Insights into the pathogenesis of eosinophilic esophagitis using mouse models
Anish Dsilva
Ariel Munitz
*

*Correspondence to:
Ariel Munitz, Department of Clinical Microbiology and Immunology, Gray Faculty of Medical and Health Sciences, Tel Aviv University, Ramat Aviv 69978, Israel.
E-mail: arielm@tauex.tau.ac.il
Myeloid Cells. 2026;1:202604. 10.70401/mc.2026.0003
Received: March 24, 2026Accepted: May 18, 2026Published: May 18, 2026
Abstract
Eosinophilic esophagitis (EoE) is a chronic, food antigen-driven, type 2 immune-mediated disease of the esophagus characterized by eosinophil-predominant mucosal inflammation, epithelial remodeling, and subepithelial fibrosis. Although patient biopsies have established the EoE transcriptome and identified key cellular and molecular mediators, biopsy-based research is inherently correlative and cannot resolve causal disease mechanisms, temporal disease progression or the functional hierarchy of immune cell interactions. Thus, animal models are indispensable tools for addressing these limitations. In this Review, we examine the landscape of experimental EoE models. We assess the capacity of each model to recapitulate key disease features including lamina propria and intraepithelial eosinophilia, subepithelial fibrosis, basal cell hyperplasia, epithelial barrier dysfunction, and angiogenesis. We further map these models to human EoE transcriptomic overlap and disease endotype relevance. Across this analysis, we highlight mechanistic insights that were obtained from these models including the eosinophil-independence of IL-13-driven esophageal remodeling, the respective roles of thymic stromal lymphopoietin and IL-33, and the critical role of epithelial-expressed IL-13Rα1. We further highlight the profibrotic functions of amphiregulin-producing T helper 2 cells and colony-stimulating factor 1-dependent macrophages. We discuss the anatomical, genetic and functional limitations of current models and outline directions for the next generation of EoE preclinical systems.
Keywords
Eosinophilic gastrointestinal diseases, eosinophilic esophagitis, allergy, IL-13, IL-13Rα1, thymic stromal lymphopoietin, IL-33
1. Introduction
Eosinophilic esophagitis (EoE) is a chronic, food antigen-induced T helper type 2 (Th2)-associated, allergic- and immune-mediated disease. It is characterized histologically by eosinophil-predominant mucosal inflammation and clinically by esophageal dysfunction[1]. EoE is diagnosed by the combination of esophageal dysfunction (e.g., dysphagia, food impaction, vomiting, abdominal pain) and a histological hallmark of eosinophil-predominant mucosal inflammation exceeding a threshold of at least 15 eosinophils per high-power field (eos/hpf), in the absence of alternative etiologies such as gastroesophageal reflux disease. Since the first description of EoE, its prevalence has dramatically increased, especially over the past two decades[2,3]. Current estimates suggest an overall prevalence of approximately 1 in 700 to 1,500 people in Western countries. EoE is frequently considered part of the “atopic march”, where patients develop a sequence of allergic conditions often starting with eczema and gradually developing additional co-morbidities such as EoE, asthma, allergic rhinitis or food allergy[4]. Importantly, EoE incidence rates continue to rise in a pattern that cannot be attributed to improved disease recognition alone.
The immunopathogenesis of EoE is driven by aberrant type 2 immune responses to dietary antigens such as milk, wheat, egg and soy in genetically susceptible individuals[5]. Epithelial cell-associated alarmins such as IL-33 and thymic stromal lymphopoietin (TSLP) are perceived to serve as early sensors of barrier disruption and allergen exposure[6-9]. Consequently, IL-33 and TSLP can initiate and amplify a type 2 inflammatory cascade involving IL-4, IL-5 and IL-13[10-12]. The latter orchestrate the recruitment of eosinophils to the esophageal mucosa, reprogram epithelial gene expression, activate mast cells and basophils, and ultimately drive a remodeling process that encompasses basal cell hyperplasia, epithelial thickening, subepithelial fibrosis, angiogenesis and smooth muscle hypertrophy. Genetic risk for EoE is associated, at least in part, by variants in various immune-related and barrier-associated pathways including CAPN14, TSLP, STAT6, DSG1 and IL-33[13-17,18,19]. The genetic associations identified in EoE are not necessarily unique to EoE and are often a shared genetic architecture between EoE and broader atopic/allergic disease.
EoE is diagnosed through endoscopic sampling of the esophagus[20]. Thus, a substantial amount of knowledge regarding the pathophysiology of EoE has been derived from the analysis of esophageal biopsies obtained during diagnostic or monitoring endoscopy. Gene expression profiling of biopsy tissue established the EoE transcriptome, a disease-specific molecular signature dominated by dysregulation of epithelial differentiation genes (e.g., DSG1, FLG, CAPN14), type 2 effector molecules (e.g., CCL26/eotaxin-3, POSTN, SPINK7) and remodeling mediators (e.g., TGFB1, COL1A1)[1,21,22]. The EoE Diagnostic Panel, derived from this transcriptome, has provided molecular precision to diagnosis and treatment monitoring[23]. More recently, single-cell RNA sequencing (scRNAseq), spatial transcriptomics and proteomics applied to biopsy material have revealed remarkable cellular heterogeneity within the EoE esophagus[24-26]. These approaches have identified distinct cellular subsets, epithelial cell states associated with barrier dysfunction and proliferation, activated fibroblast populations, and the spatial organization of inflammatory microenvironments.
Despite these advances, the reliance on biopsy-derived tissue carries fundamental limitations that constrain mechanistic understanding of EoE. Biopsies represent cross-sectional snapshots that cannot resolve the temporal progression from initial sensitization to established fibrosis. Their invasive nature limits sampling frequency, precluding longitudinal resolution at the cellular level. Most critically, biopsies cannot establish causality. The observation that a molecule or cell type is elevated in EoE tissue does not identify it as a driver rather than a consequence of inflammation. Translating correlative biopsy findings into therapeutic hypotheses requires an experimental system in which specific molecular circuits can be interrogated through genetic or pharmacological perturbation, such as those obtained by experimental animal models of human disease.
Over the past two decades, several animal models of EoE have been developed[27]. These span allergen sensitization procedures, transgenic cytokine overexpression systems, conditional knockout approaches and environmental exposure protocols. Collectively, these models have provided causal mechanistic data that substantially increased our understanding of disease pathogenesis. In this review, we examine the landscape of available EoE animal models, assess their capacity to recapitulate the defining pathological features of human disease, and discuss the mechanistic knowledge that has been uniquely enabled by preclinical work. We further address the limitations of current models and outline directions for the next generation of EoE preclinical systems.
2. Preclinical Models as Tools for Resolving Mechanistic Gaps in EoE
Several areas of EoE biology are particularly suited for preclinical investigation. The selection of a given model should be guided by the specific biological question being addressed (Figure 1). For instance, indirect cytokine administration models and direct epithelial cytokine overexpression systems (Figure 1) are best suited for dissecting downstream effector mechanisms including IL-13-driven epithelial remodeling and type 2 inflammatory pathways. Antigen- and hapten-driven models including oxazolone (OXA), MC903, and house dust mite (HDM) (Figure 1), are well suited for studying disease initiation, atopic sensitization, and epithelial-immune crosstalk. Hybrid models combining transgenic cytokine expression with hapten challenge (Figure 1), such as L2-IL5/OXA, provide robust systems to investigate chronic inflammation and fibrosis, although the relative contribution of each component should be carefully addressed. Spontaneous genetic models enable the study of disease endotypes and genetic susceptibility in the absence of external sensitization (Figure 1), whereas environmental exposure models are particularly useful for interrogating barrier disruption and antigen-independent disease initiation (Figure 1). No single model recapitulates all aspects of EoE, and the experimental design should align with the capability of each model to address specific questions including:
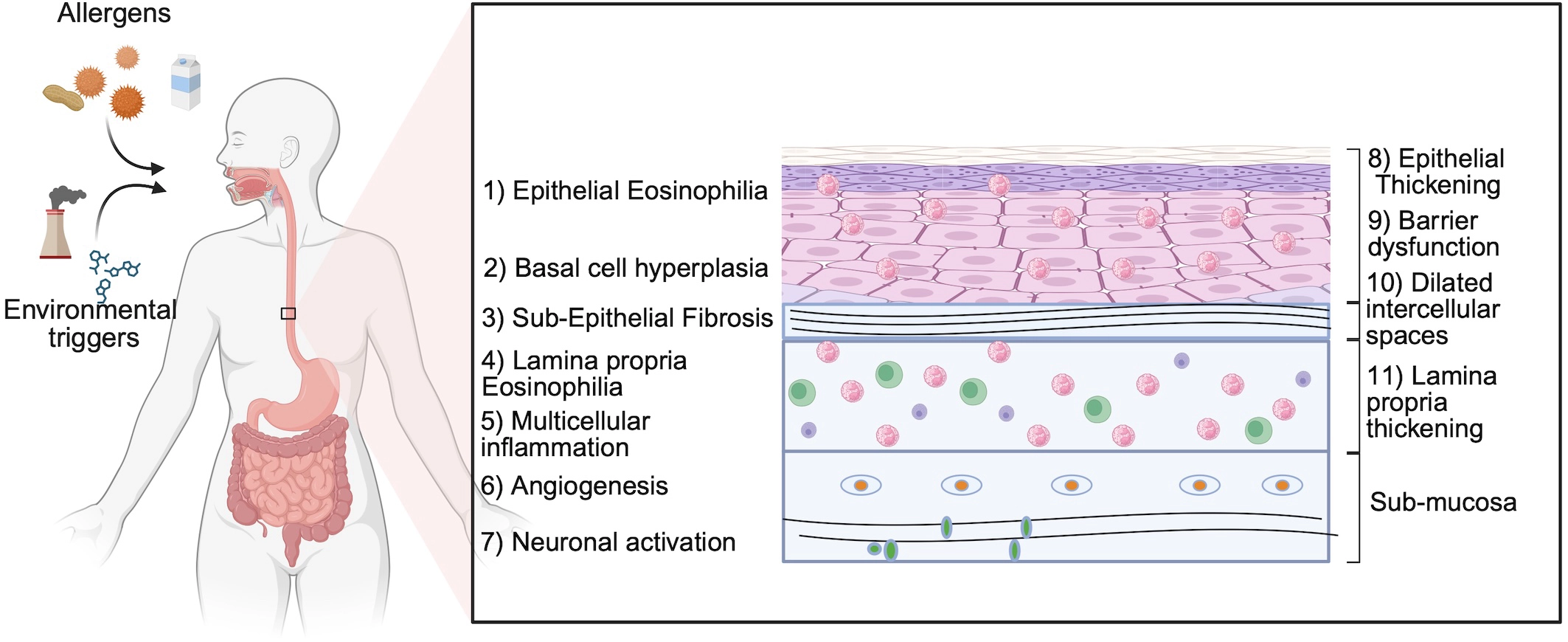
Figure 1. Key histopathological and functional features of human eosinophilic esophagitis. Schematic representation of the major pathological features observed in the esophagus of patients with EoE. Environmental triggers and food allergens initiate epithelial barrier dysfunction and type 2 inflammation. Hallmark features include intraepithelial eosinophilia, basal cell hyperplasia, and subepithelial fibrosis. Inflammation extends into the lamina propria with recruitment of multiple immune cell populations, including eosinophils, mast cells, and lymphocytes. Structural remodeling is accompanied by angiogenesis and neuronal activation, which contribute to disease symptoms. These features collectively define the tissue landscape that experimental models aim to recapitulate. Created in BioRender. Dsilva, A. (2026) https://app.biorender.com/illustrations/69bebe2c0501740c79d91728?slideId=aa068b63-5a8d-46e5-8319-216658020d4a. EoE: eosinophilic esophagitis.
