observed between different enantiomers of chiral drugs[14,15]. This notable distinction poses a significant challenge in the synthesis of chiral molecules within the realm of synthetic chemistry research. At the same time, given the extensive utilization of chiral heterocyclic compounds in scientific and technological applications, the development of effective methods for synthesizing functional heterocyclic structures has remained a primary focus in organic chemistry for over a century. Among the many strategies for constructing chiral heterocyclic compounds, traditional methods such as asymmetric cyclization and chiral resolution can achieve the synthesis of target compounds to some extent, but they often have limitations such as complicated reaction steps, low enantioselectivity and narrow substrate applicability. Transition-metal-catalyzed denitrogenation cyclization has emerged as a potent strategy for generating valuable chiral heterocycles compared toother established methodologies. Denitrogenation cyclization has outstanding advantages: it is a single-step process with high efficiency and atomic economy, and the only by-product is environmentally friendly nitrogen[16]. Additionally, this approach offers a distinctive pathway to transform easily accessible raw materials into crucial molecular frameworks, thereby placing denitrogenation-cyclized compounds at the forefront of chiral organic molecule synthesis. The active centers of transition metals cooperate with chiral ligands (e.g., bisphosphines, oxazolines), defining structures and controlling chirality through steric hindrance and electronic effects. The oxidation state of the metal regulates intermediate activity, while substrate directing groups lock the transition state, achieving multi-dimensional stereocontrol[17]. With its precise stereoselectivity, the nitrogen removal strategy enables the efficient construction of the core heterocyclic skeletons in pharmaceuticals like lenvatinib and cabozantinib. Moreover, it facilitates the total synthesis of natural products while supporting the preparation of core heterocyclic units for both pharmaceutical and functional material applications[18,19]. This review outlines and analyzes the recent advancements in the synthesis of chiral heterocyclic compounds via denitrogenation reactions based on underlying reaction mechanisms.
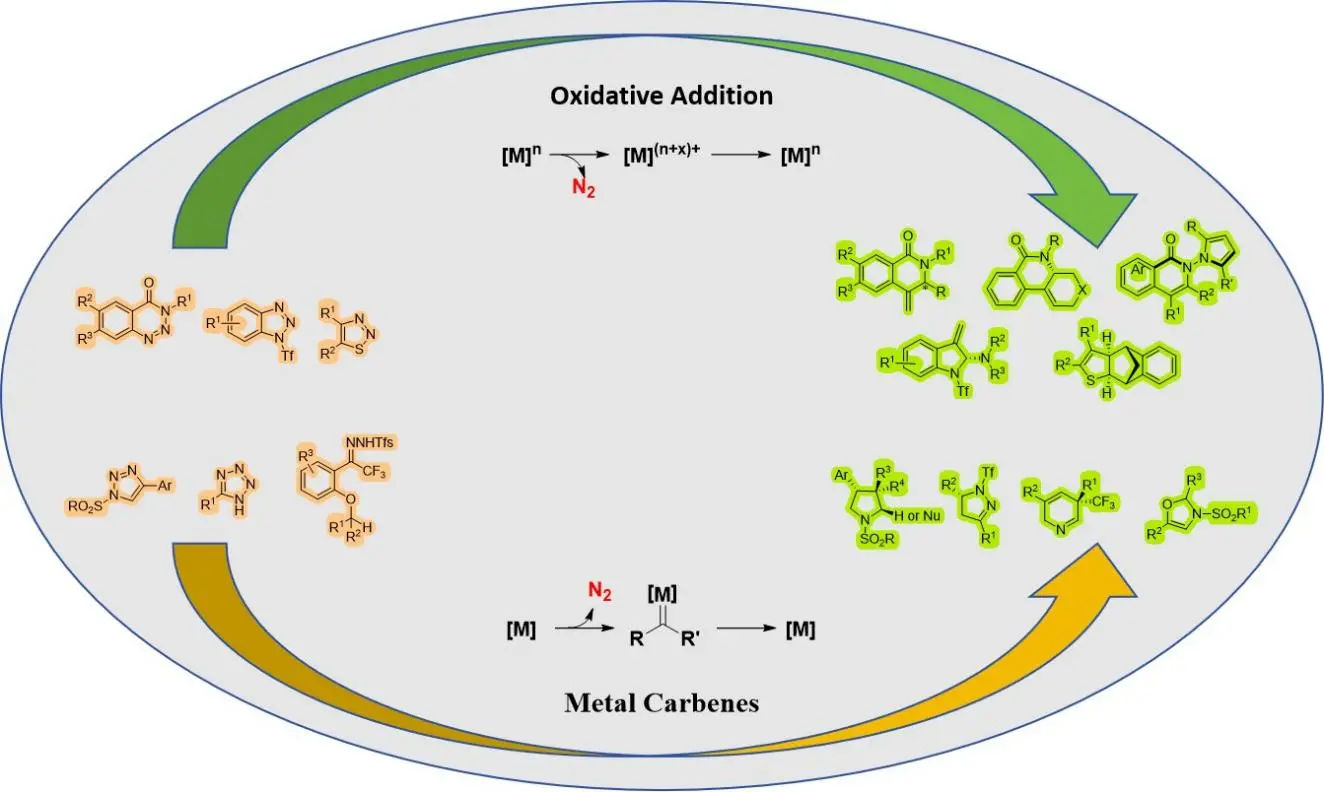
2. Denitrogenative Transannulation via Oxidative Addition
Oxidative addition involves a coordinated electron transfer process between the metal and ligand, which can be facilitated by enhancing the electron richness of the metal center using additional ligands. Typically, electron-rich metal centers and ligands that donate electrons are favored in oxidative addition reactions, but there are exceptions. Electron-donating ligands increase the electron density around metal centers, promoting metal-to-ligand charge transfer, whereas electron-withdrawing ligands decrease this density, favoring ligand-metal charge transfer mechanisms[20]. Nickel (Ni)[21], Palladium (Pd)[22], Rhodium (Rh)[23], and Iridium (Ir)[24] serve as primary catalysts for generating chiral heterocyclic compounds through denitrogenation and oxidative addition processes (Table 1).
Table 1. Denitrogenative transannulation via oxidative addition.
| Substrate | Type of Reaction | Catalyst | Ligand | Product | Reference |
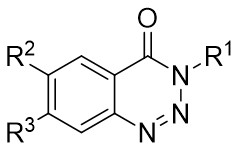 | Intermolecular Annulation | Ni(COD)2 | 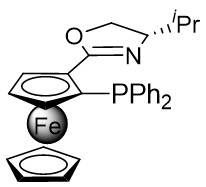 | 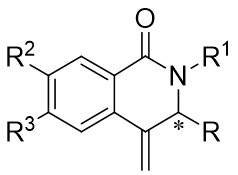 | [29] |
 | Intermolecular Annulation | Ni(COD)2 |  | 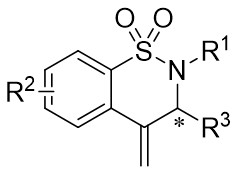 | [30] |
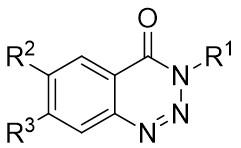 | Intermolecular Annulation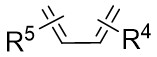 | Ni(COD)2 | 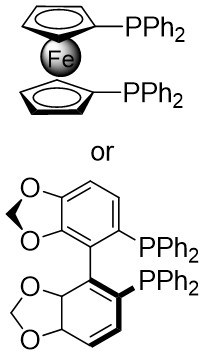 | 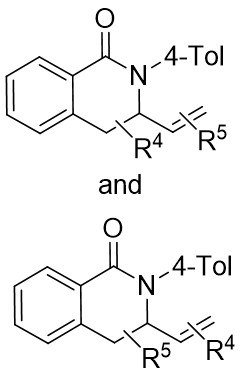 | [31] |
 | Intermolecular Annulation | Ni(COD)2 |  | 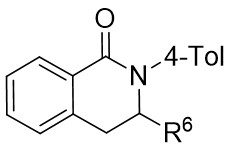 | [31] |
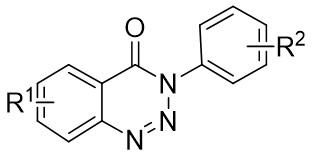 | Intermolecular Annulation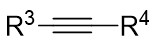 | Ni(COD)2 | 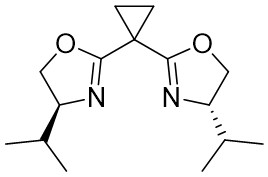 | 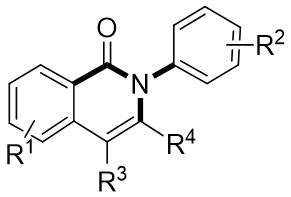 | [32] |
 | Intermolecular Annulation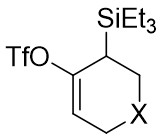 | Ni(COD)2 | 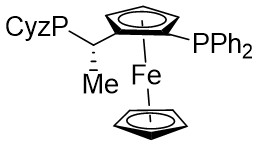 | 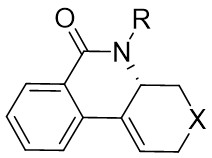 | [33] |
 | Intermolecular Annulation | Ni(COD)2 | 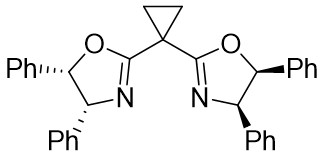 | 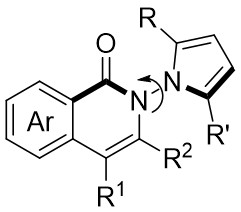 | [34] |
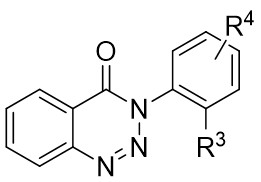 | Intermolecular Annulation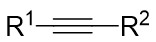 | Ni(COD)2 | 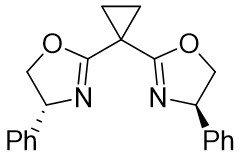 | 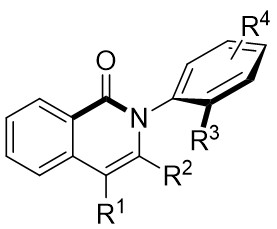 | [35] |
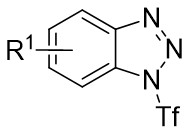 | Intermolecular Annulation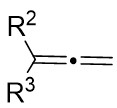 | Pd2(dba)3 | 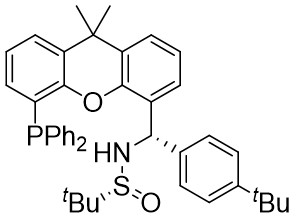 | 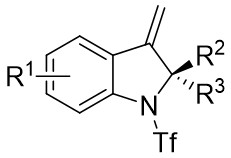 | [22] |
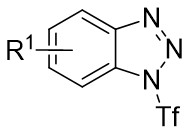 | Intermolecular Annulation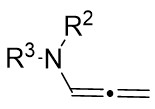 | Pd2(dba)3 |  | 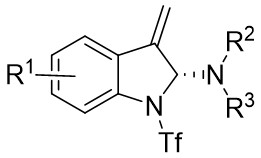 | [22] |
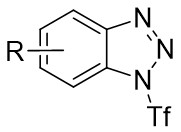 | Intermolecular Annulation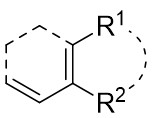 | Pd2(dba)3·CHCl3 | 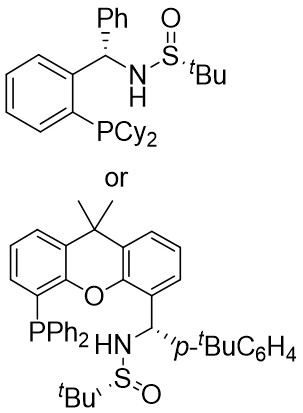 | 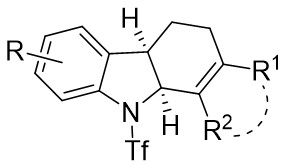 | [39] |
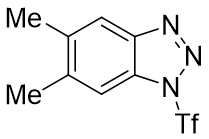 | Intermolecular Annulation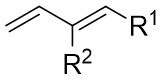 | Pd2(dba)3·CHCl3 |  | 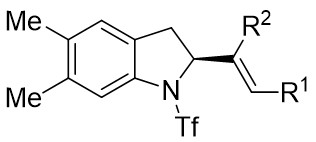 | [39] |
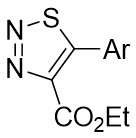 | Intermolecular Annulation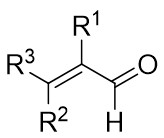 | [Rh(COD)Cl]2 |  | 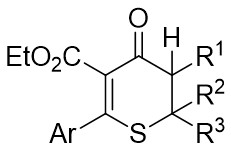 | [23] |
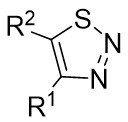 | Intermolecular Annulation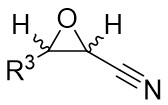 | [Rh(COD)Cl]2 | 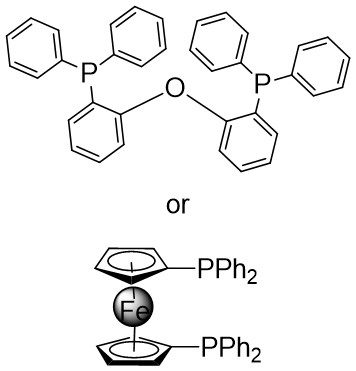 | 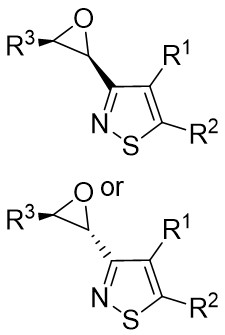 | [43] |
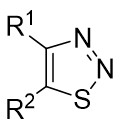 | Intermolecular Annulation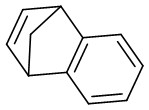 | [Rh(COD)Cl]2 |  | 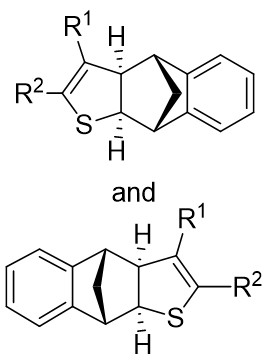 | [45] |
 | Intermolecular Annulation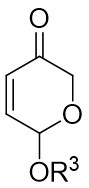 | [Rh(COD)Cl]2 | 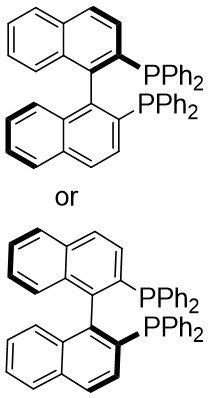 | 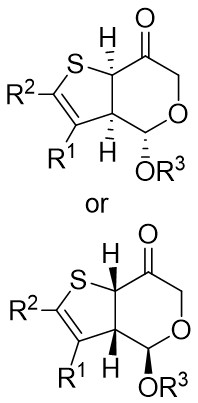 | [46] |
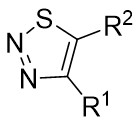 | Intermolecular Annulation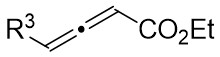 | [Rh(COD)Cl]2 | 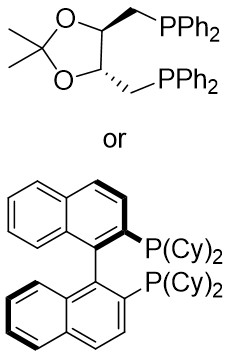 | 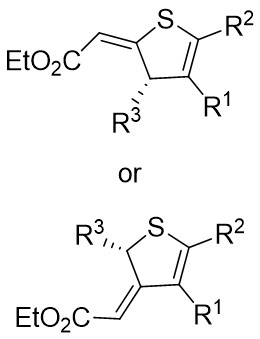 | [47] |
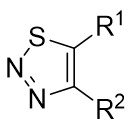 | Intermolecular Annulation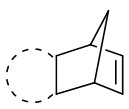 | [Ir(COD)Cl]2 | 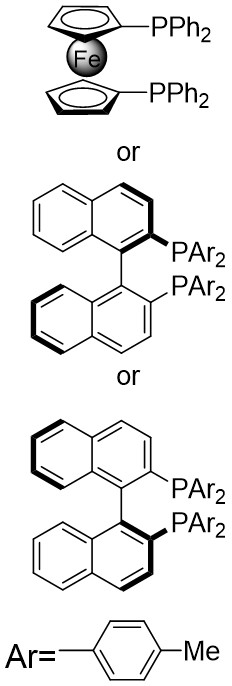 | 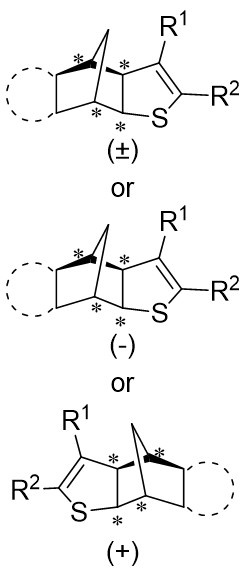 | [24] |
2.1 Nickel-catalyzed denitrogenative annulation reactions
Nickel exhibits a small nuclear radius, along with low electronegativity and reduction potential. These characteristics enable it to readily form relatively stable paramagnetic intermediates while effectively suppressing the occurrence of β-elimination. Additionally, nickel complexes can exist in both high-spin and low-spin configurations, and nickel maintains relatively good stability across all oxidation states from 0 to IV. Consequently, nickel-catalyzed reactions are versatile, capable of proceeding via either single-electron or double-electron mechanisms[25-28].
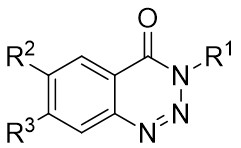
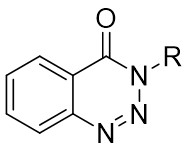
In 2009, Murakami and colleagues published a study on the denitrogenation of 1,2,3-benzotriazinones 1.1 using alkenes 1.2, resulting in the formation of substituted 3,4-dihydroisoquinoline-1(2H)-ones 1.3 with specific regioselectivity and enantioselectivity patterns (Figure 1)[29]. The selection of ligands played a crucial role in shaping the reaction outcomes. Chiral bidentate hydrogen phosphate ligand was adopted to carry out the reaction of the obtained product with high enantioselectivity. Although (R,R)-Me-DuPhos and (S,S,R,R)-TangPhos showed reasonable enantioselectivity, their regioselectivity was insufficient. Conversely, the use of (S,S)-i-Pr-FOXAP improved both regioselectivity and enantioselectivity to desirable levels, facilitating the incorporation of various functional groups in the reaction.
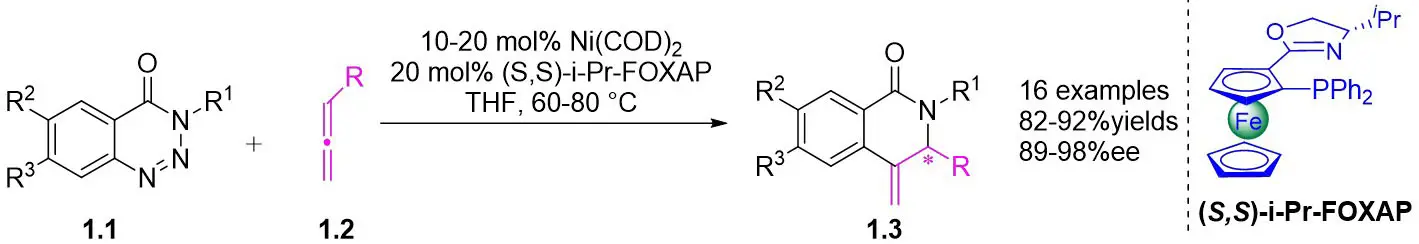
Figure 1. Denitrogenative annulation of 1,2,3-benzotriazinones 1.1 with allenes. THF: tetrahydrofuran.
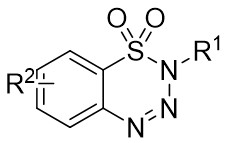
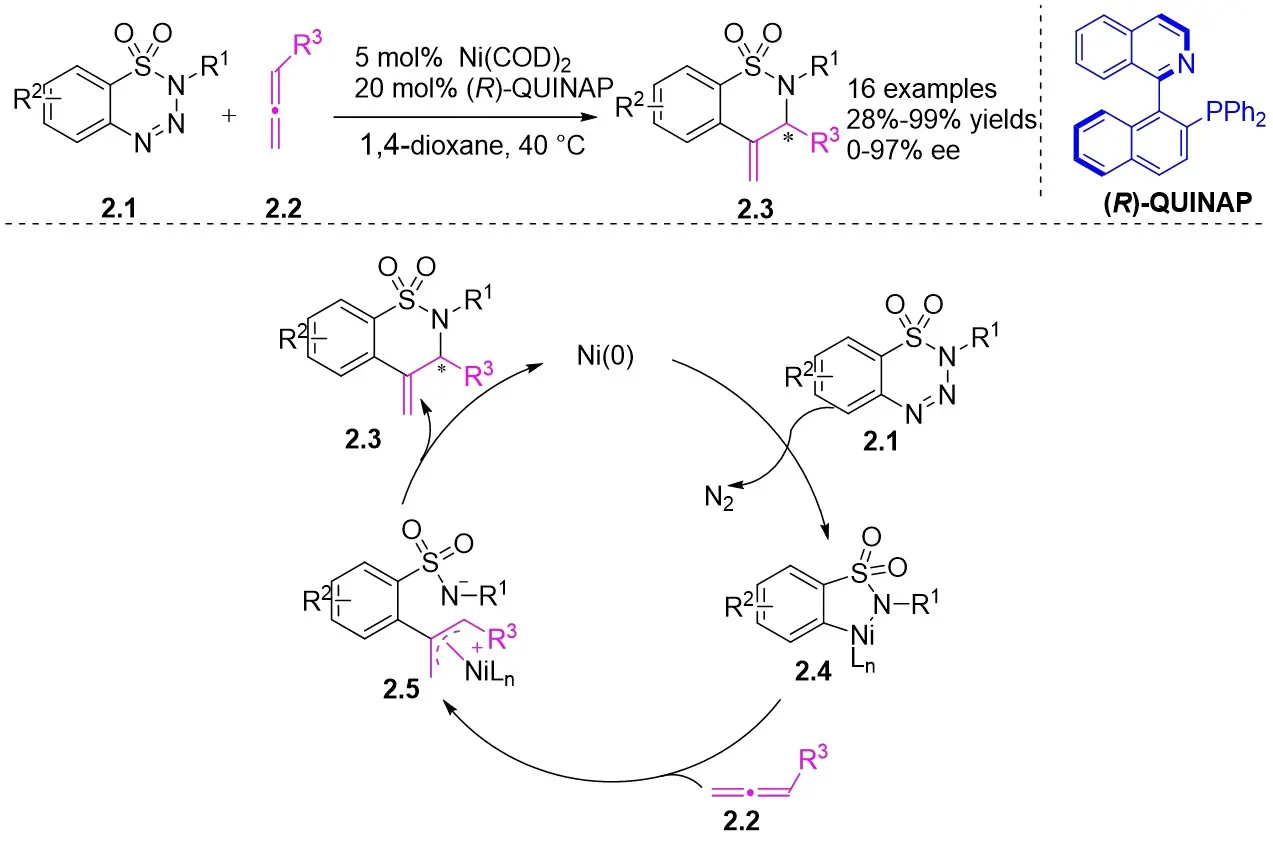
In 2010, Murakami et al. introduced a nickel-catalyzed method for enantioselective cyclization using 1,2,3,4-benzothiazinones 2.1 and allenes 2.2 to synthesize 1,2,3,4-benzothiazine-1,1(2H)-dioxide derivatives 2.3 (Figure 2)[30]. Most 1,2,3,4-benzothiazinones 2.1 and enynes 2.2, featuring primary or secondary alkyl groups as R1 and electron-donating or electron-withdrawing substituents as R2, can undergo denitrocyclization to yield 1,2,3,4-benzothiazine-1,1(2H)-dioxide derivatives 2.3 with high yield and enantioselectivity. Additionally, alkylalkynes with siloxy, benzyloxy, and N-phthalimide groups were also compatible with the reaction, albeit resulting in slightly lower enantiomeric purity. The reaction utilized Ni(COD)2 as a catalyst and (R)-QUINAP as a ligand to optimize enantioselectivity. The proposed cyclization mechanism includes: 1) oxidative addition of nickel(0) to the N-N bond of 1,2,3,4-benzothiazinone 2.1, 2) formation of a five-membered azacyclic ring 2.4 via nitrogen elimination from benzothiazinone, 3) allene insertion to form intermediate 2.5, and 4) allylamidation at the more substituted carbon for product generation and regeneration of the nickel(0).
