Multi-class pattern discovery for bacterial secretory effectors
Jing Li
1

,
Xi Su
2
,
Qing Liu
3
,
Youyu Wang
4,*
*Correspondence to:
Youyu Wang, Department of Thoracic Surgery, Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu 610072, Sichuan, China.
E-mail: syywyy123@126.com
Comput Biomed. 2026;1:202522. 10.70401/cbm.2026.0012
Received: October 31, 2025Accepted: March 02, 2026Published: March 05, 2026
Abstract
Aims: EffecTri aims to develop a comprehensive, multi-class prediction framework to accurately identify bacterial effector proteins secreted by Type III, IV, and VI secretion systems. Current methodologies often employ binary classifications, overlooking the complexity and interactions among multiple effector classes.
Methods: EffecTri integrates deep contextual embeddings from Evolutionary Scale Modeling and handcrafted descriptors, including Amino Acid Composition and Dipeptide Composition. The performance of the model was rigorously evaluated through comparative descriptor analyses and optimized feature combinations, complemented by Uniform Manifold Approximation and Projection visualization for interpretability.
Results: EffecTri outperformed traditional machine learning methods, achieving a weighted F1-score of 0.850 on an independent test dataset. The fusion of Evolutionary Scale Modeling embeddings with handcrafted descriptors demonstrated superior predictive performance, clearly distinguishing effector classes in UMAP visualizations.
Conclusion: EffecTri represents a robust, interpretable, and accurate computational tool, enhancing the multi-class identification of bacterial secretory effectors and contributing valuable insights into bacterial pathogenic mechanisms.
Keywords
Bacterial secretory effectors, multi-class classification, pattern discovery
1. Introduction
Bacterial secretion systems mediate interactions between bacteria and their hosts, allowing bacteria to manipulate host physiology or outcompete rival microbes by delivering effector proteins, DNA, or protein complexes into host or bacterial cells[1,2]. Among them, type III (T3SS), type IV (T4SS), and type VI secretion systems (T6SS) are the most extensively studied, and are intricately linked to bacterial virulence[3]. T3SS is commonly found in gram-negative pathogens such as Salmonella, Shigella, and Yersinia, and facilitates the direct injection of T3SEs into host cells to disrupt immune responses and cytoskeletal structures[4]. T4SS, by contrast, is a versatile system present in both gram-negative and certain gram-positive bacteria. It enables the secretion of T4SEs as well as DNA-protein complexes, thereby contributing to pathogenesis and horizontal gene transfer[5,6]. T6SS, first identified in Vibrio cholerae and Pseudomonas aeruginosa, plays a dual role in host manipulation and interbacterial competition by injecting T6SEs into neighboring cells[7]. These effector proteins are crucial for immune evasion, colonization, and bacterial fitness. For example, T3SEs can suppress innate immunity by interfering with Toll-like receptor signaling and cytoskeletal dynamics[8]. T4SEs modulate host cell proliferation and impair DNA repair mechanisms to promote intracellular survival[9]. T6SEs assist pathogens in dominating ecological niches by killing competitors or dampening the phagocytic activity of host immune cells[10,11].
Most existing studies focus on binary classification tasks, such as distinguishing T3SEs from non-T3SEs, whereas comprehensive multiclass frameworks that simultaneously predict T3SE, T4SE, and T6SE remain limited. Secretion-related prediction has also been studied beyond bacterial effectors; for example, SecretP proposed a support vector machine (SVM)-based framework using pseudo-amino-acid-composition-derived features to predict secreted proteins[12]. Recently, a wide array of computational methods has been proposed to predict bacterial effectors secreted via type III, IV, and VI secretion systems. These methods leverage sequence-derived features, structural motifs, and machine learning frameworks to improve predictive performance[13]. For T3SEs, Samudrala[14] integrated the GC content, amino acid composition (AAC), and N-terminal motifs to uncover conserved secretion signals. Wagner[15] developed the Effectidor framework, employing protein language model embeddings to accurately distinguish T3SEs and achieving precision–recall AUCs exceeding 0.98. Ensemble models such as EP3[16] and T3SEpp[17] further enhanced the prediction accuracy through multi-feature integration. In the realm of T4SEs, Wang’s Bastion4[18] utilised stacked ensemble classifiers based on diverse sequence-derived features, whereas Xiong’s PredT4SE-Stack[19] adopted PSSM-composition with stacking to improve performance. Acici[20] explored clustering patterns of T4SEs using unsupervised learning, revealing structural trends. More recently, T4Seeker[21] integrated handcrafted and language model-derived features for robust performance. For T6SEs, Durand[22] emphasised the diversity and delivery mechanisms of T6SEs, although few prediction tools directly target this class. Several efforts, such as DeepT3_4[23], have been attempted to develop unified models covering multiple effector types using deep learning architectures. However, most of these methods still depend on binary settings or lack balanced performance across effector subtypes. In this work, we propose EffecTri, a unified multiclass prediction framework that leverages Evolutionary Scale Modeling (ESM) embeddings and handcrafted features through a deep neural network, achieving superior and well-balanced performance across T3SE, T4SE, and T6SE categories. More recently, EDIFIER integrated structural cues with pretrained protein language models in a structure-aware graph learning framework for T3SS effector characterization, highlighting the ongoing shift toward representation learning for secretion-related prediction[24].
Although these studies have significantly advanced effector prediction, the majority focus on binary classification within individual secretion systems. Comprehensive multiclass models that can simultaneously classify T3SEs, T4SEs, and T6SEs remain underexplored. Therefore, a pressing need exists for integrated frameworks that can distinguish among multiple effector types using unified representations and interpretable learning strategies. We developed an efficient multi-classification model for identifying and classifying T3SE, T4SE, and T6SE. We first conducted a systematic analysis of protein characteristics at different levels, including sequence, structural, and functional characteristics, to capture the full diversity of effector proteins. These feature analyses were used to develop an efficient multi-classification model that accurately identifies and classifies T3SE, T4SE, and T6SE. Our model uses complementary ESM, AAC, and dipeptide composition (DPC) features to train a multiclass classifier. It achieved a weighted F1-score of 0.850, providing a powerful tool for distinguishing T3SE, T4SE, and T6SE proteins, and highlighting the potential of integrative feature representation in predicting secretory effectors.
2. Methods
2.1 Data description
We collected effector protein sequences corresponding to T3SE, T4SE, and T6SE from a wide range of publicly available computational tools and databases to build a high-performance multi-class classification model. For T4SEs, data were integrated from nine representative sources, including DeepT3_4[23], Bastion4[18], iT4SE-EP[25], OPT4e[26], DeepSecE[27], T4SE-XGB[28], T4SEfinder[28], T4SEpp[29] and TSE-ARF[30]. Among them, T4SEfinder, T4SEpp and DeepSecE primarily drew their samples from the SecReT4 database, whereas DeepT3_4 and Bastion4 utilised SecretEPDB. In addition, iT4SE-EP, T4SE-XGB and TSE-ARF expanded their datasets by integrating experimentally verified T4SEs from 10 bacteria reported in the literature, such as Agrobacterium, Brucella and Legionella, and OPT4e focused on effectors from four representative gram-negative pathogens. In addition, part of the TSE-ARF dataset incorporated sequences from the BastionHub platform. After merging and redundancy removal using CD-HIT at 80% sequence identity[31], we retained 730 non-redundant T4SEs. For T3SEs, sequences were collected from DeepT3_4, Bastion3, DeepT3-Keras, and TSE-ARF, based on entries from the NCBI Protein[32] and UniProt databases[33]. CD-HIT clustering reduced the T3SE dataset to 606 unique sequences. Similarly, T6SEs were collected from Bastion6, SecReT6 and TSE-ARF, with most entries originally curated from the SecretEPDB database[34], resulting in a final set of 308 non-redundant T6SEs. These processed datasets formed the basis for constructing a balanced and high-quality benchmark for multi-class effector protein classification (Figure 1).
