The caspase-2 paradox in liver polyploidy and cancer risk
Loretta Dorstyn
*
,
Sharad Kumar
*

*Correspondence to:
Loretta Dorstyn, Centre for Cancer Biology, College of Health, Adelaide University, Adelaide, SA 5005, Australia.
E-mail: loretta.dorstyn@adelaide.edu.au
Sharad Kumar, Centre for Cancer Biology, College of Health, Adelaide University, Adelaide, SA 5005, Australia. E-mail: sharad.kumar@adelaide.edu.au
Sharad Kumar, Centre for Cancer Biology, College of Health, Adelaide University, Adelaide, SA 5005, Australia. E-mail: sharad.kumar@adelaide.edu.au
EXO. 2026;1:202609. 10.70401/EXO.2026.0006
Received: February 25, 2026Accepted: April 13, 2026Published: April 15, 2026
This manuscript is made available in its unedited form to allow early access to the
reported findings. Further editing will be completed before final publication. As
such,
the content may include errors, and standard legal disclaimers are applicable.
Abstract
Caspase-2 is a key genome-surveillance protease that functions to kill or arrest cells with abnormal chromosome content, via
Keywords
Caspases, cell death, polyploidy, aneuploidy, pathogenic ploidy, genomic instability, ferroptosis, inflammation, metabolism, liver disease, hepatocellular carcinoma
1. Introduction
Caspase-2 is one of the most evolutionarily conserved caspases, with established pro-apoptotic functions as both an initiator and effector caspase[1]. Diverse functions for caspase-2 beyond apoptosis are well described, including roles in genomic stability, DNA repair, oxidative stress, metabolism, and tumour suppression[1,2]. Recent work shows that caspase-2 activation during mitotic catastrophe[3] is a pivotal safeguard mechanism to eliminate cells with aberrant chromosome content. This includes aneuploid cells generated through chromosome mis-segregation and polyploid cells formed after failed cytokinesis[4]. Our recent work using caspase-2 knockout (Casp2-/-) and catalytically inactive (Casp2C320S) mice highlights the physiological importance of this mechanism in maintaining liver homeostasis during ageing. We demonstrate that caspase-2-dependent ploidy sensing mechanisms are essential for maintaining hepatic genome integrity, that protects against liver damage, chronic disease and the development of age-associated hepatocellular carcinoma (HCC)[5].
A central protective function of caspase-2 is its ability to eliminate cells with aneuploidy[6,7], polyploidy[8], or pathogenic ploidy states, by triggering apoptosis[5]. This acts as a critical mitotic surveillance function to limit chromosome instability[1,6,7]. Activation of caspase-2 is mediated by homodimerization via its conserved amino-terminal, caspase activation and recruitment domain followed by subsequent autocatalytic cleavage[2]. Following genotoxic or mitotic stress, caspase-2 dimerizes and becomes activated through recruitment to the PIDDosome complex comprising the C-terminal cleaved fragment of p53-induced death domain protein 1 (PIDD1) and receptor interacting protein-associated protein with a death domain (RAIDD)[2,9,10]. Importantly, caspase-2 can also be activated independently of the PIDDosome[2]. Similarly, PIDD1 and RAIDD can function independently of caspase-2 under certain stress conditions and cellular contexts[11-13].
The mechanisms that activate caspase-2 in response to either aneuploidy or polyploidy are thought to be distinct and drive different cellular fates[1]. Aneuploidy, caused by spindle assembly checkpoint (SAC) defects or chromosome missegregation, triggers
Polyploidy is tightly governed by the PIDDosome–caspase-2–MDM2-p53 signalling axis that is activated in the presence of supernumerary centrosomes and/or acytokinetic mitosis[4,9,15,16]. Following failed cytokinesis, Ankyrin Repeat Domain 26 recruits PIDD1 to the distal appendages of centrioles, facilitating PIDDosome assembly, caspase-2 activation and consequent MDM2 cleavage, thus stabilising p53[17,18]. This mechanism serves as a critical checkpoint to prevent hyperpolyploidization and limit proliferation of genomically unstable cells[4]. These data collectively establish caspase-2 activation in response to different mitotic failure signals, as a key mechanism for both aneuploidy and polyploidy surveillance.
