Distilling genomic knowledge into pathology slides for robust cancer survival prediction
*Correspondence to:
Runming Wang, Institute of Biopharmaceutical and Health Engineering, Shenzhen International Graduate School, Tsinghua University, Shenzhen 518055, Guangdong, China.
E-mail: runmingwang@sz.tsinghua.edu.cn
Yongbing Zhang, School of Computer Science and Technology, Harbin Institute of Technology (Shenzhen), Shenzhen 518055, Guangdong, China. E-mail: ybzhang@hit.edu.cn
Yongbing Zhang, School of Computer Science and Technology, Harbin Institute of Technology (Shenzhen), Shenzhen 518055, Guangdong, China. E-mail: ybzhang@hit.edu.cn
Comput Biomed. 2026;1:202527. 10.70401/cbm.2026.0015
Received: December 23, 2025Accepted: April 24, 2026Published: April 29, 2026
This manuscript is made available in its unedited form to allow early access to the
reported findings. Further editing will be completed before final publication. As
such,
the content may include errors, and standard legal disclaimers are applicable.
Abstract
Aims: To develop a robust and clinically feasible framework for cancer survival prediction using only histopathology images while leveraging transcriptomic knowledge during training.
Methods: The study proposed Adaptive Multi-modality Knowledge Distillation (AMKD), a framework designed to transfer complementary molecular-level information from transcriptomic data to pathology-based models. The AMKD framework consists of two essential elements. First, a gene-guided pathology enhancement module is designed to inject genomics-aware information from a multimodal teacher into pathology features. Second, an adaptive redundancy reduction loss is introduced to regulate knowledge distillation by accounting for prediction discrepancies between teacher and student models. This design allows the student model to retain biologically meaningful knowledge during training and remain effective with only histopathology data at inference.
Results: Comprehensive experiments on four The Cancer Genome Atlas (TCGA) cancer cohorts demonstrate that AMKD achieves
Conclusion: The proposed AMKD framework provides a clinically practical solution for robust cancer survival analysis when transcriptomic data are unavailable. By adaptively distilling multi-modal knowledge into a pathology-based model, AMKD bridges the gap between research and clinical applicability, enabling scalable and cost-effective prognostic prediction in real-world settings.
Keywords
Survival analysis, missing modality, knowledge distillation, cancer
1. Introduction
Cancer has become the leading cause of death worldwide and remains a major obstacle to improving human life expectancy[1]. As predicted by the World Health Organization (WHO), the global cancer burden is expected to rise to 28.4 million cases in 2040, a increase from 2020[2]. Survival analysis is to model the time until an event of interest occurs, such as death or disease recurrence[3]. Accurate survival prediction is a critical task in oncology, as it enables risk stratification, prognosis estimation, and treatment planning[4-6], which ultimately improves patient outcomes and optimizes healthcare resources. Therefore, developing robust survival analysis models is of paramount importance in cancer research and clinical practice.
The methods for survival analysis can be broadly categorized into traditional statistical models and modern machine learning approaches. Among all the statistical models, the Cox proportional hazards model[7] is the most widely used one due to its interpretability and ability to handle censored data. In addition, machine learning methods such as survival trees[8,9], support vector machine (SVM)[10-12], and Bayes classifier[13] have achieved better performance in capturing complex relationships among prognostic factors compared with statistical models. However, these methods often fail to capture complex non-linear relationships and interactions among prognostic factors.
With the rapid growth of high-throughput biomedical data, survival analysis has increasingly moved beyond traditional statistical and early machine learning methods toward deep learning-based approaches capable of modeling complex nonlinear relationships[14]. Deep neural networks, particularly those designed for imaging and sequence data, have demonstrated strong capacity in capturing intricate patterns that are difficult to represent using Cox models or classical machine learning[15,16]. At the same time, the availability of multi-omics and whole-slide imaging data has further expanded the scope of survival prediction, prompting the development of modality specific and multimodal frameworks that better exploit the prognostic signals embedded in modern biomedical
Recent studies have demonstrated that patients with different survival outcomes exhibit substantial heterogeneity across multiple biological levels, including pathological morphology, transcriptomic expression, and genetic alterations[19-21]. Accordingly, both histopathology and transcriptomic data have been widely incorporated into survival prediction models, yielding remarkable improvements in identifying prognostic biomarkers and capturing patient-specific risk patterns[19,21,22]. These findings underscore the importance of leveraging diverse biological modalities to accurately model the complex determinants of cancer prognosis.
Multi-modal learning frameworks that integrate pathology images and transcriptomic profiles have consistently shown superior predictive accuracy over unimodal methods[22-24]. This improvement arises from their ability to capture complementary biological information and aggregate diverse prognostic signals across modalities. However, despite their promising results, these multi-modal approaches are often constrained by their dependence on complete modality availability. In real-world clinical practice, missing modalities are common due to the high cost and technical difficulty of acquiring transcriptomic data[25]. In contrast, histopathology slides are routinely available and cost-effective[25]. Therefore, it is of great clinical value to develop models that can be trained with both pathology and transcriptomic data but perform robust survival prediction using only pathology slides during inference.
Addressing this challenge requires an effective strategy for cross-modal knowledge transfer, enabling pathology-based models to benefit from transcriptomic information even when the latter is unavailable at deployment. Although several methods have explored missing-modality learning[26-28], most approaches enforce rigid cross-modal alignment throughout training under the assumption that multi-modal supervision always provides superior guidance[29,30]. This assumption often introduces irrelevant or redundant features from non-informative modalities, thereby diluting survival-specific signals. Moreover, conventional survival analysis frameworks commonly rely on temporal discretization[26,31], which undermines temporal granularity and limits the ability to model dynamic risk variations, particularly in cancers with complex patterns.
To overcome these limitations, we introduce an Adaptive Multi-modality Knowledge Distillation (AMKD) framework grounded in knowledge distillation[32]. AMKD learns genomics-aware pathological representations from whole-slide images (WSIs), enabling accurate survival prediction even when transcriptomic data are unavailable at inference time. While related to knowledge distillation paradigms, AMKD addresses a distinct problem setting: multimodal survival prediction under missing modalities at inference time, with biologically structured supervision. Extensive experiments on four cancer cohorts from The Cancer Genome Atlas (TCGA) demonstrate that AMKD consistently outperforms existing survival analysis approaches. Furthermore, external validation on an independent cohort from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) confirms the robustness and cross-dataset generalization of the proposed framework.
The main contributions of this work can be summarized as follows:
1) We present a novel adaptive knowledge distillation framework that transfers genomic prognostic information into a pathology-only survival model, allowing genomic-informed prediction using WSIs alone during deployment.
2) We design a gene-guided pathology enhancement module that aligns student pathology representations with genomics-informed features learned by a multimodal teacher, facilitating effective cross-modal knowledge transfer without requiring direct genomic input.
3) We introduce an adaptive redundancy reduction loss that dynamically adjusts the distillation process by measuring prediction discrepancies between teacher and student models, thereby mitigating conflicting gradients in survival optimization.
2. Methods
2.1 Survival analysis formulation
Predicting the survival time of cancer patients plays a crucial role in understanding disease progression and guiding clinical decision-making. Survival analysis enables researchers to identify key factors influencing patient outcomes, thereby providing deeper insights into the mechanisms of cancer development and supporting strategies to extend patient life expectancy. Moreover, by evaluating the effectiveness of different therapeutic strategies, survival modeling facilitates the discovery of prognostic biomarkers and the integration of multi-omics data to uncover underlying biological patterns.
A unique challenge in survival analysis lies in the presence of censored data, where the event of interest (typically death) cannot always be observed, either because patients withdraw from the study or the event occurs beyond the study period. Proper handling of censored observations is therefore essential to ensure reliable estimation and avoid biased results.