1) Establishing causal hierarchies among cell types and cytokines.
Human EoE biopsies reveal simultaneous activation of eosinophils, mast cells, Th2 lymphocytes, basophils, ILC2s, epithelial cells, fibroblasts and macrophages[26]. Yet, the sequential relationship between these cells and the complexity of their interactions cannot be addressed. Furthermore, identification of which cells and/or molecules contribute to disease initiation and perpetuation is limited. Conditional knockout and transgenic mouse models enable specific ablation of distinct cell populations or molecular pathways in a tissue-specific and temporally controlled manner. This methodology can provide direct causal evidence. For example, conditional deletion of Il13ra1 from esophageal epithelial cells demonstrated that the type 2 IL-4 receptor on the epithelium is critically involved in EoE pathogenesis[28]. Similarly, diphtheria toxin receptor (DTR)-mediated depletion of basophils demonstrated their Involvement in disease initiation[29].
2) Modeling disease initiation and the atopic march.
EoE frequently presents in the context of atopic comorbidities, including asthma, atopic dermatitis and food allergy[4,30]. This suggests that sensitization through the skin or mucosal barriers precedes and primes for esophageal disease. The mechanisms by which skin sensitization to food antigens leads to esophageal eosinophilia cannot be studied prospectively in humans. Skin sensitization models have been particularly informative for defining the mechanisms that link barrier disruption, allergic sensitization and subsequent esophageal inflammation. Such models demonstrate that skin sensitization is sufficient to drive esophageal eosinophilia, and that the downstream signaling pathways involved depend on the nature and route of sensitization. For example, skin sensitization of filaggrin-null mice with ovalbumin (OVA) demonstrated that a genetically impaired skin barrier is sufficient to drive EoE in an IL-33-ST2-basophil-dependent manner[31].
3) Dissecting the mechanisms of fibrosis.
Subepithelial fibrosis is a major clinical long-term complication of EoE. It drives esophageal stricture and food impaction, is largely irreversible, and is found in many EoE patients[32,33]. Defining the cellular mediators of fibrosis requires longitudinal models of sufficient duration to generate established fibrotic disease. Chronic allergen challenge models and specific transgenic mouse lines have enabled the identification of amphiregulin-producing Th2 cells[34], colony-stimulating factor 1 (CSF1)-dependent macrophages[35], and provisional basal lamina membrane signatures that precede irreversible structural remodeling. These findings have direct implications for early intervention.
4) Validating therapeutic targets before clinical trials.
Preclinical validation in animal models is often important to establish proof-of-concept efficacy, dose-response relationships and target specific activity. For instance, the identification of IL-13 as a potent driver of EoE pathology in humans and mice and its preclinical targeting provided rationale for neutralization of the IL-4/IL-13 receptor pathway[36-39]. Furthermore, studies using Il13ra1-/- mice provide mechanistic explanation for the efficacy of dupilumab in EoE. Finally, animal models continue to provide the first line of evidence for emerging targets, including anti-TSLP and anti-IL-33/ST2 strategies[40-42].
3. Animal Models of EoE
To date, experimental EoE animal models are exclusively murine and can be broadly classified into five categories based on their induction strategy: (a) models relying on indirect cytokine administration, in which a cytokine is delivered systemically or expressed distally and accumulates in the esophagus; (b) models based on direct, tissue-specific cytokine overexpression in the esophageal epithelium; (c) antigen- and hapten-driven sensitization models, which recapitulate the allergen-driven etiology of the human disease; (d) hybrid models that combine transgenic cytokine overexpression with hapten challenge; (e) spontaneous genetic loss-of-function models; and f) environmental exposure models, which model non-allergen-based triggers of esophageal disease (Figure 2, Table 1). While no animal model can fully mimic human disease, each experimental category/model has distinct characteristics enabling it to address different gaps of knowledge. Furthermore, findings from one model category do not always replicate in others. Such discrepancies between models are important to discuss since they are likely to be informative especially in terms of translation to human disease.
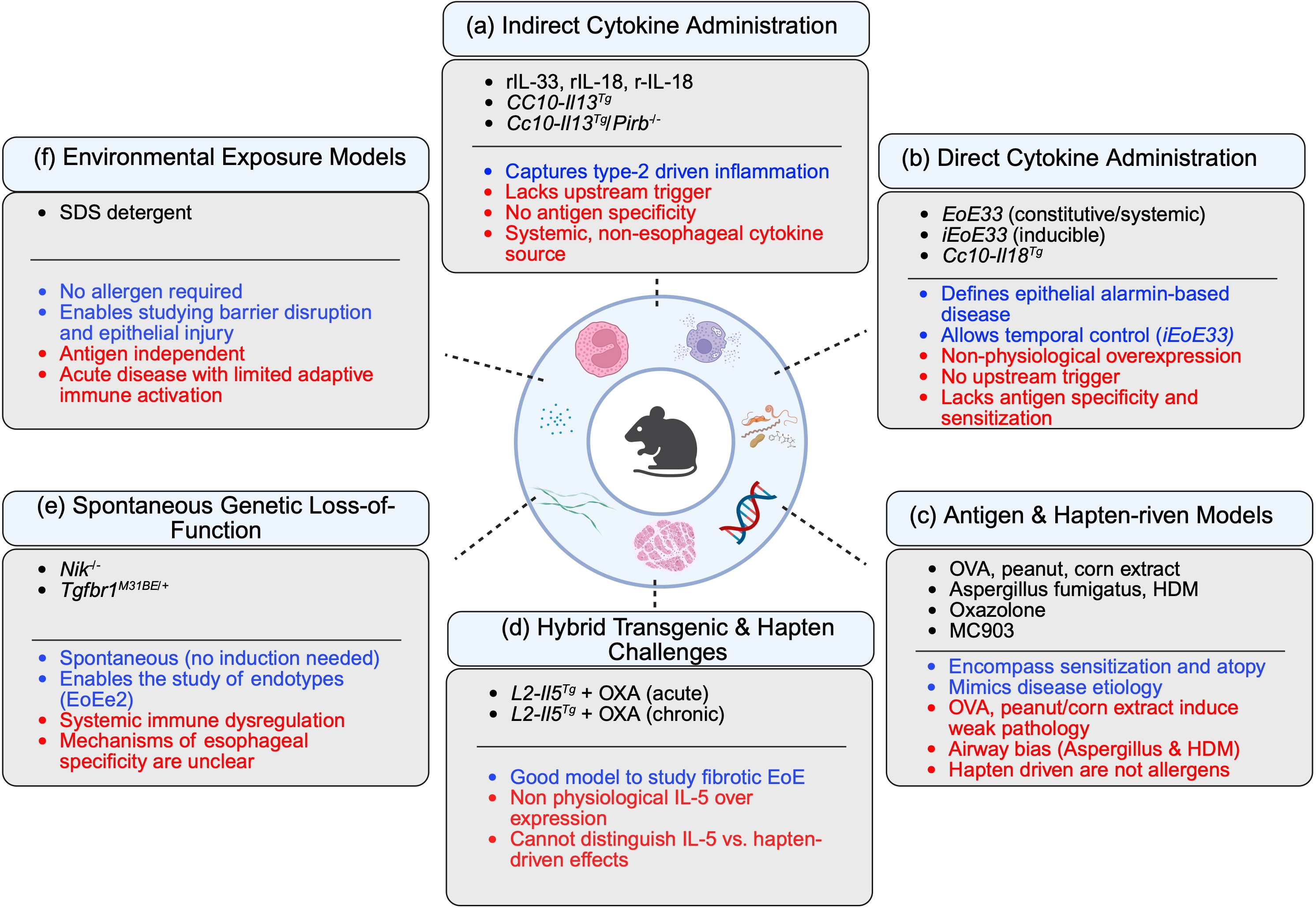
Figure 2. Experimental models of eosinophilic esophagitis categorized by induction strategy. Schematic overview of the major classes of experimental EoE models organized according to their primary mode of induction: (a) Indirect cytokine administration; (b) Direct esophageal cytokine overexpression; (c) Antigen- and hapten-driven sensitization models; (d) Hybrid transgenic–hapten models; (e) Spontaneous genetic loss-of-function models; (f) Environmental exposure models. Each category is annotated with representative systems and their principal strengths (blue text) and limitations (red text). Indirect cytokine models capture downstream type 2 effector pathways but lack upstream triggers and antigen specificity. Direct epithelial cytokine overexpression models define the role of epithelial-derived alarmins with temporal control but bypass physiological sensitization. Antigen- and hapten-driven models recapitulate allergen sensitization and atopy but vary in disease severity and may exhibit airway bias. Hybrid models enable the study of chronic and fibrotic disease but rely on non-physiological cytokine expression. Spontaneous genetic models provide insight into disease endotypes and genetic susceptibility but involve systemic immune dysregulation. Environmental exposure models isolate barrier disruption as a primary trigger independent of allergen sensitization. Collectively, these models address complementary aspects of EoE pathogenesis. Created in BioRender. Dsilva, A. (2026) https://app.biorender.com/illustrations/69bcfbd4b4fe7f6f47ada8d1?slideId=aa639fc0-b385-433a-bdf2-9cba96020c77. EoE: eosinophilic esophagitis; SDS: sodium dodecyl sulfate; OXA: oxazolone; OVA: ovalbumin; HDM: house dust mite.