Caspase-2’s mitotic surveillance role presents a physiological paradox in the liver, a tissue that largely depends on high levels of polyploidy to support regeneration and ageing homeostasis[5,19]. While caspase-2 activity normally constrains hepatocyte polyploidy, our work shows that caspase-2 loss disrupts the hepatocyte ploidy balance by permitting uncontrolled aneuploidy and pathogenic ploidy accumulation. These aberrant ploidy states drive liver inflammation, ferroptosis, fibrosis, and ultimately promote the development of HCC (Figure 1)[5]. Together, these findings provide a direct physiological link between caspase-2 deficiency, pathogenic liver polyploidy and age-associated liver cancer. They also highlight how impaired mitotic surveillance determines
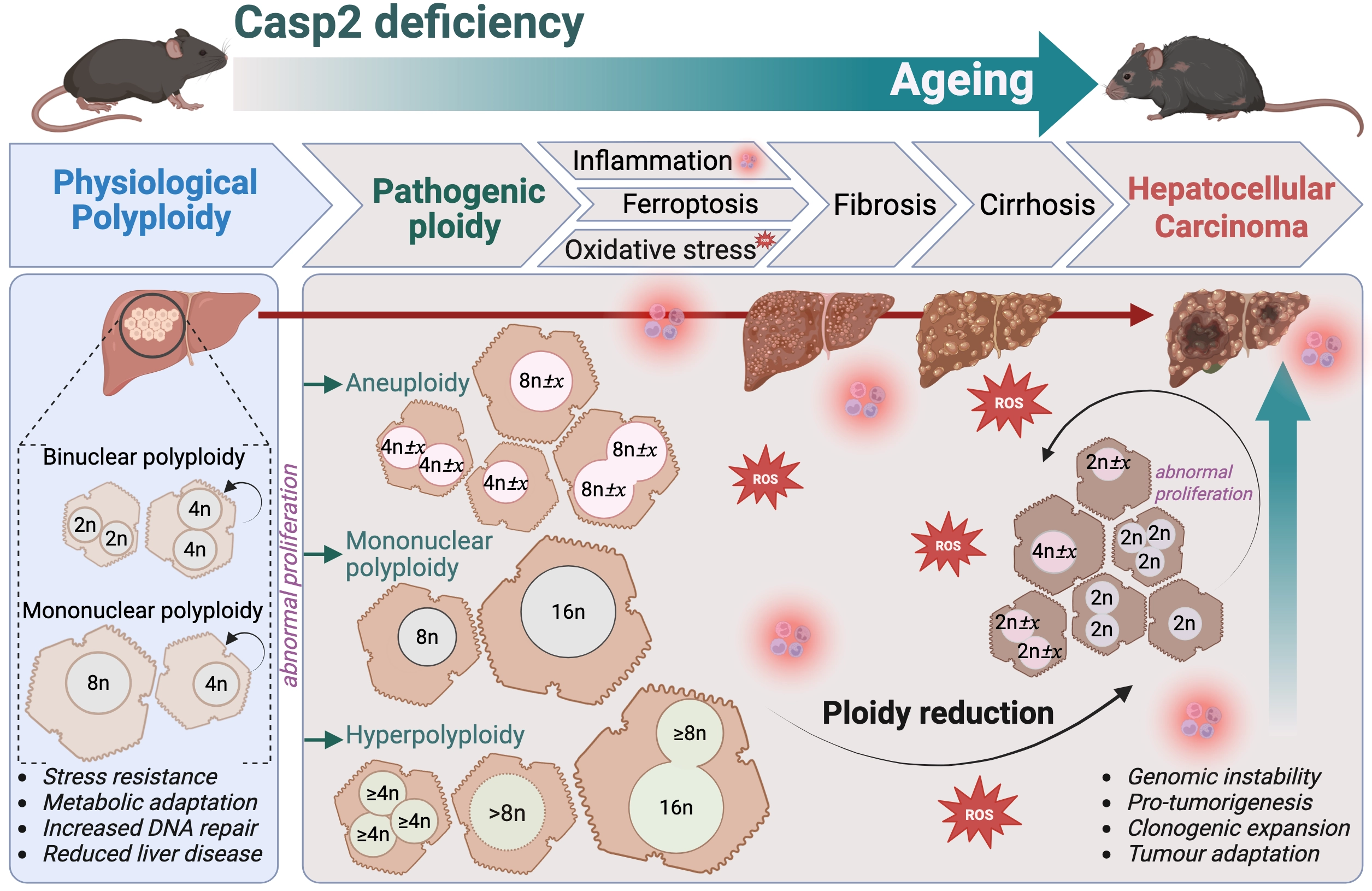
Figure 1. Caspase-2 deficiency drives pathogenic polyploidy-induced liver disease and HCC during ageing. Loss of caspase-2 activity enhances hepatocyte polyploidy that fosters short-term physiological adaptation but also enables survival of cells with aberrant ploidy states, including aneuploidy, mononuclear polyploidy and hyperpolyploidy. Accumulation of these abnormal highly polyploid hepatocytes together with ploidy reduction events in proliferating polyploid cells promotes inflammation, oxidative injury and ferroptosis. These processes collectively drive chronic liver disease (fibrosis, cirrhosis) and create a pro-tumorigenic microenvironment that elevates HCC susceptibility during ageing. Created in BioRender.com. HCC: hepatocellular carcinoma.
2. Polyploidy: A Double-Edged Sword in Liver Biology
Liver polyploidy, a feature described over a century ago, is one of the best characterized mammalian polyploid tissues[19]. Polyploidy primarily arises through endoreplication, where DNA duplication occurs in the absence of cell division or cytokinesis[22]. Polyploidization provides genomic buffering and redundancy[15,19], enabling hepatocytes to tolerate DNA damage, oxidative stress and metabolic overload[19,23]. These functions become increasingly important as regenerative capacity declines with ageing. Previous studies have demonstrated that hepatocyte aneuploidy is also selectively amplified in liver disease as a protective adaptation or “resistance” mechanism to chronic insults[24]. This has led to the established “ploidy conveyor” model, describing the dynamic relationship between polyploidy