In the same year, Murakami and colleagues detailed a process in which 1,2,3-benzotriazin-4(3H)-ones 3.1 react with 1,3-dienes 3.2 and alkenes 3.3 in the presence of nickel(0)/phosphine complexes, yielding a variety of 3,4-dihydroisoquinoline-1(2H)-ones (Figure 3)[31]. Specifically, they examined the reaction of 3.1 with 1,3-dienes (3.2, 2 equiv.) in tetrahydrofuran (THF) at 60 °C utilizing in-situ-generated nickel(0) catalysts derived from Ni(COD)2 and PMe3. Investigations into chiral phosphine ligands revealed enhanced yield and enantioselectivity with ligands such as (S)-Segphos. However, the enantioselectivity was noted to be suboptimal. The proposed mechanism for this nickel-catalyzed cyclization bears close resemblance to the one described for the nickel-catalyzed cyclization of benzotriazinones 2.1 with allenes 2.2.
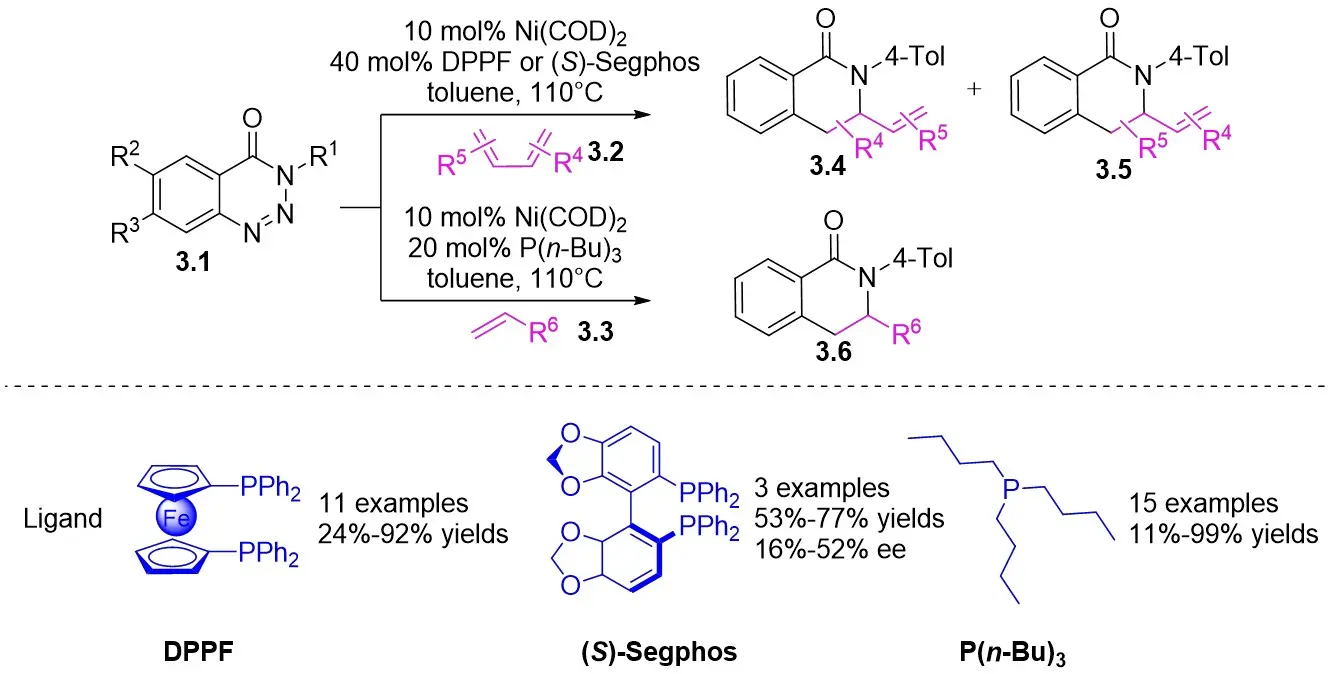
Figure 3. Denitrogenative annulation of 1,2,3-benzotriazin-4(3H)-ones 3.1 with 1,3-dienes and alkenes. DPPF: 1,1’-bis(diphenylphosphino)ferrocene.
In 2015, Liu et al. made a significant advancement by pioneering the Ni-catalyzed asymmetric denitrogenative transannulation of 1,2,3-benzotriazine-4(3H)-ones 4.1 with bulky internal alkynes 4.2 (Figure 4)[32]. This method resulted in the creation of a new class of axially chiral isoquinolones with high yield, remarkable enantioselectivity, and nearly complete regioselectivity, all achieved under mild reaction conditions. The study revealed the universal nature of the R1 substituent, showing that various alkynes with diverse substituents at different positions and benzene ring substituents with electron-neutral or electron-deficient groups at various nitrogen atom positions could afford product 4.3 with good yield and high enantiomeric excess (ee) values. The reaction mechanism involves the triazole’s interaction with neutral Ni(COD)2 in the presence of excess trimethylphosphine, leading to the formation of intermediate 4.4, which further evolves into a seven-membered ring 4.5. Intermediate 4.5 undergoes C-N bond cleavage and Ni–C bond formation, resulting in the formation of a five-membered ring complex 4.6. Subsequent reactions involve 4.7 interacting with Ni complexes 4.6 via oxidative addition of the alkyne C≡C bond, forming the carbon-metallated species 4.8. This is succeeded by the insertion reaction of the Ni–C bond to generate the seven-membered Ni heterocyclic complex 4.9. Further steps lead 4.9 through a closed-loop reaction to produce intermediate 4.10, which upon reductive elimination of 4.10 yields the final product. This cyclic process also regenerates the catalyst, enabling it to reinitiate the catalytic cycle. Additionally, the study highlighted the preference for (aR) configured products due to their passage through energetically more stable states, where the naphthyl substituent and chiral ligand ring adopt opposing positions through atropselectivity, optimizing energy utilization[17,32].
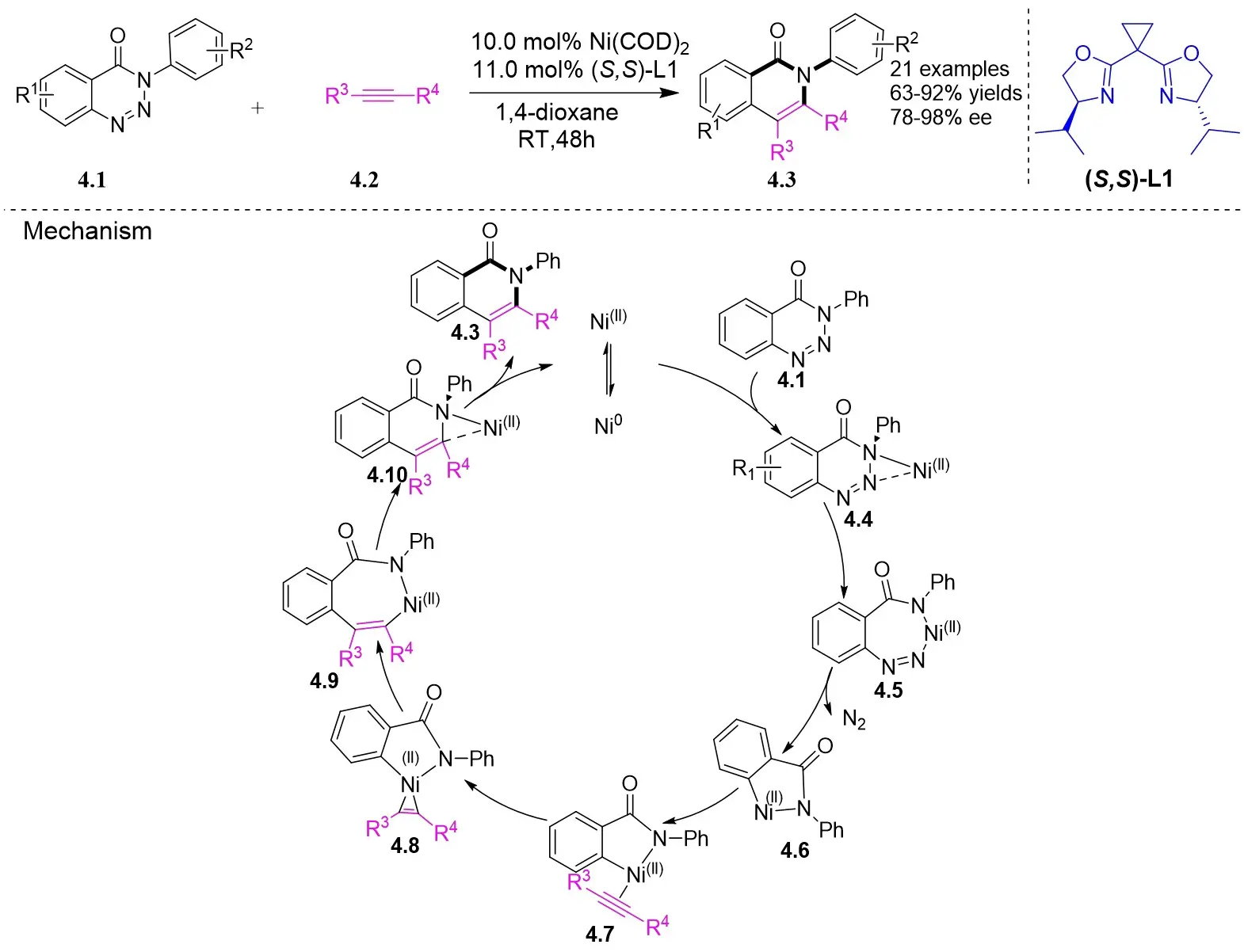
Figure 4. Asymmetric synthesis of axially chiral isoquinolones 4.3. RT: room temperature.
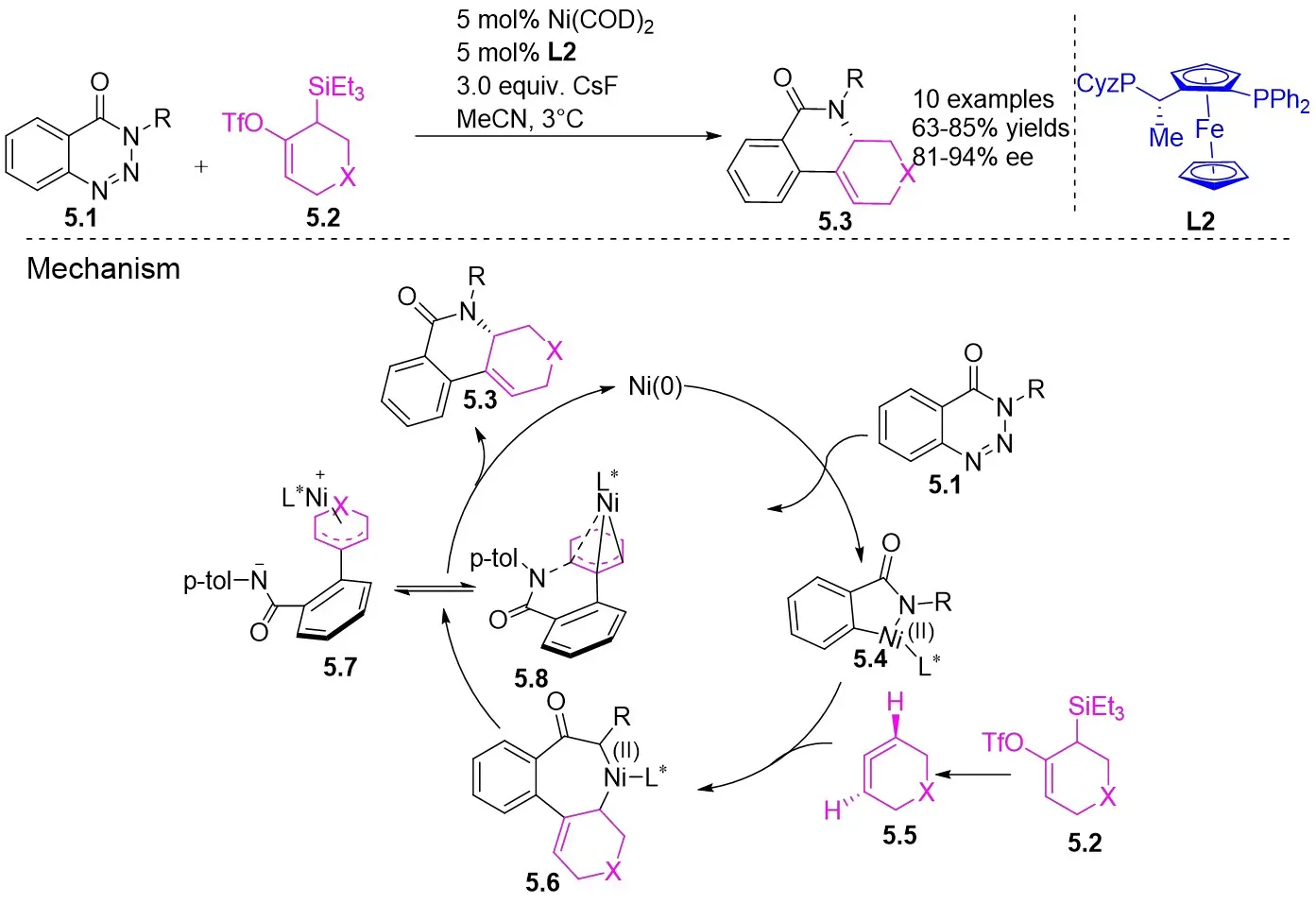
In 2020, Garg and Houk developed a denitrogenation strategy to cyclize phenyltriazinones 5.1 with acetylene precursors 5.2, resulting in chiral tricyclic compounds 5.3 (Figure 5)[33]. Initially, they investigated racemic variants by reacting different benzotriazinones with cyclic olefins at 35 °C using Ni(COD)2 as a precatalyst, 1,1’-bis(diphenylphosphino)ferrocene (DPPF) as a ligand, and cesium fluoride as a fluoride source in acetonitrile, achieving high-yield racemic products that showcased the broad applicability of the method. Subsequent utilization of the L2 chiral ligand led to a highly enantioselective product with excellent yield. The catalytic mechanism involved the rapid and irreversible oxidative addition of benzotriazinones, leading to the formation of metal ring 5.4, followed by olefin insertion to generate 5.6, ultimately producing the final product and regenerating the catalytic cycle.
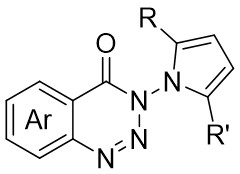
In 2024, Liu and colleagues reported the first discovery that 1,2,3-benzotriazones 6.1 and internal alkynes 6.2 undergo oxidative addition to form N–N diastereomeric isoquinolinone derivatives substituted at the 2-position under nickel catalysis (Figure 6)[34]. They determined that an in situ generated chiral Ni(0) catalyst using Ni(COD)2 and L3 in THF at 25 °C was optimal for the reaction of benzotriazones with alkynes in terms of yield, enantioselectivity, and regioselectivity. Various asymmetric alkynes and substituents were found to be universally applicable, yielding the corresponding N–N isomers with excellent yields (65-94%) and high enantioselectivities (80-99%), all being single regioisomers. The reaction involves the conversion of 6.1 to 6.4 under specific conditions. Subsequently, 6.4 transforms into the nickelacycle species 6.5. N2 extrusion occurs through 6.5, followed by the coordination of alkynes 6.2 to give 6.7. Migratory insertion via 6.8 leads to the formation of intermediate 6.9, which then undergoes reductive elimination to yield 6.10. The catalytic cycle is completed with the interchange between substrate 6.1 and product 6.3, initiating the next reaction.
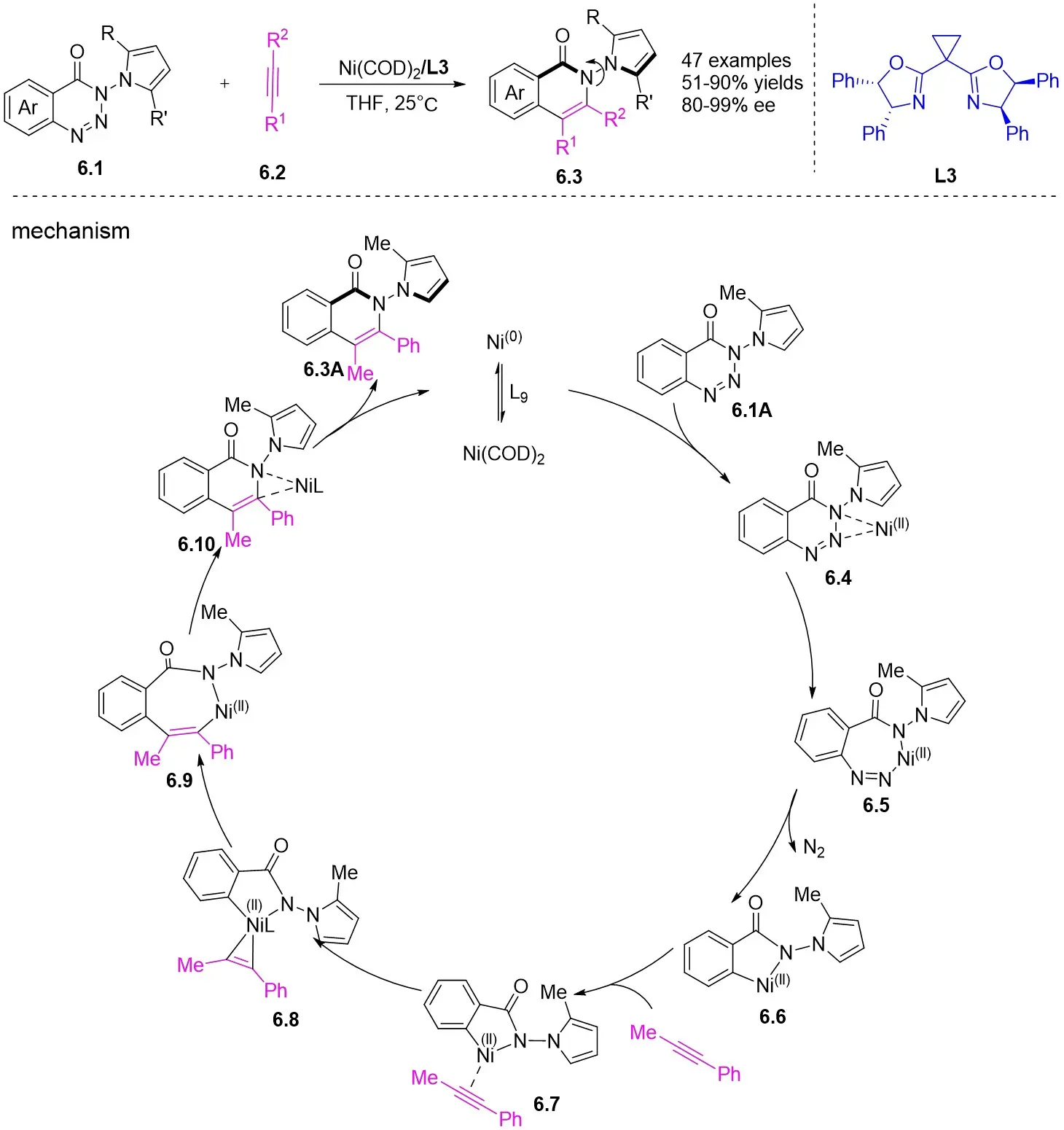
Figure 6. Asymmetric synthesis of N–N atropisomers isoquinolinone derivatives 6.3. THF: tetrahydrofuran.
In 2025, Guo et al. developed an efficient enantioselective nickel-catalyzed cyclization reaction using a nitrogen removal strategy to synthesize new enantiomers under mild conditions (Figure 7)[35]. Experimental results demonstrated the formation of a series of axial chiral enantiomers with chiral C–N bonds, exhibiting exceptional enantioselectivity and regioselectivity. Notably, this method exhibited broad tolerance toward various alkynes (excluding diphenyl substitution) and 1,2,3-benzotriazinone substrates. Mechanistically, the initial step involves the oxidative addition reaction of nickel(0) and 1,2,3-benzotriazinones 7.1, resulting in the release of nitrogen and the formation of chiral five-membered intermediate 7.4. Subsequently, alkyne 7.2 inserts into the C–Ni bond at a specific angle precisely controlled by the chiral box ligand L4, leading to the formation of intermediate 7.5 through cleavage of one of the π bonds of the alkyne. The steric hindrance imposed by ligand L1 prevents the free rotation of the nitrogen atom substituent, playing a crucial role in controlling enantioselectivity. Furthermore, the cooperation of phenyl (Ph) and carbonyl (CO) groups inhibited the rotation of the substituent on the nitrogen atom, ultimately resulting in the optical activity of the newly formed atropisomers 7.3. This nickel-catalyzed asymmetric denitrogenation method offers an effective pathway for synthesizing chiral C–N atopic isomers.
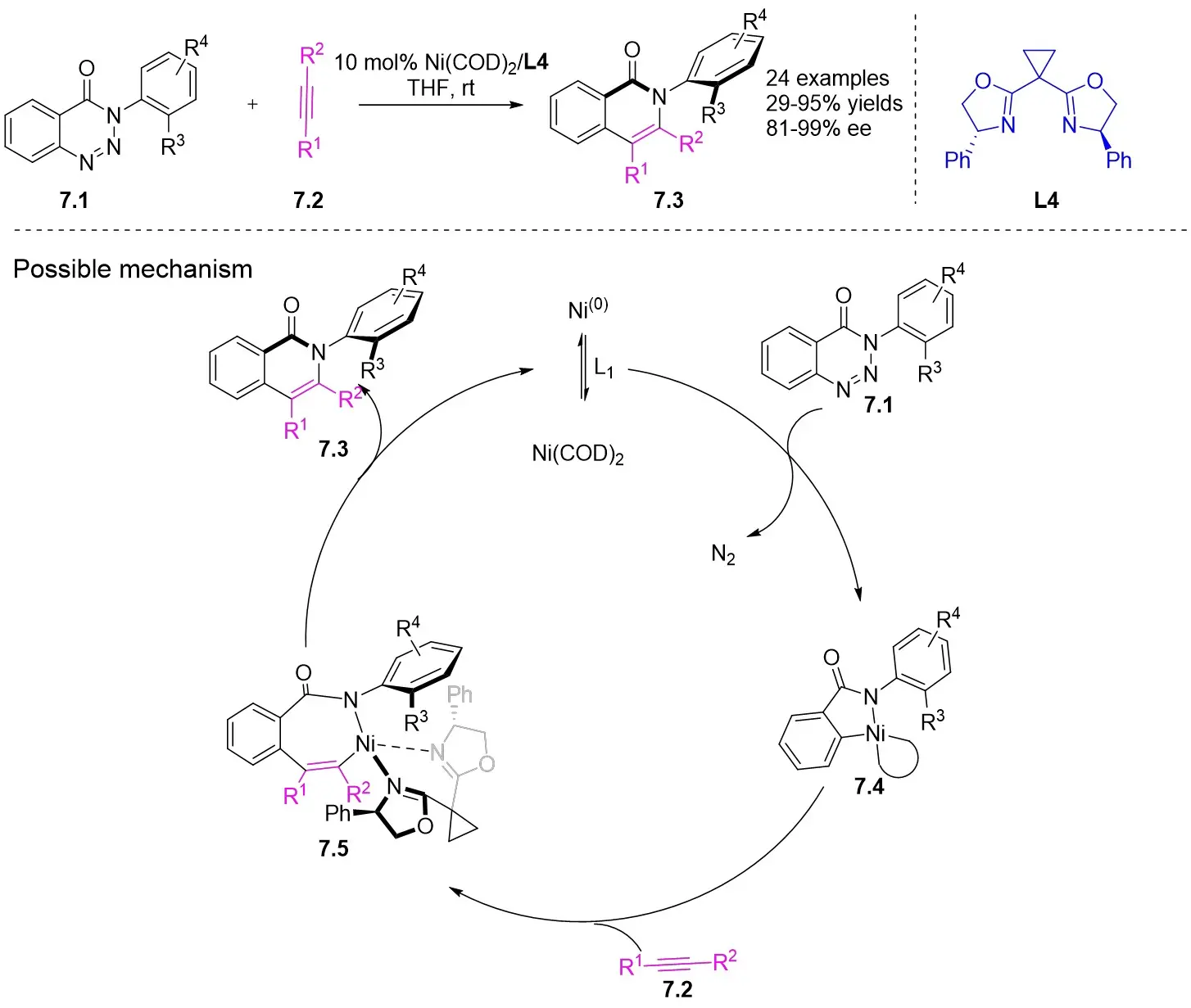
Figure 7. Asymmetric synthesis of C–N atropisomers isoquinolinone derivatives 7.3. THF: tetrahydrofuran.