Table 1. Comprehensive overview of eosinophilic esophagitis animal models.
| Model Name | Induction Method | Trigger | Duration | Key Disease Features | Key Reference(s) |
| A. Antigen- and Hapten-Driven Sensitization Models | |||||
| OVA i.p. + intranasal | i.p. OVA/alum sensitization; intranasal OVA challenge x3-5 | OVA | 4-6 weeks | LP & IE eosinophilia; BCH; Th2 cytokine upregulation; eotaxin-1 dependent; IL-5 required | [49,54] |
| Epicutaneous OVA/filaggrin-null ft/ft | Topical OVA on intact or tape-stripped skin; intranasal OVA challenge | OVA + disrupted FLG skin barrier | 4-6 weeks | LP & IE eosinophilia; IL-33–ST2–basophil axis; skin-to-esophagus atopic march link | [31] |
| MC903/OVA skin + intraesophageal OVA | MC903/OVA topical ear sensitization; intragastric OVA challenge | OVA + MC903 (calcipotriol) | 5-6 weeks | IE eosinophilia; microabscesses; BCH; LP remodeling; TSLP-basophil axis; exacerbated by epithelial IKKβ loss | [29,62] |
| Aspergillus fumigatus intranasal | i.p. sensitization; intranasal A. fumigatus challenge x3/week for 3-4 weeks | Aspergillus fumigatus extract | 4-6 weeks | IE & LP eosinophilia; Th2 inflammation; TSLP-basophil axis identified; TRAIL/MID1 pathway | [29,54] |
| Chronic HDM intranasal | Repeated intranasal HDM x3/week for 7-10 weeks after s.c. sensitization | HDM | 8-12 weeks | IE & LP eosinophilia; subepithelial fibrosis; Th2 cell infiltration; amphiregulin-Th2 axis | [34,59] |
| OXA ear/intraesophageal | Topical OXA on ear x5 (sensitization); intraesophageal OXA gavage x5 (challenge) | OXA | 35 days | IE & LP eosinophilia; BCH; LP thickening; angiogenesis; mast cell signature; TSLP dominant; IL-33 dispensable for remodeling; epithelial IL-13Rα as primary transducer | [28,40,60] |
| B. Indirect Cytokine Administration Models | |||||
| Exogenous rIL-33 (i.p./s.c.) | Daily i.p. or s.c. recombinant IL-33 for 7 days | IL-33 | 7 days | Transmural eosinophilia; BCH; ILC2/M2 macrophage activation; Th2 cytokines; IL-13 dependent; accessible model | [43] |
| Exogenous rIL-18 (intranasal) | Intranasal recombinant IL-18 in a time- and dose-dependent manner | IL-18 | 1-2 weeks | Esophageal eosinophilia; mast cell infiltration; IL-5- and iNKT-dependent axis; dose- and time-dependent | [48] |
| CC10-Il13Tg | Lung-specific IL-13 overexpression (CC10 promoter); constitutive/DOX-inducible | IL-13 | Constitutive or 2-8 weeks DOX | IE & LP eosinophilia; epithelial hyperplasia; angiogenesis; collagen deposition; eosinophil-independent remodeling; overlaps human EoE transcriptome; VDR/STAT6 interactome | [44,46] |
| CC10-Il13Tg/Pirb-/- | CC10-Il13Tg crossed to PIR-B knockout | IL-13 | Constitutive | Exaggerated IE eosinophilia; fibrosis; angiogenesis; pro-remodeling eosinophil transcriptome with EMT/pro-fibrotic genes | [45] |
| rtTA-Cc10-IL18 (inducible lung IL-18 Tg) | DOX-inducible IL-18 overexpression in airway epithelium (CC10 promoter) | IL-18 | 2-3 weeks DOX | Transmural eosinophilic and mast cell infiltration; esophageal inflammation; increased LP thickness; establishes sustained IL-18 sufficient to drive disease | [48] |
| C. Direct Cytokine Overexpression Models | |||||
| EoE33 | Esophageal epithelium-specific constitutive IL-33 overexpression (ED-L2 promoter) | IL-33 | Constitutive | IE & LP eosinophilia; CD4+ infiltration; BCH; DIS; epithelial thickening; IL-13-dependent; steroid responsive | [41] |
| iEoE33 | Tet-on: doxycycline-inducible esophageal IL-33 (iSophagus × TRE33 cross) | IL-33 | 7-14 days DOX chow | IE & LP eosinophilia; BCH; DIS; weight loss; esophageal thickening; onset day 7; steroid responsive; IL-13-dependent | [42] |
| TslpTg (esophageal - unpublished) | Esophageal-specific TSLP overexpression (proposed) | TSLP | Not reported | Not yet characterized; would test whether esophageal TSLP alone drives structural EoE; rationale supported by dominant TSLP role in OXA model | Not published |
| D. Hybrid Models: Transgenic Cytokine + Hapten Challenge | |||||
| L2-IL5OXA (acute) | Esophageal IL-5 overexpression (ED-L2 promoter) + OXA epicutaneous/intraesophageal challenge | IL-5 + OXA | 6-8 weeks | Pan-esophageal eosinophilia; microabscesses; BCH; elevated IL-13/eotaxin-1; corticosteroid responsive; dose-dependent | [63] |
| L2-IL5OXA (chronic) | Extended L2-IL5OXA protocol with prolonged/repeated OXA challenges | IL-5 + chronic OXA | 16+ weeks | Full fibrotic EoE; CSF1-macrophage axis orchestrates fibrosis; KRT16/TNC biomarkers; ECM remodeling; HIF-1α/CD73/ADORA2B barrier axis; pharmacological ADORA2B restoration | [35,64] |
| E. Spontaneous Genetic Loss-of-Function Models | |||||
| Nik-/- | Global knockout of NIK (MAP3K14), upstream activator of non-canonical NF-κB signaling | Nik deficiency (spontaneous) | EoE by ~12 weeks | Spontaneous esophageal eosinophilia; eosinophil degranulation; mucosal thickening; BCH; fibrosis; elevated TSLP; T cell-dependent; mimics human EoE gene signature including EoE-associated cytokines | [67] |
| Tgfbr1M318R/+ variant knock-in | Heterozygous knock-in of TGFβR1 loss-of-function missense variant M318R into endogenous Tgfbr1 locus | TGFβR1 hypomorphic variant (spontaneous) | EoE by 6 months | Spontaneous EoE; esophageal dilation; food impaction; eosinophilic infiltration; hyperproliferative de-differentiated epithelium; elevated TSLP/TNFAIP6; transcriptome matches EoEe2 endotype; lymphocyte-independent; epithelial-intrinsic | [68] |
| F. Environmental Exposure and Barrier Disruption Models | |||||
| SDS detergent drinking water | 0.5% SDS ad libitum in drinking water for 14 days; no allergen sensitization required | SDS | 14 days | IE eosinophilia; eotaxin-3 upregulation; barrier dysfunction (↓ DSG1/FLG; ↑ permeability); inflammatory transcriptome; no allergen required; models environmental epithelial disruption | [70] |
Models grouped by induction strategy. BCH: basal cell hyperplasia; DIS: dilated intercellular spaces; DOX: doxycycline; IE: intraepithelial; LP: lamina propria; OVA: ovalbumin; OXA: oxazolone; Tg: transgenic; VD: vitamin D; HDM: house dust mite; SDS: sodium dodecyl sulfate; TSLP: thymic stromal lymphopoietin; EoE: eosinophilic esophagitis; ECM: extracellular matrix; NIK: NFκB-inducing kinase; ↓: decreased; ↑: increased.
3.1 Indirect cytokine administration models
These models probe the sufficiency of individual type 2 cytokines to drive esophageal pathology by delivering them systemically or expressing them at distal sites. As such, they are particularly useful for defining downstream effector pathways, although they bypass upstream events such as epithelial barrier dysfunction and allergen sensitization.
3.1.1 Exogenous recombinant IL-33 (rIL-33) administration
Short-term administration of rIL-33 to naïve mice induces transmural esophageal eosinophilia, epithelial hyperproliferation, ILC2 activation, Th2 cytokine production, and an M2 macrophage signature[43]. The pathological response is IL-13-dependent, as it is abolished in Il13-/- mice. This model has been important in delineating the IL-33-IL-13-eosinophil axis. However, its acute nature and systemic cytokine delivery raise questions regarding the relative contribution of local versus systemic immune activation, and it produces only modest epithelial pathology without clear barrier disruption.