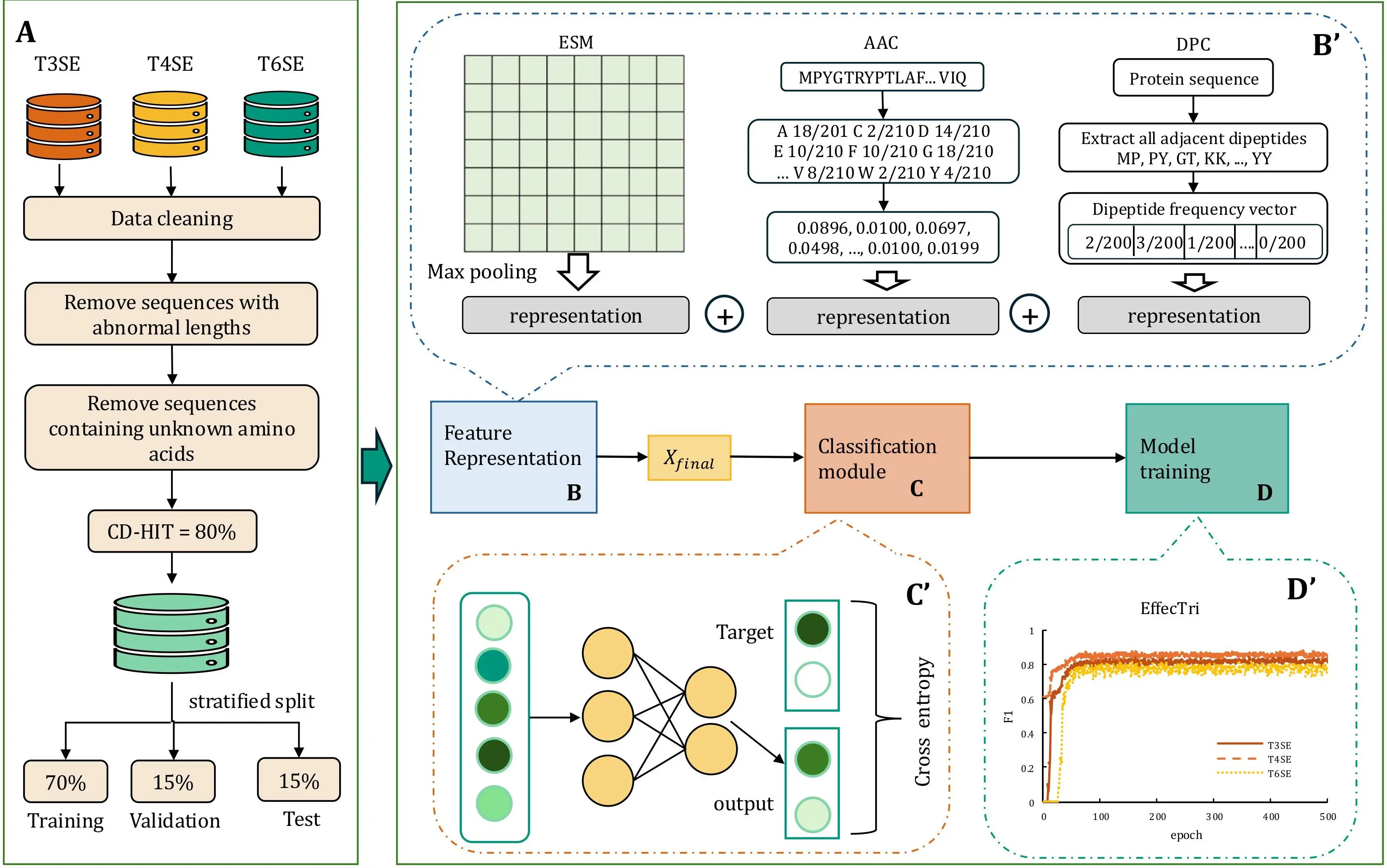
Figure 1. Overview of the EffecTri framework. (A) Data preprocessing pipeline including sequence cleaning, redundancy removal (CD-HIT, 80% identity), and stratified training/validation/test split; (B) Feature representation using ESM embeddings, AAC, and DPC, followed by vector concatenation; (B′) Detailed view of ESM max pooling, AAC frequency calculation, and DPC vector construction; (C) Classification module using a MLP; (C′) Training with cross-entropy loss; (D) Model training; (D′) Model convergence curves showing stable training and consistent F1 performance across classes. ESM: Evolutionary Scale Modeling; AAC: amino acid composition; DPC: dipeptide composition; MLP: multi-layer perceptron.
After data preprocessing was completed, we constructed a multi-classification task using T3SE, T4SE, and T6SE samples. Unlike previous studies, which largely focused on binary classification (such as distinguishing effector proteins from non-effector proteins), this work focuses on the multiple classification of three secretory effector proteins, T3SE, T4SE, and T6SE. This multi-classification task is not only more challenging but also can more fully reflect the characteristics of different secretory system effector proteins. We adopted a random segmentation method to divide the dataset into a training set, verification set and test set, with proportions of 70%, 15%, and 15% respectively, to ensure the training and evaluation effects of the model. This partitioning method helps to better evaluate the generalization ability of the model during training and provides reliable data support for the subsequent performance optimization. As shown in Table 1, the final dataset consisted of 606 T3SEs, 730 T4SEs and 308 T6SEs. Among them, 424, 511 and 215 samples were assigned to the training set for T3SEs, T4SEs and T6SEs, respectively. The validation set included 91 T3SEs, 109 T4SEs and 47 T6SEs, whereas the test set contained 91 T3SEs, 110 T4SEs and 46 T6Ses (Table 1), demonstrating that both subsets maintained a consistent and balanced class distribution. This balanced partitioning ensured that the model was exposed to sufficient examples from each class during training while also allowing independent and unbiased evaluation on held-out data. The resulting data foundation was well-suited for evaluating multi-class classification performance and generalization across secretory effector types. We focused on T3SEs, T4SEs and T6SEs because these classes currently provide the most comprehensive and experimentally supported sequence collections in the public literature. We also attempted to compile T1SEs and T2Ses, but the available experimentally supported samples were much smaller, and including them as additional classes would introduce severe class imbalance in a multi-class setting. For T7SEs, we were unable to identify a sufficiently large and reliable set of experimentally validated substrates from publicly available sources.