In summary, constructing accurate survival models is fundamental for predicting patient outcomes, comparing treatment efficacy, and advancing personalized cancer therapy. To build such models, it is essential to first formalize the patient-level clinical data, which can be represented as follows, equation 1:
where Pi denotes the set of whole slide images, Gi represents the corresponding genomic profiles, ci∈{0,1} indicates the right-censoring status, and ti∈R+ denotes the overall survival time of the patient. To estimate the survival distribution, we aim to model the conditional hazard function, defined as equation 2:
where T is a random variable representing the time of event occurrence, and t is a specific time point at which the death event may occur. This function describes the instantaneous risk of experiencing the event at time t, given that the individual has survived up to that time.
Based on the hazard function, the survival probability at time t can be expressed as the cumulative product of survival over previous time steps:
This formulation intuitively captures the probability that the patient survives beyond time t.
To train the survival prediction model, we employ the negative log-likelihood (NLL) loss[33], which is defined as
where ND denotes the total number of patients in the training set. This loss encourages the model to assign higher survival probabilities to censored patients and to accurately estimate the hazard for those who experienced the event.
2.2 Overall framework
As shown in Figure 1, our proposed AMKD framework implements a hierarchical teacher-student paradigm consisting of a multimodal Transformer teacher and a pathology only Transformer student. The teacher model jointly processes transcriptomic profiles and whole slide images to learn genomics aware prognostic representations, and is trained using the survival objective LtSURV.
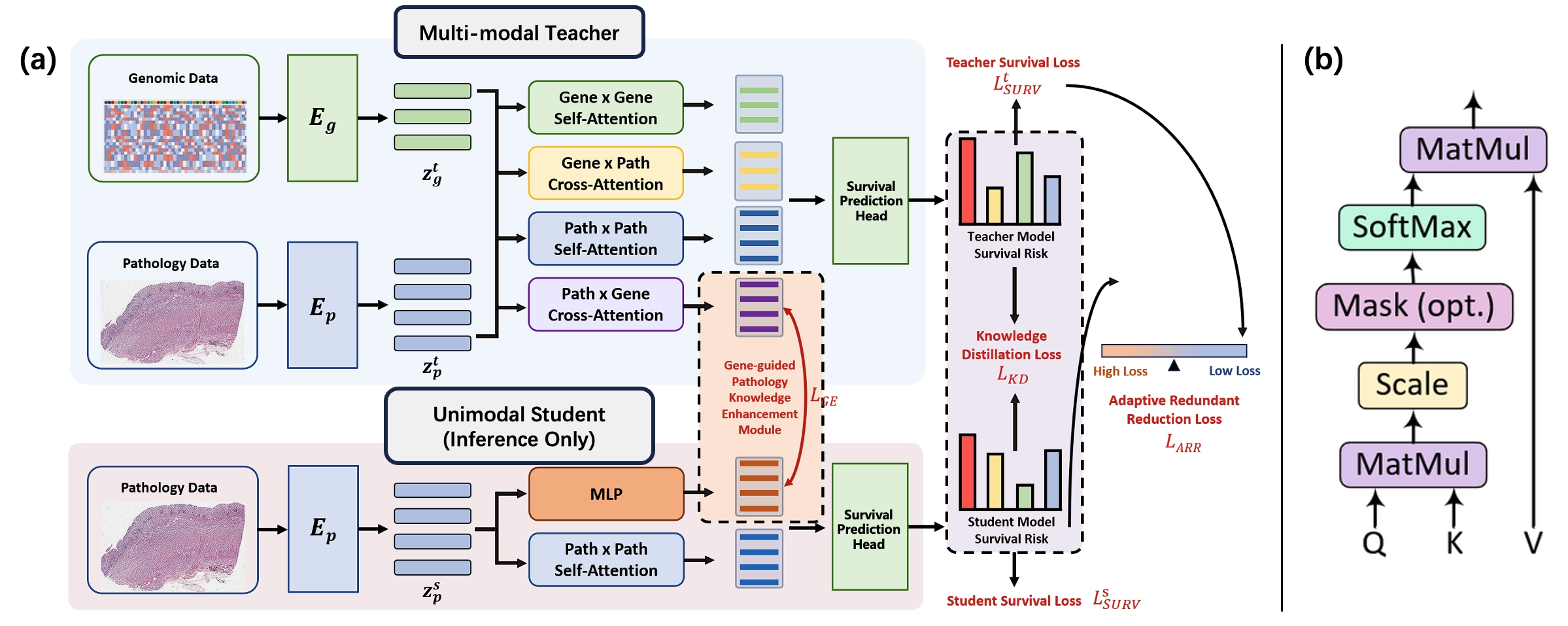
Figure 1. Overview of the proposed AMKD framework. (a) A multimodal teacher learns joint genomic–pathology representations using self-attention and cross-attention modules for survival prediction. A pathology only student model distills cross-modal knowledge from the teacher through a gene-guided enhancement module and is trained with survival, distillation, and redundancy reduction losses. The student operates without genomic data during inference; (b) Scaled dot-product attention mechanism shared by all attention modules. AMKD: Adaptive Multi-modality Knowledge Distillation; MLP: multilayer perceptron.
The student model is specifically designed for practical clinical settings in which only pathology slides are accessible at test time. During training, the student extracts prognostic knowledge from the frozen teacher through two complementary supervision signals:
1) a Gene-guided Pathology Knowledge Enhancement Module regulated by the gene enhancement loss LGE.
2) a standard knowledge distillation objective LKD that aligns survival predictions between teacher and student.
To address diminishing returns in later training stages where the student may outperform the teacher, we introduce the Adaptive Redundant Reduction loss LARR, which dynamically suppresses conflicting gradients arising from teacher-student prediction discrepancies. During inference, the student operates solely on pathology slides while retaining distilled cross-modal prognostic intelligence, achieving multi-modal comparable survival prediction without genomic dependency.
2.3 Multi-modal teacher model
Within the proposed AMKD framework, the multi-modal teacher model is designed to jointly exploit molecular and morphological information for survival modeling. Specifically, the teacher follows a structured three-stage pipeline, beginning with
2.3.1 Feature extraction
For genomic feature extraction, transcriptomic profiles are first transformed into biologically informed pathway-level representations ztg through a Sparse Multi-Layer Perceptron (S-MLP)[34], which serves as our gene encoder Eg. Inspired by SurvPath[22], we organize gene inputs according to predefined biological pathways, enabling the model to preserve functional structure and reduce the noise inherent in high-dimensional gene expression data.
Raw transcriptomic data were obtained from the TCGA cohorts via the UCSC Xena platform[35], where each sample contains expression levels for 60,499 genes. To incorporate biological prior knowledge and reduce dimensionality, genes were organized into functional pathways using curated databases from Reactome[36] and MSigDB-Hallmark[37]. Reactome provides detailed signaling and metabolic pathways, while the Hallmark collection summarizes core biological processes.
To ensure reliable coverage in the TCGA dataset, only pathways with at least of genes present in the transcriptomic profiles were retained. After filtering, 281 pathways from Reactome and 50 pathways from MSigDB-Hallmark were selected, resulting in a total of
The S-MLP encoder maps genes to pathway-level representations through biologically constrained sparse connections, where each pathway node aggregates signals from its member genes. The output dimensionality d = 1,024 corresponds to the embedding size of each pathway representation and is treated as a learnable feature dimension, chosen to balance representational capacity and computational efficiency. This dimensionality is also aligned with the pathology feature dimension to facilitate multimodal fusion.