2.2 Palladium-catalyzed denitrogenative annulation reactions
Palladium catalysts represent a class of crucial metal catalysts that have garnered significant attention in the field of chemistry. They can form stable complexes with a diverse range of ligands, efficiently facilitate reactions under relatively mild conditions, and exhibit excellent product selectivity. Furthermore, these catalysts possess distinctive reaction versatility—being capable of catalyzing various types of reactions—and high modifiability. These combined features have led to extensive research and application on palladium catalysts by chemists[36-38].
In 2019, Zhang et al. first reported the enantioselective denitrogenative cyclization of benzotriazoles 8.1 catalyzed by Pd/PC-Phos (Figure 8)[22]. Benzotriazoles 8.1 reacted with alkenes 8.4 at 40 °C in the presence of catalytic amounts of Pd2(dba)3 and (Sc,Rs)-L5, using dimethyl sulfoxide (DMSO) as the solvent, to yield 3-methylindololines 8.3. The reaction showed tolerance to both electron-donating and electron-withdrawing groups on the aryl unit of the allenes, leading to high yields between 89% and 97% and enantiomeric excesses of 89% to 98%. For expanding this method in synthesizing 2-substituted 3-methylindololine derivatives, N-acrylamides 8.4 were used as the cycloaddition partner in conjunction with (Sc,Rs)-L6 as the chiral ligand. The corresponding indololine products were obtained in yields ranging from 88% to 95%. Importantly, numerous sensitive functional groups were shown to be compatible with these optimized reaction conditions. Benzotriazoles 8.1 undergo chain isomerization to give rise to 8.6. Insertion of C–Pd bond of alkynenes 8.2 or alkynamines 8.4, a p-allyl palladium complex 8.7 can be formed. After an allyl substitution reaction, a cyclization product 8.3 or 8.5 will be generated, and a palladium catalyst will be regenerated.
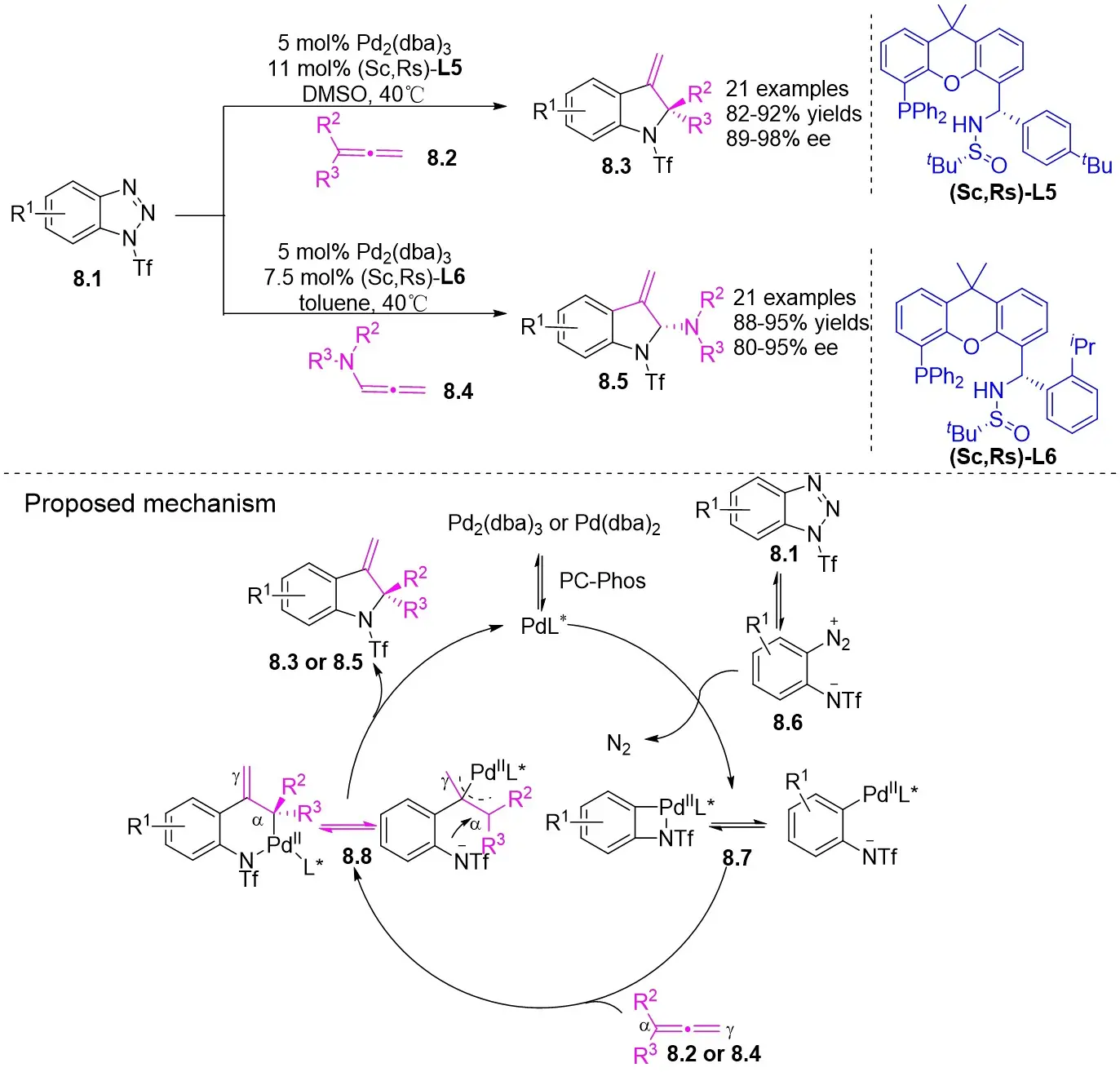
Figure 8. Denitrogenative cyclization of benzotriazoles 8.1 with Allenes and N Allenamides. DMSO: dimethyl sulfoxide.
In 2021, the team conducted a chemically selective, regio- and enantioselective denitrocycloaddition Heck/Tsuji-Trost reaction of benzotriazoles 9.1 with cyclic and acyclic 1,3-dienes, employing palladium catalysis and chiral sulfonamide phosphine ligands (Figure 9)[39]. This method offered a straightforward pathway for producing a range of substituted hexahydrocarbazoles and indolines. Benzotriazoles 9.1 was treated with cycloene-1,3-dienes 9.2 using Pd2(dba)3·CHCl3 as a catalyst, leading to the production of hexahydrocarbazoles 9.3. The specific products formed was influenced by the choice of chiral sulfonamide phosphine ligand in the reaction. Treatment with phosphine ligand L7 in n-hexane solvent yielded hexahydrocarbazoles. Conversely, the use of phosphine ligand L8 resulted in the formation of the corresponding enantiomer. The reaction shown good compatibility with benzotriazoles containing different functional groups R at various positions on the phenyl ring, as well as substituted cyclohexa-1,3-dienes, cyclohepta-1,3-dienes, and non-terminal 1,3-dienes, leading to high yields and exceptional enantioselectivity. To enhance the synthesis of indoline derivatives 9.4 and 1,3-dienes 9.5 underwent a reaction with L9 as the chiral ligand, resulting in the production of the respective indolines with satisfactory yields.
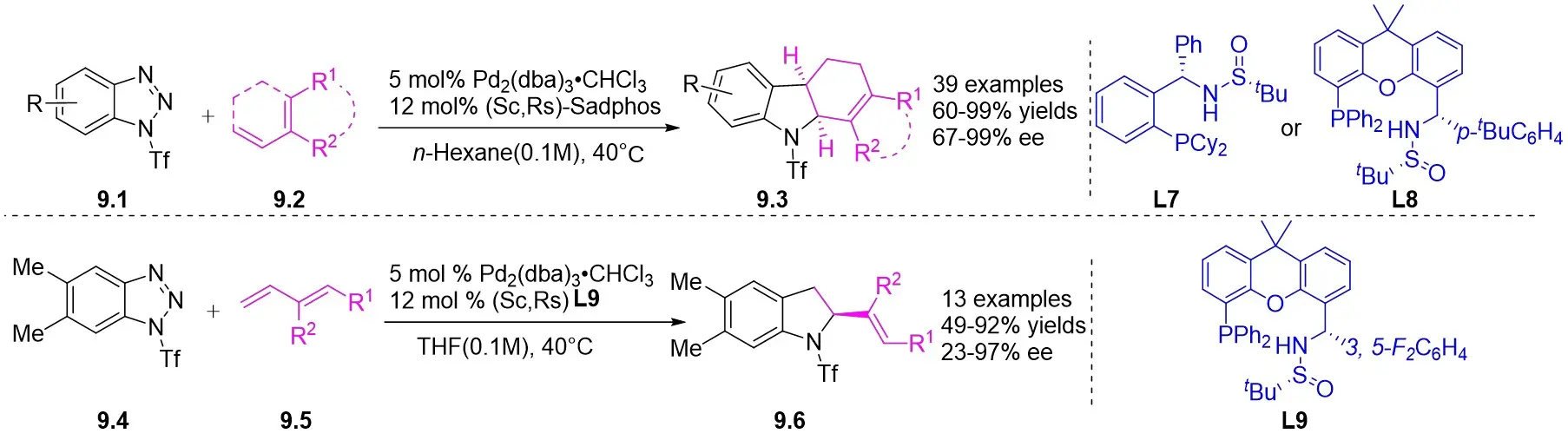
Figure 9. Asymmetric tandem Heck/Tsuji-Trost of benzotriazoles with 1,3-dienes. THF: tetrahydrofuran.
2.3 Rhodium-catalyzed denitrogenative annulation reactions
Rhodium, a noble metal catalyst characterized by high activity and stable chemical properties, exhibits significantly higher catalytic activity than common catalysts such as palladium, nickel, and copper. Furthermore, rhodium catalysts boast prominent advantages, including a broad substrate scope and excellent selectivity, making them widely employed in diverse key fields like organic synthesis, pharmaceuticals, and chemical pharmaceutical manufacturing[40-42].
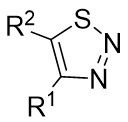
In 2021, Yang’s team reported the Rh(I)-catalyzed [3+3] cyclization of 1,2,3-thiadiazoles 10.1 with a variety of alk-2-enals 10.2, producing 2,3-dihydrothio-4-ones 10.3 in moderate to excellent yields (Figure 10)[23]. The use of [Rh(COD)Cl]2 and DPPF ligands demonstrated a broad substrate scope and tolerance for various functional groups under optimized conditions. However, the presence of an alpha substituent on enals 10.2 hinders the desired [3+3] cyclization, resulting in reduced yields. Notably, the use of (R)- or (S)-BINAP ligands did not yield optically active products. Mechanistically, ligand exchange between DPPF and [Rh(COD)Cl]2 generated the active catalyst [Rh]. Thiadiazoles 10.1 decompose to thioimide 10.5. The oxidative addition of 10.5 to enals 10.2 facilitates intramolecular 1,2-hydrogen transfer of 10.6, 10.6 to form intermediate 10.7, which then reductively eliminates the regenerated catalyst and produces β-thioketene 10.8. Tautomerization of 10.8 to 10.9, followed by intramolecular sulfur-Michael addition, yield the desired product 10.3. Reversible oxidative additions between carbene-coordinated metals and alkenes governed their selectivity for six-membered rings.
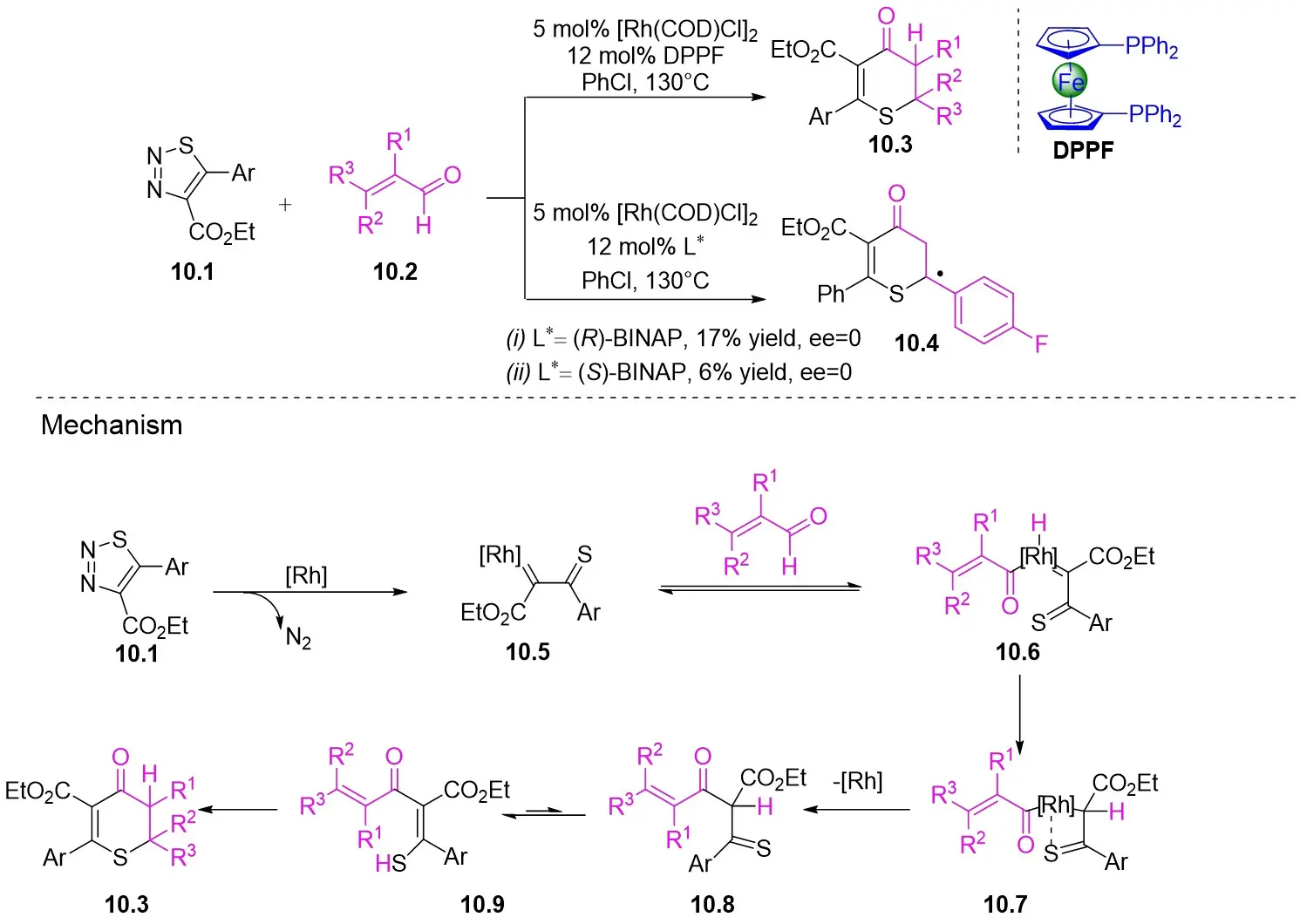
Figure 10. [3+3] transannulation of 1,2,3-thiadiazoles 10.1 with alk-2-enals. DPPF: 1,1’-bis(diphenylphosphino)ferrocene; BINAP: 2,2’-bis(diphenylphosphino)-1,1’-binaphthyl.
In the same year, the Yang team published on the Rh(I)-catalyzed (3 + 2) cyclization of 1,2,3-thiadiazoles 11.1 with cyanoepoxides 11.2, resulting in epoxy-functionalized isothiazoles exhibiting high chemoselectivity and diastereoselectivity (Figure 11)[43]. The reaction yielded either cis- or trans-epoxyisothiazoles, determined by the choice of ligand, from cis- or trans-epoxynitriles. Specifically, using the bis[(2-diphenylphosphino)phenyl]ether (DPEPhos) ligand resulted in cis-epoxyisothiazole formation from the cyclization of cis-cyanoepoxide, while employing the DPPF ligand generated epoxyisothiazole through cyclization. This observation can be attributed to the kinetic competition between the direct cyclization of cis cyano epoxide and the ligand-catalyzed isomerization of the cis product. With the use of DPEPhos, ring cyclization occurred much more rapidly than the isomerization process; rapid formation of the cis product prompted cooling to inhibit isomerization, enabling the exclusive isolation of the cis product. When DPPF was employed, the low ratio of cyclization to isomerization rates caused the initially formed cis product to isomerize to equilibrium, leading to the predominance of the thermally stable trans product. Implementing trans-cyanoepoxide under condition C enhances trans selectivity. The dependency of this outcome on the ligand can be explained by the thermal isomerization from cis to trans products, as previously elucidated by Bao et al.[44]. The transformation initiates with the coordination of the cyano nitrogen in cyanoepoxides 11.2 to form Rh(III) complex 11.6, generating intermediate 11.7. The subsequent migration of nitriles to the C1–C3 bond, along with the isomerization of 11.8 to the more stable pentacoordinate Rh(III) complex 11.9, followed by reductive elimination, results in the production of epoxyisothiazoles 11.3, 11.4, and 11.5. Due to the thermodynamic instability of intermediate 11.8, highlighted by computational research conducted by Bao[44], an alternative pathway was suggested. This pathway involves the direct migratory insertion of CN bonds into the Rh–C3 bond as a supplementary approach to generate 11.9. The preference for reacting with cyano groups rather than epoxy groups is attributed to the steric hindrance effect around the oxygen atoms in epoxy groups. Additionally, the electron-withdrawing nature of epoxy groups enhanced the remarkably high reactivity of cyano groups.
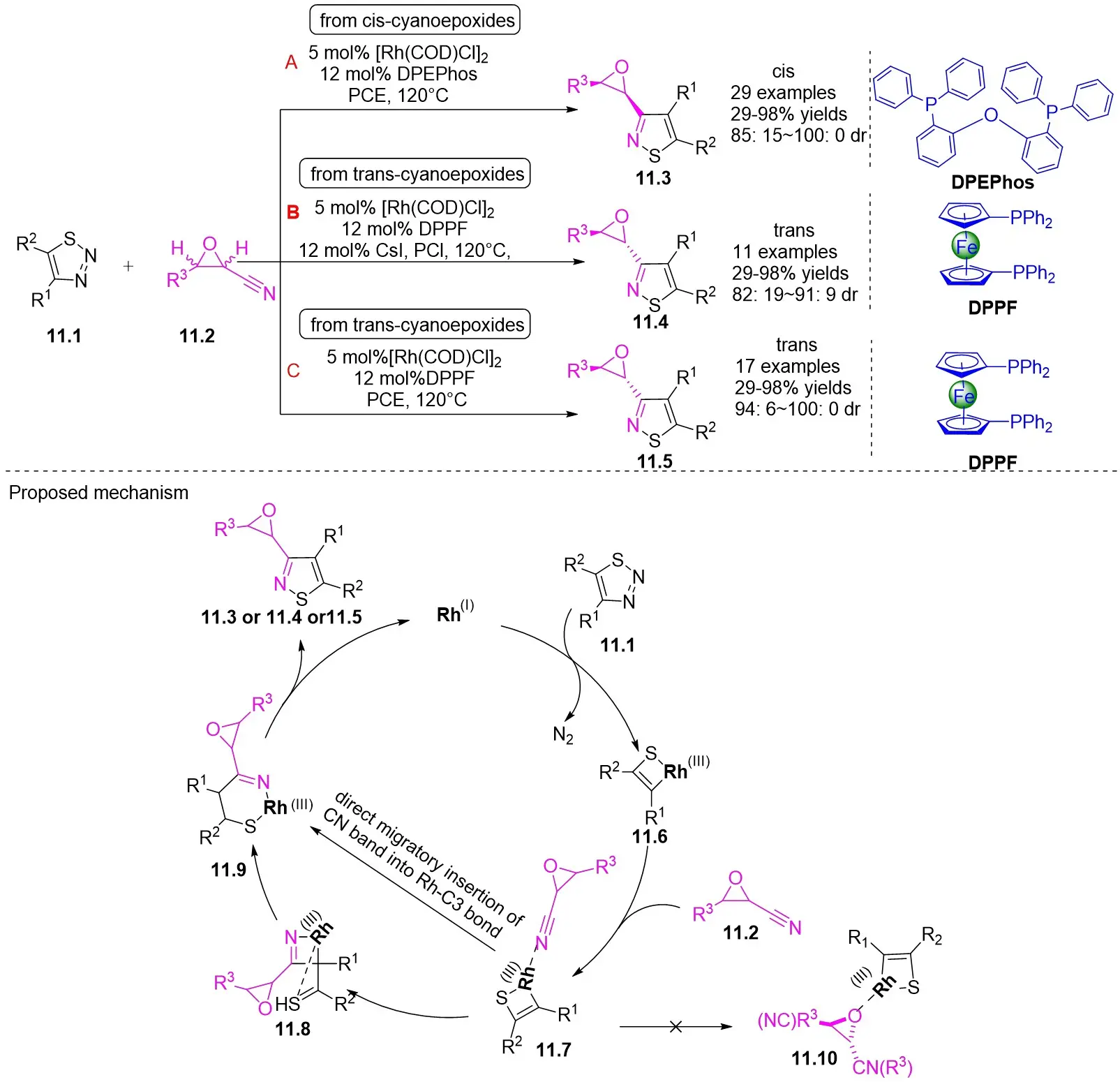
Figure 11. [3+2] cyclization of 1,2,3-thiadiazoles with cyanoepoxides. DPEPhos: bis[(2-diphenylphosphino)phenyl]ether; DPPF: 1,1’-bis(diphenylphosphino)ferrocene; PCE: power conversion efficiency.
In 2023, a study presented a method for the (3 + 2) transcyclization of 1,2,3-thiadiazoles 12.1 with bicycloalkenes 12.2, employing [Rh(COD)Cl]2 as the catalyst and P-chiral monodentate phosphide as the ligand to yield polycyclic dihydrothiophenes with four newly formed stereocenters (Figure 12)[45]. This study confirmed that monodentate phosphine could be a beneficial ligand for facilitating the (3 + 2) transcyclization of 1,2,3-thiadiazoles. Yield and enantioselectivity were strongly influenced by the electronic characteristics of the 5-aryl group in 1,2,3-thiadiazoles. More electron-deficient 1,2,3-thiadiazoles exhibited increased yields and improved enantioselectivity, while more electron-rich ones led to lower yields and enantioselectivity. Mechanistically, the process begins with ligand exchange to form the diphosphine-coordinated Rh(I) species L2RhCl (L=L10), succeeded by catalytic denitrogenation oxidative addition to produce α-sulfurylcarbenes 12.4, proposed to exist as a resonance structure of rhodium ring 12.5. The planar quaternion ring is orthogonal to the P–Rh–P plane. Exchange of bicycloalkene 12.2 from its less exposed side to the enantiomeric plane of 12.5 results in the formation of complexes 12.6 and ent-12.6. Transferring the C=C bond into the Rh–C bond in 12.6 and ent-12.6 generates the respective four-membered rhodium rings 12.7 and ent-12.7, which subsequently undergo reductive elimination to yield the (3 + 2) transcyclization products 12.3 and ent-12.3 and regenerate the catalyst.