and aneuploidy in providing genetic diversity in the liver[25]. Our findings refine this model by showing that hepatocyte polyploidy and aneuploidy are not inherently protective. Instead, ploidy balance determines whether genomic heterogeneity promotes adaptation or shifts toward pathological instability that increases age-related liver damage and tumour susceptibility[5].
Polyploidization during liver development and regeneration is tightly regulated by the E2F-PIDDosome-p53 axis[16]. Mice lacking Pidd1, Casp2 or Tp53 show markedly elevated hepatocyte polyploidy[5,16]. Our recent work further demonstrates that the role of caspase-2 is essential for maintaining ploidy homeostasis during physiological ageing in the mouse liver[5]. Instead of promoting beneficial adaptation, loss of Casp2 gene or persistent loss of caspase-2 enzymatic activity disrupts the liver polyploidy/aneuploidy balance, leading to early-onset pathological ploidy in young mice[5]. Critically, caspase-2-deficient livers accumulate both highly aneuploid and hyperpolyploid hepatocytes, mononuclear and binuclear, with octaploid (8c) or higher chromosome content, detectable by 3-4 months of age[5]. This pathogenic ploidy is the earliest detectable hallmark of caspase-2 deficiency. It appears before, and likely contributes to, increased inflammatory and oxidative tissue damage during ageing[5,26].
Caspase-2 deficiency also leads to aberrant proliferation of hepatocytes and hepatic stellate cells. This is accompanied by dysregulated expression of G2/M cell cycle proteins and a proteomic signature strongly correlated with pathogenic polyploidy and chromosome instability[5]. By 12-months of age, caspase-2-deficient livers show increased cellular senescence, a driver of hepatic steatosis, hyperpolyploidy, fibrosis and poor outcome in metabolic dysfunction-associated liver disease (MAFLD)[5,27]. Significantly, mononucleated polyploid cells, enriched in young caspase-2 deficient livers, are an established hallmark of chronic liver diseases, particularly MAFLD, viral hepatitis and cirrhosis[23,28]. The presence of these cells is indicative of compensatory regeneration and/or aberrant proliferation associated with the severity of chronic liver disease progression in caspase-2-deficient mice. These findings highlight how early cell cycle disruption and adverse ploidy changes converge to establish a pathological state in the liver[5].