This process yields structured pathway embeddings that provide a biologically meaningful and noise-resistant genomic representation f or downstream multimodal survival modeling.
where Ng is the number of pathways and d (1024) is the feature dimension.
WSIs are preprocessed using the clustering-constrained-attention multiple-instance learning (CLAM) toolbox[38] to extract 256 × 256 patches at 20 × magnification. Each patch is then encoded using UNI[39], a foundation model trained on large-scale histopathological datasets, to obtain semantically rich patch-level embeddings. UNI is used as a frozen feature extractor to provide standardized pathology representations. Formally, the resulting pathology features are represented as
where Np denotes the total number of extracted patches and d is the embedding dimension. Notably, d is aligned with the dimensionality of the genomic pathway representations to ensure compatibility in the subsequent multimodal fusion stage. This unified feature space enables the model to effectively integrate morphological and molecular information for survival prediction.
2.3.2 Multi-modal fusion
To effectively integrate genomic and pathological information, we employ a hierarchical attention–based fusion module that models both intra-modal contextual structure and inter-modal interactions. This design enables the model to first learn modality-specific representations and then capture complementary signals across modalities, reflecting the biological interplay between molecular pathways and tissue morphology.
Within each modality, we apply a Transformer-style self-attention layer that aggregates contextual information across genes or patches:
where Wqg,Wkg,Wvg∈Rd×d and Wqp,Wkp,Wvp∈Rd×d are learnable projection matrices for queries, keys, and values in genomics and pathology, respectively.
To enable information flow across modalities, we introduce a symmetric cross-attention mechanism. We apply this in two directions to capture complementary biological signals:
• Genomics-to-Pathology attention (Gene × Path): genomic features query pathological patches to produce:
• Pathology-to-Genomics attention (Path × Gene): pathological features query genomic pathways to produce:
Here, Wqp2p and Wqp2g are learnable projection matrices in the cross-attention module, randomly initialized and optimized jointly with the rest of the network during training.
2.3.3 Survival prediction
To jointly leverage both intra-modal information and cross-modal interactions, we concatenate the self-attention and
where || denotes feature-wise concatenation. Since Htatt is still a sequence-level representation, we further apply pooling to obtain a fixed-length patient-level representation before feeding it into the multilayer perceptron (MLP) prediction head.
Since Htatt is still a sequence-level representation whose length depends on the number of genomic pathways and image patches, we apply a pooling operation along the sequence dimension to obtain a fixed-length patient-level representation.
The pooled representation is then fed into a task-specific MLP to produce the survival risk prediction:
The predicted risk ŷsurv is subsequently optimized using the Cox partial likelihood loss, enabling end-to-end learning of both
2.4 Gene-guided pathology knowledge enhancement module
The student model mirrors the teacher’s pathology processing architecture but operates solely on pathology slides during inference. It first extracts patch-level features using the frozen UNI encoder:
which are then refined through a self-attention layer to capture intra-slide contextual relationships:
To distill cross-modal knowledge, the student employs the Gene-guided Pathology Knowledge Enhancement Module. This module projects hsp into a latent space that mimics the teacher’s genomics-informed pathology features htg2p ∈RNp×d producing:
Let
where the softmax is applied along the feature dimension. The Gene Enhancement loss LGE is defined as
where DKL(∙‖∙) denotes the KL Divergence. This enables the student network to implicitly reconstruct genomic-correlated pathological signatures, bridging the modality gap without requiring direct genomic input.
The refined pathology features hsp are then concatenated with the gene-guided features
where σ(∙) is the sigmoid function, Ls and Lt are the student and teacher logits, and is a temperature hyperparameter controlling soft probabilities.
2.5 Adaptive redundant reduction loss
To address the well-known issue that arises when the student model surpasses the teacher, which could cause the traditional distillation objective to impose harmful constraints. We incorporate an adaptive redundant reduction (ARR) loss. This loss dynamically modulates the influence of the teacher based on the mismatch between teacher and student predictions. Formally, ARR is defined as:
where σ(∙) denotes the sigmoid function, and LsSURV, LtSURV represent the student’s and teacher’s Cox negative loglikelihood losses. To ensure numerical stability, a small constant ε = 10-6 is added to the logarithm arguments. The first term increases the reliance on the teacher when the distillation discrepancy LKD is small, indicating that the student is still underperforming and can safely mimic the teacher. In contrast, the second term adaptively down-weights teacher supervision when the student demonstrates stronger survival prediction ability, thereby preventing the teacher from forcing the student back toward an inferior solution. Through this balancing mechanism, ARR effectively reduces redundant or harmful teacher signals while preserving beneficial guidance, enabling more robust and flexible knowledge transfer.
The overall student training objective integrates the standard distillation losses with the adaptive ARR component:
where α∈[0,1] balances the contribution of conventional distillation losses and the adaptive loss. This composite loss ensures stable optimization and enables the student to retain teacher knowledge without being constrained by its limitations.
3. Results
3.1 Datasets and implementation details
The proposed AMKD framework is evaluated on four cancer cohorts obtained from TCGA[40], including breast invasive carcinoma (BRCA), stomach adenocarcinoma (STAD), head and neck squamous cell carcinoma (HNSC), and colon and rectum adenocarcinoma (CRAD). To obtain reliable and unbiased performance estimates, we employ a stratified five-fold cross-validation strategy, ensuring that the proportions of uncensored and censored cases are preserved in each fold. Within each split, samples are partitioned into training, validation, and test sets with ratios of 64%, 16%, and 20%, respectively.
To further assess the generalization ability of the proposed framework beyond TCGA, we additionally introduce an independent external validation cohort from the CPTAC, namely the CPTAC-HNSC dataset. Unlike the TCGA cohorts used for model development, the CPTAC-HNSC cohort is used exclusively for external testing and is not involved in model training, hyperparameter tuning, or cross-validation. This setting provides a more rigorous evaluation of the robustness and transferability of AMKD under cross-dataset distribution shifts. All experiments are conducted using PyTorch 2.5.1 on a single NVIDIA RTX 3090 GPU. Model parameters are optimized with the Adam optimizer, using an initial learning rate of 5 × 10-5 and a weight decay of 1 × 10-5. The AMKD framework is not a pre-training scheme but a task-specific knowledge distillation framework trained end-to-end on the target dataset. The multimodal teacher is first trained using both genomic and pathology modalities. Subsequently, the teacher parameters are frozen, and the pathology-only student is trained under the proposed distillation objectives. Unless otherwise specified, models are trained for 100 epochs, and early stopping based on validation performance is applied.
To ensure fair comparison across methods, all models use the same WSI preprocessing pipeline. Patches are extracted using the CLAM toolbox, and patch-level embeddings are obtained from the UNI foundation model. The UNI encoder is kept frozen, and the resulting features are precomputed and shared across all methods.
Survival prediction performance is primarily assessed using the concordance index (C-index). In addition, Kaplan-Meier survival curves combined with log-rank tests are used to evaluate the statistical significance of risk stratification between predicted low-risk and high-risk patient groups. For the external validation experiment, the model is trained on the TCGA-HNSC cohort and directly evaluated on CPTAC-HNSC without any fine-tuning. The detailed statistics of all datasets are summarized in Table 1.
Table 1. Statistics of the TCGA and CPTAC cohorts used in this study.
| Dataset | Patients | WSIs | Patches |
| TCGA-BRCA | 871 | 929 | 2,608,105 |
| TCGA-STAD | 319 | 343 | 1,089,079 |
| TCGA-HNSC | 394 | 414 | 1,183,322 |
| TCGA-CRAD | 298 | 301 | 981,941 |
| CPTAC-HNSC | 106 | 364 | 1,069,112 |
TCGA: The Cancer Genome Atlas; CPTAC: Clinical Proteomic Tumor Analysis Consortium; WSIs: whole-slide images; BRCA: breast invasive carcinoma; STAD: stomach adenocarcinoma; HNSC: head and neck squamous cell carcinoma; CRAD: colon and rectum adenocarcinoma.