Table 1. Dataset distribution of type III, IV and VI secretory effectors for training, validation and test sets.
| T3SEs | T4SEs | T6SEs | |
| Training | 424 | 511 | 215 |
| Validation | 91 | 109 | 47 |
| Test | 91 | 110 | 46 |
2.2 Description of the proposed EffecTri
EffecTri is a multi-class classification framework specifically designed to distinguish between Type III, Type IV, and Type VI secretory effectors (T3SE, T4SE, and T6SE). The overall workflow of EffecTri is illustrated in Figure 1, consisting of four major components: data preprocessing, feature extraction and fusion, classification module, and model training and evaluation. In the data preprocessing stage (Figure 1A), annotated protein sequences corresponding to T3SE, T4SE, and T6SE classes were collected and subjected to a series of quality control steps. We retained sequences with lengths between 30 and 3500 amino acids and containing only canonical amino acids (sequences with any non-canonical residue such as B/J/O/U/X/Z, were excluded). CD-HIT was employed with an 80% sequence identity threshold to reduce sequence redundancy and potential bias. The resulting non-redundant dataset was subsequently stratified into training, validation and test sets in a 70%:15%:15% ratio, ensuring balanced class distribution across subsets.
For feature representation (Figure 1B), EffecTri integrated three complementary types of protein features to capture different levels of sequence information. The first type of feature was a pretrained protein language model, Evolutionary Scale Modeling[35,36] (ESM-2, esm2_t6_8M_UR50D), which produces contextualized embeddings for each amino acid residue. We extracted residue embeddings from layer 6 and then applied sequence-level max pooling over the residue dimension to obtain a fixed-length 320-dimensional representation for each protein. The second and third types of features were based on handcrafted descriptors: AAC encoded the normalised frequency of each of the 20 amino acids, whereas DPC captured the frequency of all possible adjacent residue pairs, thereby modeling local patterns in the sequence[37]. In addition, we evaluated a broader set of commonly used protein descriptors to provide complementary and interpretable representations, including distance-based residue (DR), k-mer frequencies (2 mer and 3 mer), CTD-based features, Composition–Transition–Distribution–Composition (CTDC) and Composition–Transition–Distribution–Distribution (CTDD), and pseudo amino acid composition descriptors Parallel Correlation Pseudo Amino Acid Composition (PC-PseAAC), Generalized Parallel Correlation Pseudo Amino Acid Composition (PC-PseAAC-General), Series Correlation Pseudo Amino Acid Composition (SC-PseAAC), and Generalized Series Correlation Pseudo Amino Acid Composition (SC-PseAAC-General). Together with the pretrained language model representation (ESM-2), these descriptors cover global composition, local motif-like patterns, physicochemical distribution, and sequence-order correlation information, enabling a systematic comparison of different feature families. All three features were concatenated to form a unified feature vector that reflected both high-level semantic and low-level compositional characteristics. The concatenated feature vector was subsequently passed through a multi-scale feature aggregation module, which was designed to enhance cross-feature interaction and allow the model to adaptively learn from heterogeneous inputs. This module incorporated a small network of parallel fully connected layers with different hidden dimensions, enabling the model to capture information at multiple resolutions. The pooled ESM embedding and the handcrafted AAC and DPC descriptors are each represented as fixed-length vectors. We fuse these features by concatenation to form a single input vector, which is then fed into the multi-layer perceptron (MLP) classifier. Since the classifier consists of fully connected layers and does not model any positional structure across the concatenated dimensions, the concatenation order does not introduce ordering bias.
In the classification stage (Figure 1C), Xfinal was fed into a MLP consisting of two hidden layers and a softmax output layer. The MLP was trained using the categorical cross-entropy loss function to classify each protein into one of the three effector categories. As shown in Figure 1D, the EffecTri model converged rapidly and exhibited stable training behaviour across different classes. The final model, trained with the combination of ESM, AAC and DPC features, achieved the best performance among all tested feature settings. This demonstrated the effectiveness of multi-view feature integration in enhancing discriminative power and robustness. EffecTri provides a powerful and interpretable framework for effector classification in bacterial secretion systems by combining deep contextual embeddings with handcrafted sequence descriptors.
2.3 Hyperparameter settings
We conducted a series of controlled experiments to evaluate the impact of key hyperparameters on classification performance and to select a well-performing hyperparameter configuration based on validation weighted F1-score within the predefined ranges. Each experiment varied one hyperparameter while keeping the others fixed at default values, and the performance was assessed using the weighted F1-score on the validation set. The results are summarised in Figure 2.
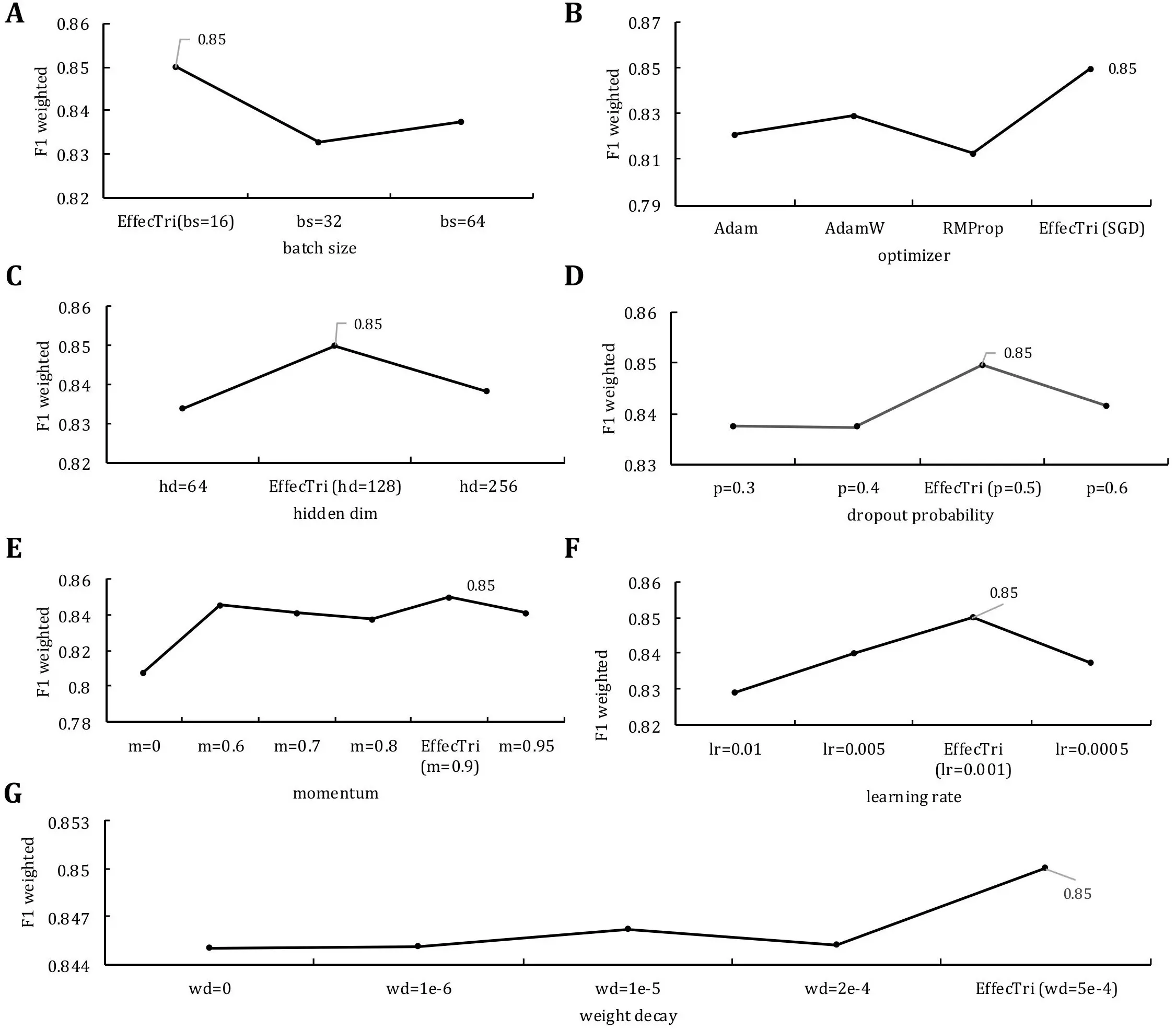
Figure 2. The performance of EffecTri was evaluated under varying hyperparameter settings using weighted F1-score on the validation set. (A) Batch size comparison (16, 32, 64); (B) Optimiser comparison (Adam, AdamW, RMSProp, SGD); (C) Hidden layer dimensions (64, 128, 256); (D) Dropout probability (0.3-0.6); (E) Momentum values (0.0-0.95); (F) Learning rate (0.01-0.0005); (G) Weight decay (0 to 5e-4). The best performance (weighted F1 = 0.85) was consistently achieved with batch size = 16, hidden dim = 128, dropout = 0.5, momentum = 0.9, learning rate = 0.001, optimiser = SGD and weight decay = 5e-4. SGD: stochastic gradient descent.