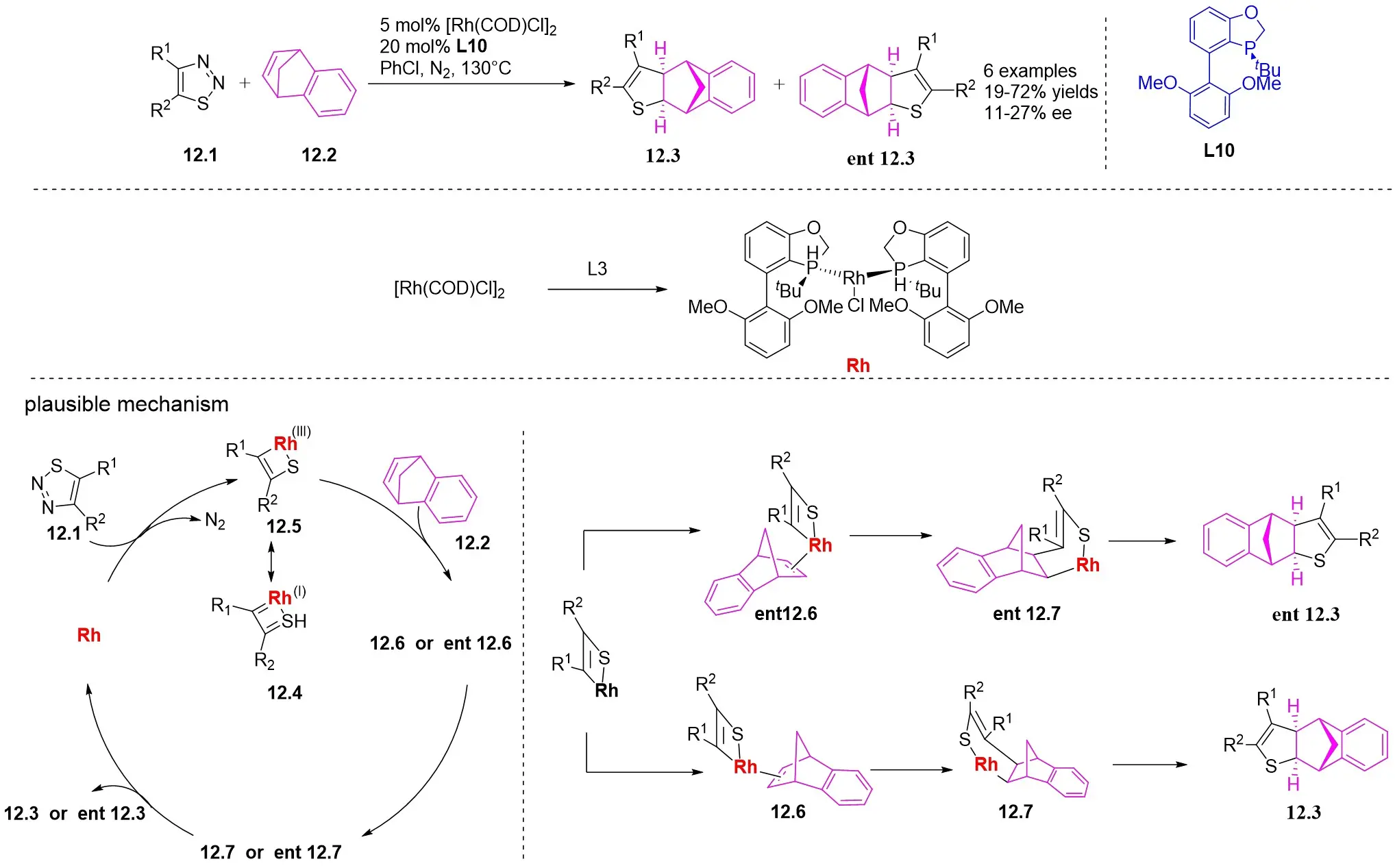
Figure 12. (3 + 2) transannulations of 1,2,3-thiadiazoles 12.1 with bicycloalkenes.
In 2024, they published findings on a significantly enantioselective, diastereoselective, and regioselective (3 + 2) cyclization of azimatowicz rearrangement products utilizing nucleophilic thioacylRh(I)-carbene within a Rh(I)/BINAP catalytic system (Figure 13)[46]. This procedure resulted in the formation of bicyclic dihydrothiophenes containing various functional groups, three contiguous chiral centers, and five prochiral centers, achieving yields as high as 97% and enantiomeric ratios of up to 99:1. This developed technique provided a novel route for synthesizing structurally diverse and highly enantiomerically pure bicyclic dihydrothiophenes, serving as sulfur analogs of natural iridoids. Additionally, the outstanding compatibility of this reaction laid the groundwork for the structural alteration of enantiomeric natural products and pharmaceutical compounds. Mechanistically, the initial step involved the reaction of 1,2,3-thiadiazoles 13.1 with (R)-BINAP-RhCl through denitrooxidative addition to generate a quaternary thioacyl Rh(I)-carbene species (13.5↔13.6). Subsequent anticoordination with 13.2 occurred from the opposite face of the exocyclic acetoxy group, leading to the migration of the carbon-carbon double bond into the Rh–C bond, forming a bicyclic 1-thia-2-rhodium heterocyclic ring 13.8 with all stereocenters in place. The reductive elimination process yielded the enantiomerically pure product 13.3 or 13.4 and regenerated the catalyst.
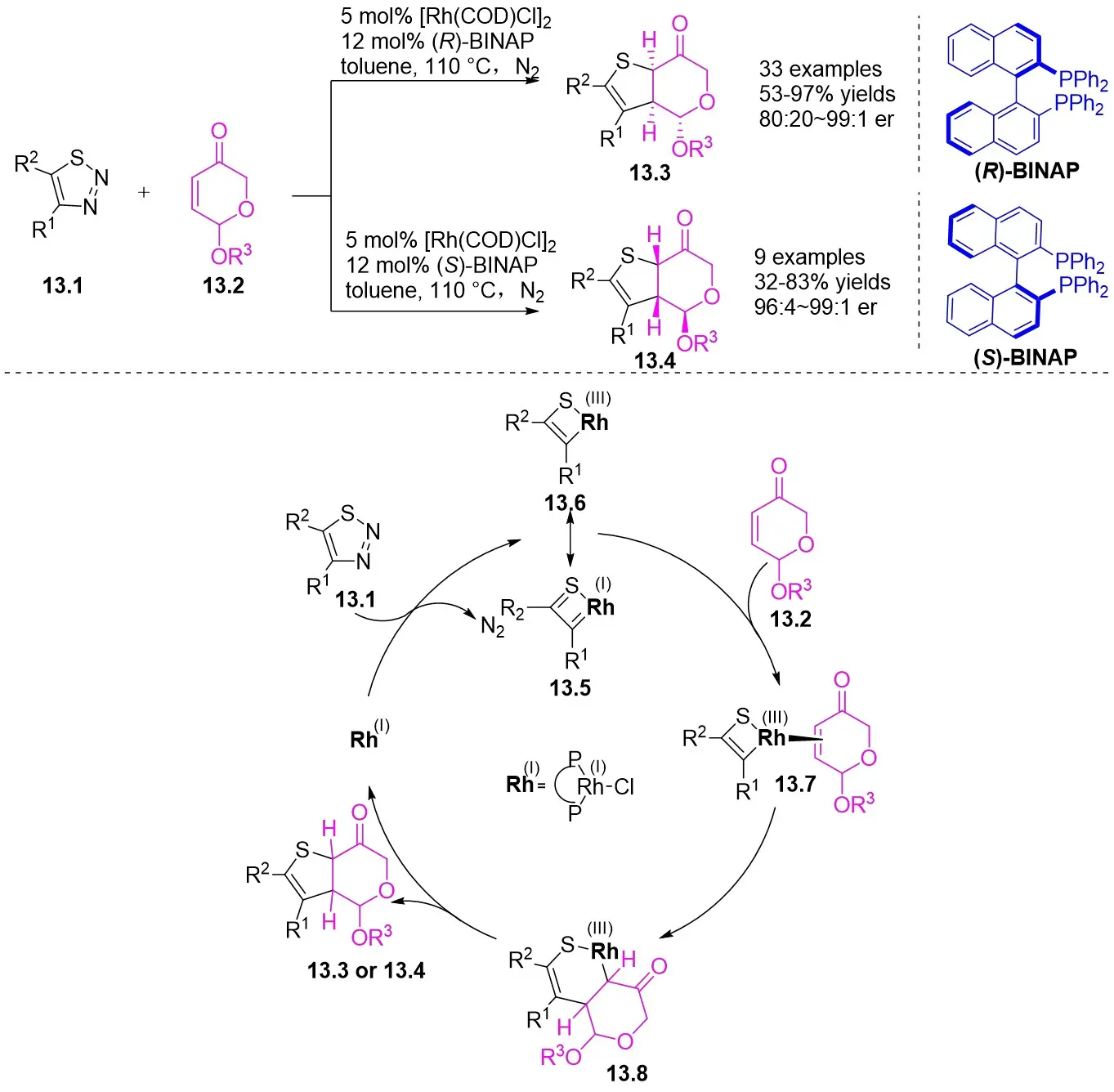
Figure 13. (3 + 2)Annulations of 1,2,3-thiadiazoles 13.1 with ARPs. ARPs: azimatowicz rearrangement products; BINAP: 2,2’-bis(diphenylphosphino)-1,1’-binaphthyl.
In 2025, the team successfully developed a method controlled by ligands for the rhodium-catalyzed synthesis of alkylene-2,3-dihydrothiophenes with notable regioselectivity and enantioselectivity (Figure 14)[47]. This accomplishment stemmed from diatomic skeleton reactions between 1,2,3-thiadiazoles 14.1 and allenoates 14.2. The resulting compounds demonstrated outstanding yields (up to 99%) and ee levels reaching 99%. Researchers discovered that 5-aryl-substituted 1,2,3-thiadiazole-4-carboxylic acids, irrespective of the aryl ring’s functional group, produced the desired compounds with satisfactory yields and high ee values. Altering the ester group or substituting the methyl group of allyl compounds with phenyl and propyl groups had no impact on the regioselectivity or enantioselectivity. Moreover, the team successfully synthesized more complex chiral compounds utilizing this technique, providing significant support for future efforts in drug exploration and advancement. Chiral diphosphine ligands dictated the alignment of coordinated allenes. By studying the reaction mechanism with (4R, 5R)-DIOP ligand, the oxidative addition of 14.1 to monomeric Rh(I) through denitrogenation produced α-thioacylRh(I)-carbene species 14.5↔14.6, which selectively interacted with the electron-rich carbon-carbon double bond plane of the allene. (S)-14.2 participated in forming intermediate 14.7, followed by inserting migration to generate a six-membered 1-thio-2-rhodium heterocyclic ring 14.8, ultimately resulting in the enantiomerically pure product 14.3 through reductive elimination. However, employing (R)-Cy-BINAP as a ligand caused the enyne (R)-14.2 to react with the opposing geometric configuration. The steric hindrance effect of the ligand is proposed as the primary determinant of regioselectivity.
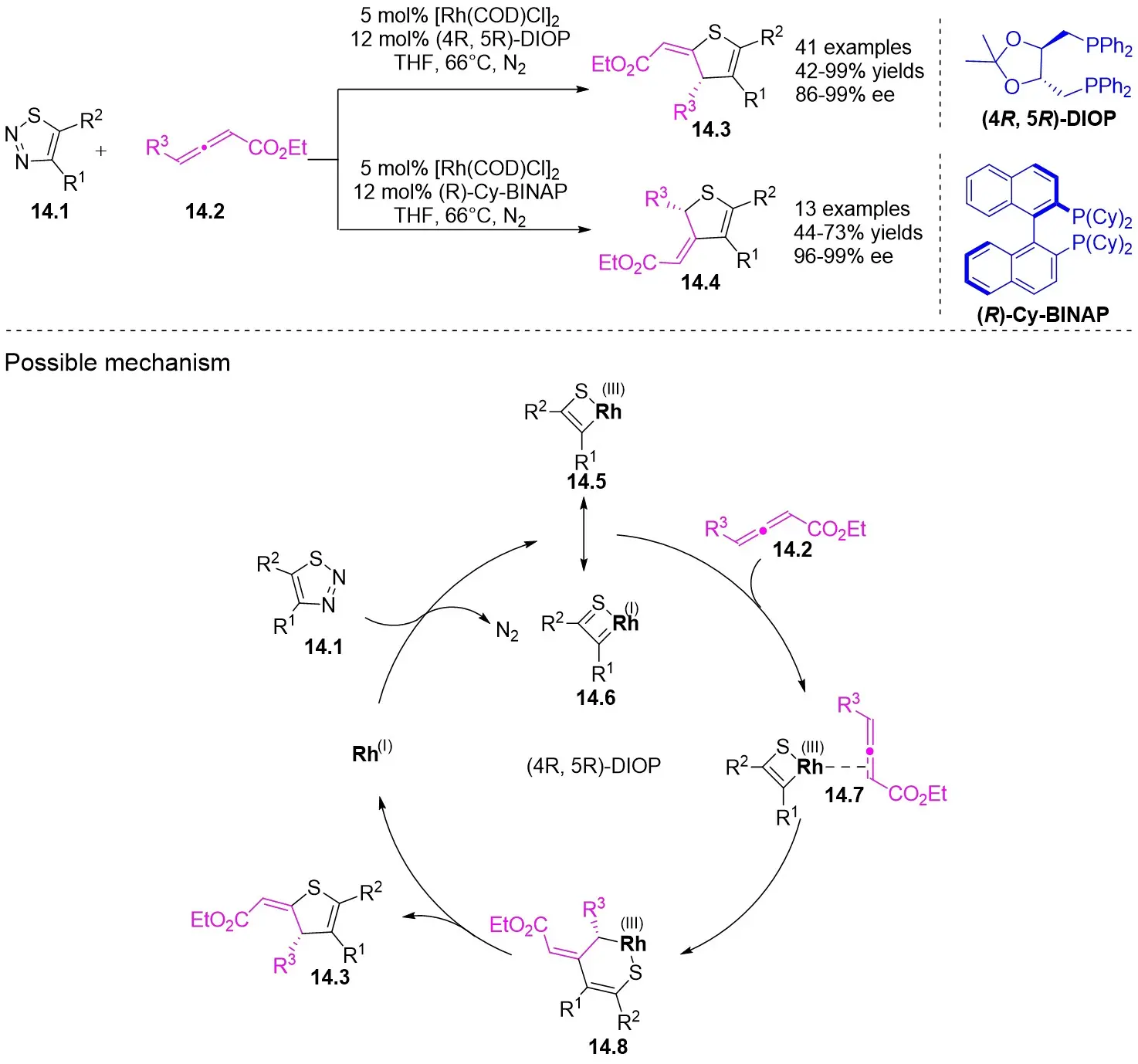
Figure 14. Transformations of 1,2,3-thiadiazoles 14.1 to alkylidene dihydrothiophenes. DIOP: (4R,5R)-2,2-dimethyl-1,3-dioxolane-4,5-diyl)bis(diphenylphosphine); BINAP: 2,2’-bis(diphenylphosphino)-1,1’-binaphthyl; THF: tetrahydrofuran.
2.4 Iridium-catalyzed denitrogenative annulation reactions
Iridium is not only chemically stable, acid-resistant, and corrosion-resistant, but also ranks among the most corrosion-resistant metals known to date. Additionally, it exhibits a strong tendency for coordination. Leveraging these exceptional properties, research on the catalytic synthesis and conversion of various functional chemicals using iridium has been extensively reported[48-50].
In 2022, Yang and colleagues investigated the denitrogenation (3 + 2) cyclization of 1,2,3-thiadiazoles 15.2 with polysubstituted olefins 15.1 (Figure 15)[24]. Initially, disubstituted, trisubstituted, and tetrasubstituted linear olefins failed to react with either terminal or internal counterparts. The steric hindrance of the C=C plane was thought to impede its interaction with the α-sulfurylrhodium carbene intermediate. To address this steric deactivation, the researchers developed a strategy by employing cyclic olefins in place of linear ones. Experimental findings revealed that norbornene, as a bicycloolefin with a ring strain of 19.2 kcal/mol, displayed significant reactivity in the targeted (3 + 2) cross-modulation. This was attributed to the alleviation of ring strain, which served as the primary driving force behind the reaction. Enantioselective and diastereoselective (3 + 2) cyclization reactions catalyzed by iridium were observed in the reaction between 1,2,3-thiadiazoles and cyclohexene derivatives. Employing DPPF as a ligand led to the specific generation of the sole diastereomer 2,3-dihydrothiophene 15.3. Replacing achiral DPPF ligands with chiral (R)-TolBINAP and (S)-TolBINAP ligands facilitated the asymmetric synthesis of optically active 2,3-dihydrothiophenes 15.4 and 15.5. A range of 1,2,3-thiadiazoles demonstrated high reactivity, with the exceptions of 5-(4-iodophenyl), 5-(4-methoxyphenyl), and 5-(4-nitrophenyl) 1,2,3-thiadiazoles. Cyclopentadiene-fused imides exhibited lower reactivity compared to bicycloalkenes, and their reactivity showed some sensitivity to N-aryls, likely due to subtle electronic differences. This study investigates the mechanism using (R)-TolBINAP. The reaction of 1,2,3-thiadiazoles 15.2 with phosphine-linked iridium catalysts formed a quaternary species that demonstrates resonance between sulfuryl Ir(I) carbene 15.6 and cyclometallated Ir(III) complex 15.7. The external coordination of bicycloolefin 15.1, taking into account its steric disparity relative to the enantiomeric plane of 15.7, led to the formation of complex 15.8. The subsequent migration of the C=C bond promoted the insertion of the Ir-C bond, resulting in reductive elimination and the formation of (3 + 2) straddle products, thus regenerating the catalyst. When (S)-TolBINAP was employed, coordination occurred from the right side, resulting in the enantioselective formation of the final product 15.5.
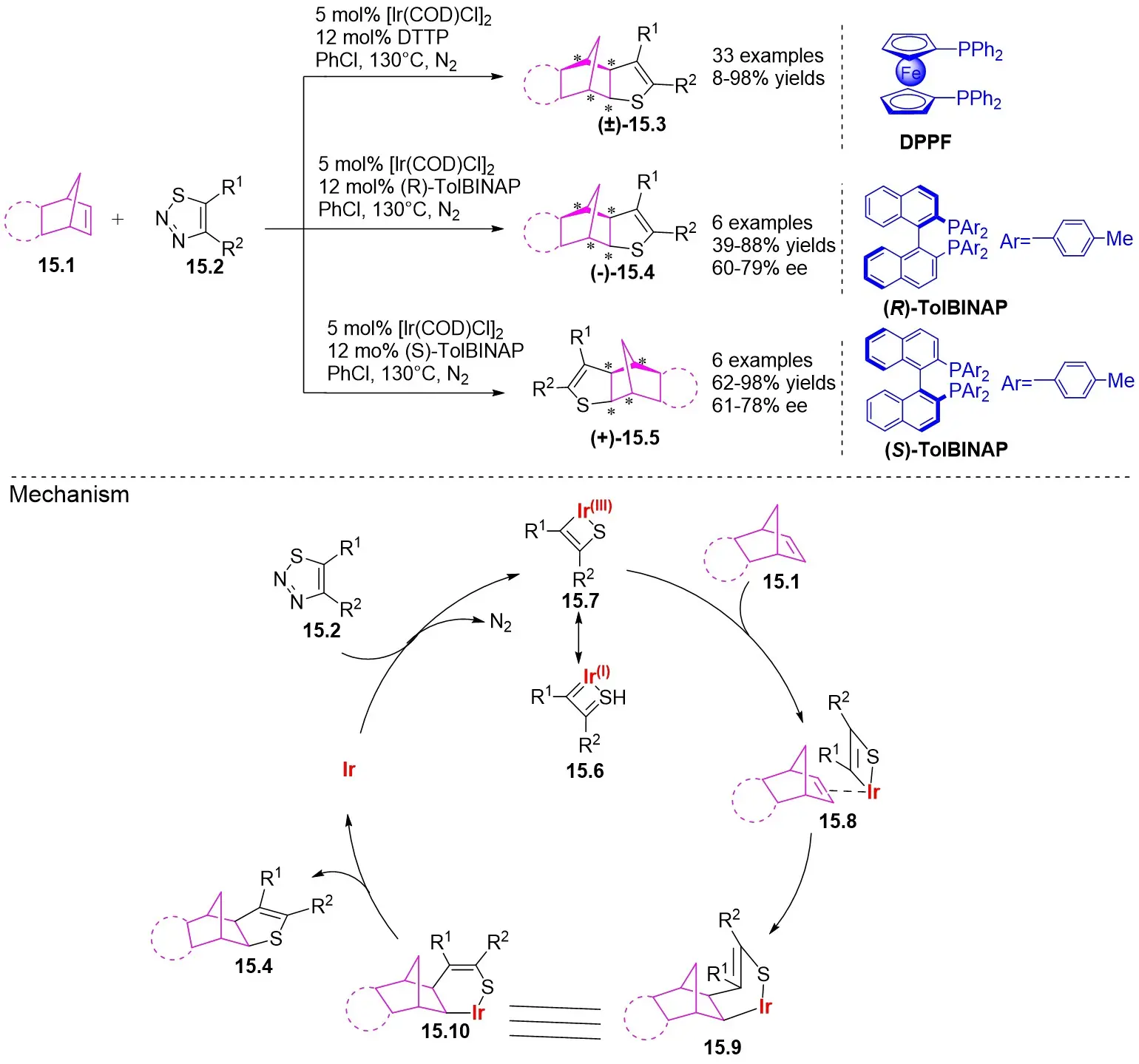
Figure 15. (3 + 2) Cycloaddition of 1,2,3-thiadiazoles 15.2 with strained cyclohexene derivatives. BINAP: 2,2’-bis(diphenylphosphino)-1,1’-binaphthyl; DPPF: 1,1’-bis(diphenylphosphino)ferrocene.
3. Denitrogenative Transannulation via Metal Carbenes
Metal carbenes, as highly reactive organic reaction intermediates, hold significant utility in organic synthesis methodology[51]. They partake in various reaction mechanisms, like cyclopropanation, migration insertion, carbon/heterohydrogen bond insertion, and interactions with nucleophiles. Publications regarding metal carbenes are abundant due to their distinctive reactivity, which enhances the eco-friendliness and efficiency of numerous metal carbene-involved reactions, particularly in the synthesis of heterocyclic compounds[52-54]. Metal carbenes are synthesized from carbene precursors and metal catalysts. Rhodium serves as the primary catalyst in the production of chiral heterocyclic compounds through the transition metal-catalyzed liberation of nitrogen from diazo compounds to form metal carbenes (Table 2).