Polyploid cells are typically more resistant to DNA damage, having acquired mechanisms to suppress p53- and checkpoint protein kinase, CHK1-mediated apoptotic responses[29,30]. In the absence of caspase-2, reduced levels of p53 are associated with reduced MDM2 cleavage, which stabilises and enhances MDM2 function following cytokinesis failure[8,31]. The caspase-2-MDM2-p53 network is an established integrated quality control pathway that limits pathological ploidy, and disruption in any component of this axis leads to genomic instability and fosters tumorigenesis[5,32]. Although MDM2 primarily acts to negatively regulate p53 through its ubiquitin ligase activity, it also regulates multiple additional substrates involved in cell cycle progression, DNA repair, chromatin organisation and metabolism[32,33]. Consistent with these broad functions, MDM2 amplification or overexpression that is frequently observed in MAFLD and across multiple cancer types including HCC (The Cancer Genome Atlas (TCGA))[32], has oncogenic effects that functionally mimics p53 loss, (and caspase-2 deficiency) to drive cell cycle progression, genome instability, and the persistence of polyploid, tumorigenic cell states[15,32].
Interestingly, caspase-2 deficiency does not completely abrogate MDM2 cleavage in the ageing mouse liver, nor does it markedly alter p53 activation[5]. The persistence of MDM2 cleavage in caspase-2 deficient ageing livers, likely reflects broader compensatory responses to chronic DNA damage. We have previously shown that prolonged loss of caspase-2 augments DNA damage[7] and this can be independent of the MDM2-p53 axis[1]. Alternatively, cleavage of MDM2 can be induced by other proteases, such as caspase-3, that influence its diverse functions[31,34]. It also suggests additional layers of MDM2 regulation, including p53-independent tumour signalling pathways. Together, these findings support a model in which pathogenic hepatocyte ploidy promotes resistance to DNA damage-induced apoptosis, that enables the accumulation of oncogenic alterations and facilitates the emergence of
Our data suggest that excessive hyperploidy combined with aberrant ploidy reduction events, promotes the formation of diploid, and aneuploid clones. These clones are more susceptible to genomic instability and neoplastic transformation (Figure 1)[5,15]. Although polyploid hepatocytes proliferate at a slower rate, they undergo reductive divisions that predispose them to aneuploidy, caused by mitotic errors such as centrosome amplification, multipolar spindle formation, prolonged SAC and chromosome misalignment[19,23,35]. Importantly, caspase-2 activation following each of these aberrant mitosis events is critical to prevent survival of cells with aberrant ploidy through both PIDDosome-dependent and independent mechanisms[1,6,7]. In the absence of caspase-2, these unstable polyploid and aneuploid cells persist and continue to proliferate with errors. Collectively, our new findings underscore the importance of caspase-2 in limiting progressive pathogenic polyploidy and aneuploidy to prevent age-related liver damage, liver disease and HCC development. From a significance perspective, the findings provide insight into liver homeostasis mechanisms and enhance our understanding of the physiological effects of pathological polyploidy during ageing.
3. Polyploidy, Inflammation and Liver Disease
Adaptive and pathogenic polyploidy, have distinct biological consequences, with the latter closely linked to inflammation, fibrosis, and increased age-associated HCC[5]. Hepatocyte polyploidy and inflammation are often interdependent: increased polyploidy can induce DNA damage, which subsequently triggers lymphocytic infiltration and leads to fibrosis[19]. Immune cells, such as megakaryocytes and macrophages, also undergo polyploidization, during differentiation or inflammatory responses, generally as protective or defence mechanisms against physiological stress[15,23].
In caspase-2-deficient mice, hyperpolyploidy and aneuploidy arise before overt signs of immune infiltration, cytokine production and inflammatory signalling[5]. Interestingly, proteomic analysis of caspase-2-deficient livers also shows early upregulation of innate immune markers, associated with increased lymphocyte, macrophage, and neutrophil infiltration. These findings emphasise a tight link between early ploidy alterations and immune activation and highlight how ploidy dynamics shape the hepatic microenvironment.