The batch size was first evaluated using values of 16, 32, and 64 (Figure 2A). The model achieved the highest weighted F1 score (0.85) with a batch size of 16, whereas performance dropped slightly for larger batch sizes, suggesting that smaller batches may facilitate better gradient updates and generalization in our setting[38]. We subsequently explored different optimizers (Figure 2B), including Adam, AdamW, RMSProp and stochastic gradient descent (SGD). We compared SGD (with momentum) and commonly used adaptive optimizers (Adam, AdamW, RMSProp) as they represent standard choices with different update dynamics. Adaptive methods often accelerate early convergence, whereas SGD with momentum can provide more stable optimization and sometimes better generalization in feature-based classifiers. Among them, SGD led to the best performance (F1 = 0.85), indicating that, despite its simplicity, SGD may offer more stable convergence for this classification task compared to adaptive methods. For the hidden dimension size (Figure 2C) of the classification MLP, we tested values of 64, 128 and 256. The best result was obtained with 128 hidden units, suggesting a trade-off between model capacity and overfitting[39]. The effect of dropout probability (Figure 2D) was evaluated across values of 0.3, 0.4, 0.5 and 0.6. A dropout rate of 0.5 provided the best performance, indicating an effective level of regularization[40]. In addition, we evaluated the role of momentum (Figure 2E) in the SGD optimizer, testing values from 0 to 0.95. The model displayed consistently high performance when momentum was set to 0.9, reaching an F1 score of 0.85, which reflected improved optimization dynamics through gradient accumulation. The learning rate (Figure 2F) varied from 0.0005 to 0.01. The best performance was achieved at a learning rate of 0.001, with both lower and higher values showing degraded performance, indicating that 0.001 served as a sweet spot for balancing convergence speed and stability. Finally, we investigated weight decay values (Figure 2G) ranging from 0 to 5e-4. The model performed best with a weight decay of 5e-4, suggesting that proper L2 regularization helps reduce overfitting and improve generalization.
Our tuning strategy follows a one-factor-at-a-time protocol and therefore may not fully capture interactions among hyperparameters (e.g., learning rate vs. batch size). In addition, the selected values are dataset- and protocol-dependent and may require adjustment when applying EffecTri to datasets with different sequence length distributions, feature combinations, or class imbalance. In practice, users may tune batch size under different GPU memory constraints, adjust learning rate/optimizer/momentum when optimization becomes unstable or converges slowly, and tune dropout/weight decay to mitigate overfitting. When minority-class performance is prioritized, alternative selection criteria (e.g., F1) can also be considered. In summary, the best-performing configuration for EffecTri was: batch size = 16, optimizer = SGD, hidden dim = 128, dropout = 0.5, momentum = 0.9, learning rate = 0.001 and weight decay = 5e-4. These hyperparameters were used in all final experiments reported in this study.
2.4 Evaluation metrics
We conducted a per-class analysis by calculating the precision, recall, and F1-score individually for each effector type: T3SE, T4SE, and T6SE[41-43]. This allowed us to examine how well the model distinguished each specific class and to identify any potential bias or imbalance in classification performance[44,45]. TPc, FPc, and FNc denoted the number of true positives, false positives, and false negatives for class c, respectively[46]. C referred to the number of classes. Precision, Recall, and F1-score for each class c were defined as follows:
We adopted multiple standard classification metrics, with a primary focus on the weighted F1-score due to its ability to account for class imbalance while combining both precision and recall[47], to comprehensively evaluate the performance of the EffecTri model. This metric served as our main evaluation criterion for model selection and for comparison across different feature combinations and hyperparameter settings.
In addition to the weighted F1-score, we reported the precision-weighted and recall-weighted scores, which provided insights into the overall precision and recall behaviour of the model, respectively, while weighting each class according to its prevalence in the dataset. We also computed macro-averaged metrics, including macro precision, macro recall, and macro F1-score, to further evaluate the model’s ability to perform consistently across all classes. Unlike their weighted counterparts, macro metrics treat each class equally, offering a more balanced view of performance regardless of class distribution.
3. Results
3.1 Analysis of different descriptors
We compared 11 descriptor families to assess their effectiveness for multi-class classification of T3SE, T4SE, and T6SE. Brief definitions of each descriptor are provided in Section 2.2. The evaluated descriptors include AAC, DPC, DR, k-mer (k = 2, 3), CTDC, CTDD, PC-PseAAC, PC-PseAAC-General, SC-PseAAC, SC-PseAAC-General, and ESM[48-53]. Brief definitions and the rationale for all descriptors are provided in Section 2.2. These descriptors were selected from the iFeature/iLearn feature sets to provide an interpretable and comprehensive coverage of sequence characteristics. Specifically, AAC/DPC summarize global residue composition, k-mer features (2-mer/3-mer) capture local motif-like patterns, CTD (CTDC/CTDD) encodes composition and positional distribution of physicochemical property groups, and PC-/SC-PseAAC (and generalized variants) model sequence-order correlation beyond simple composition. DR describes distance-related residue statistics. In addition, we included ESM embeddings as a representative pretrained protein language model feature to capture contextual and evolutionary signals. The performance of each descriptor was evaluated using the weighted F1 average and the weighted F1 scores for each category (T3SE, T4SE, and T6SE). The results are shown in Figure 3A.