3.1.2 The rtTA-CC10-Il13 transgenic model
In rtTA-CC10-Il13 transgenic mice, lung-derived IL-13 reaches the esophagus via airway drainage and induces marked remodeling, including epithelial hyperplasia, angiogenesis, and fibrosis, with strong overlap to the human EoE transcriptome[44]. Notably, remodeling occurs independently of eosinophils, as demonstrated by crossing rtTA-CC10-Il13 transgenic mice with ΔdblGATA mice. These data indicate direct effects of IL-13 on structural cells in the tissue. This model also identified paired immunoglobulin-like receptor B (PIR-B) as an eosinophil-intrinsic checkpoint. Deletion of PIR-B exacerbates fibrosis and angiogenesis and promotes pro-fibrotic transcriptional programs[45]. In addition, vitamin D signaling was shown to modulate IL-13-driven pathology, with supplementation reversing a large fraction of transcriptional and epigenetic changes in a VDR/STAT6-dependent manner and restoring barrier integrity[46]. While highly informative for IL-13 effector biology, this model bypasses upstream epithelial and immune activation events due to its pulmonary origin of cytokine expression.
3.1.3 Exogenous recombinant IL-18 administration and rtTA-CC10-Il18 transgenic model
Elevated IL-18 and IL-18Rα expression in EoE patients prompted the development of complementary acute and inducible models[47]. Recombinant IL-18 administration induces esophageal eosinophilia and mast cell infiltration in an IL-5- and invariant natural killer T (iNKT) cell-dependent manner[48]. Sustained expression in rtTA-Cc10-Il18 mice recapitulates these features and additionally drives tissue inflammation and lamina propria expansion. Compared to IL-13 and IL-33 systems, epithelial remodeling is less extensively characterized, but these models establish IL-18 as sufficient to induce mixed eosinophilic–mast cell inflammation[48]. Similar to the CC10-Il13Tg mice, this model is most informative for answering questions about IL-18 effector functions in EoE.
3.2 Direct cytokine overexpression models
This category includes models in which a cytokine is overexpressed directly and specifically in the esophageal epithelium, providing a more physiologically relevant source of cytokine that mirrors the epithelial production documented in human EoE.
3.2.1 The EoE33 constitutive IL-33 transgenic model
EoE33 mice constitutively express active IL-33 in the esophageal epithelium, resulting in early-onset, ST2-dependent pathology characterized by eosinophilia, CD4+ T cell and mast cell infiltration, basal cell hyperplasia, and dilated intercellular spaces. Esophageal dysmotility is also evident. Disease is strictly IL-13-dependent in vivo, and organoid studies further support an IL-33-IL-13 epithelial activation axis. Responsiveness to corticosteroids underscores the translational relevance of this model[41].
3.2.2 The iEoE33 inducible IL-33 transgenic model
The iEoE33 system enables temporal control of epithelial IL-33 expression, inducing EoE-like pathology within 7 days, including eosinophilia, epithelial remodeling and tissue thickening[42]. As in the constitutive model, disease is IL-13-dependent and steroid-responsive. The principal advantage of this system is its ability to interrogate defined disease stages and therapeutic timing. Together, the EoE33 models provide complementary platforms for studying epithelial-derived IL-33, although they do not address disease initiation mechanisms.
3.2.3 TSLP transgenic models
While TSLP is strongly implicated in human EoE and across experimental systems, esophageal-specific overexpression models have not yet been reported. Given the availability of anti-TSLP therapeutics in clinical development, generation of such models represents an important unmet need with clear translational relevance.
Collectively, these cytokine-driven models adhere to human EoE by recapitulating IL-13 response and type-2-driven biology, which are central components of human disease progression (Table 2).
Table 2. Disease feature matrix: Recapitulation of key EoE histopathological and functional parameters across animal models.
| Model | Eosinophils | Fibrosis | BCH | Epi Thickening | Multi-cell Inflammation | Barrier Dysfunction | Angiogenesis | Neuronal Remodeling | Notes/Caveats |
| A. Antigen- and Hapten-Driven Sensitization Models | |||||||||
| OVA i.p. + intranasal | ± | - | ± | ± | ± | ± | - | ND | Limited tissue remodeling |
| Epicutaneous OVA/ft/ft filaggrin-null | ++ | - | ++ | ± | + | ± | - | ND | IL-33-T2-basophil axis; skin barrier genetics |
| MC903/OVA skin + intraesophageal OVA | + | - | ± | ± | + | ± | ± | ND | IkkbΔEEC greatly exacerbates all features; TSLP-basophil axis |
| Aspergillus fumigatus intranasal | + | - | + | ± | + | ± | ± | ND | TSLP-basophil axis; TRAIL/MID1; limited fibrosis |
| Chronic HDM intranasal | + | ++ | + | + | + | ± | + | ND | Fibrosis established; amphiregulin-Th2 axis; human validation |
| OXA ear/intraesophageal | +++ | +++ | +++ | +++ | +++ | ++ | ++ | ND | TSLP dominant; IL-33 dispensable for remodeling; epithelial IL-13Rα1 transducer |
| B. Indirect Cytokine Administration Models | |||||||||
| Exogenous rIL-33 (i.p./s.c.) | + | - | + | ± | + | ± | - | ND | Short duration; ILC2/M2 activation; IL-13-dependent; accessible model |
| Exogenous rIL-18 (intranasal) | + | - | ± | ± | ± | - | - | ND | Dose/time-dependent; IL-5- and iNKT-dependent; mast cell infiltration; limited structural characterization |
| Exogenous rIL-13 (intranasal/intratracheal) | ± | - | ± | - | ± | ± | - | ND | Minimal eosinophilia; BCH; STAT6/IL-5/eotaxin-1 axis |
| CC10-I13Tg | ++ | ++ | ++ | ++ | ++ | + | + | ND | Eosinophil-independent remodeling; overlaps human EoE transcriptome |
| CC10-Il13Tg/Pirb-/- | +++ | +++ | +++ | +++ | +++ | ND | +++ | ND | Exaggerated vs. CC10-IL13 Tg alone; PIR-B as eosinophil checkpoint |
| rtTA-Cc10-IL18 (inducible lung IL-18 Tg) | + | + | ± | ± | + | + | ND | ND | |
| C. Direct Cytokine Overexpression Models | |||||||||
| EoE33 (constitutive esoph. IL-33 Tg) | +++ | +++ | +++ | +++ | +++ | +++ | ND | ND | IL-13-dependent; DIS present; steroid responsive; esophageal contractility impaired |
| iEoE33 (inducible esoph. IL-33 Tg) | + | +++ | +++ | +++ | +++ | +++ | ND | ND | Temporally controllable; onset day 7; steroid responsive; IL-13-dependent |
| D. Hybrid Models: Transgenic Cytokine + Hapten Challenge | |||||||||
| L2-IL5OXA (acute, ≤ 8 weeks) | ++++ | ++ | +++ | +++ | + | ++ | ND | ND | Microabscesses; dose-dependent; corticosteroid responsive |
| L2-IL5OXA (chronic fibrotic, ≥ 16 weeks) | ++++ | ++++ | ++++ | ++++ | + | ++ | ND | ND | Full fibrotic EoE; CSF1-macrophage axis; KRT16/TNC biomarkers validated in human EoE |
| E. Spontaneous Genetic Loss-of-Function Models | |||||||||
| Nik-/- (NIK knockout) | ++ | + | + | + | + | ND | ND | ND | Spontaneous; esophageal-localized; T cell-dependent; elevated TSLP; NIK paradox in human EoE biopsies |
| Tgfbr1M318R/+ variant knock-in | ++ | + | + | + | + | ND | ND | ND | Spontaneous; lymphocyte-independent; epithelial-intrinsic; EoEe2 endotype transcriptome; food impaction |
| F. Environmental Exposure and Barrier Disruption Models | |||||||||
| SDS detergent drinking water | + | - | - | - | + | + | ND | ND | No allergen needed; models environmental barrier disruption; eotaxinupregulation; BCH absent |
+: present/demonstrated; -: absent; ±: partially present or variable across studies/strains; ND: not determined or not reported; Sub-epi: subepithelial; LP: lamina propria; IE: intraepithelial; BCH: basal cell hyperplasia; Multi-cell Infl.: multi-cellular inflammation (mast cells, Th2, ILC2, macrophages, basophils); Barrier Dysf.: epithelial barrier dysfunction; Epi: epithelial; EoE: eosinophilic esophagitis; OVA: ovalbumin; TSLP: thymic stromal lymphopoietin; HDM: house dust mite; DIS: dilated intercellular spaces; NIK: NFκB-inducing kinase.
3.3 Antigen- and hapten-driven sensitization models
This category encompasses all models in which experimental EoE is induced by sensitization to an exogenous antigen or hapten, followed by esophageal challenge. These models recapitulate, to some extent the allergen-driven etiology of human disease and are suited for studying immune sensitization pathways, the atopic march and the contributions of specific immune cell populations to disease initiation and propagation.