Pathological polyploidy is a defined clinical feature and hallmark of chronic liver diseases, associated with stress-adaptability, tumour cell survival and therapy resistance[15,19,23,25]. In MAFLD, hepatocyte polyploidization correlates with dysregulated lipid metabolism, inflammation, mitochondrial dysfunction and ferroptosis[19]. Omics profiling has previously suggested that highly polyploid hepatocytes have reduced mitochondrial oxidative phosphorylation (indicative of glycolytic metabolism) and decreased lipogenesis to buffer from metabolic stress and reactive oxygen species (ROS)[36,37]. However, our analysis of ageing caspase-2-deficient livers shows that the metabolic demands of highly polyploid and giant cells (i.e. increased DNA/RNA synthesis and metabolism), elevate ROS production, promoting fibrosis and pathogenic features of liver disease[5]. Notably, pathogenic hyperploidization in human liver disease, is also associated with increased β-oxidation[5], a major driver of mitochondrial ROS and ferroptosis, particularly when antioxidant defences are compromised[38]. Thus, the caspase-2-deficient mouse model closely mirrors the key features of pathological hyperploidization characteristic of human liver disease.
Earlier reports demonstrated that loss of caspase-2 promotes oxidative stress by impairing antioxidant defence systems (e.g. SOD2, GSH-Px, FOXO1, FOXO3)[26,39]. As a result, caspase-2 deficiency enhances age-associated ROS accumulation, DNA damage, lipid and protein oxidation that drive liver damage and accelerated ageing phenotypes in mice[5,26]. We demonstrate that oxidative damage is associated with heightened ferroptosis in both caspase-2 knockout and catalytic mutant mouse livers, characterised by increased lipid peroxidation, increased hepatic iron levels and reduced levels of glutathione peroxidase 4 (GPX4)[5,40]. These findings are consistent with a previous study that discovered a direct role for caspase-2 in ferroptosis inhibition via regulation of
Ferroptosis-inhibitors have shown promise for treating liver disease; however, ferroptosis itself also acts as a tumour suppressive mechanism[41,42]. Liver cancer cells commonly develop ferroptosis resistance by upregulating antioxidant defence pathways (p62/KEAP1/NRF2), and altering tumour suppressive networks (such as WNT/β-catenin; HIPPO/YAP axis, PI3K/AKT/mTOR)[43,44]. Therefore, despite their high iron load and elevated metabolic activity that should predispose cells to ferroptotic stress, tumours remain highly refractory to ferroptosis-inducing therapies, and add increasing complexity to therapeutically target in HCC[42]. In
Despite extensive research, the exact contribution of polyploidization and ploidy reduction to liver disease and HCC is still largely unresolved[19,23,25,49]. Liver cancer is often characterised by an increased proportion of proliferating diploid hepatocytes and elevated mononucleated polyploid hepatocytes that correlate with greater oxidative injury, higher HCC incidence and poorer prognosis[30]. Mouse models of chemically-induced hepatocarcinogenesis further illustrates that caspase-2 loss increases karyomegaly and hepatocyte polyploidy, yet two independent studies reported opposite outcomes: one showing enhanced HCC onset[21] and the other reduced HCC onset[20]. These differences suggest that hepatocyte polyploidy tolerance has defined limits that may depend on
4. Caspase-2 in Liver Disease and Therapeutic Targeting
Caspase-2 has an important role in lipid metabolism, ER stress responses, and the progression of MAFLD/MASH[50-54]. Earlier studies demonstrated that loss of caspase-2 reduces steatosis and protects against lipotoxicity, obesity and high-fat or