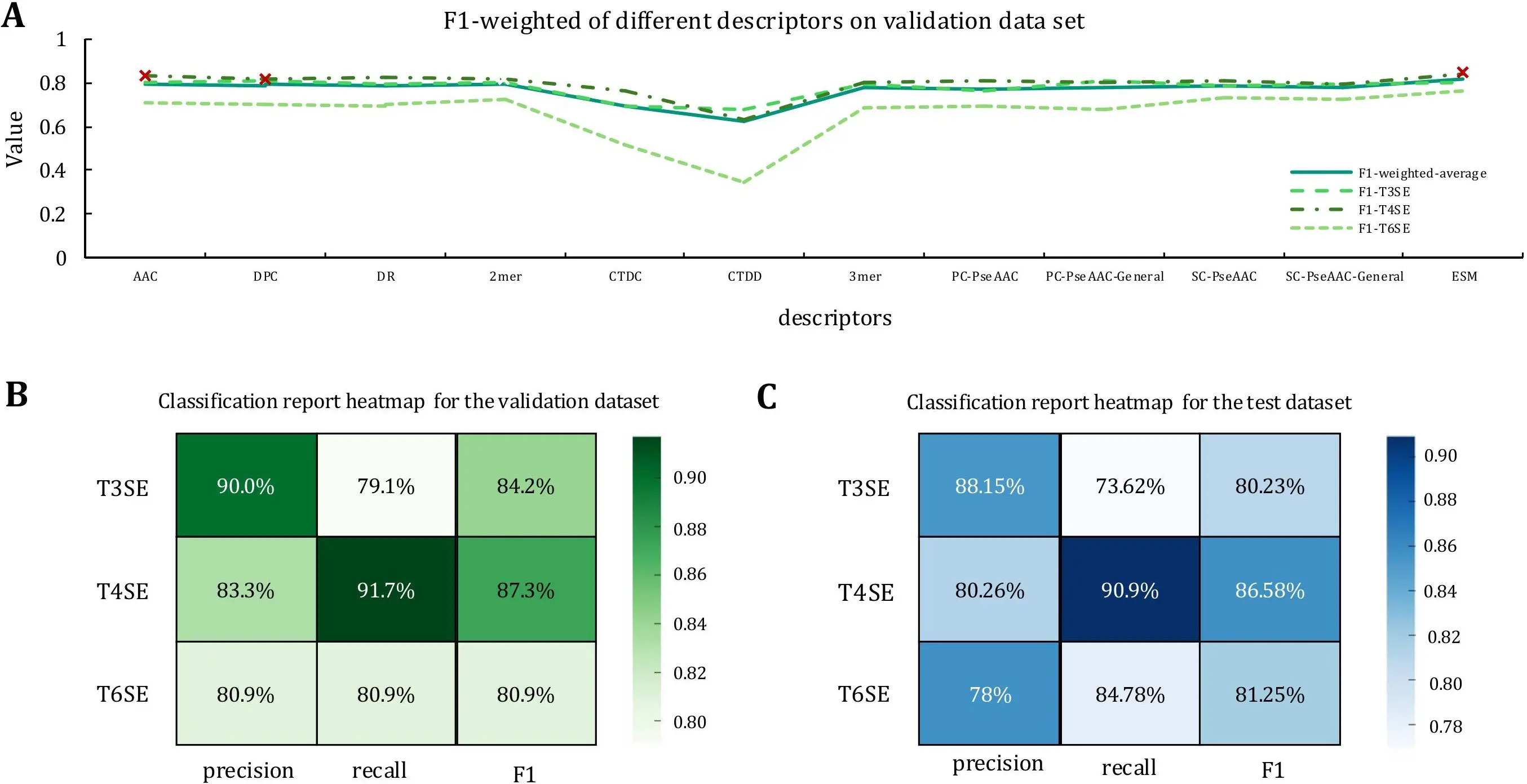
Figure 3. Performance of single-feature models and classification results of EffecTri. (A) Weighted F1 scores of models trained with individual descriptors on the validation set. ESM, AAC, and DPC (marked with red crosses) achieved the best performance and were selected for feature combination; (B) Classification metrics on the validation set; (C) Classification metrics on the test set. ESM: Evolutionary Scale Modeling; AAC: amino acid composition; DPC: dipeptide composition.
ESM is the most prominent performer, with a weighted F1 average of 0.814 and excellent performance across all categories (T3SE: 0.804, T4SE: 0.843, T6SE: 0.764)[35]. This suggests that ESM can effectively capture evolutionary and functional information of effector proteins and is the most robust descriptor for the current task. In addition, AAC and DPC displayed strong performance, with weighted F1 averages of 0.797 and 0.792, respectively. Although both performed well on the T3SE and T4SE classifications, they were relatively weak on the T6SE. The weighted F1 averages of SC-PseAAC, SC-PseAAC-General, PC-PseAAC, PC-PseAAC-General, DR, 2-mer and 3-mer were 0.782, demonstrating the same performance on T3SE and T4SE. However, the classification effect on T6SE was limited. Other descriptors, such as CTDC and CTDD, performed moderately, with weighted F1 averages between 0.62 and 0.69. We selected the top three descriptors based on the validation weighted F1 average (ESM, AAC and DPC) for further analysis. These descriptors not only performed well overall but also showed strong consistency across multiple categories, especially T3SE and T4SE. However, their relatively low performance on the T6SE suggests that further optimization is required to improve classification accuracy for this category.
3.2 Evaluation of EffecTri on validation and test sets
We developed EffecTri, a deep learning-based classifier that combines three complementary protein representations, to evaluate the effectiveness of multi-view feature integration: ESM embeddings, AAC, and DPC. These features are processed through a unified architecture that captures both global semantic and local compositional information. Experimental results demonstrate that EffecTri achieved strong overall performance, with a weighted F1-score of 0.850 and a macro F1-score of 0.841, indicating accurate and balanced classification across all effector categories. EffecTri learns robust and discriminative features by jointly leveraging complementary representations, thus contributing to improved performance, particularly for challenging classes such as T6SEs. We further assessed class-specific behaviour by visualising per-class classification metrics using heatmaps for both the validation and test sets (Figure 3B,C). EffecTri achieved F1-scores of 84.2%, 87.3%, and 80.9% for T3SE, T4SE, and T6SE, respectively, on the validation set. Performance remained consistent on the test set, with F1-scores of 80.23%, 86.58%, and 81.25% for T3SE, T4SE, and T6SE, respectively. These results confirm that EffecTri not only excels in aggregated metrics but also delivers stable, well-balanced predictions across individual effector types on both validation and unseen test data.
3.3 Ablation study
We conducted a comparative analysis using individual features (DPC, AAC, and ESM) and their combinations for the multiclass classification of T3SE, T4SE, and T6SE (Figure 4) to evaluate the effectiveness of different feature representations. In this feature fusion experiment, we conducted a comparative analysis of the performance of multiple feature combinations, including single features (DPC, AAC, ESM, Figure 4A) and their combinations (AAC + DPC, ESM + AAC, ESM + DPC, and ESM + AAC + DPC, Figure 4B). The results indicate that feature fusion does not necessarily guarantee improvement: the handcrafted-only fusion (AAC + DPC) provides limited gains, whereas fusing ESM with AAC and/or DPC leads to clear performance improvements, with ESM + AAC + DPC achieving the best overall results. According to the primary descriptor analysis (Section 3.1), AAC, DPC, and ESM were selected as the top three descriptors based on the validation weighted F1 average and were therefore chosen for subsequent ablation and fusion experiments. Among them, ESM is significantly better than DPC and AAC in all indicators. The weighted F1 score for the ESM was 0.814 and the macro F1 score was 0.804, whereas the weighted F1 scores for the DPC and AAC were 0.795 and 0.794, respectively, and the macro F1 scores were 0.776 and 0.779, respectively. This indicates that ESM, as a deep learning-based protein sequence representation method, can capture complex semantic information in sequences more effectively, thus performing well in classification tasks.