3.2 Comparison with state-of-the-art methods
We benchmark AMKD against a diverse set of representative survival analysis approaches, including unimodal, multimodal, and missing-modality methods, as summarized in Table 2. Across all evaluated TCGA cohorts, AMKD consistently achieves superior performance compared to WSI-only and omics-only baselines, highlighting its ability to effectively distill genomic information into a pathology-based survival model.
Table 2. C-Index (mean ± std) performance of different survival models on four TCGA cancer datasets.
| Modality | Methods | BRCA (↑) | STAD (↑) | HNSC (↑) | CRAD (↑) |
| WSI | ABMIL[41] | 0.589 ± 0.107 | 0.565 ± 0.078 | 0.526 ± 0.065 | 0.607 ± 0.175 |
| TransMIL[42] | 0.617 ± 0.108 | 0.538 ± 0.052 | 0.551 ± 0.057 | 0.629 ± 0.132 | |
| DSMIL[43] | 0.575 ± 0.102 | 0.525 ± 0.031 | 0.544 ± 0.053 | 0.603 ± 0.080 | |
| Omics | SNN[44] | 0.513 ± 0.063 | 0.533 ± 0.052 | 0.563 ± 0.053 | 0.567 ± 0.111 |
| MLP | 0.587 ± 0.120 | 0.485 ± 0.066 | 0.543 ± 0.089 | 0.534 ± 0.059 | |
| SMLP[34] | 0.581 ± 0.050 | 0.570 ± 0.054 | 0.496 ± 0.016 | 0.608 ± 0.140 | |
| Multi-Modal | ABMIL (Cat) | 0.644 ± 0.055 | 0.589 ± 0.089 | 0.567 ± 0.061 | 0.613 ± 0.164 |
| TransMIL (Cat) | 0.616 ± 0.054 | 0.539 ± 0.082 | 0.562 ± 0.025 | 0.624 ± 0.133 | |
| DSMIL (Cat) | 0.568 ± 0.051 | 0.513 ± 0.065 | 0.529 ± 0.047 | 0.615 ± 0.136 | |
| MCAT[24] | 0.520 ± 0.108 | 0.541 ± 0.071 | 0.536 ± 0.068 | 0.575 ± 0.131 | |
| SurvPath[22] | 0.641 ± 0.079 | 0.581 ± 0.068 | 0.546 ± 0.056 | 0.638 ± 0.097 | |
| POMP[45] | 0.700 ± 0.030 | 0.651 ± 0.035 | 0.570 ± 0.060 | 0.645 ± 0.120 | |
| Missing Modal | G-HANet[46] | 0.653 ± 0.048 | 0.603 ± 0.073 | 0.562 ± 0.065 | 0.634 ± 0.105 |
| AMKD (Ours) | 0.693 ± 0.101 | 0.716 ± 0.057 | 0.598 ± 0.032 | 0.668 ± 0.081 |
The best results are shown in bold, and the second best is underlined. TCGA: The Cancer Genome Atlas; WSI: whole-slide image; BRCA: breast invasive carcinoma;
To evaluate the statistical significance of the proposed risk stratification, we conducted log-rank tests between the predicted
Table 3. Log-rank test p-values between the predicted high-risk and low-risk groups for AMKD on all TCGA cohorts.
| Dataset | Log-rank p-value |
| TCGA-BRCA | 7.20 × 10-39 |
| TCGA-STAD | 1.48 × 10-3 |
| TCGA-HNSC | 2.80 × 10-5 |
| TCGA-CRAD | 4.40 × 10-2 |
AMKD: Adaptive Multi-modality Knowledge Distillation; TCGA: The Cancer Genome Atlas; BRCA: breast invasive carcinoma; STAD: stomach adenocarcinoma; HNSC: head and neck squamous cell carcinoma; CRAD: colon and rectum adenocarcinoma.
Figure 2 presents the Kaplan-Meier survival curves across all evaluated cohorts. The corresponding log-rank tests reveal a clear and statistically significant separation between the predicted high-risk and low-risk patient groups, indicating that AMKD provides consistent and reliable prognostic stratification across diverse cancer types.
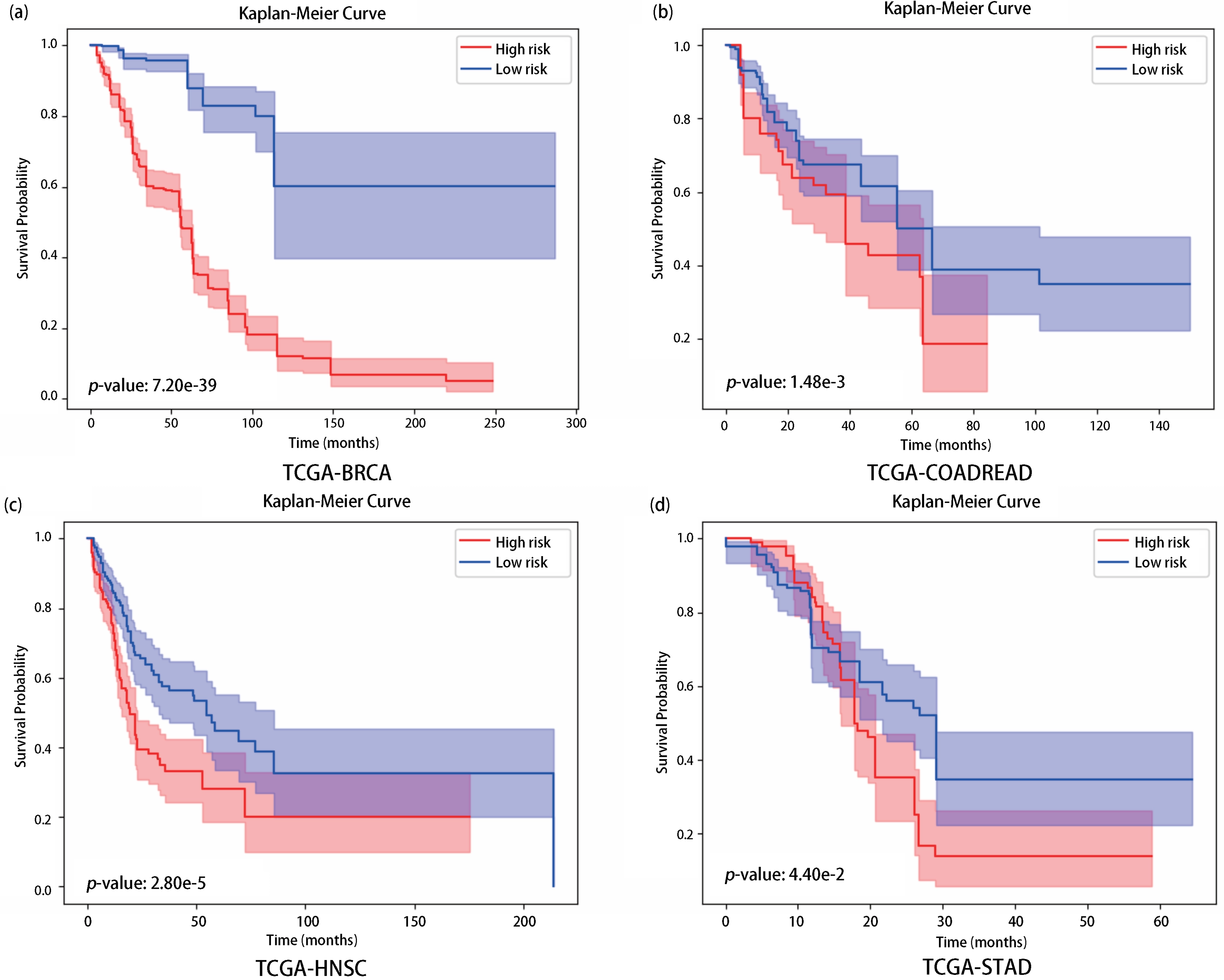
Figure 2. Kaplan-Meier survival analysis on four TCGA datasets: (a) BRCA; (b) CRAD; (c) HNSC; (d) STAD. Logrank tests were performed to assess statistical differences between low-risk (blue) and high-risk (red) groups predicted by AMKD. A p-value < 0.05 indicates significant separation. TCGA: The Cancer Genome Atlas; BRCA: breast invasive carcinoma; CRAD: colon and rectum adenocarcinoma; HNSC: head and neck squamous cell carcinoma; STAD: stomach adenocarcinoma; AMKD: Adaptive
To further examine whether the performance gains of AMKD generalize beyond the TCGA cohorts, we additionally conduct an independent external validation experiment on CPTAC-HNSC, as described in the following subsection.