Table 2. Denitrogenative transannulation via metal carbenes.
| Substrate | Type of Reaction | Catalyst | Product | Reference |
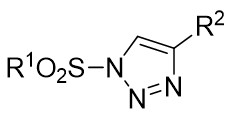
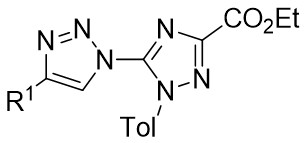 | Intermolecular Annulation | Rh2(S-NTTL)4 | 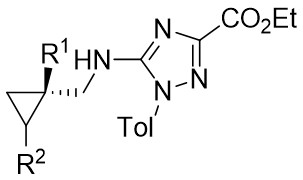 | [58] |
 | Intermolecular Annulation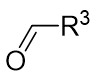 | Rh2(S-nttl)4 | 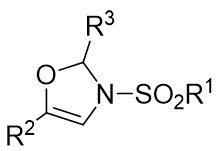 | [59] |
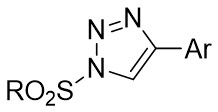 | Intermolecular Annulation | Rh2(S-PTAD)4 | 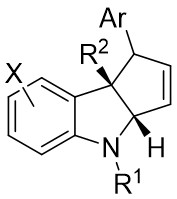 | [60] |
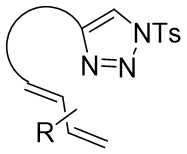 | Intramolecular Annulation | Rh2(OAc)4 | 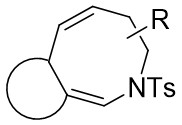 | [61] |
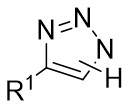 | Intermolecular Annulation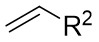 | Rh2(S-NTTL)4 | 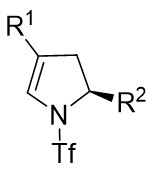 | [62] |
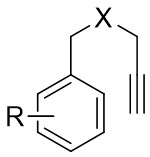 | Intermolecular AnnulationTs-N3 | Rh2(OCOtBu)4orRh2(S-TCPTTL)4 | 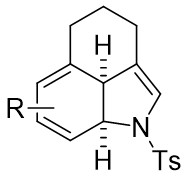 | [63] |
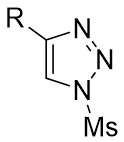 | Intermolecular Annulation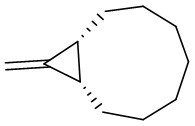 | Rh2(S-NTTL)4 | 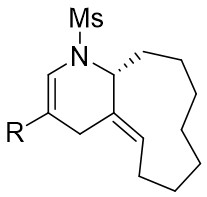 | [64] |
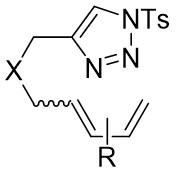 | Intramolecular Annulation | Rh2(S-ptad)4 | 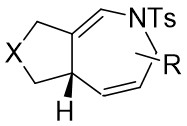 | [65] |
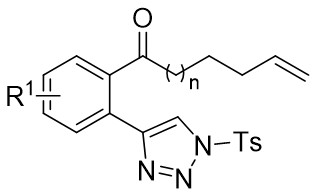 | Intramolecular Annulation | Rh2(OAc)4orRh2(S-DOSP)4 | 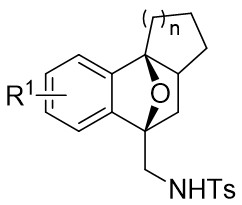 | [66] |
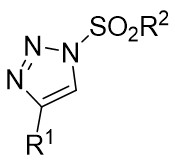 | Intermolecular Annulation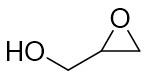 | Rh2(tBuCO2)4andMg(OtBu)2 | 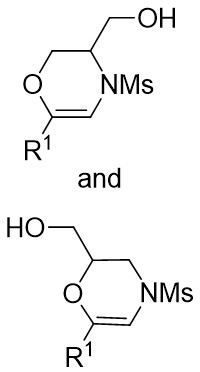 | [67] |
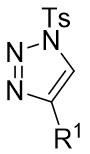 | Intermolecular Annulation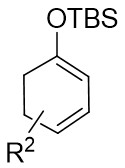 | Rh2(oct)4orRh2(S-NTTL)4orRh2(S-PTTL)4 | 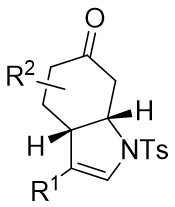 | [68] |
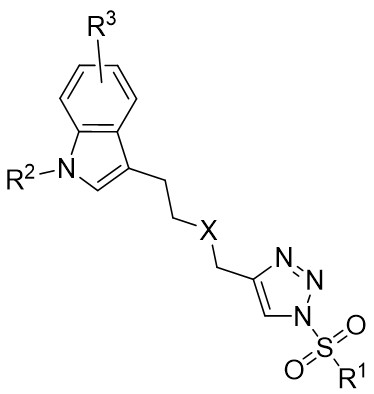 | Intramolecular Annulation | Rh2(S-PTTL)4 | 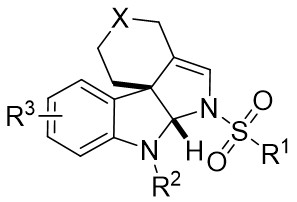 | [69] |
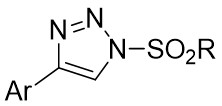 | Intermolecular Annulation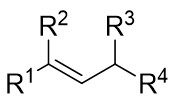 | Rh2(S-NTTL)4 | 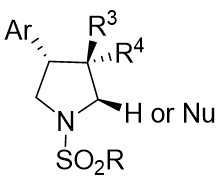 | [70] |
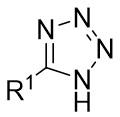 | Intermolecular Annulation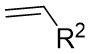 | Rh2(S-TCPTTL)4 | 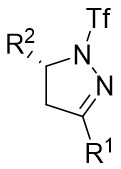 | [71] |
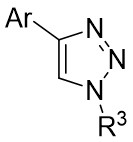 | Intermolecular Annulation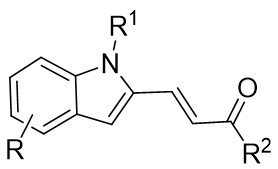 | Rh2(OAc)4 | 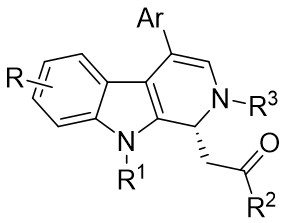 | [72] |
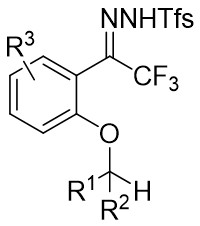 | Intramolecular Annulation | Rh2(R-BTPCP)4 | 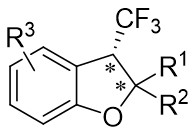 | [73] |
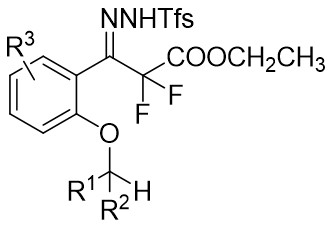 | Intramolecular Annulation | Rh2(R-BTPCP)4 | 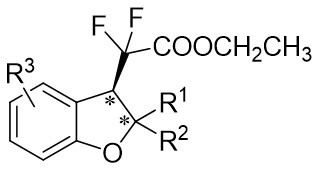 | [73] |
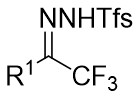 | Intermolecular Annulation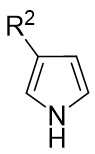 | Rh2(R-PTTL)4 | 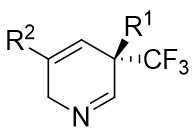 | [74] |
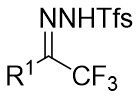 | Intermolecular Annulation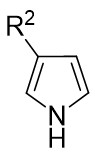 | Rh2(S-PTTL)4 | 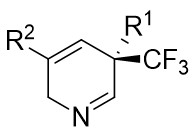 | [74] |
 | Intermolecular Annulation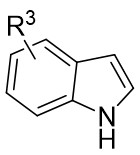 | Rh2(R-3,5-diPhTPCP)4 | 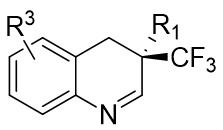 | [74] |
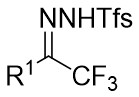 | Intermolecular Annulation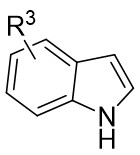 | Rh2(S-3,5-diPhTPCP)4 | 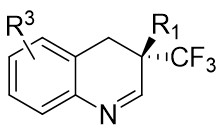 | [74] |
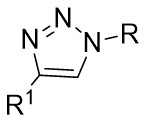 | Intermolecular Annulation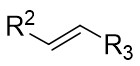 | [Rh] | 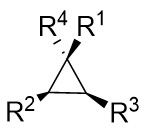 | [75-84] |
Dinuclear rhodium(II) complexes act as weak Lewis acid catalysts, featuring the ability to stabilize carbenes and exhibiting excellent catalytic activity—properties that render them widely applicable in diverse organic catalytic reactions[55,56]. Structurally, dinuclear rhodium catalysts typically comprise Rh-Rh metal bonds, four bridging ligands, and two axial empty orbitals. Their catalytic activity and selectivity can be modulated by adjusting the bridging ligands; furthermore, coordination modification of the axial empty orbitals enables tuning of the electronic properties of the dinuclear centers, thereby fostering the emergence of novel catalytic behaviors[57].
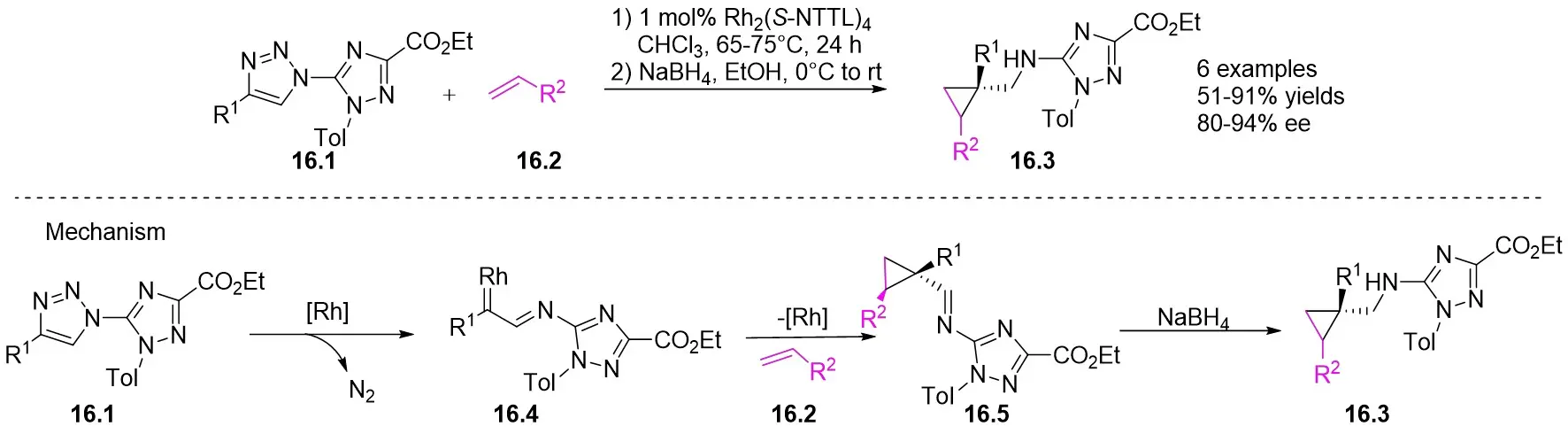
In 2011, Fokin and Zibinsky introduced a Rh(II)-catalyzed method for synthesizing cyclopropanes 16.3 from N-(1,2,4-triazolyl)-substituted 1,2,3-triazoles 16.1 and olefins 16.2 in CHCl3 using Rh2(S-NTTL)4 at 65 °C (Figure 16)[58]. The process involved the subsequent reduction of the resulting imine with NaBH4 to produce cyclopropane with notable enantioselectivity and diastereoselectivity. Triazoles 16.1 featuring an N-1 carbon substituent were observed to promote the ring-chain isomerization of triazoles. The proposed reaction mechanism involves the easy formation of azavinyl carbene 16.4 from 1,2,4-triazolyl-1,2,3-triazolethyl esters. This carbene can subsequently react with olefins to form imine 16.5, which can then be reduced with NaBH4 to afford the final product.
In 2012, they described a rhodium(II)-catalyzed denitrogenation process that utilized 1-sulfonyl-1,2,3-triazoles 17.1 and aldehydes 17.2 to produce 4-oxazolidine derivatives 17.3 (Figure 17)[59]. The highest enantioselectivity was attained through the reaction of 1-methanesulfonyl-1,2,3-triazoles 17.1 with a Rh2(S-nttl)4 catalyst at ambient temperature. Prolonged reaction times were observed to markedly decrease the enantioselectivity of the majority of products. Additionally, imines derived from aliphatic aldehydes underwent irreversible ring opening at the C-O bond of aminoaldehydes under high temperatures, followed by β-proton elimination. The denitrogenation of triazoles 17.1 by the catalyst produces carbene 17.4, which then reacts with the carbonyl to generate imine salt 17.5. This intermediate undergoes cyclization to form oxazoline 17.3.
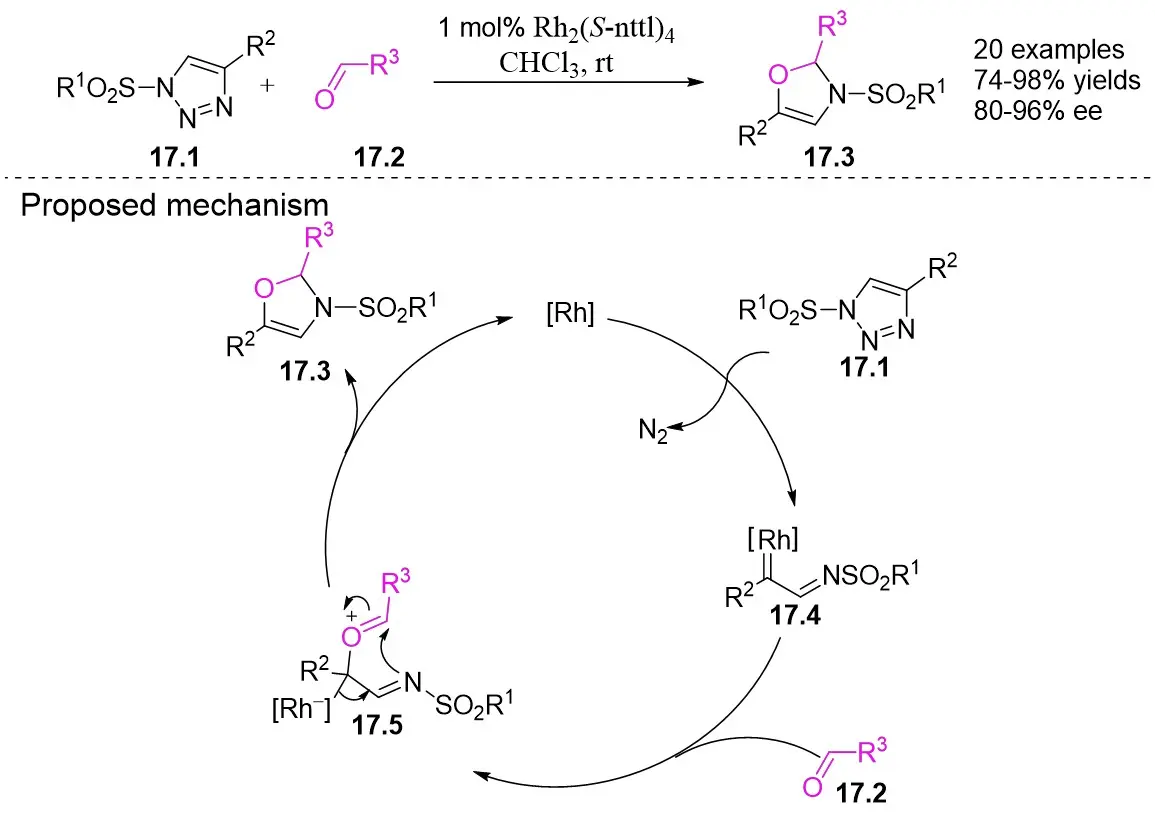
Figure 17. Synthesis of 4-oxazoline from 1-sulfonyl-1,2,3-triazoles 17.1 and aldehydes.
In 2013, Davies and Spangler reported a [3+2] cycloaddition process involving 4-aryl-1-sulfonyl-1,2,3-triazoles 18.1 and C3-substituted indoles 18.2 in cyclohexane at 65°C with Rh2(S-PTAD)4 as the catalyst (Figure 18)[60]. The reaction produced pyrroloindolines 18.3 with notable enantioselectivity and satisfactory yields. The research showed that performing the cyclization in a nonpolar hydrocarbon solvent led to exceptional conversion rates and enantiomeric control. The reaction exhibited sensitivity to steric hindrance, resulting in the inability to obtain the target products when utilizing larger 1-sulfonyl-1,2,3-triazoles. However, the target product can be achieved when R1 was hydrogen or allyl; larger volumes or electron-withdrawing groups in R1 hinder product formation. Substituting the indole aryl ring significantly increased the yield of the desired product. The authors proposed a plausible pathway for product formation, which includes the initial cyclopropanation of the C2–C3 bond of the indoles, followed by ring opening and complexation of the strained cyclopropylindoline intermediate, ultimately leading to the formation of the observed pyrrolindolines.
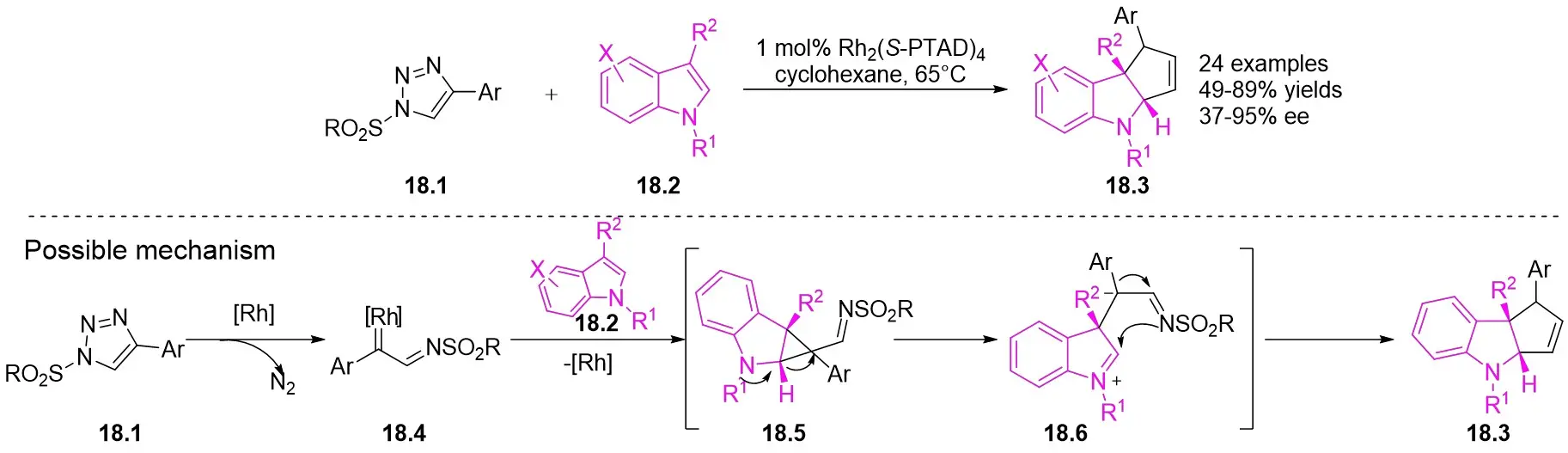
Figure 18. [3+2] Cycloaddition of 1,2,3-thiadiazoles 18.1 with substituted indoles.
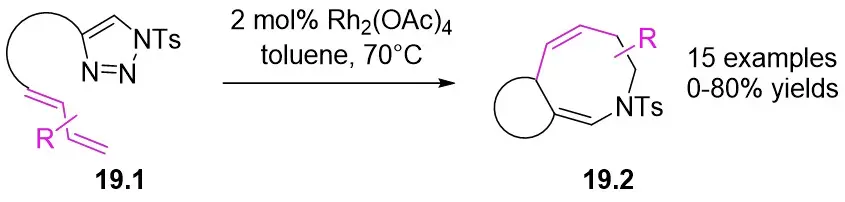
In 2014, Shen and Xu introduced a Rh2(OAc)4 catalyst for diverse dienyltriazoles 19.1, utilizing a carbene-cyclopropanation-aza-cope rearrangement cascade to produce dihydroazepanes 19.2 (Figure 19)[61]. This process involved strain relief of the three-membered ring, with substitution at position 7 providing exceptional diastereoselectivity. Diene triazoles substituted with 1-phenyl groups can be effectively converted into the respective dihydroazepanes with high yields. However, the addition of a second substituent (such as a methyl group at position 4 of the diene structure) could potentially have a negative impact on the reaction outcomes. Minor modifications to the reactive dienetriazole resulted in intricate reaction mixtures without any predominant products being isolated.
In 2014, Fokin’s team published a study on the one-pot asymmetric synthesis of 2,3-dihydropyrroles 20.3 using trifluorotriazoles 20.1 and olefins 23.2 under rhodium catalysis (Figure 20)[62]. The study found that the choice of catalyst influenced the enantioselectivity of the reaction, with Rh2(S-NTTL)4 demonstrating the highest enantioselectivity at 72% ee. Enantioselectivity was significantly affected by both the electronic and steric effects of the substrate, exhibiting a cumulative influence. The reaction mechanism involves the interaction of triazoles 20.1 with the Rh2(S-NTTL)4 catalyst to form rhodium carbene 20.6, which then reacts with electron-rich olefins to produce cyclopropylaldimine 20.7. The strong electron-withdrawing effect of trifluoromethanesulfonic acid, along with the electron-donating effect of the methoxy group, facilitates the cleavage of the cyclopropane ring and the formation of the stable diionic intermediate 20.8, which subsequently transforms into 2,3-dihydropyrroles 20.3A. In this mechanism, the chiral rhodium catalysts exert stereocontrol over the final product by influencing the enantioselectivity of the intermediate cyclopropane. Subsequent rearrangements lead to a decrease in the enantiomeric excess of the final product, the degree of which is influenced by the relative rates of Cα–Cβ bond rotation and the ring closure of open-chain intermediate 20.8.
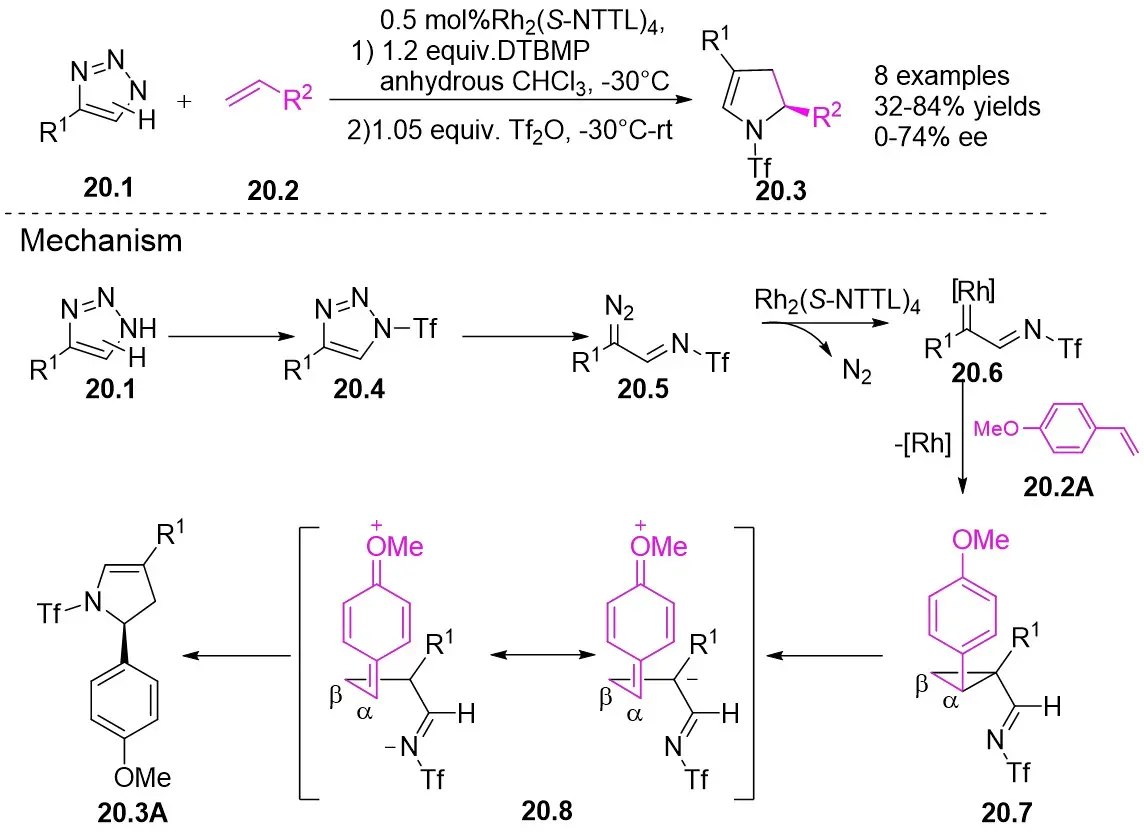
Figure 20. Transannulation of NH-1,2,3-triazoles 20.1 with olefins. DTBMP: 2,6-di-tert-butyl-4-methylpyridine.
In 2014, Murakami, Miura and Funakoshi presented a rhodium-catalyzed intramolecular dearomatizing [3+2] cyclization converting 4-(3-arylpropyl)-1,2,3-triazoles into 3,4-fused indoles 21.2 using basic 5-aryl-1-ynes 21.1 in a one-pot synthesis (Figure 21)[63]. The reaction mixture was initially stirred at room temperature for 12 hours, resulting in the conversion to triazoles. Subsequently, the mixture was stirred at 80 °C for 3 hours, leading to the isolation of the products in high yield. A significant enantioselective induction (81% ee) was noted with the utilization of the chiral rhodium(II) complex Rh2(S-TCPTTL)4. The seamless one-pot process demonstrated minimal interference between the reagents and catalysts necessary for individual steps. The reaction mechanism involves the initial reaction of alkynes 21.1 with azide to generate triazoles 21.3, which undergo partial reversible isomerization into α-diazimine 21.4, subsequently reacting with rhodium(II) to liberate molecular nitrogen, forming α-iminorhodium carbene 21.5. The electrophilic carbene carbon of 21.5 facilitates an intramolecular attack on the benzene ring, forming diionic intermediate 21.6. Anionic rhodium releases bonding electrons, leading to cyclization at the imino nitrogen to produce the desired products 21.2.