3.3.1 Allergen driven models involving epicutaneous and intraperitoneal sensitization
Epicutaneous sensitization of BALB/c mice with OVA (or Aspergillus fumigatus) followed by subsequent intranasal antigen challenge resulted in the development of esophageal eosinophilia in association with skin eosinophilia, accelerated eosinophilopoiesis and elevated antigen-specific IgG1/IgE. Using this model with gene-targeted mice, IL-5 was shown to be required for esophageal eosinophilia, while IL-4, IL-13 and STAT6 contributed to a lesser extent[49]. Allergen driven OVA-induced models have been studied also for therapeutic testing. For instance, anti-Siglec-F administration (the mouse analog of Siglec-8) reduced esophageal eosinophilia, angiogenesis, basal cell hyperplasia (BCH) and fibronectin deposition but did not influence tissue fibrosis[50]. Pharmacologic inhibition of acid mammalian chitinase with allosamidin inhibited OVA-induced esophageal eosinophilia and OVA-induced esophageal remodeling (e.g., fibrosis, epithelial basal zone hyperplasia, extracellular matrix deposition of fibronectin). Inhibition of eosinophilic inflammation in this study was associated with reduced eotaxin-1 expression in the esophagus[51]. Intraperitoneal sensitization with peanut extract and subsequent intranasal and intragastric challenge has also been also shown to induce experimental EoE[52]. Using this model, stem cell factor (SCF) was targeted using anti-SCF antibodies. Neutralization of SCF resulted in decreased accumulation of mast cells and eosinophils that correlated with decreased expression of IL-5 and IL-13 in restimulated mediastinal lymph node cells[52]. Furthermore, peanut allergen-induced EoE was shown to be dependent on eotaxin and iNKT cells, as CD1d and eotaxin-1/2 gene-deficient mice were protected from disease induction[53].
The role of defects in skin barrier and its relations with EoE was explored using filaggrin-null flaky tail (ft/ft) mice[31]. Topical application of OVA to intact, unstripped ft/ft skin was sufficient to induce experimental EoE without mechanical barrier disruption. This effect was abolished in ft/ft×Il1rl1-/- (St2-/-) mice and upon selective depletion of basophils using Mcpt8DTR mice. Bone marrow reconstitution experiments demonstrated that ST2 expression on basophils was required for their accumulation in the esophagus and for induction of experimental EoE. IL1RL1/ST2 mRNA was elevated in esophageal biopsies from pediatric EoE patients, supporting the translational relevance of these findings. These data collectively established that a genetically impaired skin barrier is sufficient to prime for EoE through the IL-33-ST2-basophil axis.
While these data provided important mechanistic insights, they have several limitations. First, the extent of esophageal pathology appears to vary. Often, these models do not develop substantial EoE-like histopathology except for the influx of eosinophils. Furthermore, some of these rely on intranasal delivery of OVA and not direct intragastric/intraesophageal OVA administration.
3.3.2 Aspergillus fumigatus and HDM-dependent models
Intranasal delivery of Aspergillus fumigatus was shown to induce EoE-like disease[54]. Subsequent studies using this model have shown a role for mast cells in EoE pathogenesis[55]. Mechanistically, TNF-related apoptosis-inducing ligand and E3 ubiquitin-ligase midline-1 were shown to mediate esophageal eosinophil and mast cell levels, and their silencing protected against esophageal circumference enlargement[56]. In contrast, Sharpin mice, lacking a key component of the linear ubiquitin chain assembly complex, regulating inflammation and cell death, displayed increased eosinophilic inflammation and epithelial thickening[57]. This model has also been used to evaluate probiotic interventions[58], demonstrating that therapeutic administration of Lactococcus lactis NCC 2287 reduced esophageal and bronchoalveolar eosinophilia in a strain-specific and timing-dependent manner.
Repeated intranasal HDM administration over 8 to 12 weeks was reported to generate acute eosinophilic inflammation and progressive subepithelial fibrosis[34]. Studies using this model demonstrated that amphiregulin-producing, ST2+ Th2 cells preferentially accumulate in the fibrotic zone of the inflamed esophagus. T cell-specific deletion of amphiregulin in Cd4Cre/Aregfl/fl mice ameliorated fibrosis without substantially reducing eosinophilia. Single cell RNA-sequencing and immunohistochemistry of human EoE biopsies confirmed that amphiregulin-producing CD4+ T cells are more abundant in EoE than in controls and that their numbers correlate with the degree of subepithelial fibrosis, identifying the amphiregulin-EGFR axis as a profibrotic pathway driven by a specific Th2 cell subset.
A complementary study examining esophageal ILC2s demonstrated that KLRG1+ ILC2s reside in the healthy mouse esophagus and that they expand in EoE. ILC2, but not eosinophils, were shown to mediate the epithelial defects in this model. ILC2s produced amphiregulin in the esophagus, and exogenous amphiregulin induced epithelial defects via phosphorylation of EGFR. Therapeutic targeting of amphiregulin and EGFR attenuated the development of EoE highlighting the therapeutic potential of targeting the amphiregulin-EGFR axis in EoE[59]. Together, these two studies suggest that amphiregulin produced by both Th2 cells and ILC2s is a shared driver of epithelial remodeling in EoE, representing a convergence point between adaptive and innate type 2 immunity.
3.3.3 The OXA model
The OXA model of EoE employs topical OXA applied to the ear for skin sensitization, followed by repeated intraesophageal OXA challenges over a 35-day protocol[60]. This model generates intraepithelial and lamina propria eosinophilia, BCH, lamina propria thickening, angiogenesis, and a prominent mast cell gene signature[60]. Since this model does not require transgenic animals for its induction, it is broadly accessible for interrogating the cellular and molecular mechanisms of EoE. Studies in this model using Crlf2-/- mice demonstrated that TSLP deficiency confers near-complete protection from all structural EoE features, including eosinophilia, BCH, lamina propria thickening and vascularization[40]. TSLP neutralization by antibody treatment similarly reduced basal cell proliferation and vascularization. In contrast, Il33-/- mice displayed reduced eosinophilia but no significant protection from tissue remodeling. Thus, in this model, TSLP and IL-33 have non-redundant and distinct roles in EoE, where TSLP is the dominant alarmin for structural remodeling, while IL-33 amplifies, to some extent, eosinophilic inflammation. Importantly, studies using anti-IL-5 antibodies have shown that depletion of eosinophils after skin sensitization with OXA had no preclinical benefit. RNA sequencing in Crlf2-/- mice revealed that TSLP regulates key EoE-associated transcripts including eotaxins, Klk5, Flg, and Il1 signaling genes, positioning TSLP as a broad regulator of both epithelial differentiation and immune activation[40]. The OXA model has also been used to define the cell-intrinsic function of the epithelial type 2 IL-4 receptor. Global deletion of Il13ra1 rendered OXA-challenged mice completely protected from EoE. Furthermore, mice harboring a conditional deletion of Il13ra1 in their esophageal epithelial cells (K14Cre-Il13rafl/fl), OXA-induced EoE was markedly attenuated, with reduced eosinophilia and BCH. These findings established the esophageal epithelial cell as the primary cellular source responding to IL-13 in EoE. They further provided cellular-level mechanistic support for the therapeutic efficacy of dupilumab and cendakimab[28,39]. Importantly, the transcriptional and structural features observed in this model display overlap with human EoE, supporting its relevance to both EoE1 and EoE2 endotypes (Table 2).
3.3.4 The vitamin D analogue/MC903 model
Topical application of the vitamin D analogue, calcipotriol (MC903) has been described to elicit atopic dermatitis-like inflammation. When combined with OVA sensitization and intragastric challenge, this MC903/OVA model can induce EoE-like disease. Initial studies using this model demonstrated that it develops independently of IgE but is dependent on TSLP and basophils. Using DTR-transgenic Mcpt8DTR mice, selective basophil depletion abolished esophageal eosinophilia. TSLP-primed (but not naïve) basophils could transfer experimental EoE, establishing the TSLP-basophil axis as a key innate-adaptive interface in EoE. This model also demonstrated that therapeutic neutralization of TSLP or basophil depletion can ameliorate established EoE-like disease[29]. Earlier work from the same group showed that TSLP promotes IL-3-independent basophil hematopoiesis and elicits a functionally distinct basophil population that drives Th2 inflammation, providing the cellular basis for the TSLP-basophil EoE axis[61]. The model was recently used to dissect the contribution of the IKKβ-NFκB signaling pathway in EoE. EoE was induced in mice harboring a conditional deletion of IKKβ in their esophageal epithelial cells (using K14Cre-Ikkbfl/fl mice). Mice lacking epithelial IKKβ displayed exacerbated EoE, with more severe eosinophilia, microabscesses, BCH, and lamina propria remodeling compared to wild-type controls. Single cell RNA sequencing revealed disruption of multiple epithelial cell states, including differentiated suprabasal populations and barrier programs, as well as augmented type 2 responses and peptidase activity. These findings established that IKKβ/NFκB signaling in the esophageal epithelium is paradoxically protective, and that its loss exacerbates disease[62]. These models capture aspects of allergen driven disease initiation observed in human EoE and in some cases show overlap with defined human EoE endotypes (Table 2).
3.5 Hybrid models: Transgenic cytokine combined with hapten challenge
3.5.1 The L2-IL5OXA model
The L2-IL5 transgenic mouse overexpresses IL-5 under the Epstein-Barr virus ED-L2 squamous epithelial promoter, producing IL-5 constitutively in the esophagus. L2-IL5 mice do not develop spontaneous esophageal eosinophilia at baseline, but upon OXA challenge, they develop dose-dependent pan-esophageal eosinophilia, including microabscess formation and degranulation, BCH, and upregulation of IL-13 and eotaxin-1. The model is sensitive to corticosteroid treatment[63]. The L2-IL5OXA model is therefore a hybrid: transgenic IL-5 creates a permissive eosinophil-rich environment, while hapten-driven local inflammation provides the esophageal recruiting signal.
The chronic variant of the L2-IL5OXA model, generated by extended or repeated OXA challenges, develops subepithelial fibrosis and was proposed to serve as an experimental system to study fibrotic EoE[35]. Unbiased ECM-focused proteomic analysis of the chronic L2-IL5 OXA-challenged esophagus identified a provisional basal lamina membrane signature and a wound-healing/hemostasis gene set that preceded fibrosis. Macrophage accumulation was shown to precede fibrotic remodeling, and CSF1R-dependent macrophages were identified as critical orchestrators of this process[35]. Recently, the L2-IL5OXA model was used to study the HIF-1α/CD73-adenosine axis. In vitro studies demonstrated that defective HIF-1α expression in EoE epithelium led to impaired CD73-mediated adenosine production and downstream ADORA2B signaling, resulting in impaired epithelial wound healing and barrier dysfunction. In vivo, treatment of mice with the ADORA2B agonist (BAY606583) decreased the inflammatory infiltrate in both the epithelium and lamina propria, alongside improved histological architecture and organization of the epithelial layer[64].