3.3 External validation on CPTAC-HNSC
To further evaluate the out-of-distribution generalization ability of AMKD, we conduct an independent external validation experiment on the CPTAC-HNSC cohort. Specifically, the model is trained on the TCGA-HNSC cohort and then directly tested on CPTAC-HNSC without any additional fine-tuning. This experimental design is more challenging than in-domain cross-validation, as the two cohorts may differ in patient composition, slide preparation protocols, and data distribution. Table 4 reports the external validation results. AMKD achieves the best performance among all compared methods on CPTAC-HNSC, demonstrating that the proposed adaptive multi-modality knowledge distillation strategy generalizes well beyond the TCGA dataset. Notably, although AMKD relies only on pathology slides at inference time, it still maintains competitive prognostic performance under cross-cohort evaluation, indicating that the genomics-informed knowledge learned from the teacher model is transferable and robust.
Table 4. External validation results on the CPTAC-HNSC cohort. Models are trained on TCGA-HNSC and directly tested on CPTAC-HNSC.
| Methods | CPTAC-HNSC C-index (↑) |
| ABMIL | 0.533 |
| TransMIL | 0.542 |
| DSMIL | 0.521 |
| SNN | 0.518 |
| MLP | 0.525 |
| SMLP | 0.530 |
| ABMIL (Cat) | 0.538 |
| TransMIL (Cat) | 0.535 |
| DSMIL (Cat) | 0.525 |
| MCAT | 0.522 |
| SurvPath | 0.539 |
| POMP | 0.545 |
| G-HANet | 0.540 |
| AMKD (Ours) | 0.558 |
The best results are shown in bold, and the second best is underlined. CPTAC: Clinical Proteomic Tumor Analysis Consortium; HNSC: head and neck squamous cell carcinoma; ABMIL: attention-based multiple instance learning; TransMIL: transformer-based multiple instance learning; DSMIL: dual-stream multiple instance learning;
These results provide additional evidence that AMKD does not merely fit the TCGA data distribution but can also capture prognostically meaningful histopathological patterns that remain effective on an independent external cohort.
3.4 Ablation studies
We conduct a series of ablation experiments across all four TCGA datasets to systematically assess the impact of key components in the proposed framework, including the Adaptive Redundant Reduction loss (LARR), the Gene Enhancement loss (LGE), and the
3.4.1 Effect of LARR and LGE
The ablation results reported in Table 5 demonstrate that jointly removing LARR and LGE causes a pronounced degradation in performance (ΔC-index = -10.6%), underscoring the critical role of the gene-guided pathology knowledge enhancement mechanism in transferring genomic information to the pathology-only student model. When LARR alone is excluded, the performance decline is less severe (ΔC-index = -3.0%), suggesting that the adaptive redundancy reduction loss effectively mitigates conflicting optimization signals between the teacher and student networks.
3.4.2 Effect of multi-modal teacher model
To further assess the importance of the multi-modal teacher design, we replace the proposed teacher with alternative
3.4.3 Ablation study on the balancing coefficient α
We further examine the influence of the balancing coefficient α, which regulates the trade-off between the standard supervision terms (LGE + LKD + LsSURV) and the adaptive redundancy reduction loss LARR. To this end, we conduct ablation experiments with
As reported in Table 6, extreme values of lead to inferior performance, indicating that relying exclusively on either regular supervision or adaptive modulation is insufficient. The best results are obtained when α = 0.75, implying that a balanced combination of conventional distillation objectives and redundancy-aware adjustment yields the most effective learning behavior.
Table 6. C-Index (mean ± std) performance of ablation study on the balancing coefficient α across all datasets.
| α | BRCA | STAD | HNSC | CRAD | Average |
| 0.25 | 0.632 ± 0.095 | 0.664 ± 0.072 | 0.587 ± 0.048 | 0.631 ± 0.088 | 0.629 |
| 0.5 | 0.673 ± 0.087 | 0.654 ± 0.057 | 0.592 ± 0.047 | 0.644 ± 0.107 | 0.641 |
| 0.75 | 0.693 ± 0.101 | 0.716 ± 0.057 | 0.598 ± 0.032 | 0.668 ± 0.081 | 0.669 |
| 1 | 0.666 ± 0.042 | 0.694 ± 0.083 | 0.583 ± 0.036 | 0.654 ± 0.118 | 0.649 |
The best results are shown in bold. BRCA: breast invasive carcinoma; STAD: stomach adenocarcinoma; HNSC: head and neck squamous cell carcinoma; CRAD: colon and rectum adenocarcinoma.
When α = 1, the weight of LARR is effectively zero, so the model does not apply redundancy-aware adjustments. As a result, it relies solely on standard supervision, which weakens its ability to handle redundant information and leads to decreased performance.
In contrast, when α = 0.5 or α = 0.25, LARR exerts excessive influence, dominating over standard supervision. This imbalance can cause the model to overfit redundant features in the data, reducing its overall effectiveness.
3.4.4 Computational efficiency analysis
As shown in Table 7, AMKD has the smallest parameter count (803 K) and low computational cost (1.69 G floating-point operations (FLOPs)), demonstrating its high efficiency. The reported parameter count corresponds to the deployable pathology-only student model. The UNI foundation model is used as a frozen feature extractor to generate patch embeddings, and its parameters are not included. Likewise, the multimodal teacher is used only during training and is not required during inference. In comparison, SurvPath requires 24.8 M parameters and 3.48 G FLOPs, indicating substantially higher computational demands. Both AMKD and SurvPath operate on pathway-level gene features (n = 331), while MCAT, despite using fewer input features, contains more parameters (2.64 M) and slightly lower FLOPs (1.08 G). These results suggest that AMKD achieves an effective balance between representational capacity and computational efficiency while maintaining a compact deployable model footprint.
Table 7. Computational complexity comparison of different models in terms of parameter count and FLOPs.
| Methods | Model Size | FLOPs |
| AMKD | 803 K | 1.69 G |
| SurvPath | 24.8 M | 3.48 G |
| MCAT | 2.64 M | 1.08 G |
FLOPs: floating-point operations; AMKD: Adaptive Multi-modality Knowledge Distillation; MCAT: multimodal co-attention transformer.
3.5 Visualization and interpretability analysis
After completing the result analysis of the AMKD framework, this paper further conducts a comprehensive visualization study to gain deeper insights into the underlying working mechanism of AMKD. In addition, we explore more prognosis-related information to uncover the interactions between histopathological features and genomic patterns, providing valuable insights that may contribute to the advancement of precision medicine in clinical practice.