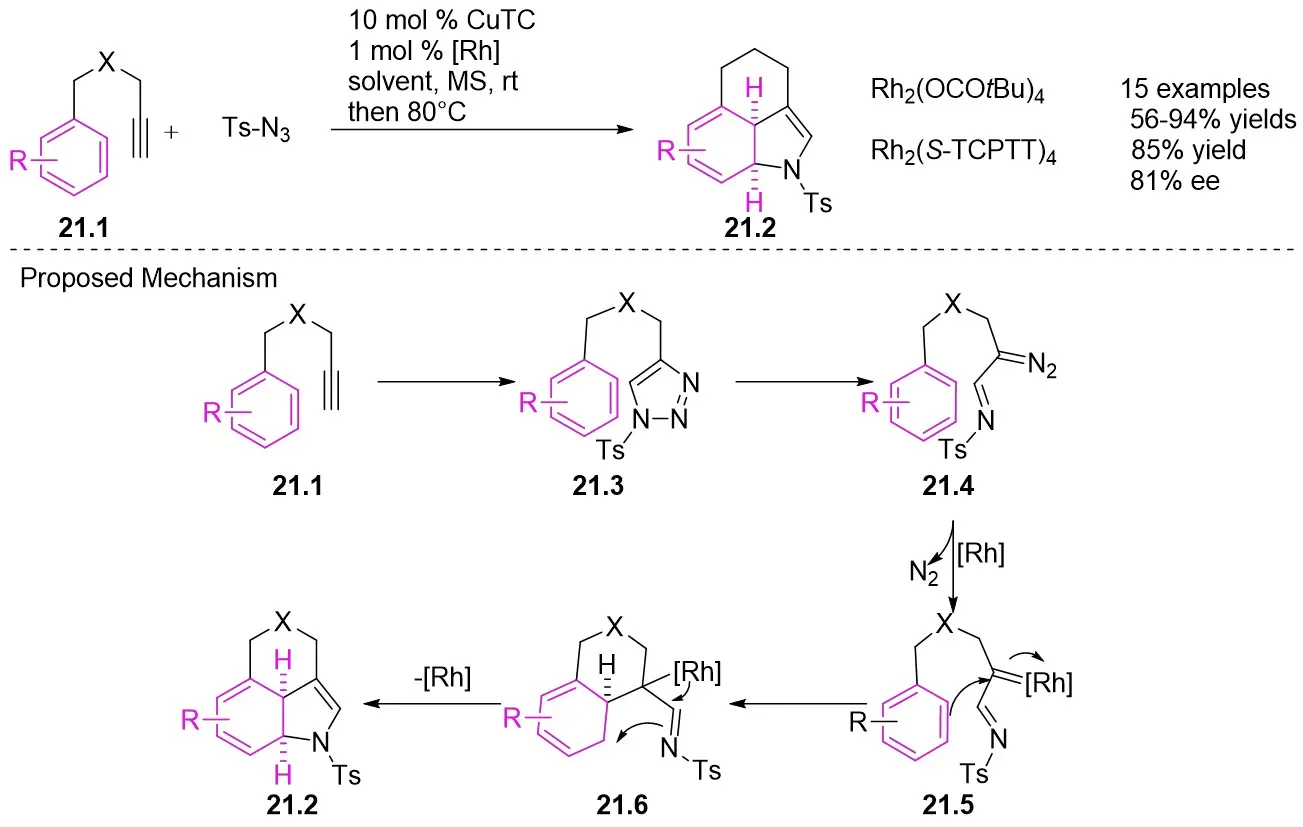
Figure 21. Intramolecular dearomatization [3+2] cyclization reaction to form 3,4-fused indoles 21.2. MS: mass spectrometry; rt: room temperature.
In 2014, Murakami’s research team published an effective one-pot, two-step asymmetric synthesis method for piperidine-fused trans-cyclic olefins 22.3 utilizing methylcyclopropanes 22.2 and N-sulfonyl-1,2,3-triazoles 22.1 (Figure 22)[64]. The process involved the enantioselective cyclopropanation of methylcyclopropanes with α-iminorhodium carbene, succeeded by a skeletal rearrangement. The primary driving force behind the skeletal rearrangement could be linked to the ring strain of the spiro structure. Reacting cyclic olefins 22.3 with trans-PtCl2(2,4,6-piperidine) (η2-ethylene) resulted in the formation of platinum(II) complexes in crystalline form. Single crystal X-ray analysis validated the absolute stereochemistry of the trans-olefins. Alternatively, this synthesis can be achieved through a one-pot three-component approach starting directly from terminal alkynes.
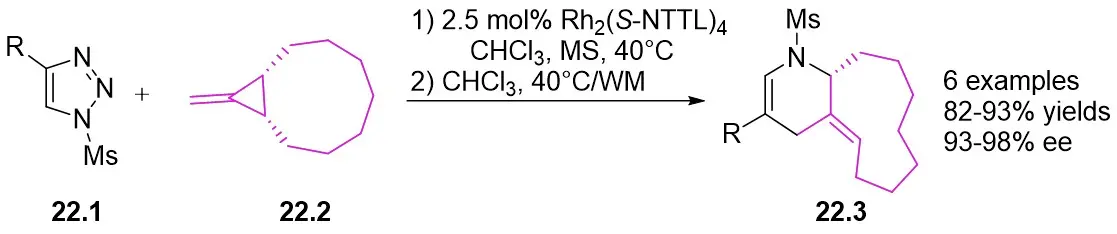
Figure 22. Asymmetric synthesis of piperidine-fused trans-cycloalkenes 22.3. MS: mass spectrometry.
In 2015, Tang and co-workers devised a Rh2(S-ptad)4-catalyzed intramolecular [4+3] cycloaddition method using diverse dienyltriazoles 23.1 to produce fused 2,5-dihydroazepines 23.2 (Figure 23)[65]. Dialkenyl triazoles bearing aryl substituents and various linking groups were effectively transformed into the respective dihydroazepanes with satisfactory yields. However, 1,1,4-trialkenyl triazoles did not yield the anticipated [4+3] products. Regrettably, this reaction only resulted in low ee values ranging from 5% to 10%. The reaction mechanism involves the formation of Rh(II)-iminocarbene 23.3 from dienetriazole in the presence of Rh catalyst, followed by its initial [2+1] cycloaddition to yield 23.4 (from the E isomer) and/or 23.5 (from the Z isomer). Subsequently, 23.4 and/or 23.5 can be transformed into azepane 23.2 via the aza-cope rearrangement.
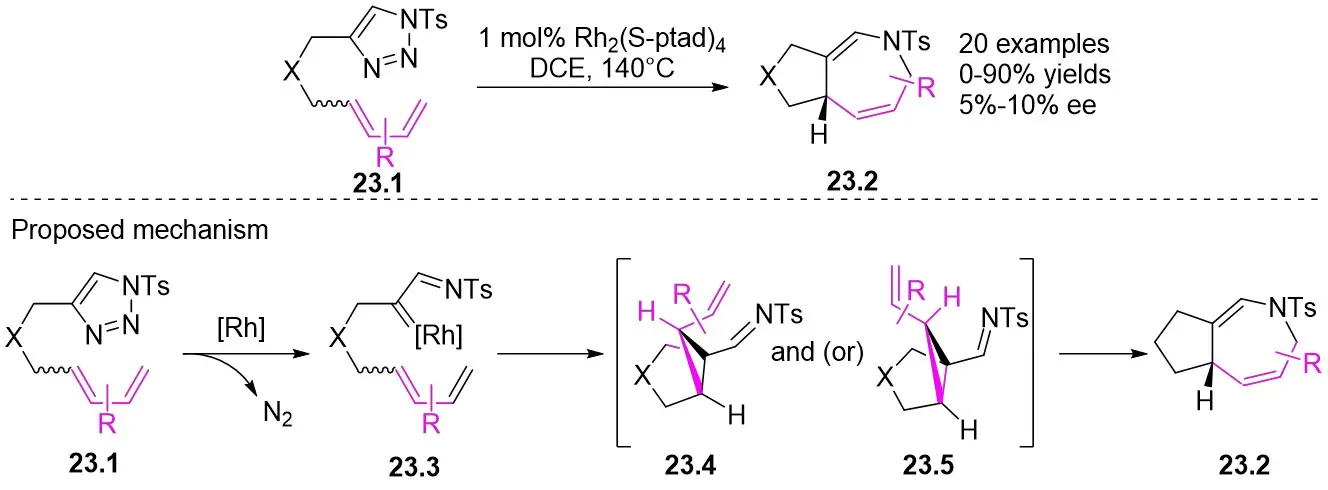
Figure 23. Intramolecular [4+3] cycloaddition reactions of dienetriazoles 23.1. DCE: 1,2-dichloroethane.
In 2016, Gong et al. investigated a Rh(II)-catalyzed tandem reaction involving the deazotization of N-sulfonyl-1,2,3-triazoles 24.1 followed by a [3+2] cycloaddition withvariously substituted oxabicyclo[2.2.1]heptenes 24.2 (Figure 24)[66]. Their findings indicated that the reaction exhibited diastereoselectivity when using Rh2(OAc)4 and displayed acceptable enantioselectivity (83% ee) with Rh2(S-DOSP)4. Substrates containing either electron-withdrawing or electron-donating groups on the aromatic ring all produced the desired cyclization products. However, substrates with substituents at the C3 position of the benzene ring exhibited improved yields. Ketone substituents were found to be compatible with a diverse range of alkyl, alkynyl, and aryl groups. Notably, when the substituent was a hydrogen atom (triazole of benzaldehyde), the reaction achieved the highest yield. This reaction involved Rh-catalyzed denitrogenation cyclization to generate the 5-membered oxonium ylide intermediate 24.4, which subsequently underwent an intramolecular [3+2] cycloaddition.
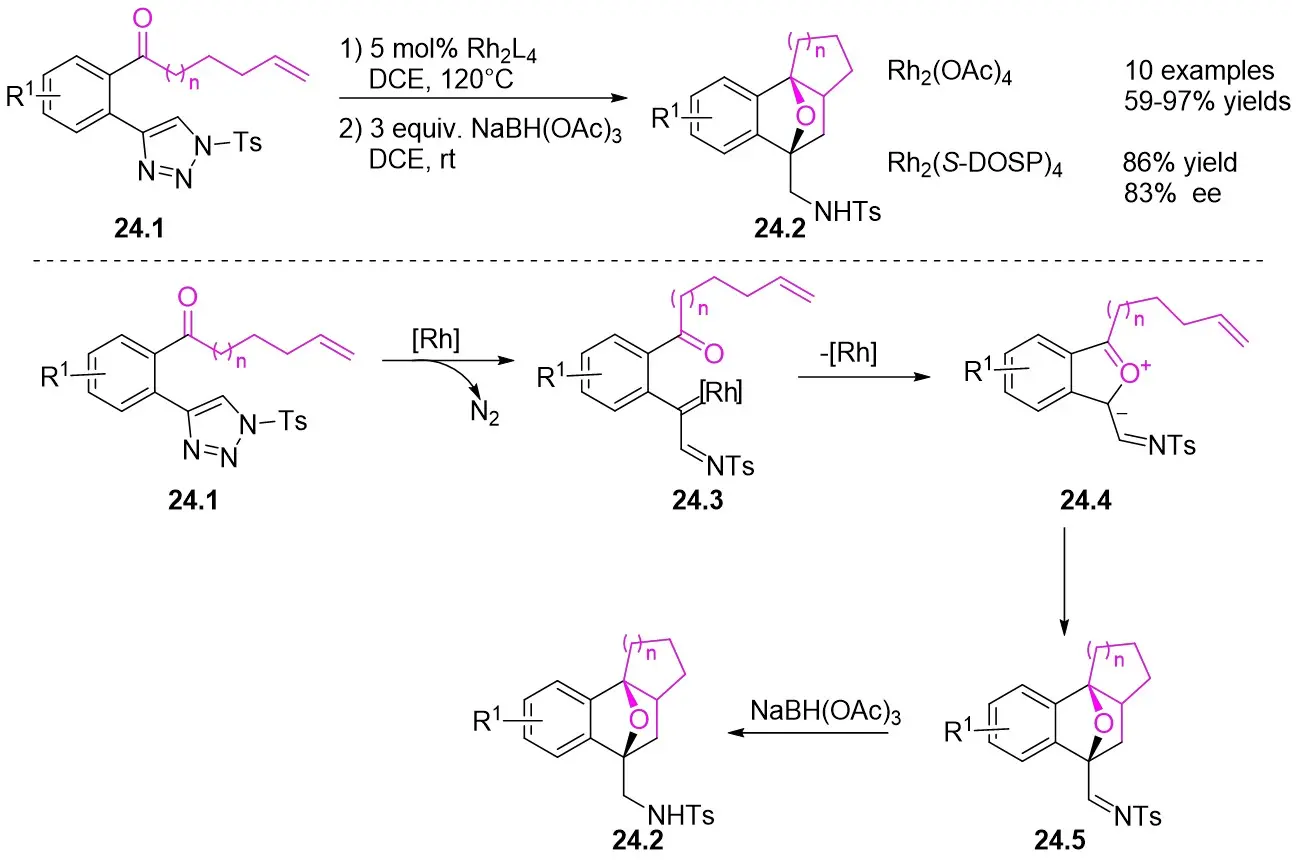
Figure 24. [3+2] Cycloaddition synthesis of oxabicyclo [2.2.1] heptenes 24.2. DCE: 1,2-dichloroethane; rt: room temperature.
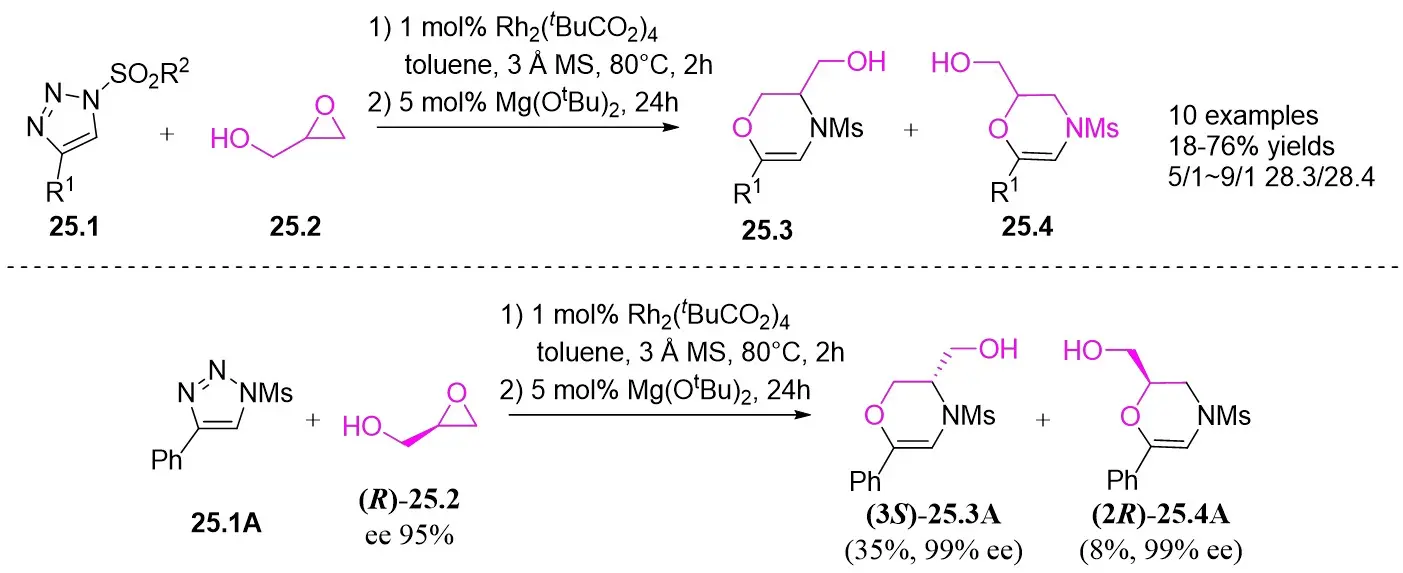
In 2016, Lee and co-workers introduced a novel Rh(II)/Mg(OtBu)2-catalyzed one-pot tandem reaction involving N-sulfonyl-1,2,3-triazoles 25.1 and epoxy alcohols 25.2 to yield derivatives of inseparable regioisomeric oxazines 25.3 and 25.4 (Figure 25)[67]. The reaction involved the O-H insertion of a rhodium(II) carbene, succeeded by a regioselective epoxy ring opening reaction sequence catalyzed by a Lewis acid. Positively charged α-imine rhodium(II)-carbene intermediates exhibited higher reactivity toward alcohol groups, facilitating O–H bond insertion for 1,3 insertions. Unsubstituted epoxy alcohols demonstrated direct reactivity with the epoxy ring when employed in the reaction. Furthermore, it was found that employing chiral epoxy alcohol (R)-25.2 and 25.1A as the starting materials led to complete chiral transfer during the SN2 reaction of epoxy ring opening. The product yield was moderate, and the enantiomers could be isolated.
In 2017, Zhai’s team described a rhodium-catalyzed synthesis of indolones 26.3 via Denitrogenative [3+2] cyclization addition reactions between 1-sulfonyl-1,2,3-triazoles 26.1 and cyclic silicon-based dienol ethers 26.2 (Figure 26)[68]. The substrates exhibited broad applicability and displayed tolerance toward various functional groups. The optimized reaction conditions involved 1,2-dichloroethane (DCE) as the solvent and Rh2(oct)4 as the catalyst at 100 °C. Experimental results demonstrated that the target products displayed specific enantioselectivity with Rh2(S-NTTL)4 and Rh2(S-PTTL)4 as catalysts, whereas the Rh2(S-TBSP)4 catalyst with a proline-derived ligand did not exhibit enantioselectivity. Mechanistically, the process begins with the reaction of N-sulfonyl-1,2,3-triazoles 26.1 with the rhodium catalyst to generate an α-iminocarbene intermediate 26.5. Subsequently, this intermediate 26.5 is captured by a cyclic silylglycol ether, leading to the production of cyclopropylimine 26.6. Finally, intramolecular nitrogen-Michael addition-mediated ring opening, followed by hydrolysis yields the intended bicyclohydroxyindolone 26.7.
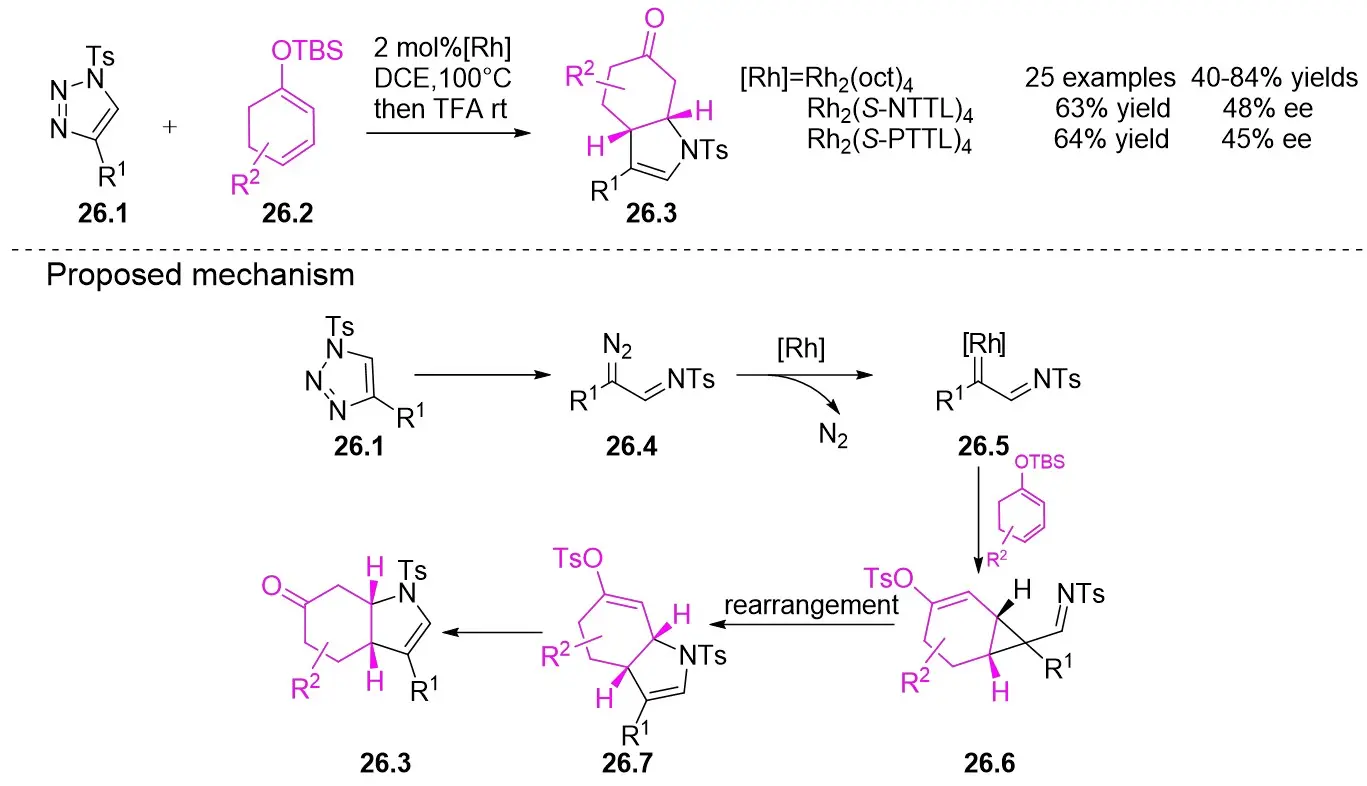
Figure 26. [3+2] cycloaddition of 1-sulfonyl-1,2,3-triazoles 26.1 with cyclic silyl dienol ethers. DCE: 1,2-dichloroethane; rt: room temperature; TFA: trifluoroacetic acid; rt: room temperature.
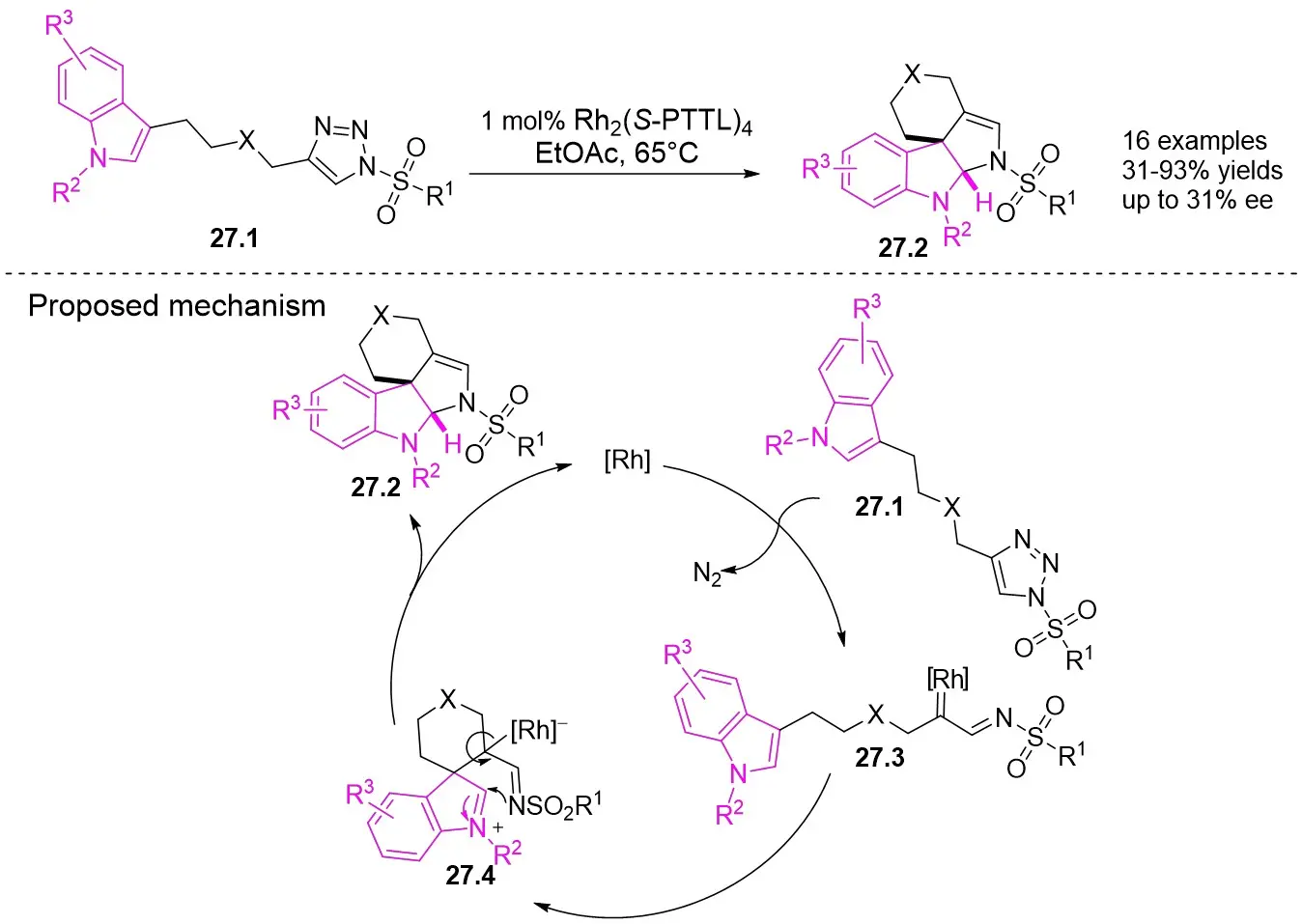
In 2017, Davies and Fu described the intramolecular [3+2] cyclization or C-H functionalization of connected N-sulfonyl 1,2,3-triazoles 27.1 for the synthesis of polycyclic spiroindoles 27.2 (Figure 27)[69]. The utilization of the Rh2(S-PTTL)4/EtOAc system resulted in a high product yield and typically excellent diastereoselectivity (> 19:1). However, the chiral catalyst exhibited relatively low enantioselectivity (up to 31% ee with Rh2(S-PTTL)4). Various substituents on N1 or in the linking nitrogen of the indole ring and triazoles were found to result in good yields. The proposed reaction mechanism involves the initial nucleophilic attack of indole C3 on Rh(II) carbene 27.3 to generate a zwitterionic intermediate 27.4, which then undergoes rearrangement to yield the final [3+2] cycloadduct 27.2.