While the L2-IL5-OXA model induces substantial disease and is likely relevant to model fibrostenotic human EoE (Table 2), it is unclear whether the fibrosis in this model is driven by the high levels of eosinophilia sustained by transgenic IL-5, by oxazolone-specific hapten responses, or by both. This is important since clinical studies have shown that depletion of eosinophils was not beneficial for EoE patients[65].
3.6 Spontaneous ghenetic loss-of-function models
Two additional genetic models are notable because they generate EoE-like disease spontaneously or near-spontaneously, without antigen sensitization or cytokine delivery. Both arise from genetic dysregulation of pathways that normally suppress esophageal allergic inflammation, and both have human disease correlates, making them particularly relevant for studying EoE endotypes.
3.6.1 The Nik-/- model
NFκB-inducing kinase (NIK; MAP3K14) is an essential upstream activator of the non-canonical NFκB signaling pathway. Nik-/- mice were previously reported to develop hypereosinophilic syndrome in peripheral blood in a T cell-dependent manner[66]. Nik-/- mice develop progressive inflammation that leads to loss of subcutaneous fat and severe dermatitis. Characterization of the gastrointestinal tract of these mice demonstrated that Nik-/- mice develop significant, localized EoE that closely mimics the human disease[67]. Histological features included severe eosinophil accumulation, degranulation, mucosal thickening, BCH, and fibrosis. Interestingly, this effect was localized to the esophagus since other segments of the gastrointestinal tract were unaffected. Esophageal tissue from Nik-/- mice displayed increased mRNA expression of type 2 and inflammatory cytokines (e.g., IL-4, IL-13, IFN-γ, TNF-α) as well as elevated TSLP protein expression. Analysis of human EoE biopsy datasets revealed a paradoxical finding where NIK mRNA was upregulated in EoE patients, yet NIK protein was destabilized by concurrent upregulation of NIK-associated negative regulators (i.e., cIAP1/2, CYLD, NLRP12, HSP1 and CHIP)[67]. These data suggest that post-translational suppression of NIK activity may be a feature of human EoE pathogenesis. A key advantage of this model is spontaneous disease development, which avoids the artificial induction procedures that characterize most EoE models. Its limitation is that NIK deficiency affects multiple tissues and immune compartments beyond the esophagus, and the mechanism by which NIK loss leads to esophageal-specific eosinophilia has not been fully resolved.
3.6.2 The Tgfbr1M318R variant model
Loss-of-function variants in TGFβ receptor genes are associated with increased prevalence of allergic disorders, including EoE. Multiple EoE-associated epithelial gene variants encode for proteins that are involved in TGFβ signaling. In attempt to study the role of TGFβ variants, the Tgfbr1M318R missense variant, which eliminates TGFβR1 kinase activity, was introduced into the endogenous Tgfbr1 locus[68]. Heterozygous Tgfbr1M318R/+ mice, spontaneously developed EoE-like disease by 6 months of age that clinically, immunologically, histologically and transcriptionally resembles human EoE. Clinical and histopathological features of EoE included dilation of the esophagus, food impaction, eosinophilic infiltration, hyperproliferative and de-differentiated epithelial cells, and overexpression of innate proinflammatory mediators including TSLP and TNFAIP6[68]. Transcriptome analysis of the esophagus suggested that this model resembles the EoEe2 endotype, which is characterized by an inflammatory and steroid-refractory phenotype[69]. Interestingly, Tgfbr1M318R/+-driven EoE was independent of T cells, and esophageal organoids derived from these mice displayed the EoEe2 gene signature and hyperproliferative basal cells. These data establish that TGFβR1-mediated signaling has an epithelial cell-intrinsic and lymphocyte-independent role in maintaining esophageal homeostasis. While this model is not triggered by allergen challenge and displays low involvement of adaptive immunity, it is one of the only models to date that is driven by a human disease-associated variant, making well-positioned for studying genetic predisposition and early disease mechanisms in distinct EoE patient subsets/endotypes.
The models discussed in this section display transcriptomic and phenotypic overlap with human EoE and provide key insights into disease endotypes driven by genetic susceptibility.
3.7 Environmental exposure and barrier disruption models
This category includes models that test the hypothesis that non-allergen environmental exposures can initiate or perpetuate esophageal inflammation.
3.7.1 The sodium dodecyl sulfate (SDS) detergent model
Exposure of esophageal epithelial cells to SDS reduces transepithelial electrical resistance, increases FITC-dextran permeability, and downregulates barrier genes including Dsg1 and Flg. In vivo, mice receiving 0.5% SDS in drinking water for 14 days developed esophageal eosinophilia and an inflammatory gene expression signature without any allergen sensitization[70]. This model provides proof-of-concept that barrier-disrupting environmental chemicals can initiate EoE-like esophageal inflammation, and positions environmental epitheliotoxic exposures as potential primary EoE triggers rather than modifiers of allergen-driven disease. Collectively, these models support emerging evidence that epithelial barrier disruption may act as an initiating factor in a subset of human EoE cases.
3.8 Comparative analysis of disease feature recapitulation
A central purpose of EoE animal models is to faithfully recapitulate the major histopathological and functional disease features observed in human biopsies (Figure 1). These encompass: (1) lamina propria eosinophilia; (2) intraepithelial eosinophilia; (3) subepithelial fibrosis; (4) basal cell hyperplasia; (5) epithelial thickening; (6) multi-cellular inflammation; (7) epithelial barrier dysfunction; (8) angiogenesis; (9) neuronal remodeling; and (10) epithelial transcriptomic remodeling. Systematic assessment of these features across models reveals that no single model fully recapitulates all parameters. The choice of model must therefore be matched to the scientific question. Table 2 summarizes the histopathological readouts established in each model, while Table 3 maps the relevant models to human transcriptomic overlap and EoE endotype relevance.
Table 3. Transcriptomic overlap and endotype mapping in mouse EoE models.
| Model | Human transcriptome overlap | EoE endotype relevance |
| OVA (i.p. + intranasal) | ND | ND |
| Epicutaneous OVA/filaggrin-null | ND | ND |
| MC903 + OVA | ND | EoE1 (TSLP-driven) |
| Aspergillus | ND | ND |
| Chronic HDM | ND | EoE2 (fibrotic) |
| OXA | Yes | EoE1/EoE2 |
| rIL-33 | ND | ND |
| rIL-18 | ND | ND |
| CC10-IL13Tg | Yes | EoE1 |
| CC10-IL13Tg/Prb-/- | ND | ND |
| rtTA-IL18 | ND | ND |
| EoE33 | ND | EoE1 |
| iEoE33 | ND | EoE1 |
| L2-IL5 + OXA (acute) | ND | ND |
| L2-IL5 + OXA (chronic) | Yes | EoE2 |
| NIK-/- | Yes | EoE1 |
| Tgfbr1M318R/+ | Yes | EoE2 |
| SDS model | ND | ND |
ND: not determined; EoE: eosinophilic esophagitis; OVA: ovalbumin; HDM: house dust mite; OXA: oxazolone; SDS: sodium dodecyl sulfate.
3.9 Mechanistic insights uniquely enabled by animal models
3.9.1 Eosinophil-independent remodeling
One of the most important findings to emerge from EoE animal models is that tissue remodeling, including epithelial hyperplasia, angiogenesis, and collagen deposition, can proceed in the complete absence of eosinophils. Studies using eosinophil lineage-deficient (dblGATA) mice crossed to the CC10-Il13Tg background demonstrated that IL-13-driven esophageal remodeling is preserved in eosinophil-deficient mice[44]. Similar results were obtained in the EoE33 mouse model and in the OXA model, which were IL-13-dependent and eosinophil-independent[40,41]. These data establish that IL-13 acts directly on esophageal epithelial and mesenchymal cells. They also provide a mechanistic framework for understanding why eosinophil depletion alone may be insufficient to prevent structural disease progression, while explaining why IL-4Rα blockade with dupilumab yields broader histological improvement[39,65,71,72].
3.9.2 IL-33 and TSLP activities
IL-33 drives EoE in multiple independent systems. Esophageal-specific overexpression of IL-33 (EoE33 and iEoE33) generates an impressive EoE pathology in an IL-13-dependent manner[41,42]. Exogenous rIL-33 generates transmural eosinophilia and mucosal hyperproliferation. Epicutaneous sensitization in filaggrin-null mice is abolished in Il1rl1-/- animals, placing the IL-33-ST2 axis as an important orchestrator of the skin-esophageal atopic march[31]. Yet, in the OXA-based model, Il33-/- mice displayed reduced eosinophilia but intact structural remodeling[40], demonstrating that IL-33 was dispensable for esophageal tissue remodeling in this context.
Several factors may account for this discrepancy. First, the source and context of IL-33 production differ between models. In EoE33, IL-33 is produced constitutively and at high levels by the esophageal epithelium itself. In the OXA model, IL-33 is produced as part of a broader hapten-driven inflammatory response and its contribution may be partially redundant with TSLP, which is simultaneously induced. Second, the genetic backgrounds and sensitization routes differ, altering the relative cytokine contributions. Third, and perhaps most importantly, this may reflect genuine heterogeneity in human EoE. A subset of patients may be primarily IL-33-driven, while others may be predominantly TSLP-driven. If this notion is true, then the different models may be assessing different patient endotypes. Prospective endotyping of EoE patients based on alarmin signatures, combined with matched animal model validation, will be required to resolve this question.