3.5.1 Comparison of gene-guided pathology knowledge enhancement heatmaps
The Gene-guided Pathology Knowledge Enhancement Module is one of the core components of the AMKD framework. By learning gene-guided pathological representations, this module enhances the student model’s ability to express pathological features, thereby improving its survival prediction performance. To gain a deeper understanding of the working mechanism of this module, we compare the pathological feature representations learned by the Gene-guided Pathology Knowledge Enhancement Module in the AMKD framework with those of the multi-modal teacher model. This comparison allows us to evaluate the extent to which the student model successfully learns from the teacher through this module.
In the STAD dataset, we selected a representative patient’s whole slide image for visualization. Figure 3 presents the patient’s WSI along with the corresponding ground-truth survival time. Based on the pathological features extracted through the Gene-guided Pathology Knowledge Enhancement Module, we generated two heatmaps that illustrate the gene-guided pathological feature representations obtained from the student and teacher models, respectively. As shown in Figure 3, these heatmaps visualize the differences in gene-guided feature expression between the student and the multimodal teacher model.
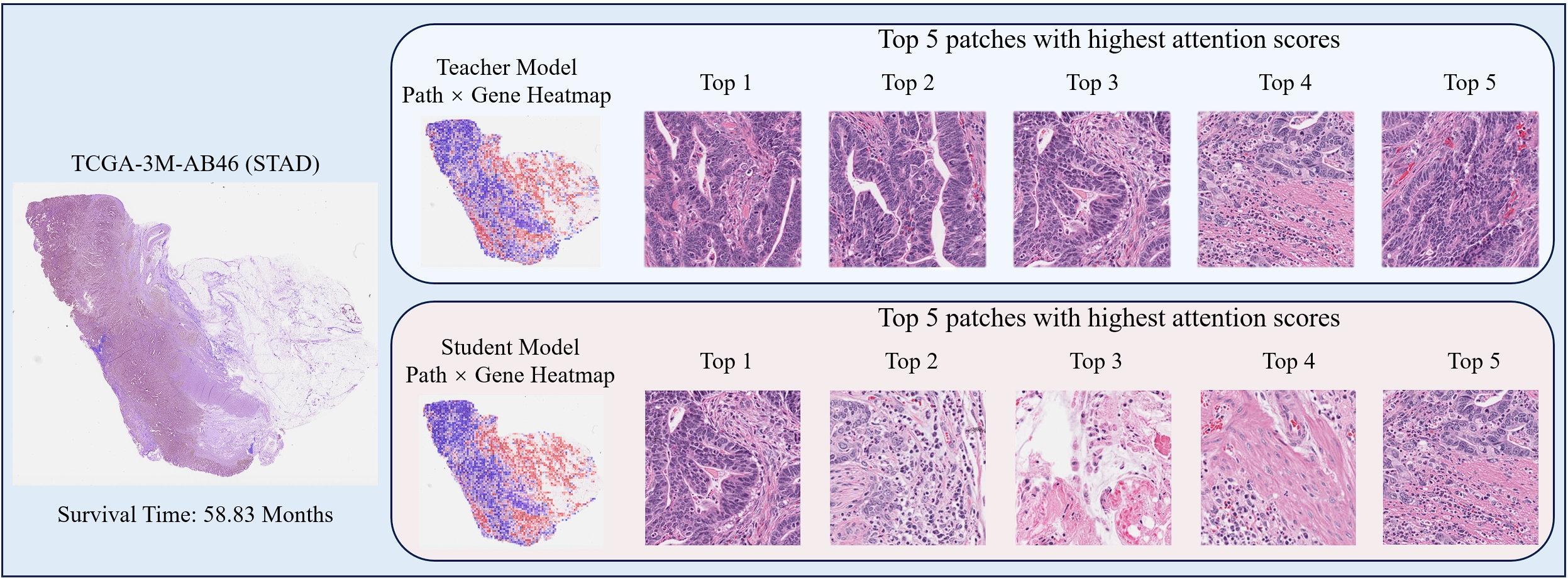
Figure 3. Comparison of the teacher model co-attention weights (top) and learned weights from gene-guided pathology knowledge enhancement module (bottom) of student.
By comparing the two gene-guided pathological feature heatmaps, it can be observed that the gene-guided pathological feature representations of the student model in the AMKD framework are highly similar to those of the teacher model. This indicates that the Gene-guided Pathology Knowledge Enhancement Module effectively enables the student model to learn teacher-like gene-guided pathological representations, thereby improving its pathological feature expressiveness and enhancing survival prediction performance. These results validate the effectiveness of the proposed module.
Furthermore, based on the attention scores, the top five most critical instances identified by both the student and teacher models in the Gene-guided Pathology Knowledge Enhancement Module were selected, as shown in Figure 3. It can be observed that the key instances attended to by the student model are partly consistent with those of the teacher model. Specifically, the teacher’s Top-3 and Top-4 instances overlap with the student’s Top-1 and Top-5 instances. This further demonstrates that the module helps the student model capture gene-guided pathological features similar to those of the teacher, thus enhancing its survival prediction capability.
In addition, the student model attends to a more diverse set of key instances than the teacher model, suggesting that the AMKD framework improves the student’s ability to capture richer prognostic information from pathology images, which further contributes to the performance gains in survival prediction and confirms the effectiveness of the Gene-guided Pathology Knowledge Enhancement Module.
3.5.2 Key gene pathway discovery
Beyond achieving more accurate survival prediction using a single image modality, the AMKD framework also enables the identification of key gene pathways by leveraging the attention scores from the gene-guided pathological knowledge enhancement module. This capability allows for the discovery of additional prognostic information, providing deeper insights for clinical
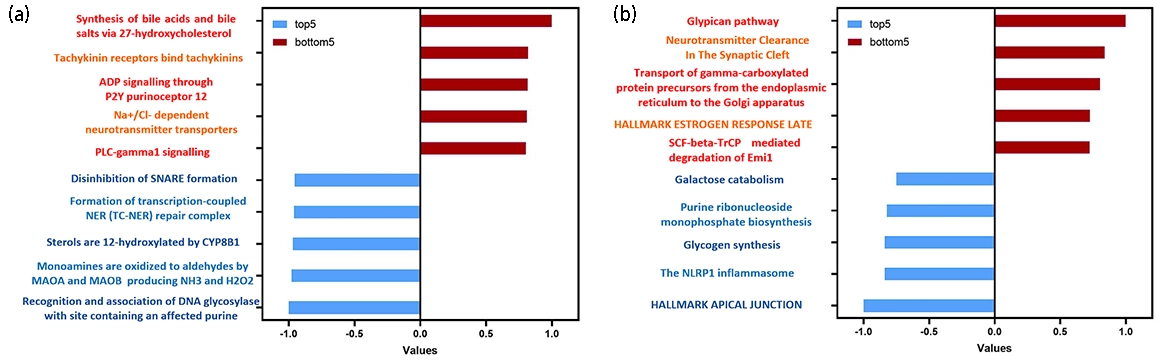
Figure 4. Key gene pathways identified by the AMKD framework. (a) Key gene pathways in the BRCA dataset; (b) Key gene pathways in the STAD dataset. The pathways in each dataset are ranked by their normalized attention scores. Red pathways represent the top five most prognosis-related pathways, while blue pathways denote the five least prognosis related ones. Higher attention scores indicate greater importance of the pathway in survival prediction, whereas lower scores suggest minimal or no relevance to prognosis. AMKD: Adaptive Multi-modality Knowledge Distillation; BRCA: breast invasive carcinoma; STAD: stomach adenocarcinoma.