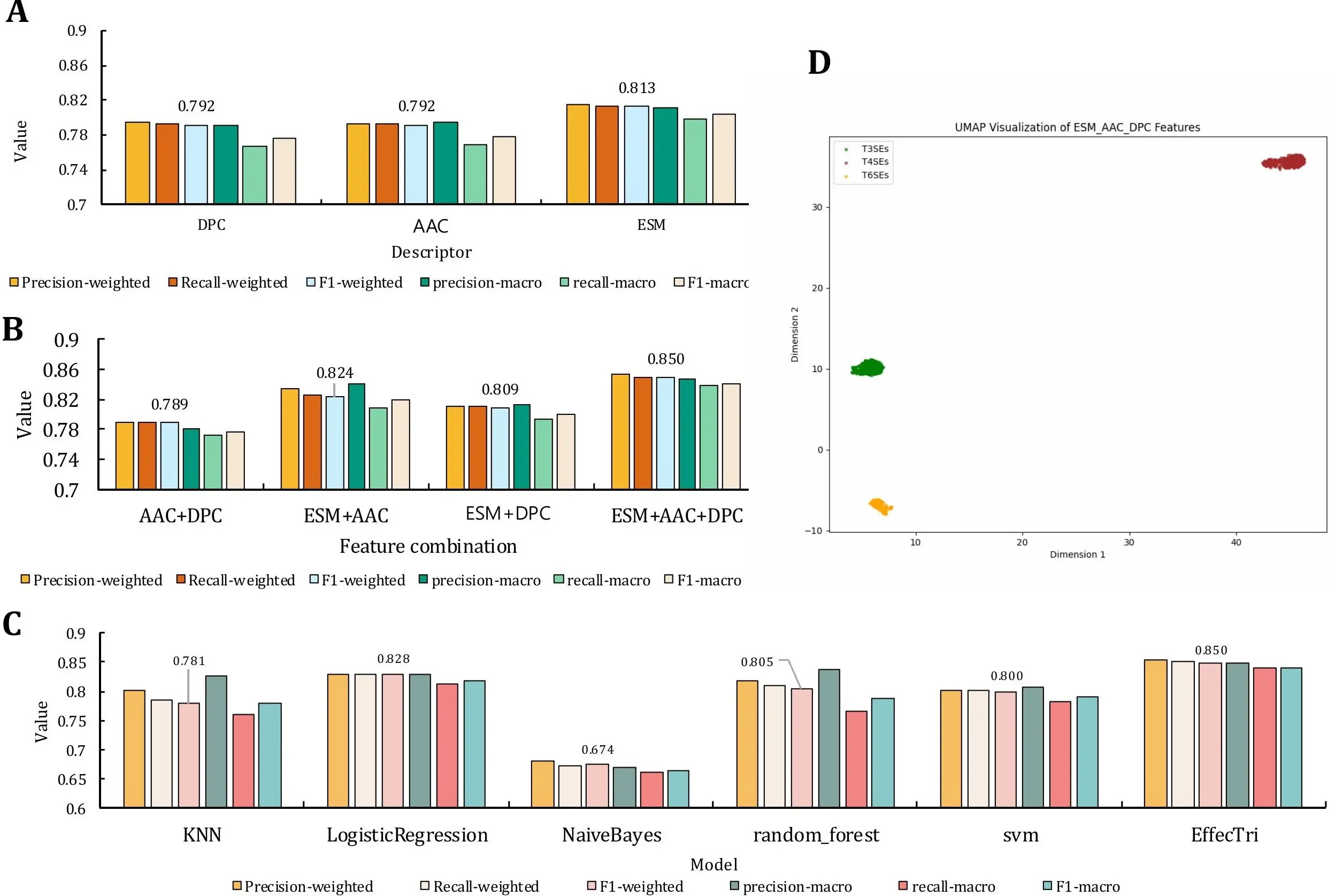
Figure 4. Performance comparison across feature types, model variants and visualization. (A) Comparison of classification performance using individual descriptors (DPC, AAC, ESM); (B) Performance of feature fusion models combining AAC, DPC, and ESM; (C) Comparison between traditional machine learning models and the proposed EffecTri model; (D) UMAP visualization of the combined ESM + AAC + DPC features used in EffecTri, demonstrating evident separation between T3SE, T4SE, and T6SE. DPC: dipeptide composition; AAC: amino acid composition; ESM: Evolutionary Scale Modeling; UMAP: Uniform Manifold Approximation and Projection.
The simple combination of AAC and DPC did not result in performance improvement, suggesting potential redundancy or limited complementarity between these handcrafted features. However, incorporating ESM with either AAC or DPC substantially boosted performance. The ESM + AAC model achieved a weighted F1-score of 0.824 and a macro F1-score of 0.819, demonstrating complementarity between global sequence embeddings and amino acid composition. Similarly, ESM + DPC attained 0.809 weighted F1 and 0.801 macro F1, also surpassing individual features. The best performance was achieved using the ESM + AAC + DPC combination, with a weighted F1-score of 0.850 and macro F1-score of 0.841. This confirmed that integrating global contextual features (ESM) with local physicochemical and compositional patterns (AAC and DPC) provides the most discriminative power. The results validate the complementary nature of these feature types and demonstrate the advantage of multi-view fusion in effector protein classification. This behaviour is common in feature engineering: adding partially redundant descriptors can increase effective dimensionality and variance, which may limit gains or mildly hurt generalization if not sufficiently regularized. In our case, AAC and DPC both encode composition-related statistics (global residue frequencies vs. dipeptide frequencies) and therefore overlap substantially, which explains the limited benefit of fusing AAC and DPC alone. By contrast, ESM embeddings capture high-level contextual and evolutionary patterns that are largely distinct from composition-based descriptors; thus, AAC/DPC provide auxiliary low-level cues that complement ESM when fused, leading to improved performance for ESM-based combinations and the best overall results for ESM + AAC + DPC.
3.4 Performance comparison with traditional classifiers and state-of-the-art models
We conducted a comparative study using multiple classical machine learning classifiers trained on the same set of concatenated features, including ESM embeddings, AAC, and DPC, to further validate the effectiveness of our model architecture. As shown in Figure 4C, we evaluated the performance of six classifiers: k-nearest neighbors[54], logistic regression[55], naive Bayes[56], random forest[57-59], SVM[60-62], and our proposed EffecTri model, which employs an MLP[63,64] architecture. EffecTri achieved the best overall performance, reaching a weighted F1-score of 0.850, surpassing all baseline methods. Among the traditional models, logistic regression and random forest performed relatively well, with weighted F1-scores of 0.828 and 0.805, respectively. In contrast, naive Bayes exhibited the weakest performance (weighted F1 = 0.674), possibly due to its limited capacity to capture complex feature interactions in high-dimensional data. These findings demonstrate that, even under identical feature representations, deep neural architectures such as MLP provide superior learning capacity and generalization ability compared to classical classifiers. This highlights the importance of combining both advanced representation learning and effective model design for accurate effector classification.