In human liver disease and hepatobiliary carcinoma (including HCC), CASP2 mutations and variants are rare; however, TP53 is the most frequently mutated gene in HCC, with a frequency between 30%-60% of cases (TCGA)[56]. Clinical datasets show a striking elevation of CASP2 expression levels across multiple liver pathologies, including viral hepatitis, alcoholic liver disease, MAFLD/MASH, cirrhosis, and solid liver tumours (TCGA). Mechanistically, higher CASP2 in liver disease would augment hepatocyte apoptosis, an established driver of fibrosis and cirrhosis[51]. The mechanisms and functional implications of increased CASP2 levels, including whether this is associated with higher protein levels or enzymatic activity, remain unknown, but may reflect genomic-context specificity or compensatory mechanisms when TP53 function is impaired. Collectively, the mouse steatosis studies, and clinical datasets strongly indicate that CASP2 expression has both diagnostic and potential therapeutic relevance that has prompted interest in targeting CASP2 in metabolic liver disease[57]. Caspase-2 inhibition offers a novel mechanistic approach to reduce hepatocyte apoptosis, while improving fat metabolism and lipolysis, thereby limiting MAFLD progression to steatohepatitis, a key driver of
Conversely, chronic dysregulation of hepatocyte ploidy control is also a key driver of progressive liver disease and markedly increases HCC risk[19,28]. Consistent with this, loss of caspase-2 activity permits the survival and expansion of genomically unstable hyperploid and aneuploid hepatocytes, creating a permissive environment for tumour initiation and subsequent cancer development. Clinical data further support this model, with low CASP2 expression associated with immune dysfunction, characterised by increased infiltration of M2 macrophages, monocyte, endothelial cell, B cells, CD4+ T cells, and common lymphoid progenitors[58]. In addition, higher CASP2 expression correlates with TP53 mutations and poorer prognosis in HCC[57,58]. Together, these findings highlight caspase-2 as a key regulator that maintains the balance between adaptive polyploidy and pathological genome instability in the liver, particularly under conditions of chronic injury and inflammatory stress.
Conceptually, these findings also place caspase-2 at a critical nexus between genome surveillance and metabolic regulation in the liver. Its functions in controlling hepatocyte ploidy and regulating lipid metabolism are mechanistically distinct but ultimately converge on maintaining long-term hepatic homeostasis. Therefore, while short-term caspase-2 inhibition may transiently alleviate steatosis, prolonged loss of caspase-2 promotes genomic instability that amplifies inflammation and oxidative tissue damage[5]. Given that these features are already elevated in liver disease, inhibition of caspase-2 may further aggravate disease progression in MAFLD patients. Together these observations underscore the importance of considering long-term genomic stability and cancer risk when evaluating therapeutic strategies that target caspase-2.
Whether polyploidy acts as a primary driver of MAFLD or instead emerges as a consequence of liver injury and disease progression remains unresolved. Despite extensive efforts to target lipogenesis, inflammation, mitochondrial and oxidative stress, ferroptosis or fibrosis, no current therapies have shown effective clinical benefit on their own and suggest combined therapies may better target HCC[15,59]. Clarifying how caspase-2 roles in genome surveillance intersect with its various functions in regulating oxidative stress, ferroptosis, senescence, inflammatory signalling, and lipid metabolism[1,2] will be essential for understanding the early events that initiate liver disease and promote tumourigenesis. In parallel, determining whether catalytically inactive caspase-2 variants differentially influence these pathways[5] will provide crucial insight into how age-related shifts in hepatocyte ploidy contribute to liver homeostasis, disease susceptibility, and long-term cancer risk.
5. Conclusion and Future Perspectives
Caspase-2 has emerged as a central coordinator of hepatic genome stability, balancing mitotic surveillance, metabolic stress responses, and the unique physiological demands of a polyploid organ. Our work identifies caspase-2 activity as a critical ‘ploidy rheostat’ that safeguards hepatic genome integrity and prevents the transition from adaptive to pathological polyploidy, thereby limiting inflammation, oxidative stress, fibrosis and tumour-promoting conditions that accumulate with age.