Furthermore, gene-pathway visualizations were generated for a representative STAD case (TCGA-3M-AB46), as shown in Figure 5. The visualization is generated from the attention scores assigned to image patches by the multimodal fusion module, providing a pathway-conditioned importance map. The pathways with the highest attention scores: bile acid and bile salt synthesis via

Figure 5. Gene pathway-level visualization of prognostic heatmaps. (a) Original WSI image; (b) WSI heatmap of the bile acid and bile salt synthesis via 27-hydroxycholesterol pathway; (c) WSI heatmap of the tachykinin receptor binding pathway; (d) WSI heatmap of the disinhibition of SNARE formation pathway. Red regions indicate high attention scores, suggesting a strong contribution to survival prediction, while blue regions indicate low attention scores, suggesting minimal or no relevance to prognosis.
These results collectively demonstrate that AMKD not only identifies biologically and prognostically meaningful gene pathways but also effectively suppresses irrelevant or redundant ones, enhancing model stability, interpretability, and clinical applicability.
4. Discussion
In this study, we present a multi-modal survival analysis framework that integrates histopathological and transcriptomic information through hierarchical attention and adaptive knowledge distillation. Our results demonstrate that combining modality specific encoders with a bidirectional cross-modal attention mechanism effectively captures complementary prognostic signals that are difficult to recover using single modality approaches. Notably, the gene-pathway encoder provides biologically structured representations inspired by pathway-level organization, while the WSI encoder preserves spatially resolved morphological cues. The synergy between these two modalities yields superior risk stratification performance across multiple cancer cohorts.
Compared with previous pathology-only survival models, our framework benefits from transcriptomic supervision during training, which helps capture biological processes that may not be directly observable from tissue morphology alone. At the same time, unlike most existing multimodal survival models that require all modalities at inference time, our method distills multimodal knowledge into a pathology-only student model, making it more practical for real clinical deployment where molecular data are often unavailable. In addition, while earlier multimodal studies commonly use simple fusion or co-attention strategies, our bidirectional cross-modal attention explicitly models the interaction between pathway-level molecular representations and histopathological features, leading to a more structured and informative joint representation. A key contribution of our framework lies in the ARR loss, which addresses a common yet overlooked issue in knowledge distillation: student models may outperform the teacher during training. Traditional distillation methods typically force the student to mimic the teacher regardless of the relative quality of their predictions, which may introduce redundant or even harmful supervision. ARR resolves this problem by dynamically adjusting teacher guidance according to the discrepancy between teacher and student outputs. This mechanism not only reduces degradation caused by inferior teacher signals but also preserves useful structural knowledge. Empirically, ARR improves both optimization stability and prognostic performance, especially in heterogeneous cancer cohorts.
Overall, compared with prior work, our study offers three main contributions: it incorporates biologically structured multimodal information into survival modeling, enables multimodal to unimodal knowledge transfer for pathology-only inference, and introduces an adaptive distillation mechanism to better handle unreliable teacher supervision. These features distinguish our method from both conventional pathology-only models and standard multimodal fusion approaches.
Despite the promising results, several limitations remain. First, the model is primarily trained and evaluated on public datasets with limited sample size and diversity, which may constrain generalizability in real-world clinical settings. Second, the current framework focuses on histopathology and transcriptomics, while other potentially informative modalities such as proteomics, metabolomics, radiology, and whole-genome sequencing are not considered. Third, our method assumes that both modalities are available during training, whereas real clinical datasets may also contain missing modalities in the training phase. Finally, the framework relies on pre-extracted features rather than end-to-end optimization, which may limit adaptability across datasets.
Future work will focus on incorporating additional modalities, developing training strategies that explicitly handle missing data, improving interpretability, and validating the framework on larger multi-center real-world cohorts. Overall, this study provides a biologically grounded and clinically practical framework for multimodal survival analysis, with the potential to support more accurate cancer prognosis in real-world settings.
5. Conclusion
In this work, we addressed the practical challenge of incomplete multimodal data in cancer survival analysis by proposing AMKD, an Adaptive Multi-modal Knowledge Distillation framework. The method transfers genomics-informed prognostic knowledge from a multimodal teacher to a pathology-only student through gene-guided cross-modal enhancement, enabling accurate survival prediction when genomic data are unavailable at inference time. To facilitate stable knowledge transfer, we further introduced the ARR loss, which dynamically adjusts teacher supervision and mitigates negative transfer during training.
Extensive experiments on four TCGA cohorts demonstrate that AMKD consistently improves survival prediction under
Overall, AMKD provides a practical and efficient solution for survival modeling in modality-missing scenarios, bridging the gap between multimodal research settings and real-world clinical deployment. Owing to its modular design, the framework can be readily extended to other multimodal biomedical applications, offering a general strategy for leveraging privileged information to enhance unimodal predictive models.
Acknowledgements
The authors used AI-assisted tools during manuscript preparation for language editing, and readability improvement. No AI tools were used for data generation, data analysis, model training, statistical testing, or autonomous interpretation of results. All scientific content, including the study design, methodology, experiments, results, and conclusions, was created, reviewed, and verified by the authors. The authors take full responsibility for the accuracy, originality, and integrity of all contents in the manuscript.
The authors would like to express their sincere gratitude to the Tsinghua Shenzhen International Graduate School (SIGS), the Institute of Biopharmaceutical and Health Engineering (iBHE) of SIGS, and the Harbin Institute of Technology, Shenzhen (HITSZ) for providing the research platforms and facilities.
Authors contribution
Xu Y: Conceptualization, methodology, software, formal analysis, investigation, visualization, writing- -original draft, writing-review & editing.
Cai L, Wang Y: Conceptualization, methodology, formal analysis.
Cheng H: Visualization, investigation, data curation.
Liu F: Formal analysis, investigation.
Li Y, Xue R: Writing-original draft, writing-review & editing.
Zhang Y, Wang R: Supervision, conceptualization, writing-review & editing.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Data supporting the findings of this study are available from the corresponding author upon reasonable request.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) Young Scientists Fund (Grant No. 22307063), the Shenzhen Medical Research Fund (Grant No. A2303013), and the Guangdong Innovative and Entrepreneurial Research Team Program (Grant No. 2023ZT10C040). This work was also supported in part by the National Natural Science Foundation of China (Grant Nos. 62031023 and 62331011).
Copyright
©The Author(s) 2026.
References
-
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209-249.[DOI]
-
3. Kartsonaki C. Survival analysis. Diagn Histopathol. 2016;22(7):263-270.[DOI]
-
5. Bulten W, Kartasalo K, Chen PC, Ström P, Pinckaers H, Nagpal K, et al. Artificial intelligence for diagnosis and Gleason grading of prostate cancer: The PANDA challenge. Nat Med. 2022;28:154-163.[DOI]
-
6. Campanella G, Hanna MG, Geneslaw L, Miraflor A, Werneck Krauss Silva V, Busam KJ, et al. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat Med. 2019;25:1301-1309.[DOI]
-
7. Cox DR. Regression models and life-tables. J R Stat Soc Ser B Stat Methodol. 1972;34(2):187-202.[DOI]
-
9. Bou-Hamad I, Larocque D, Ben-Ameur H. A review of survival trees. Statist Surv. 2011;5:44-71.[DOI]
-
10. Cortes C, Vapnik V. Support-vector networks. Mach Learn. 1995;20(3):273-297.[DOI]
-
11. Van Belle V, Pelckmans K, Suykens JA. Support vector machines for survival analysis. In: Proceedings of the third international conference on computational intelligence in medicine and healthcare (cimed2007). p. 1-8.