To evaluate the effectiveness of our proposed method, we compared it with the recently published SBSM-Pro[65], which integrates raw sequence alignment with multiple kernel learning to classify proteins based on physicochemical properties. SBSM-Pro is specifically designed for protein classification tasks and has demonstrated promising performance across multiple datasets, particularly in identifying protein functions and post-translational modifications. SBSM-Pro achieved a macro-averaged F1-score of only 0.4223 and a weighted F1-score of 0.3885 on the validation set. Its recall for class 1 (T4SE) was particularly low (9.17%), indicating poor sensitivity in detecting T4SEs. In contrast, our model attained a weighted F1-score of 0.850 and a macro F1-score of 0.841 on the validation set, demonstrating both balanced and high performance. Similarly, SBSM-Pro reached a macro F1-score of 0.4248 and a T4SE recall of 8.18% on the test set, whereas our model achieved a macro F1-score of 0.8269 and a weighted F1-score of 0.8325. These results highlight that our approach not only achieved overall better precision and recall but also provided consistently high performance across all effector types, particularly outperforming SBSM-Pro in terms of robustness and generalization.
3.5 Visualization of feature representations using Uniform Manifold Approximation and Projection (UMAP)
We employed UMAP to visualize the protein representations in a two-dimensional space and further assess the discriminative power of the extracted features. The visualization was performed on the fused features generated by the EffecTri model, which combined ESM embeddings, AAC, and DPC descriptors. UMAP is a widely used nonlinear dimensionality reduction technique that preserves both global structure and local relationships in the data, making it well-suited for analyzing high-dimensional biological representations[66-68].
As shown in Figure 4D, the projected representations of T3SEs, T4SEs, and T6SEs form well-separated and compact clusters in the two-dimensional space. Each class exhibited a distinct spatial distribution, indicating that the fused features learned by EffecTri contained sufficient class-specific information to distinguish among the three effector types. There existed minimal overlap between the clusters, which further confirmed the effectiveness of the feature combination strategy in capturing underlying biological differences. The separation among clusters suggests that the multi-view representation (ESM + AAC + DPC) not only improves classification performance but also provides biologically meaningful embeddings. These results reinforce the capability of EffecTri to learn robust and interpretable protein representations for secretory effector classification.
4. Discussion
We proposed EffecTri, a deep learning-based framework for multi-class classification of T3SE, T4SE, and T6SE effector proteins. The model achieved strong performance, with a weighted F1-score of 0.850, surpassing traditional machine learning baselines and several feature combinations. Among the descriptors evaluated, ESM embeddings exhibited the most consistent and robust performance, possibly due to their ability to capture contextual and evolutionary information from protein sequences. Although AAC and DPC performed reasonably well, particularly on T3SE and T4SE, their effectiveness on T6SE was limited, possibly reflecting the structural or functional diversity of this class. Combining AAC and DPC alone did not lead to performance improvement, suggesting limited complementarity between these handcrafted features. In contrast, integrating ESM with either AAC or DPC resulted in noticeable gains, and the full combination of ESM + AAC + DPC achieved the best overall results, confirming the benefit of multi-view representation that leverages both deep contextual and local compositional features. The superior performance of the model over classical methods further highlights the advantage of neural architectures in learning complex, nonlinear relationships in high-dimensional biological data[69]. Hyperparameter tuning also played a key role, with smaller batch sizes, appropriate dropout, and the use of SGD with momentum contributing to stable and effective training. In addition, the quality of the fused representations is supported by UMAP visualization, which showed evident separation between effector classes. Compared with most prior effector predictors that focus on binary settings within a single secretion system (T3SE vs. non-T3SE or T4SE vs. non-T4SE), EffecTri explicitly addresses a unified multi-class scenario (T3SE/T4SE/T6SE), which is closer to practical annotation, where multiple effector types may co-exist in the same organism. In our experiments, ESM embeddings provide the most robust single-descriptor performance, while AAC/DPC offer complementary low-level compositional cues, leading to the best overall results under feature fusion. Unlike structure-aware frameworks, EffecTri remains sequence-only and is thus easier to deploy at scale on large proteomes, although it may benefit from incorporating reliable structural or subtype annotations in future work. Overall, these comparisons position EffecTri as a practical and reproducible multi-class baseline that bridges interpretable handcrafted descriptors and modern protein language model representations for secretion effector identification. Despite these promising results, this work has several limitations. First, we did not explicitly distinguish injectisome T3SS from flagellar T3SS substrates because such annotations are not consistently available across the integrated sources. Future work will explore subtype-specific modeling when reliable labels become available. Our current benchmark uses a single-label taxonomy. Proteins with potential multi-system secretion are not explicitly modeled due to the lack of consistent multi-label annotations. Second, the available training data are limited and class-imbalanced (with T6SE being the smallest class), which may constrain the model’s ability to fully capture the diversity of minority-class effectors. While weighted metrics and regularization alleviate this issue, performance on rare or underrepresented subclasses may still vary across datasets. Third, because the benchmark is compiled from publicly available resources, it may be biased toward well-studied organisms and effector families, and the integrated labels can be incomplete or noisy. Finally, our evaluation is primarily based on internal splits; broader external validation on independent proteomes or newly published sequences will be an important direction for future work. In addition, the model currently depends solely on sequence-derived features, without incorporating structural, functional, or interaction-level information. Future work will explore the integration of structural and contextual features, as well as the extension of the framework to additional effector types or novel effector discovery tasks in microbial genomics and host–pathogen interaction research.
5. Conclusion
In this study, we developed EffecTri, a robust deep learning framework for accurate multi-class classification of bacterial effector proteins (T3SE, T4SE, T6SE). EffecTri outperformed traditional machine learning methods, achieving a weighted F1-score of 0.850 by effectively integrating deep contextual embeddings with handcrafted sequence descriptors. Our results underline the importance of leveraging both global evolutionary context and local compositional information. Visualization analyses further confirmed the interpretability and distinctiveness of the learned representations. While demonstrating significant performance advances, the model currently focuses on sequence-derived features. In the future, enhancements will incorporate structural and functional characteristics to broaden EffecTri’s applicability in microbial genomics and host–pathogen interaction research.
Authors contribution
Li J: Conceptualization, methodology, software, data curation, formal analysis, investigation, visualization, writing-original draft, writing-review & editing.
Su X: Data curation, investigation, validation, visualization.
Liu Q: Methodology, formal analysis, validation.
Wang Y: Conceptualization, supervision, funding acquisition, project administration, resources, writing-review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Data supporting the findings of this study are available from the corresponding author upon reasonable request.
Funding
The work was supported by the National Natural Science Foundation of China (Grant NO. 32270786 and No. 62371403), the Key Science and Technology Research Project of Foshan (Grant No. 2120001008276) and the Foshan Birth Defects Prevention and Control Engineering Technology Research Center (Grant No. 2420001000293).
Copyright
© The Author(s) 2026.