3.10 Limitations of EoE animal models
The current EoE animal models carry several limitations that must be acknowledged when interpreting preclinical data. The most fundamental is anatomical. The mouse esophagus is lined by a keratinized stratified squamous epithelium, while the human esophagus is non-keratinized[73]. The murine esophagus is also composed entirely of striated muscle, whereas the human esophagus transitions to smooth muscle in its distal third[74]. Furthermore, human esophageal epithelium features prominent papillae and 2-4 basal layers, whereas mice have a single basal proliferative layer, making human tissue more complex[75]. In fact, the adult esophagus turns over faster in mice (~3.5 days) compared to humans (~11 days) and while both share a stratified squamous structure (basal, suprabasal, superficial), human suprabasal layers express distinct keratins such as K4 and K13, while surface cells express involucrin and filaggrin[76]. These differences affect baseline epithelial transcriptomics, barrier properties and functional motor characteristics, and they require caution when extrapolating mechanistic findings to the human disease. Beyond anatomy, most existing mouse model develops EoE spontaneously and most systems require external intervention. This reflects the multifactorial and stochastic nature of human EoE, but it limits the ability to study primary disease prevention or the earliest sensitization events that precede clinically overt disease.
A further constraint is genetic homogeneity. Most EoE mouse studies are conducted in a single inbred strain, typically BALB/c or C57BL/6. Human EoE is genetically and clinically heterogeneous, encompassing fibrostenotic and inflammatory phenotypes, pediatric and adult presentations, and treatment-responsive and treatment-refractory disease[69]. This heterogeneity is almost certainly driven in part by genetic variation, yet the systematic use of gene-edited animals carrying human EoE risk variants such as those in CAPN14, CRLF2 or IL33 remains largely unexplored.
Endpoint alignment is also substantially different in humans and mice. The cardinal clinical symptoms of EoE, abdominal pain, dysphagia and feeding aversion, determine patient quality of life and serve as primary endpoints in clinical trials. These symptoms cannot be reliably assessed in rodents. Esophageal manometry, capsaicin sensitivity assays and operant conditioning-based feeding aversion paradigms have not been systematically validated in EoE mouse models, creating a significant gap between preclinical and clinical readouts. Duration of disease is also a recurring challenge. Most experimental EoE induction protocols require 3-8 weeks, whereas human EoE develops over years. Chronic fibrotic disease features require prolonged challenge protocols that are logistically demanding and resource-intensive, and only a minority of available models generate established subepithelial fibrosis.
Variability across EoE models extends to multiple pathways, including the involvement of cytokines (TSLP, IL-33, IL-18, IL-13, IL-5), basophil involvement and structural remodeling. For example, basophils are essential drivers of disease initiation in TSLP-dependent models such as MC903 and epicutaneous sensitization systems, whereas their contribution appears more limited or context-dependent in hapten-driven models such as OXA. Similarly, while TSLP is a dominant regulator of tissue remodeling in certain models, its role is less pronounced in others where alternative pathways may compensate. These discrepancies likely reflect differences in experimental design, including the route and nature of sensitization (e.g., epicutaneous vs. intragastric vs. intraesophageal), duration of exposure, and reliance on transgenic cytokine expression.
An additional limitation of current EoE models is the limited consideration of the microbiome. Emerging evidence suggests that alterations in the esophageal and gut microbiota are associated with EoE, including reduced microbial diversity and shifts in bacterial composition during active disease[77]. These changes may influence epithelial barrier function, local immune activation and antigen sensitization. Despite this, the role of the microbiome has not been systematically interrogated in most experimental models of EoE. Mice are typically housed under specific pathogen-free conditions with relatively uniform microbial communities, which do not reflect the complexity and variability of the human microbiome. In addition, commonly used inbred strains such as C57BL/6 and BALB/c differ in their baseline immune polarization and microbiota composition, which may influence susceptibility to type 2 inflammation and impact experimental outcomes. Incorporating microbiome-aware experimental designs, including gnotobiotic systems, cohousing strategies, or microbiota transfer approaches, may therefore provide important insights into how microbial and host factors interact to regulate EoE susceptibility and disease progression.
Finally, human EoE involves polyvalent sensitization to multiple food antigens, while most mouse models reduce this to a single antigen or hapten, likely underestimating the complexity of the human allergic response.
4. Future Directions
Recently, scRNAseq, spatial transcriptomics, and proteomics applied to biopsy material have revealed remarkable cellular heterogeneity within the esophagus in EoE[24-26]. Importantly, these approaches provide a high-resolution framework for interpreting findings from animal models, enabling direct comparison of cell-type-specific responses and spatial organization across species. Several directions will likely expand the translational resolution of EoE preclinical research in the coming years. Human esophageal organoids derived from esophageal progenitor cells and cultured at air-liquid interface are emerging as complementary tools to in vivo models. They enable study of human epithelial barrier dysfunction, differentiation and cytokine responsiveness under defined conditions, and their integration with in vivo mouse models will progressively narrow the translational gap between mouse and human esophageal biology. Three-dimensional esophageal-on-chip microfluidic systems that incorporate human esophageal epithelial cells, immune cells and fibroblasts in anatomically relevant spatial arrangements would offer a complementary drug screening platform.
At the genetic level, the systematic introduction of human EoE GWAS risk variants particularly in CAPN14, CRLF2 and IL33, into mouse backgrounds using CRISPR-Cas9 knock-in technology would enable direct in vivo testing of disease-associated alleles in a controlled genetic context. Such models would add insights that are beyond the current reliance on inbred wild-type strains. Validated behavioral and functional endpoints are equally needed, including neurological involvement and physiological measurements of pain[78]. Developing and validating candidate approaches, including esophageal manometry, capsaicin sensitivity assays, fluorescent in vivo barrier permeability measurements and high-resolution impedance studies, are in need. Further integration of these with standard histopathological and transcriptomic readouts, would substantially improve the alignment between preclinical models and clinical outcomes and/or characteristics. Finally, future models should incorporate multi-allergen sensitization protocols that combine responses towards several common food antigens. These would better reflect the clinical etiology of EoE. Together, these advances will position the next generation of EoE animal models as more faithful, more functional and more directly translatable than the currently available model systems.
5. Conclusions
Animal models are indispensable tools in EoE research, and their diverse nature has already generated valuable mechanistic insight regarding the cellular and molecular pathways that regulate the development of EoE. Such models also provide preclinical evidence-based approaches for approving and testing emerging therapeutics. The integration of next-generation tools, including humanized organoid systems, CRISPR-edited risk variant models and validated functional endpoints, will likely expand the translational resolution of EoE preclinical research in the coming years.
Authors contribution
Dsilva A: Visualization, writing-original draft, writing-reviewing & editing.
Munitz A: Visualization, writing-original draft, writing-reviewing & editing, supervision, resources, conceptualization.
Conflicts of interest
Ariel Munitz serves as a consultant for GSK, AstraZeneca and Sanofi. The other author declares no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the US-Israel Bi-national Science Foundation (Grant No. 2023029), the Israel Science Foundation (Grants No. 542/20), the Israel Cancer Association (Grant No. 01039502), the Food Allergy Fund, the Israel Cancer Research Fund, the Cancer Biology Research Center (TAU) and the Azrieli Foundation Canada-Israel (These programs do not have grant numbers).
Copyright
© The Author(s) 2026.
References
-
1. O’Shea KM, Aceves SS, Dellon ES, Gupta SK, Spergel JM, Furuta GT, et al. Pathophysiology of eosinophilic esophagitis. Gastroenterology. 2018;154(2):333-345.[DOI]
-
3. Zifman E, Banai H, Shamir R, Ringel-Kulka T, Zevit N. Practice differences in the diagnosis and management of eosinophilic esophagitis among adult and pediatric gastroenterologists in Israel. J Pediatr Gastroenterol Nutr. 2018;67(1):34-39.[DOI]
-
5. Spergel J, Aceves SS. Allergic components of eosinophilic esophagitis. J Allergy Clin Immunol. 2018;142(1):1-8.[DOI]
-
7. Comeau MR, Ziegler SF. The influence of TSLP on the allergic response. Mucosal Immunol. 2010;3(2):138-147.[DOI]
-
8. Stanbery AG, Smita S, Von Moltke J, Wojno EDT, Ziegler SF. TSLP, IL-33, and IL-25: Not just for allergy and helminth infection. J Allergy Clin Immunol. 2022;150(6):1302-1313.[DOI]
-
9. Martin NT, Martin MU. Interleukin 33 is a guardian of barriers and a local alarmin. Nat Immunol. 2016;17(2):122-131.[DOI]
-
12. Ziegler SF, Roan F, Bell BD, Stoklasek TA, Kitajima M, Han H. The biology of thymic stromal lymphopoietin (TSLP). Adv Pharmacol. 2013;66:129-155.[DOI]
-
13. Rothenberg ME, Spergel JM, Sherrill JD, Annaiah K, Martin LJ, Cianferoni A, et al. Common variants at 5q22 associate with pediatric eosinophilic esophagitis. Nat Genet. 2010;42(4):289-291.[DOI]
-
15. Davis BP, Stucke EM, Khorki ME, Litosh VA, Rymer JK, Rochman M, et al. Eosinophilic esophagitis-linked calpain 14 is an IL-13-induced protease that mediates esophageal epithelial barrier impairment. JCI Insight. 2016;1(4):e86355.[DOI]
-
17. Sleiman PMA, Wang ML, Cianferoni A, Aceves S, Gonsalves N, Nadeau K, et al. GWAS identifies four novel eosinophilic esophagitis loci. Nat Commun. 2014;5:5593.[DOI]
-
20. Dellon ES, Falk GW, Katzka DA, Lightdale J, Muir AB, Bredenoord AJ, et al. American Society for Gastrointestinal Endoscopy consensus recommendations on the endoscopic management of eosinophilic esophagitis-part 2: Disease assessment, monitoring, and pediatric considerations. Gastrointest Endosc. 2026;103(3):396-417.