In 2018, Davies and Kubiak successfully synthesized highly substituted and stereoenriched β-arylpyrrolidines 28.3 (Figure 28)[70]. This synthesis was accomplished via catalytic enantioselective intermolecular allylic C(sp3)-H functionalization of trans-alkenes 28.2. The reaction utilized 4-aryl-1-sulfonyl-1,2,3-triazoles 28.1 as the carbene precursor and Rh2(S-NTTL)4 as the catalyst. Subsequently, the resulting cyclic semiaminaldehyde underwent ozonation, reduction, and in situ diversification processes. The compounds were obtained in good yields with high levels of diastereoselectivity and enantioselectivity. Various triazoles with diverse electronic and steric properties on the aryl moiety were found to be suitable for the insertion reaction. The cis substitution of olefins impedes allylic C-H functionalization, leading to unsatisfactory outcomes.

Figure 28. Synthesis of highly substituted and stereoenriched β-arylpyrrolidines 28.3.
In 2018, Murakami and colleagues reported a method for synthesizing 3,5-diaryl-2-pyrazolines 29.3 (Figure 29)[71]. In the presence of a chiral rhodium(II) carboxylate dimer, 1H-tetrazoles 29.1 underwent a sulfonylation reaction with trifluoromethanesulfonic anhydride to produce the α-azorhodium(II) carbene species. This novel donor/acceptor carbene subsequently reacted readily with styrenes 29.2. Several chiral rhodium(II) catalysts and solvents were assessed to enhance the yield and enantioselectivity. Rh2(S-TCPTTL)4 was identified as the optimal catalyst, and ethyl cyclohexane as the ideal solvent based on the results. Various styrenes and substituted tetrazoles were examined, yielding between 79% to 99% with good to excellent enantioselectivity (mostly exceeding 90% ee), except for n-hexyl substituted 1H-tetrazole, benzyl ethylene, and 2-methylstyrene. The proposed reaction mechanism proceeds as follows: 1) Tetrazole 29.1, in the presence of tetrazole trianhydride, generates an isomeric mixture of tetrazole 29.4 and tetrazole 29.5, which equilibrate to form α-diazodiazo 29.6. 2) Upon the addition of a rhodium(II) catalyst, dinitrogen is released from the intermediate 29.6, leading to the formation of a Rh(II)-carbene species 29.7. This carbene species 29.7 undergoes an asymmetric reaction with styrene, yielding the E-cyclopropyl azide 29.8. 3) Subsequently, cyclopropyl azide 29.8 engages in a ring expansion process to generate 2-pyrazoline via transition state 29.9.
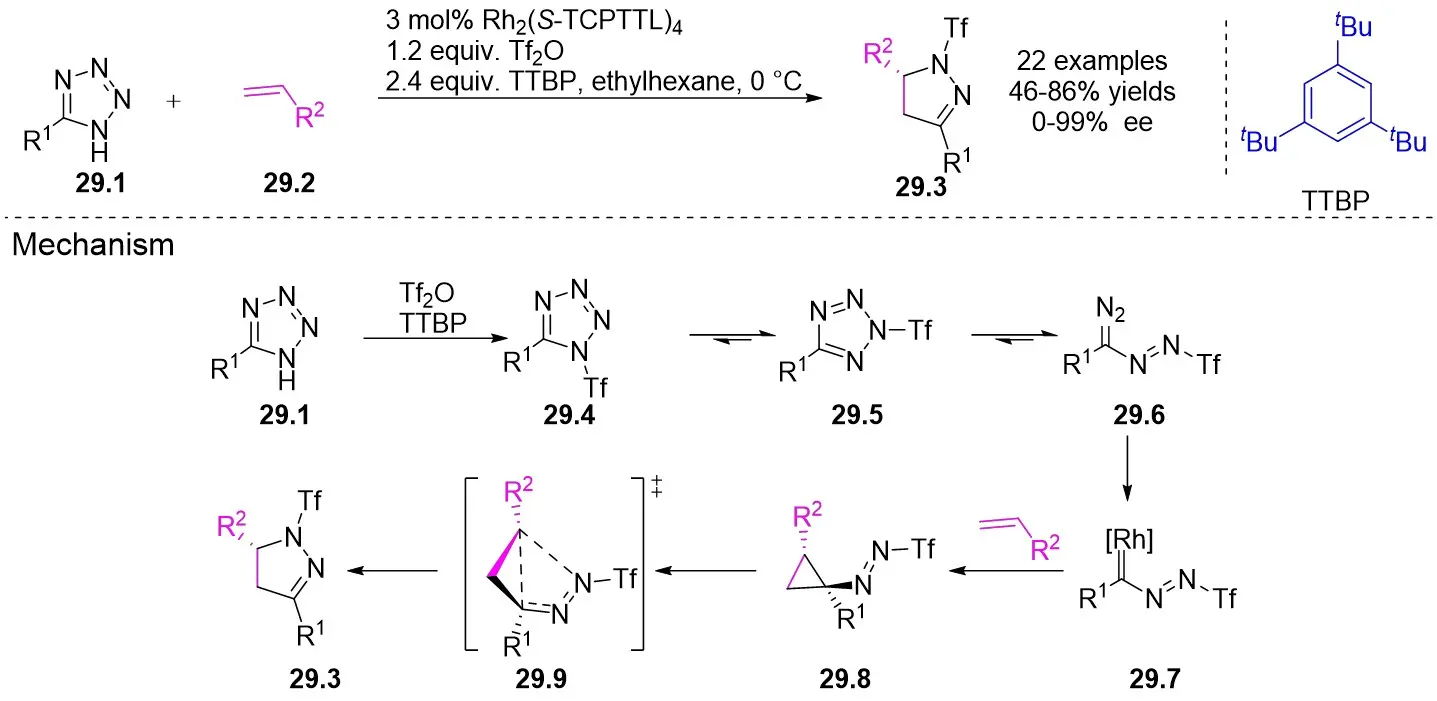
Figure 29. Denitrogenative annulation of 1H-tetrazoles 29.1 with styrenes. TTBP: 2,4,6-Tri-tert-butylphenol.
In 2019, Anbarasan and Jasekar devised an effective rhodium(II)/squaramide relay catalytic approach to synthesize diastereoselective dihydro β-carbolines 30.3 using easily obtainable, suitably substituted indole derivatives 30.2 and N-sulfonyl-1,2,3-triazoles 30.1 (Figure 30)[72]. The process entailed the selective insertion of the C3-H bond of the indole derivatives by an in situ formed imine vinyl rhodium carbene, followed by an enantioselective intramolecular imine Michael reaction facilitated by tetrahydropyranamide. Employing optimal conditions, a range of N-sulfonyl-1,2,3-triazoles and diverse substituents on the indole moiety were subjected to afford the desired dihydro-β-carbolines with high yield and excellent enantiomeric ratio.
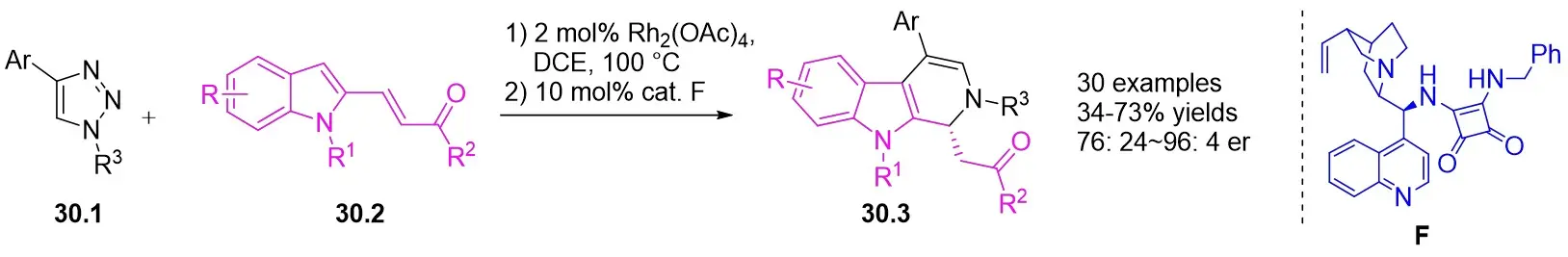
Figure 30. Tandem reaction of indole derivatives with N-sulfonyl-1,2,3-triazoles 30.1. DCE: 1,2-dichloroethane.
In 2021, the Bi group reported a novel rhodium-catalyzed enantioselective intramolecular insertion reaction of ethers 31.1 and 31.2 α-C(sp3)-H bonds under mild conditions (Figure 31)[73]. Employing fluorinated alkyl N-trifluoroformylhydrazines—safe and readily decomposable compounds—as the carbene source, they efficiently synthesized a series of chiral fluoroalkyl-substituted 2,3-dihydrobenzofuran derivatives 31.3 and 31.4 that were previously challenging to access. Among the three screened substrate types (-CF3, -CF2CF3, and -CF2R), the -CF₃ substrates exhibited the optimal catalytic performance with Rh2(R-BTPCP)4, affording a yield of 92% and an enantiomeric ratio (er) of 97:3. The -CF2CF3 substrates achieved a yield of 80% but only moderate er. For the -CF2R substrates, catalysis by Rh2(S-BTPCP)4 delivered yields of 90%-95% and er values ranging from 91:9 to 98:2. Substrates bearing cyclic alkyl groups (e.g., cyclopropyl) or linear alkyl groups (e.g., ethyl) on the ether chain reacted smoothly, with product yields of 75%-95%, a maximum er of 97:3, and a diastereomeric ratio (dr) > 20:1. Substrates containing functional groups such as phenyl, chloro, and bromo also showed good compatibility, with excellent yields and er values (93:7-98:2), and no side reactions involving alkyne or alkene moieties were observed. Aromatic substrates with different substituents afforded good to excellent yields, with only unsubstituted substrates showing a slight decrease in enantioselectivity. The practical utility of this method was validated by the concise synthesis of CF₃ analogues of natural products and bioactive molecules. Mechanistically, the reaction proceeds via two steps: rhodium carbene 31.5 formation (the rate-determining step, with an energy barrier of 11.5 kcal/mol) and intramolecular C-H insertion (1,5-hydrogen migration followed by cyclization).
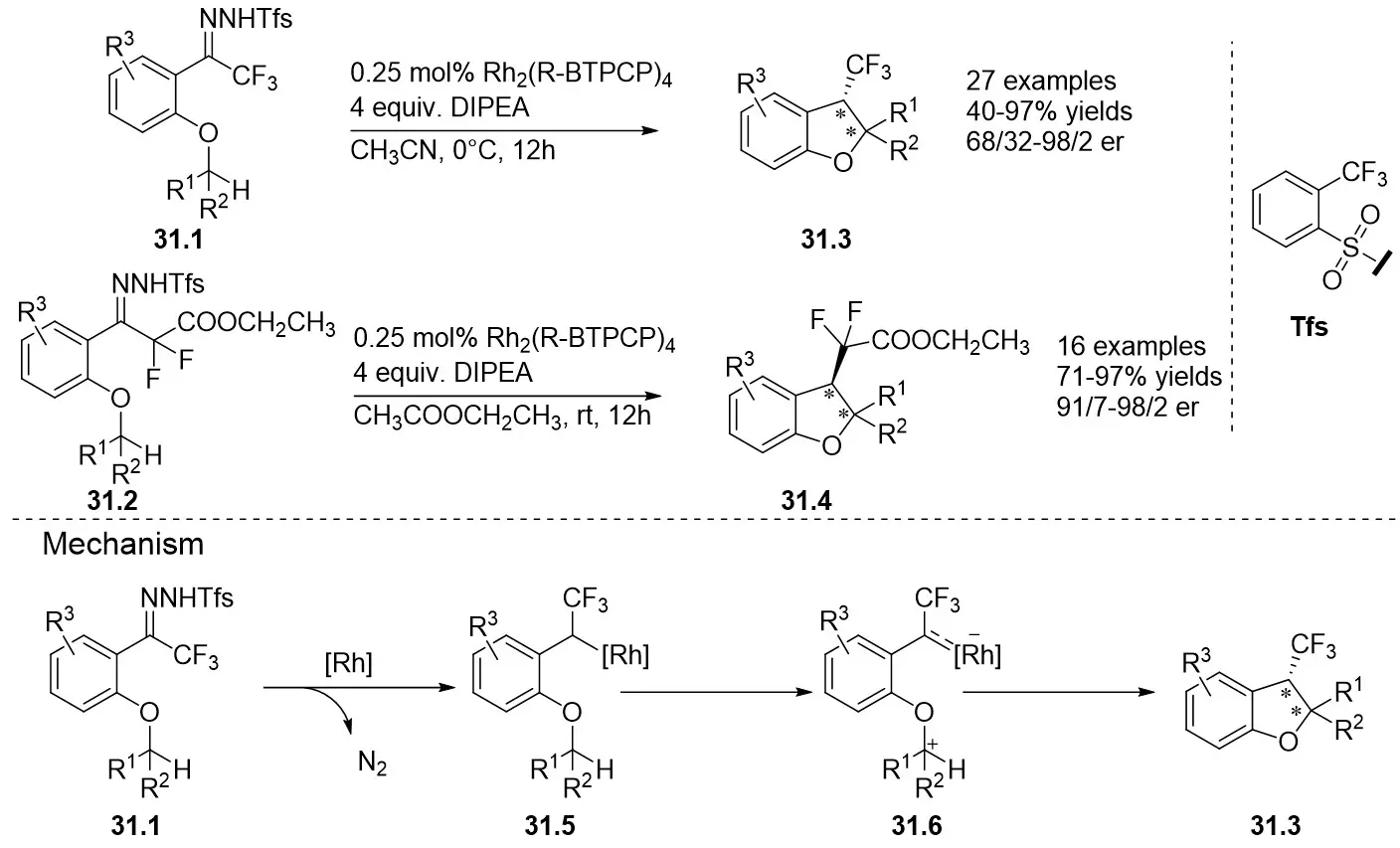
Figure 31. Intramolecular insertion reaction of ethers 31.1 and 31.2 α-C(sp3)-H bonds. DIPEA: N,N-Diisopropylethylamine.
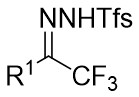
In 2025, the group developed another rhodium-catalyzed asymmetric single-carbon skeleton modification methodology, utilizing trifluoromethyl N-trifluorobarazides 32.1 as the carbene precursor for the modification of indoles 32.2 and pyrroles 32.3 (Figure 32)[74]. This reaction enables the rapid synthesis of both (S)- and (R)-enantiomers of 3,4-dihydroquinolines and 2,5-dihydropyridines, characterized by excellent chemical selectivity, regioselectivity, and enantioselectivity. For indoles, the optimal reaction system was identified as follows: Rh2(R-3,5-diPhTPCP)4 as the catalyst, NaH as the base, and trifluorotoluene as the solvent. Under these conditions, the target product was obtained with a yield of 92% and an ee of 92%. For pyrroles, the optimal catalytic system employed Rh2(R-PTTL)4 as the catalyst, N,N-Diisopropylethylamine (DIPEA) as the base, and cyclopentane as the solvent, affording the product in 84% yield with 96% ee. Notably, reversing the configuration of the catalyst enables complete enantioselectivity inversion. This methodology exhibits a broad substrate scope. Indole substrates bearing functional groups such as halogens and alkynyl groups undergo the reaction smoothly, delivering products with yields of 58~98% and ee of (88~99%. For pyrrole substrates with different substituents, the yields of 40~98% with ee of 80~99%, and gram-scale synthesis is also feasible. Furthermore, the products can be subjected to derivatization to construct various pharmaceutical molecular skeletons. Mechanistic studies revealed that the reaction proceeds via two key steps: asymmetric cyclopropanation and regioselective ring-opening, with complete retention of configuration. As depicted in the catalytic cycle, the process initiates with the formation of chiral rhodium carbene IntII. Subsequent nucleophilic attack by indole generates the zwitterionic intermediate IntIII, which then undergoes cyclopropanation to form IntIV while regenerating the rhodium catalyst. After deprotonation, IntIV undergoes ring-opening and hydration-protonation to ultimately yield the ring-expanded product.
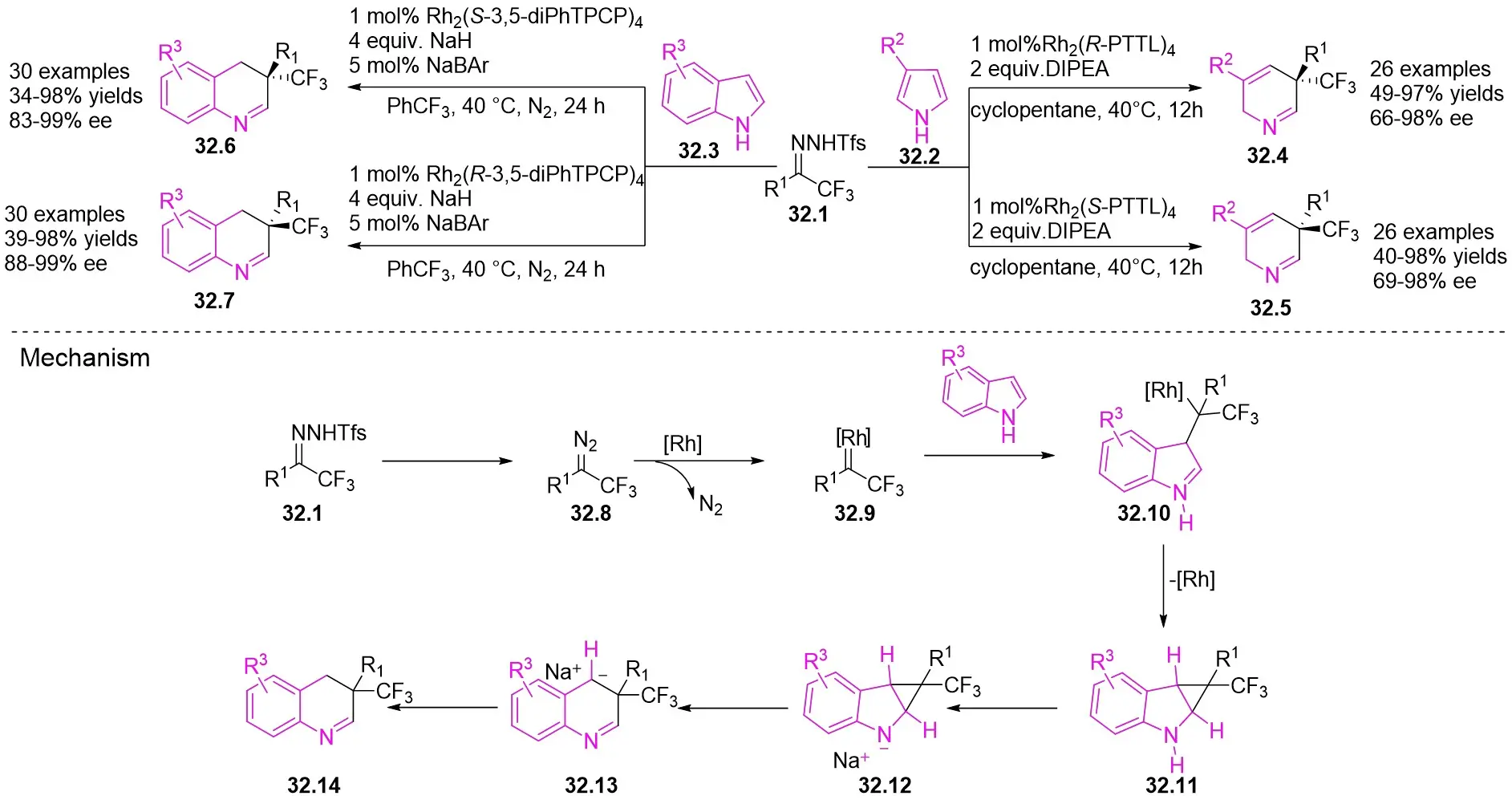
Figure 32. The asymmetric single-carbon skeleton modification methodology utilizes trifluoromethyl N-trifluorobarazides 32.1. DIPEA: N,N-Diisopropylethylamine; PTTL: N-phthaloyl-(S)-tert-leucinate.
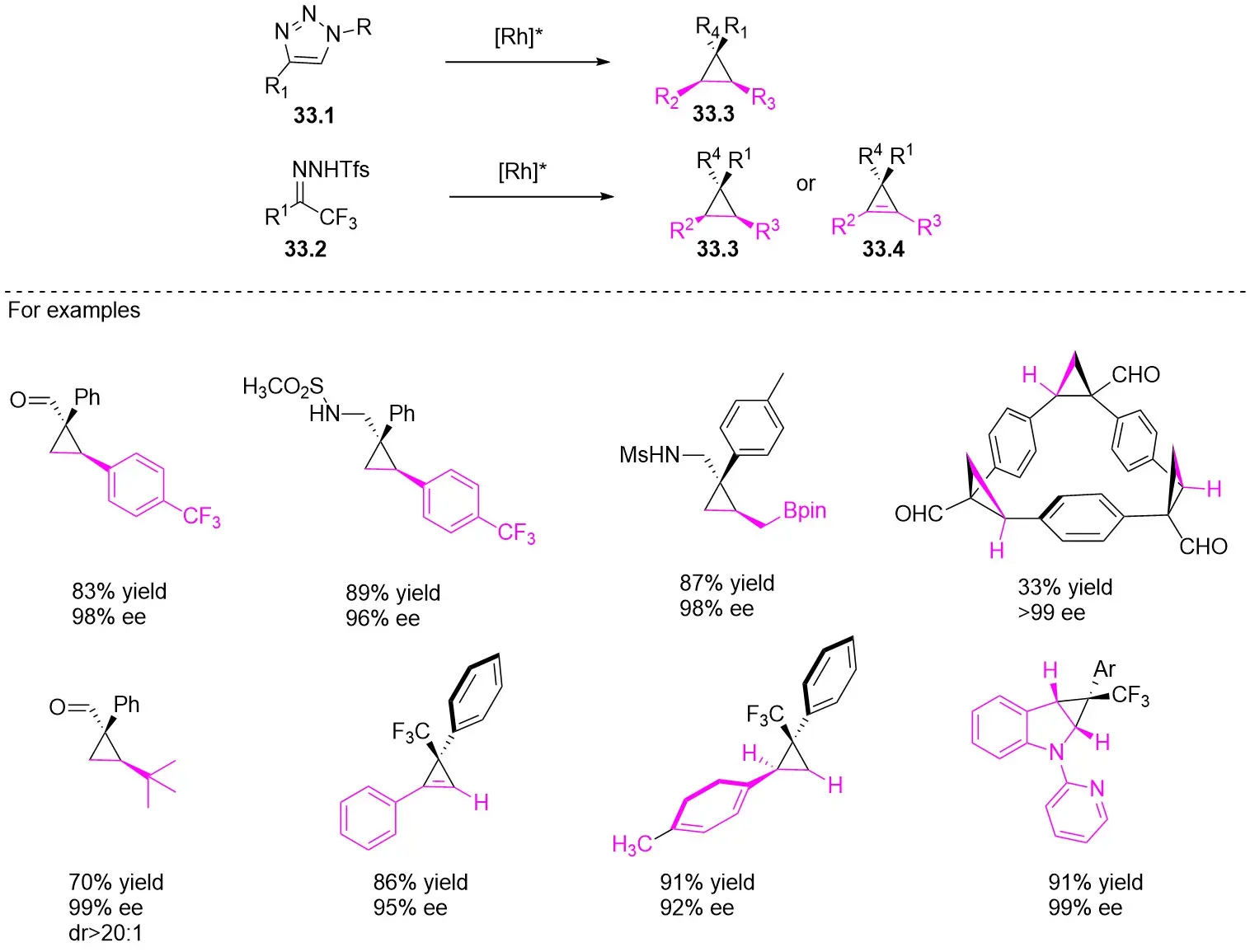
In addition, triazoles and hydrazones stand out as highly efficient carbene precursors. Under metal catalysis, they undergo denitrogenation to release nitrogen gas, concomitantly generating highly reactive carbene intermediates. These carbene intermediates readily react with unsaturated compounds (e.g., olefins) to construct cyclopropane architectures of significant application value (Figure 33). Characterized by high selectivity and broad substrate scope, this class of reactions finds widespread use in pharmaceutical synthesis—for instance, in building cyclopropane-containing drug scaffolds—and in the fabrication of functional materials, thereby offering a pivotal pathway for the efficient synthesis of complex molecules[75-84].