References
-
2. Polanco C, Uversky VN, Huberman A, Vargas-Alarcon G, Buhse T, Marquez MF, et al. Bioinformatics study of the DNA and RNA viruses infecting plants and Bacteria that could potentially affect animals and humans. Curr Bioinform. 2023;18(2):170-191.[DOI]
-
5. Christie PJ, Whitaker N, González-Rivera C. Mechanism and structure of the bacterial type IV secretion systems. Biochim Biophys Acta BBA Mol Cell Res. 2014;1843(8):1578-1591.[DOI]
-
15. Wagner N, Alburquerque M, Ecker N, Dotan E, Zerah B, Pena MM, et al. Natural language processing approach to model the secretion signal of type III effectors. Front Plant Sci. 2022;13:1024405.[DOI]
-
19. Xiong Y, Wang Q, Yang J, Zhu X, Wei DQ. PredT4SE-stack: Prediction of bacterial type IV secreted effectors from protein sequences using a stacked ensemble method. Front Microbiol. 2018;9:2571.[DOI]
-
20. Acici K, Asuroglu T. Unsupervised learning for characterizing type IV secreted effectors. In: 2024 4th International Conference on Applied Artificial Intelligence (ICAPAI); 2024 Apr 16; Halden, Norway. Piscataway: IEEE; 2024. p. 1-6.[DOI]
-
23. Yu L, Liu F, Li Y, Luo J, Jing R. DeepT3_4: A hybrid deep neural network model for the distinction between bacterial type III and IV secreted effectors. Front Microbiol. 2021;12:605782.[DOI]
-
26. Esna Ashari Z, Brayton KA, Broschat SL. Prediction of T4SS effector proteins for anaplasma phagocytophilum using OPT4e, a new software tool. Front Microbiol. 2019;10:1391.[DOI]
-
27. Zhang Y, Guan J, Li C, Wang Z, Deng Z, Gasser RB, et al. DeepSecE: A deep-learning-based framework for multiclass prediction of secreted proteins in gram-negative bacteria. Research. 2023;6:258.[DOI]
-
28. Chen T, Wang X, Chu Y, Wang Y, Jiang M, Wei DQ, et al. T4SE-XGB: Interpretable sequence-based prediction of type IV secreted effectors using eXtreme gradient boosting algorithm. Front Microbiol. 2020;11:580382.[DOI]
-
29. Hu Y, Wang Y, Hu X, Chao H, Li S, Ni Q, et al. T4SEpp: A pipeline integrating protein language models to predict bacterial type IV secreted effectors. Comput Struct Biotechnol J. 2024;23:801-812.[DOI]
-
30. Tang X, Luo L, Wang S. TSE-ARF: An adaptive prediction method of effectors across secretion system types. Anal Biochem. 2024;686:115407.[DOI]
-
31. Li W, Godzik A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658-1659.[DOI]
-
32. Coordinators NR. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2012;41(D1):D8-D20.[DOI]
-
34. An Y, Wang J, Li C, Revote J, Zhang Y, Naderer T, et al. SecretEPDB: A comprehensive web-based resource for secreted effector proteins of the bacterial types III, IV and VI secretion systems. Sci Rep. 2017;7:41031.[DOI]
-
37. Zou X, Ren L, Cai P, Zhang Y, Ding H, Deng K, et al. Accurately identifying hemagglutinin using sequence information and machine learning methods. Front Med. 2023;10:1281880.[DOI]
-
38. Keskar NS, Mudigere D, Nocedal J, Smelyanskiy M, Tang PTP. On large-batch training for deep learning: Generalization gap and sharp minima. arXiv:1609.04836 [Preprint]. 2016.[DOI]
-
39. Goodfellow I, Bengio Y, Courville A, Bengio Y. Deep learning. Cambridge: MIT Press; 2016. Available from: https://mitpress.mit.edu/9780262035613/deep-learning/
-
40. Srivastava N, Hinton GE, Krizhevsky A, Sutskever I, Salakhutdinov R. Dropout: A simple way to prevent neural networks from overfitting. J Mach Learn Res. 2014;15(1):1929-1958. Available from: https://scholar.cnki.net/GARJ2014
-
44. Sokolova M, Lapalme G. A systematic analysis of performance measures for classification tasks. Inf Process Manag. 2009;45(4):427-437.[DOI]
-
45. Zhang HQ, Arif M, Thafar MA, Albaradei S, Cai P, Zhang Y, et al. PMPred-AE: A computational model for the detection and interpretation of pathological myopia based on artificial intelligence. Front Med. 2025;12:1529335.[DOI]
-
46. Dao F, Lebeau B, Ling CCY, Yang M, Xie X, Fullwood MJ, et al. RepliChrom: Interpretable machine learning predicts cancer-associated enhancer-promoter interactions using DNA replication timing. iMeta. 2025;4(4):e70052.[DOI]
-
47. Liu M, Li C, Chen R, Cao D, Zeng X. Geometric deep learning for drug discovery. Expert Syst Appl. 2024;240:122498.[DOI]
-
48. Chou KC. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins. 2001;43(3):246-255.[DOI]
-
51. Oren Y, Smith MB, Johns NI, Kaplan Zeevi M, Biran D, Ron EZ, et al. Transfer of noncoding DNA drives regulatory rewiring in bacteria. Proc Natl Acad Sci U S A. 2014;111(45):16112-16117.[DOI]
-
55. Mentari O, Shujaat M, Tayara H, Chong KT. Toxicity prediction for immune thrombocytopenia caused by drugs based on logistic regression with feature importance. Curr Bioinform. 2024;19(7):641-650.[DOI]
-
58. Lv Z, Zhang J, Ding H, Zou Q. RF-PseU: A random forest predictor for RNA pseudouridine sites. Front Bioeng Biotechnol. 2020;8:134.[DOI]
-
61. Kumar Meher P, Hati S, Sahu TK, Pradhan U, Gupta A, Rath SN. SVM-root: Identification of root-associated proteins in plants byEmploying the support vector machine with sequence-derived features. Curr Bioinform. 2024;19(1):91-102.[DOI]
-
63. Zhao L, Song S, Wang P, Wang C, Wang J, Guo M. A MLP-Mixer and mixture of expert model for remaining useful life prediction of lithium-ion batteries. Front Comput Sci. 2023;18(5):185329.[DOI]
-
64. Li W, Wu Y, Liu Y, Pan W, Ming Z. BMLP: Behavior-aware MLP for heterogeneous sequential recommendation. Front Comput Sci. 2024;18(3):183341.[DOI]
-
65. Wang Y, Zhai Y, Ding Y, Zou Q. SBSM-Pro: Support bio-sequence machine for proteins. Sci China Inf Sci. 2024;67(11):212106.[DOI]
-
66. McInnes L, Healy J, Melville J. UMAP: Uniform manifold approximation and projection for dimension reduction. arXiv:1802.03426 [Preprint]. 2018.[DOI]
-
68. Ai C, Yang H, Liu X, Dong R, Ding Y, Guo F. MTMol-GPT: De novo multi-target molecular generation with transformer-based generative adversarial imitation learning. PLoS Comput Biol. 2024;20(6):e1012229.[DOI]
-
69. Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform. 2016;bbw068.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Li J, Su X, Liu Q, Wang Y. Multi-class pattern discovery for bacterial secretory effectors. Comput Biomed. 2026;1:202522. https://doi.org/10.70401/cbm.2026.0012
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Li J, Su X, Liu Q, Wang Y. Multi-class pattern discovery for bacterial secretory effectors. Comput Biomed. 2026;1:202522. https://doi.org/10.70401/cbm.2026.0012
copy
Share Link
copy