-
12. Khan FM, Zubek VB. Support vector regression for censored data (SVRc): A novel tool for survival analysis. In: 2008 eighth IEEE international conference on data mining; 2008 Dec 15-19; Pisa, Italy. Piscataway: IEEE; 2008. p. 863-868.[DOI]
-
13. Fard MJ, Wang P, Chawla S, Reddy CK. A Bayesian perspective on early stage event prediction in longitudinal data. IEEE Trans Knowl Data Eng. 2016;28(12):3126-3139.[DOI]
-
14. Catalano M, D’Angelo A, De Logu F, Nassini R, Generali D, Roviello G. Navigating cancer complexity: Integrative multi-omics methodologies for clinical insights. Clin Med Insights Oncol. 2025;19:11795549251384582.[DOI]
-
18. Zhou H, Zhou F, Chen H. Cohort-individual cooperative learning for multimodal cancer survival analysis. IEEE Trans Med Imaging. 2025;44(2):656-667.[DOI]
-
19. Győrffy B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput Struct Biotechnol J. 2021;19:4101-4109.[DOI]
-
20. Nagy Á, Munkácsy G, Győrffy B. Pancancer survival analysis of cancer hallmark genes. Sci Rep. 2021;11:6047.[DOI]
-
21. Wang Z, Ma J, Gao Q, Bain C, Imoto S, Liò P, et al. Dual-stream multi-dependency graph neural network enables precise cancer survival analysis. Med Image Anal. 2024;97:103252.[DOI]
-
22. Jaume G, Vaidya A, Chen RJ, Williamson DFK, Liang PP, Mahmood F. Modeling dense multimodal interactions between biological pathways and histology for survival prediction. In: 2024 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR); 2024 Jun 16-22; Seattle, USA. Piscataway: IEEE; 2024. p. 11579-11590.[DOI]
-
23. Jiang S, Gan Z, Cai L, Wang Y, Zhang Y. Multimodal cross-task interaction for survival analysis in whole slide pathological images. In: Linguraru MG, Dou Q, Feragen A, Giannarou S, Glocker B, Lekadir K, Schnabel JA, editors. Medical Image Computing and Computer Assisted Intervention – MICCAI 2024. Cham: Springer; 2024. p. 329-339.[DOI]
-
24. Chen RJ, Lu MY, Weng WH, Chen TY, Williamson DF, Manz T, et al. Multimodal co-attention transformer for survival prediction in gigapixel whole slide images. In: 2021 IEEE/CVF international conference on computer vision (ICCV). Piscataway: IEEE; 2021. p. 3995-4005.[DOI]
-
25. Lathe W, Williams J, Mangan M, Karolchik D. Genomic data resources: challenges and promises. Nat Educ. 2008;1(3):2. Available from: https://www.nature.com/scitable/topicpage/genomic-data-resources-challenges-and-promises-743721/
-
26. Xing X, Chen Z, Zhu M, Hou Y, Gao Z, Yuan Y. Discrepancy and gradient-guided multi-modal knowledge distillation for pathological glioma grading. In: Wang L, Dou Q, Fletcher PT, Speidel S, Li S, editors. Medical image computing and computer assisted intervention–MICCAI 2022. Cham: Springer; 2022. p. 636-646.[DOI]
-
30. Guetarni B, Windal F, Benhabiles H, Petit M, Dubois R, Leteurtre E, et al. A vision transformer-based framework for knowledge transfer from multi-modal to mono-modal lymphoma subtyping models. IEEE J Biomed Health Inform. 2024;28(9):5562-5572.[DOI]
-
31. Liu P, Fu B, Ye F, Yang R, Ji L. DSCA: A dual-stream network with cross-attention on whole-slide image Pyramids for cancer prognosis. Expert Syst Appl. 2023;227:120280.[DOI]
-
32. Hinton G, Vinyals O, and Dean J. Distilling the knowledge in a neural network. arXiv:1503.02531 [Preprint]. 2015.[DOI]
-
34. Elmarakeby HA, Hwang J, Arafeh R, Crowdis J, Gang S, Liu D, et al. Biologically informed deep neural network for prostate cancer discovery. Nature. 2021;598:348-352.[DOI]
-
35. Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38:675-678.[DOI]
-
37. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database hallmark gene set collection. Cell Syst. 2015;1(6):417-425.[DOI]
-
38. Lu MY, Williamson DFK, Chen TY, Chen RJ, Barbieri M, Mahmood F. Data-efficient and weakly supervised computational pathology on whole-slide images. Nat Biomed Eng. 2021;5:555-570.[DOI]
-
40. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333-339.[DOI]
-
41. Ilse M, Tomczak J, Welling M. Attention-based deep multiple instance learning. In: Proceedings of the 35th International Conference on Machine Learning; 2018 Jul 10-15; Stockholm, Sweden. Cambridge: Proceedings of Machine Learning Research; 2018. p. 2127-2136. Available from: http://proceedings.mlr.press/v80/ilse18a.html
-
42. Shao Z, Bian H, Chen Y, Wang Y, Zhang J, Ji X, et al. Transmil: Transformer based correlated multiple instance learning for whole slide image classification. In: Ranzato M, Beygelzimer A, Dauphin Y, Liang PS, Wortman Vaughan J, editors. Proceedings of the 35th International Conference on Neural Information Processing Systems; 2021 Dec 6-14; Online. United States: Curran Associates Inc.; 2021. p. 2136-2147. Available from: https://dl.acm.org/doi/abs/10.5555/3540261.3540425
-
43. Li B, Li Y, Eliceiri KW. Dual-stream multiple instance learning network for whole slide image classification with self-supervised contrastive learning. In: 2021 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR); 2021 Jun 20-25; Nashville, USA. Piscataway: IEEE; 2021. p. 14313-14323.[DOI]
-
44. Klambauer G, Unterthiner T, Mayr A. Self-normalizing neural networks. arXiv:1706.02515 [Preprint]. 2017.[DOI]
-
45. Wang S, Zhang S, Lai H, Huo W, Zhang Q. POMP: Pathology-omics multimodal pre-training framework for cancer survival prediction. In: Kwok J, editor. Proceedings of the Thirty-Fourth International Joint Conference on Artificial Intelligence; 2025 Aug 16-22; Montreal, Canada. Karlsruhe: International Joint Conferences on Artificial Intelligence Organization; 2025. p. 7813-7821.[DOI]
-
46. Wang Z, Zhang Y, Xu Y, Imoto S, Chen H, Song J. Histo-genomic knowledge association for cancer prognosis from histopathology whole slide images. IEEE Trans Med Imaging. 2025;44(5):2170-2181.[DOI]
-
47. Grillo PK, Győrffy B, Götte M. Prognostic impact of the glypican family of heparan sulfate proteoglycans on the survival of breast cancer patients. J Cancer Res Clin Oncol. 2021;147(7):1937-1955.[DOI]
-
49. Alshammari FO, Al-Saraireh YM, Youssef AM, AL-sarayra YM, Alrawashdeh HM. Glypican-1 overexpression in different types of breast cancers. OncoTargets Ther. 2021;14:4309-4318.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Xu Y, Cai L, Wang Y, Cheng H, Liu F, Li Y, et al. Distilling genomic knowledge into pathology slides for robust cancer survival prediction. Comput Biomed. 2026;1:202527. https://doi.org/10.70401/cbm.2026.0015
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Xu Y, Cai L, Wang Y, Cheng H, Liu F, Li Y, et al. Distilling genomic knowledge into pathology slides for robust cancer survival prediction. Comput Biomed. 2026;1:202527. https://doi.org/10.70401/cbm.2026.0015
copy
Share Link
copy