4. Conclusion and Perspective
Transition metal-catalyzed denitrocyclization has significantly advanced the synthesis of chiral heterocyclic compounds, establishing a novel pathway for their construction. This method enables the efficient transformation of simple raw materials into complex chiral heterocyclic frameworks, particularly for the synthesis of pharmaceutical intermediates and natural products. Despite significant advancements, several bottlenecks remain to be addressed. First, most systems must be conducted under high temperatures and inert gas conditions, which restricts their applicability to substrate with sensitive functional group. Second, the high cost of catalysts and the complex synthesis pathways of certain ligands pose additional challenges. Limitations in the final substrate scope are also apparent, as the most effective reactions predominantly focus on specific types of heterocyclic precursors. Future efforts should aim to optimize the reaction systems, develop a more efficient, cost-effective, and environmentally friendly catalytic framework, conduct in-depth exploration of the reaction mechanisms, and create a structurally diverse array of substrates to facilitate the precise construction of various chiral heterocyclic compounds, promoting sustainable development in related fields.
Authors Contribution
Guo WG: Literature search and writing, review and revise.
Liu RR: Checking the design of the paper, review and revise.
Conflicts of interest
Ren-Rong Liu is an Associate Editor of Chiral Chemistry. The other author declares no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
The article was supported by the National Natural Science Foundation of China (22371152), National Natural Science Foundation of Shandong (ZR2023JQ006) and the Science and Technology Innovation Project of Colleges and Universities in Shanxi Province (2022L383).
Copyright
© The Author(s) 2025.
References
-
1. Gul S, Aslam K, Pirzada Q, Rauf A, Khalil AA, Semwal P, et al. Xanthones: A class of heterocyclic compounds with anticancer potential. Curr Top Med Chem. 2022;22(23):1930-1949.[DOI]
-
2. Kerru N, Gummidi L, Maddila S, Gangu KK, Jonnalagadda SB. A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules. 2020;25(8):1909.[DOI]
-
3. Reen GK, Kumar A, Sharma P. Recent advances on the transition-metal-catalyzed synthesis of imidazopyridines: An updated coverage. Beilstein J Org Chem. 2019;15(1):1612-1704.[DOI]
-
4. Sharma PC, Bansal KK, Sharma A, Sharma D, Deep A. Thiazole-containing compounds as therapeutic targets for cancer therapy. Eur J Med Chem. 2020;188(9):112016.[DOI]
-
5. Devendar P, Yang GF. Sulfur-containing agrochemicals. Top Curr Chem. 2017;375(6):82.[DOI]
-
6. Smejkal T, Hachisu S, Scutt JN, Willetts NJ, Sayer D, Wildsmith L, et al. Herbicidal aryldiones incorporating a 5-methoxy-[1,2,5]triazepane ring. Bioorg Med Chem Lett. 2018;28(3):339-343.[DOI]
-
7. Kong X, Zare N, Quchan Atigh ZB, Fayazi M, Karimi-Maleh H, Sadeghifar H, et al. Unveiling the interactions between biomaterials and heterocyclic dyes: A sustainable approach for wastewater treatment. Chemosphere. 2023;338(1):139625.[DOI]
-
8. Pina F, Melo MJ, Laia CA, Parola AJ, Lima JC. Chemistry and applications of flavylium compounds: A handful of colours. Chem Soc Rev. 2012;41(2):869-908.[DOI]
-
9. Choi S, Guo MC, Coombs GM, Miller SJ. Catalytic asymmetric synthesis of atropisomeric N-aryl 1,2,4-triazoles. J Org Chem. 2023;88(12):7815-7820.[DOI]
-
10. Lenz M, Borlinghaus N, Weinmann L, Nestl BM. Recent advances in imine reductase-catalyzed reactions. World J Microbiol Biotechnol. 2017;33(11):199.[DOI]
-
11. Rani Kumar N, Agrawal AR. Advances in the chemistry of 2,4,6-tri(thiophen-2-yl)-1,3,5-triazine. ChemistryOpen. 2023;12(1):e202200203.[DOI]
-
12. Taylor RD, MacCoss M, Lawson AD. Rings in drugs. J Med Chem. 2014;57(14):5845-5859.[DOI]
-
13. Vitaku E, Smith DT, Njardarson JT. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J Med Chem. 2014;57(24):10257-10274.[DOI]
-
14. Roy S, Das SK, Khatua H, Das S, Chattopadhyay B. Road map for the construction of high-valued N-heterocycles via denitrogenative annulation. Acc Chem Res. 2021;54(23):4395-4409.[DOI]
-
15. Chattopadhyay B, Gevorgyan V. Transition-metal-catalyzed denitrogenative transannulation: Converting triazoles into other heterocyclic systems. Angew Chem Int Ed Engl. 2012;51(4):862-872.[DOI]
-
16. Das SK, Roy S, Chattopadhyay B. Transition-metal-catalyzed denitrogenative annulation to access high-valued N-heterocycles. Angew Chem Int Ed. 2023;62(2):e202210912.[DOI]
-
17. Wang N, Zheng SC, Zhang LL, Guo Z, Liu XY. Nickel(0)-catalyzed denitrogenative transannulation of benzotriazinones with alkynes: Mechanistic insights of chemical reactivity and regio- and enantioselectivity from density functional theory and experiment. ACS Catal. 2016;6(6):3496-3505.[DOI]
-
18. Cheng JK, Xiang SH, Tan B. Imidodiphosphorimidates (IDPis): Catalyst motifs with unprecedented reactivity and selectivity. Chin J Chem. 2023;41(6):685-694.[DOI]
-
19. Zhang J, Huo X, Liu Y, Zhu C. Dynamic kinetic resolution and dynamic kinetic asymmetric transformation of atropisomeric biaryls. Chem Catal. 2025;5(4):101329.[DOI]
-
20. Gopakumar K, Samantaray V, Prusty MK, Swain L, Ramanan R. Internal charge-transfer in a metal-catalyzed oxidative addition reaction turns an inhibitive electric field stimulus to catalytic. Chem Commun. 2023;59(87):13054-13057.[DOI]
-
21. Wang P, Hu L, Wang F, Li XX, Huang G, Lu G, et al. Atroposelective de novo access to biaryls via nickel-catalyzed C–C activation of benzocyclobutenols: Aryl sources powered by dual ring scissions. ACS Catal. 2025;15(16):14432-14442.[DOI]
-
22. Zhang PC, Han J, Zhang J. Pd/PC-Phos-catalyzed enantioselective intermolecular denitrogenative cyclization of benzotriazoles with allenes and N-allenamides. Angew Chem Int Ed. 2019;58(33):11444-11448.[DOI]
-
23. Wu Q, Dong Z, Xu J, Yang Z. Sulfur-controlled and rhodium-catalyzed formal (3 + 3) transannulation of thioacyl carbenes with alk-2-enals and mechanistic insights. Org Biomol Chem. 2021;19(14):3173-3180.[DOI]
-
24. Chen C, Fang S, Dong Z, Xu J, Yang Z. Catalytic diastereospecific and enantioselective (3 + 2) transannulations of 1,2,3-thiadiazoles with strained norbornene derivatives. Org Lett. 2022;24(11):2110-2114.[DOI]
-
25. Diccianni J, Lin Q, Diao T. Mechanisms of nickel-catalyzed coupling reactions and applications in alkene functionalization. Acc Chem Res. 2020;53(4):906-919.[DOI]
-
26. Weix DJ. Methods and mechanisms for cross-electrophile coupling of Csp2 halides with alkyl electrophiles. Acc Chem Res. 2015;48(6):1767-1775.[DOI]
-
27. Kariofillis SK, Doyle AG. Synthetic and mechanistic implications of chlorine photoelimination in nickel/photoredox C(sp3)-H cross-coupling. Acc Chem Res. 2021;54(4):988-1000.[DOI]
-
28. Wang Y, He Y, Zhu S. NiH-catalyzed functionalization of remote and proximal olefins: New reactions and innovative strategies. Acc Chem Res. 2022;55(23):3519-3536.[DOI]
-
29. Yamauchi M, Morimoto M, Miura T, Murakami M. Enantioselective synthesis of 3,4-dihydroisoquinolin-1(2H)-ones by nickel-catalyzed denitrogenative annulation of 1,2,3-benzotriazin-4(3H)-ones with allenes. J Am Chem Soc. 2010;132(1):54-55.[DOI]
-
30. Miura T, Yamauchi M, Kosaka A, Murakami M. Nickel-catalyzed regio- and enantioselective annulation reactions of 1,2,3,4-benzothiatriazine-1,1(2H)-dioxides with allenes. Angew Chem Int Ed. 2010;49(29):4955-4957.[DOI]
-
31. Miura T, Morimoto M, Yamauchi M, Murakami M. Nickel-catalyzed denitrogenative annulation reactions of 1,2,3-benzotriazin-4(3H)-ones with 1,3-dienes and alkenes. J Org Chem. 2010;75(15):5359-5362.[DOI]
-
32. Fang ZJ, Zheng SC, Guo Z, Guo JY, Tan B, Liu XY. Asymmetric synthesis of axially chiral isoquinolones: Nickel-catalyzed denitrogenative transannulation. Angew Chem. 2015;54(33):9528-9532.[DOI]
-
33. Yamano MM, Kelleghan AV, Shao Q, Giroud M, Simmons BJ, Li B, et al. Intercepting fleeting cyclic allenes with asymmetric nickel catalysis. Nature. 2020;586(7828):242-247.[DOI]
-
34. Ge FB, Lu CJ, Chen X, Yao W, An M, Jiang YK, et al. Enantioselective nickel-catalyzed denitrogenative transannulation en route to N–N atropisomers. Angew Chem. 2024;63(26):e202400441.[DOI]
-
35. Chen Y, Xiao L, Wan Y, Liu W, Li J, Guo Y. Asymmetric nickel-catalyzed denitrogenative transannulation to construct axially chiral C–N atropisomers. Org Lett. 2025.[DOI]
-
36. Sabu S, Saranya PV, Anilkumar G. An overview of palladium-catalyzed N-alkylation reactions. Org Biomol Chem. 2025;23(7):1533-1551.[DOI]
-
37. Yang Y, Miao C, Wang R, Zhang R, Li X, Wang J, et al. Advances in morphology-controlled alumina and its supported Pd catalysts: Synthesis and applications. Chem Soc Rev. 2024;53(10):5014-5053.[DOI]
-
38. Peddiahgari Vasu GR, Motakatla Venkata KR, Kakarla RR, Ranganath KVS, Aminabhavi TM. Recent advances in sustainable N-heterocyclic carbene-Pd(II)-pyridine (PEPPSI) catalysts: A review. Environ Res. 2023;225(17`):115515.[DOI]
-
39. Li YL, Zhang PC, Wu HH, Zhang J. Palladium-catalyzed asymmetric tandem denitrogenative heck/tsuji-trost of benzotriazoles with 1,3-dienes. J Am Chem Soc. 2021;143(33):13010-13015.[DOI]
-
40. Liu CX, Yin SY, Zhao F, Yang H, Feng Z, Gu Q, et al. Rhodium-catalyzed asymmetric C–H functionalization reactions. Chem Rev. 2023;123(16):10079-10134.[DOI]
-
41. Blaszczyk SA, Glazier DA, Tang W. Rhodium-catalyzed (5 + 2) and (5 + 1) cycloadditions using 1,4-enynes as five-carbon building blocks. Acc Chem Res. 2020;53(1):231-243.[DOI]
-
42. Zhang X, Wu X, Shi H. Mechanistic investigation on rhodium(III)-catalyzed cycloaddition of 2-vinylphenol derivatives with ethyne or carbon monoxide by DFT study. Chin J Chem Phys. 2022;35(4):685-696.[DOI]
-
43. Dong Z, Chen C, Wang J, Xu J, Yang Z. Dual roles of bisphosphines and epoxides: Rh-catalyzed highly chemoselective and diastereoselective (3 + 2) transannulations of 1,2,3-thiadiazoles with cyanoepoxides. Org Chem Front. 2021;8(23):6687-6698.[DOI]
-
44. Lv K, Bao X. Mechanistic insights into the Rh(I)-catalyzed transannulation of 1,2,3-thiadiazoles with alkenes, alkynes, and nitriles: Does the intermediacy of α-thiavinyl Rh-carbenoids play an important role? Org Chem Front. 2021;8(2):310-318.[DOI]
-
45. Wei H, Chen C, Gao A, Yang Z. Rh(I)-catalyzed enantioselective (3 + 2) transannulations of 1,2,3-thiadiazoles with P-chiral monodentate phosphines. Phosphorus, Sulfur, Silicon Relat Elem. 2023;199(2):157-161.[DOI]
-
46. Chen C, Lv K, Chen Y, Du H, Xu J, Liu T, et al. Catalytic enantioselective (3 + 2) annulations of nucleophilic thioacyl Rh(I)-carbenes with achmatowicz rearrangement products. ACS Catal. 2024;14(11):8949-8957.[DOI]
-
47. Chen C, Chen Y, Du H, Xu J, Yang Z. Enantioselective and regiodivergent skeletal transformations of 1,2,3-thiadiazoles to alkylidene dihydrothiophenes. Org Lett. 2025;27(16):4337-4342.[DOI]
-
48. Wang D, Astruc D. The golden age of transfer hydrogenation. Chem Rev. 2015;115(13):6621-6686.[DOI]
-
49. Jimenez MV, Fernández-Tornos J, Pérez-Torrente JJ, Modrego FJ, Winterle S, Cunchillos C, et al. Iridium(I) complexes with hemilabile N-heterocyclic carbenes: Efficient and versatile transfer hydrogenation catalysts. Organometallics. 2011;30(20):5493-5508.[DOI]
-
50. Wang C, Lan F, He Z, Xie X, Zhao Y, Hou H, et al. Iridium-based catalysts for solid polymer electrolyte electrocatalytic water splitting. ChemSusChem. 2019;12(8):1576-1590.[DOI]
-
51. Jia M, Ma S. New approaches to the synthesis of metal carbenes. Angew Chem Int Ed. 2016;55(32):9134-9166.[DOI]
-
52. Ardkhean R, Caputo DF, Morrow SM, Shi H, Xiong Y, Anderson EA. Cascade polycyclizations in natural product synthesis. Chem Soc Rev. 2016;45(6):1557-1569.[DOI]
-
53. Chen JR, Hu XQ, Lu LQ, Xiao WJ. Formal [4+1] annulation reactions in the synthesis of carbocyclic and heterocyclic systems. Chem Rev. 2015;115(11):5301-5365.[DOI]
-
54. Guo XX, Gu DW, Wu Z, Zhang W. Copper-catalyzed C–H functionalization reactions: Efficient synthesis of heterocycles. Chem Rev. 2015;115(3):1622-1651.[DOI]
-
55. Weichen X, Xinfang X. Dirhodium-catalyzed enantioselective double cyclopropanation using alkynes as carbene precursors. Chin J Org Chem. 2023;43(8):2977.[DOI]
-
56. Dong K, Fan X, Pei C, Zheng Y, Chang S, Cai J, et al. Transient-axial-chirality controlled asymmetric rhodium-carbene C(sp2)-H functionalization for the synthesis of chiral fluorenes. Nat Commun. 2020;11(1):2363.[DOI]
-
57. Kornecki KP, Briones JF, Boyarskikh V, Fullilove F, Autschbach J, Schrote KE, et al. Direct spectroscopic characterization of a transitory dirhodium donor-acceptor carbene complex. Science. 2013;342(6156):351-354.[DOI]
-
58. Zibinsky M, Fokin VV. Reactivity of N-(1,2,4-triazolyl)-substituted 1,2,3-triazoles. Org Lett. 2011;13(18):4870-4872.[DOI]
-
59. Zibinsky M, Fokin VV. Sulfonyl-1,2,3-triazoles: Convenient synthones for heterocyclic compounds. Angew Chem Int Ed. 2012;52(5):1507-1510.[DOI]
-
60. Spangler JE, Davies HML. Catalytic asymmetric synthesis of pyrroloindolines via a rhodium(II)-catalyzed annulation of indoles. J Am Chem Soc. 2013;135(18):6802-6805.[DOI]
-
61. Xu HD, Xu K, Zhou H, Jia ZH, Wu H, Lu XL, et al. Ring-strain-driven catalytic carbene formation–cyclopropanation–aza-cope rearrangement cascade: A facile entry to fused dihydroazepines from 1,3-dienyltriazoles. Synthesis. 2014;47(5):641-646.[DOI]
-
62. Kwok SW, Zhang L, Grimster NP, Fokin VV. Catalytic asymmetric transannulation of NH-1,2,3-triazoles with olefins. Angew Chem. 2014;53(13):3452-3456.[DOI]
-
63. Miura T, Funakoshi Y, Murakami M. Intramolecular dearomatizing [3+2] annulation of α-imino carbenoids with aryl rings furnishing 3,4-fused indole skeletons. J Am Chem Soc. 2014;136(6):2272-2275.[DOI]
-
64. Miura T, Nakamuro T, Liang CJ, Murakami M. Synthesis of trans-cycloalkenes via enantioselective cyclopropanation and skeletal rearrangement. J Am Chem Soc. 2014;136(45):15905-15908.[DOI]
-
65. Tian Y, Wang Y, Shang H, Xu X, Tang Y. Rhodium(II)-catalyzed intramolecular formal [4+3] cycloadditions of dienyltriazoles: Rapid access to fused 2,5-dihydroazepines. Org Biomol Chem. 2015;13(2):612-619.[DOI]
-
66. Yuan H, Gong J, Yang Z. Stereoselective synthesis of oxabicyclo[2.2.1]heptenes via a tandem dirhodium(II)-catalyzed triazole denitrogenation and [3+2] cycloaddition. Org Lett. 2016;18(21):5500-5503.[DOI]
-
67. Ko YO, Jeon HJ, Jung DJ, Kim UB, Lee SG. Rh(II)/mg(otBu)2-catalyzed tandem one-pot synthesis of 1,4-oxazepines and 1,4-oxazines from N-sulfonyl-1,2,3-triazoles and glycidols. Org Lett. 2016;18(24):6432-6435.[DOI]
-
68. Yang H, Hou S, Tao C, Liu Z, Wang C, Cheng B, et al. Rhodium-catalyzed denitrogenative [3+2] cycloaddition: Access to functionalized hydroindolones and the framework of montanine-type amaryllidaceae alkaloids. Chem Eur J. 2017;23(52):12930-12936.[DOI]
-
69. Fu L, Davies HML. Scope of the reactions of indolyl- and pyrrolyl-tethered N-sulfonyl-1,2,3-triazoles: Rhodium(II)-catalyzed synthesis of indole- and pyrrole-fused polycyclic compounds. Org Lett. 2017;19(7):1504-1507.[DOI]
-
70. Kubiak RW, Davies HML. Rhodium-catalyzed intermolecular C-H functionalization as a key step in the synthesis of complex stereodefined β-arylpyrrolidines. Org Lett. 2018;20(13):3771-3775.[DOI]
-
71. Nakamuro T, Hagiwara K, Miura T, Murakami M. Enantioselective denitrogenative annulation of 1H-tetrazoles with styrenes catalyzed by rhodium. Angew Chem. 2018;57(19):5497-5500.[DOI]
-
72. Rajasekar S, Anbarasan P. Tandem Rh(II) and chiral squaramide relay catalysis: Enantioselective synthesis of dihydro-β-carbolines via insertion to C-H bond and aza-michael reaction. Org Lett. 2019;21(9):3067-3071.[DOI]
-
73. Zhang X, Sivaguru P, Zanoni G, Han X, Tong M, Bi X. Catalytic asymmetric C(sp3)-H carbene insertion approach to access enantioenriched 3-fluoroalkyl 2,3-dihydrobenzofurans. ACS Catal. 2021;11(22):14293-14301.[DOI]
-
74. Zhang X, Song Q, Liu S, Sivaguru P, Liu Z, Yang Y, et al. Asymmetric dearomative single-atom skeletal editing of indoles and pyrroles. Nature Chem. 2025;17(2):215-225.[DOI]
-
75. Chuprakov S, Kwok SW, Zhang L, Lercher L, Fokin VV. Rhodium-catalyzed enantioselective cyclopropanation of olefins withn-sulfonyl 1,2,3-triazoles. J Am Chem Soc. 2009;131(50):18034-18035.[DOI]
-
76. Grimster N, Zhang L, Fokin VV. Synthesis and reactivity of rhodium(II) N-triflyl azavinyl carbenes. J Am Chem Soc. 2010;132(8):2510-2511.[DOI]
-
77. Miura T, Nakamuro T, Nikishima H, Murakami M. Asymmetric synthesis of cyclopropylmethanamines by rhodium-catalyzed cyclopropanation of pinacol allylboronate with N-sulfonyl-1,2,3-triazoles. Chem Lett. 2016;45(8):1003-1005.[DOI]
-
78. Parr BT, Davies HML. Rhodium-catalyzed tandem cyclopropanation/cope rearrangement of 4-alkenyl-1-sulfonyl-1,2,3-triazoles with dienes. Angew Chem. 2013;52(38):10044-10047.[DOI]
-
79. Shen H, Fu J, Gong J, Yang Z. Tunable and chemoselective syntheses of dihydroisobenzofurans and indanones via rhodium-catalyzed tandem reactions of 2-triazole-benzaldehydes and 2-triazole-alkylaryl ketones. Org Lett. 2014;16(21):5588-5591.[DOI]
-
80. Miura T, Nakamuro T, Stewart SG, Nagata Y, Murakami M. Synthesis of enantiopure C3‐symmetric triangular molecules. Angew Chem Int Ed. 2017;56(12):3334-3338.[DOI]
-
81. Xing S, Xia H, Wang X, Wu D, Xu X, Su Y, et al. Diastereoselective access to 2-aminoindanonesviathe rhodium(II)-catalyzed tandem reaction involving O–H insertion and michael addition. Org Chem Front. 2019;6(17):3121-3126.[DOI]
-
82. Zhang X, Tian C, Wang Z, Sivaguru P, Nolan SP, Bi X. Fluoroalkyl N-triftosylhydrazones as easily decomposable diazo surrogates for asymmetric [2+1] cycloaddition: Synthesis of chiral fluoroalkyl cyclopropenes and cyclopropanes. ACS Catal. 2021;11(14):8527-8537.[DOI]
-
83. He C, Song W, Wei D, Zhao W, Yu Q, Tang J, et al. Rhodium-catalyzed asymmetric cyclopropanation of indoles with N-triftosylhydrazones. Angew Chem Int Ed. 2024;63(50):e202408220.[DOI]
-
84. Liu S, Ning Y, Song W, Sivaguru P, Wang Y, Gan Y, et al. Synthesis of bridge-substituted bicyclo[1.1.1]pentanes by catalytic metal carbene insertion into bicyclo[1.1.0]butanes. Angew Chem Int Ed. 2025;e202514232:[DOI]
Copyright
© The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Guo W, Liu R. Transition-metal-catalyzed asymmetric denitrogenative transannulation. Chiral Chem. 2025;1:202504. https://doi.org/10.70401/cc.2025.0001
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Denitrogenative Transannulation via Oxidative Addition
- 3. Denitrogenative Transannulation via Metal Carbenes
- 4. Conclusion and Perspective
- Authors Contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- Copyright
Science Exploration Style
Guo W, Liu R. Transition-metal-catalyzed asymmetric denitrogenative transannulation. Chiral Chem. 2025;1:202504. https://doi.org/10.70401/cc.2025.0001
copy
Share Link
copy