-
22. Azouz NP, Ynga-Durand MA, Caldwell JM, Jain A, Rochman M, Fischesser DM, et al. The antiprotease SPINK7 serves as an inhibitory checkpoint for esophageal epithelial inflammatory responses. Sci Transl Med. 2018;10(444):eaap9736.[DOI]
-
23. Min S, Shoda T, Wen T, Rothenberg ME. Diagnostic merits of the Eosinophilic Esophagitis Diagnostic Panel from a single esophageal biopsy. J Allergy Clin Immunol. 2022;149(2):782-787.[DOI]
-
24. Ben-Baruch Morgenstern N, Ballaban AY, Wen T, Shoda T, Caldwell JM, Kliewer K, et al. Single-cell RNA sequencing of mast cells in eosinophilic esophagitis reveals heterogeneity, local proliferation, and activation that persists in remission. J Allergy Clin Immunol. 2022;149(6):2062-2077.
-
26. Ding J, Garber JJ, Uchida A, Lefkovith A, Carter GT, Vimalathas P, et al. An esophagus cell atlas reveals dynamic rewiring during active eosinophilic esophagitis and remission. Nat Commun. 2024;15:3344.[DOI]
-
27. Li D, Wei Y, Wang J, Wang B. Animal models of eosinophilic esophagitis, review and perspectives. Anim Models Exp Med. 2024;7(2):127-135.[DOI]
-
29. Wojno EDT, Noti M, Kim BS, Siracusa MC, Giacomin PR, Nair MG, et al. 282: TSLP-elicited basophil responses mediate the pathogenesis of eosinophilic esophagitis. Cytokine. 2013;63(3):310.[DOI]
-
30. Lutzu N, Favale A, Demurtas M, Del Giacco S, Onali S, Fantini MC. Eosinophilic esophagitis in the “atopic March”: Dupilumab as an “umbrella” strategy for multiple coexisting atopic diseases. Front Med. 2025;11:1513417.[DOI]
-
33. Visaggi P, Dellon ES. Epidemiology, natural history, and treatment of eosinophilic gastrointestinal diseases. Gastroenterology. 2026;170(3):476-494.[DOI]
-
34. Kaneko T, Iwamura C, Kiuchi M, Kurosugi A, Onoue M, Matsumura T, et al. Amphiregulin-producing TH2 cells facilitate esophageal fibrosis of eosinophilic esophagitis. J Allergy Clin Immunol Glob. 2024;3(3):100287.[DOI]
-
36. Olson C, Alp J, Wongjarupong N, Sloan JA. Lessons learned: Real-world effectiveness of dupilumab in patients with eosinophilic esophagitis. Dis Esophagus. 2025;38(6):doaf123.[DOI]
-
37. Marella S, Sharma A, Ganesan V, Ferrer-Torres D, Krempski JW, Idelman G, et al. IL-13-induced STAT3-dependent signaling networks regulate esophageal epithelial proliferation in eosinophilic esophagitis. J Allergy Clin Immunol. 2023;152(6):1550-1568.[DOI]
-
38. Dellon ES, Charriez CM, Zhang S, Falk GW, Oliva S, Ma C, et al. Cendakimab in adults and adolescents with eosinophilic esophagitis. NEJM Evid. 2025;4(10):EVIDoa2500095.[DOI]
-
40. Dsilva A, Wagner A, Itan M, Rhone N, Avlas S, Gordon Y, et al. Distinct roles for thymic stromal lymphopoietin (TSLP) and IL-33 in experimental eosinophilic esophagitis. Allergy. 2025;80(11):3095-3107.[DOI]
-
41. Masuda MY, Pyon GC, Luo H, LeSuer WE, Putikova A, Dao A, et al. Epithelial overexpression of IL-33 induces eosinophilic esophagitis dependent on IL-13. J Allergy Clin Immunol. 2024;153(5):1355-1368.[DOI]
-
42. Pyon GC, Masuda MY, Putikova A, Luo H, Gibson JB, Dao AD, et al. Tissue-specific inducible IL-33 expression elicits features of eosinophilic esophagitis. J Allergy Clin Immunol. 2024;154(6):1545-1553.[DOI]
-
43. Judd LM, Heine RG, Menheniott TR, Buzzelli J, O’Brien-Simpson N, Pavlic D, et al. Elevated IL-33 expression is associated with pediatric eosinophilic esophagitis, and exogenous IL-33 promotes eosinophilic esophagitis development in mice. Am J Physiol Gastrointest Liver Physiol. 2016;310(1):G13-G25.
-
46. Brusilovsky M, Rochman M, Shoda T, Kotliar M, Caldwell JM, Mack LE, et al. Vitamin D receptor and STAT6 interactome governs oesophageal epithelial barrier responses to IL-13 signalling. Gut. 2023;72(5):834-845.[DOI]
-
47. Venkateshaiah SU, Shukla JS, Mishra A. IL-18 is induced in food allergic eosinophilic esophagitis (EoE) patients and its overexpression promotes disease pathogenesis in mice. J Allergy Clin Immunol. 2015;135(2):AB160.[DOI]
-
50. Rubinstein E, Cho JY, Rosenthal P, Chao J, Miller M, Pham A, et al. Siglec-F inhibition reduces esophageal eosinophilia and angiogenesis in a mouse model of eosinophilic esophagitis. J Pediatr Gastroenterol Nutr. 2011;53(4):409-416.[DOI]
-
53. Rajavelu P, Rayapudi M, Moffitt M, Mishra A, Mishra A. Significance of para-esophageal lymph nodes in food or aeroallergen-induced iNKT cell-mediated experimental eosinophilic esophagitis. Am J Physiol Gastrointest Liver Physiol. 2012;302(7):G645-G654.[DOI]
-
54. Mishra A, Hogan SP, Brandt EB, Rothenberg ME. An etiological role for aeroallergens and eosinophils in experimental esophagitis. J Clin Invest. 2001;107(1):83-90.[DOI]
-
56. Collison AM, Sokulsky LA, Sherrill JD, Nightingale S, Hatchwell L, Talley NJ, et al. TNF-related apoptosis-inducing ligand (TRAIL) regulates midline-1, thymic stromal lymphopoietin, inflammation, and remodeling in experimental eosinophilic esophagitis. J Allergy Clin Immunol. 2015;136(4):971-982.[DOI]
-
57. Maskey A, Srivastava K, Soffer G, Dunkin D, Yuan Q, Li XM. Induction of severe eosinophilic esophagitis and multi-organ inflammation by airborne allergens is associated with IL-4/IL-13 and CCL11 but not IgE in genetic susceptible mice. J Inflamm Res. 2022;15:5527-5540.[DOI]
-
58. Holvoet S, Doucet-Ladevèze R, Perrot M, Barretto C, Nutten S, Blanchard C. Beneficial effect of Lactococcus lactis NCC 2287 in a murine model of eosinophilic esophagitis. Allergy. 2016;71(12):1753-1761.[DOI]
-
64. Kellett SK, Crue T, Cruz SNA, Markey GE, Ryan S, Crowe L, et al. Resolution of epithelial dysfunction in eosinophilic esophagitis is mediated by an HIF-1α-CD73-adenosine signaling axis. J Allergy Clin Immunol. 2026;157(3):693-710.[DOI]
-
65. Rothenberg ME, Dellon ES, Collins MH, Bredenoord AJ, Hirano I, Peterson KA, et al. Eosinophil depletion with benralizumab for eosinophilic esophagitis. N Engl J Med. 2024;390(24):2252-2263.[DOI]
-
67. Eden K, Rothschild DE, McDaniel DK, Heid B, Allen IC. Noncanonical NF-κB signaling and the essential kinase NIK modulate crucial features associated with eosinophilic esophagitis pathogenesis. Dis Models Mech. 2017;10(12):1517-1527.[DOI]
-
68. Laky K, Kinard JL, Li JM, Moore IN, Lack J, Fischer ER, et al. Epithelial-intrinsic defects in TGFβR signaling drive local allergic inflammation manifesting as eosinophilic esophagitis. Sci Immunol. 2023;8(79):eabp9940.[DOI]
-
71. Dellon ES, Rothenberg ME, Collins MH, Hirano I, Chehade M, Bredenoord AJ, et al. Dupilumab in adults and adolescents with eosinophilic esophagitis. N Engl J Med. 2022;387(25):2317-2330.[DOI]
-
72. Chehade M, Dellon ES, Spergel JM, Collins MH, Rothenberg ME, Pesek RD, et al. Dupilumab for eosinophilic esophagitis in patients 1 to 11 years of age. N Engl J Med. 2024;390(24):2239-2251.[DOI]
-
74. Katori Y, Cho BH, Song CH, Fujimiya M, Murakami G, Kawase T. Smooth-to-striated muscle transition in human esophagus: An immunohistochemical study using fetal and adult materials. Ann Anat. 2010;192(1):33-41.[DOI]
-
75. Alcolea MP. Oesophageal stem cells and cancer. Adv Exp Med Biol. 2017;1041:187-206.[DOI]
-
76. Grommisch D, Eenjes E, Troost ML, Genander M. Epithelial architecture and signaling activity in the adult human esophagus. Front Cell Dev Biol. 2025;13:1632255.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Dsilva A, Munitz A. Insights into the pathogenesis of eosinophilic esophagitis using mouse models. Myeloid Cells. 2026;1:202604. https://doi.org/10.70401/mc.2026.0003
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Preclinical Models as Tools for Resolving Mechanistic Gaps in EoE
- 3. Animal Models of EoE
- 4. Future Directions
- 5. Conclusions
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Dsilva A, Munitz A. Insights into the pathogenesis of eosinophilic esophagitis using mouse models. Myeloid Cells. 2026;1:202604. https://doi.org/10.70401/mc.2026.0003
copy
Share Link
copy