While its multifaceted roles in apoptosis, mitotic surveillance and metabolic stress responses likely cooperate to preserve liver homeostasis, they also raise important questions about how caspase-2 distinguishes beneficial polyploid states from the pathological ploidy states that drive tumourigenesis, and how its PIDDosome-independent functions contribute to disease. Addressing these uncertainties, particularly in the context of catalytically inactive caspase-2 and its potential scaffolding roles, will be vital for defining its apoptosis-independent functions, and how hepatocytes balance genomic adaptability to maintain long-term homeostasis in the face of diverse metabolic and oncogenic pressures that drive liver disease. Given the diverse liver cell populations that contribute to liver disease progression and shape the pro-tumourigenic microenvironment, it remains essential to define which cell types rely on caspase-2 and what their cell-specific functions are in driving chronic liver disease and age-related HCC. Addressing these questions provides a framework for future research aimed at resolving how caspase-2 coordinates mitotic fidelity, metabolic stress responses and polyploidy control to preserve liver homeostasis throughout ageing. As metabolic liver disease and HCC continue to rise globally, understanding how caspase-2 integrates these diverse pathways will be essential not only for identifying early biomarkers of liver dysfunction but also for guiding therapeutic strategies that maintain liver health without inadvertently increasing cancer risk.
Acknowledgements
We thank members of the Kumar laboratory for discussions, advice, and comments on the manuscript.
Authors contribution
Dorstyn L: Conceptualization, writing-original draft.
Kumar S: Conceptualization, writing-review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
The work was supported by the NHMRC project Grants (Grant No. APP1144500 to Loretta Dorstyn and Grant No. APP1156601 to Sharad Kumar), the Centre for Cancer Biology Impact Grant, the University of South Australia Support Package, an NHMRC Senior Principal Research Fellowship (Grant No. APP1103006 to Sharad Kumar), and an NHMRC Investigator Grant (Grant No. APP2007739 to Sharad Kumar).
Copyright
© The Author(s) 2026.
References
-
2. Miles M, Kitevska-Ilioski T, Hawkins C. Old and novel functions of caspase-2. Int Rev Cell Mol Biol. 2017;332:155-212.[DOI]
-
6. Dawar S, Lim Y, Puccini J, White M, Thomas P, Bouchier-Hayes L, et al. Caspase-2-mediated cell death is required for deleting aneuploid cells. Oncogene. 2017;36(19):2704-2714.[DOI]
-
8. Rizzotto D, Vigorito V, Rieder P, Gallob F, Moretta GM, Soratroi C, et al. Caspase-2 kills cells with extra centrosomes. Sci Adv. 2024;10(44):eado6607.[DOI]
-
9. Sladky VC, Villunger A. Uncovering the PIDDosome and caspase-2 as regulators of organogenesis and cellular differentiation. Cell Death Differ. 2020;27(7):2037-2047.[DOI]
-
19. Donne R, Saroul-Aïnama M, Cordier P, Celton-Morizur S, Desdouets C. Polyploidy in liver development, homeostasis and disease. Nat Rev Gastroenterol Hepatol. 2020;17(7):391-405.[DOI]
-
22. Shu Z, Row S, Deng WM. Endoreplication: The good, the bad, and the ugly. Trends Cell Biol. 2018;28(6):465-474.[DOI]
-
23. Müller M, May S, Bird TG. Ploidy dynamics increase the risk of liver cancer initiation. Nat Commun. 2021;12:1896.[DOI]
-
24. Duncan AW. Aneuploidy, polyploidy and ploidy reversal in the liver. Semin Cell Dev Biol. 2013;24(4):347-356.[DOI]
-
27. Gentric G, Desdouets C, Celton-Morizur S. Hepatocytes polyploidization and cell cycle control in liver physiopathology. Int J Hepatol. 2012;2012:282430.[DOI]
-
30. Bou-Nader M, Caruso S, Donne R, Celton-Morizur S, Calderaro J, Gentric G, et al. Polyploidy spectrum: A new marker in HCC classification. Gut. 2020;69(2):355-364.[DOI]
-
36. Miettinen TP, Pessa HKJ, Caldez MJ, Fuhrer T, Diril MK, Sauer U, et al. Identification of transcriptional and metabolic programs related to mammalian cell size. Curr Biol. 2014;24(6):598-608.[DOI]
-
38. Linkermann A. Key questions in ferroptosis. Ferroptosis Oxid Stress. 2025;1(1):202503.[DOI]
-
45. Liang Q, Xu S, Fang Y, Wang X, Xiao Y, Wang Y, et al. UFMylation deficiency in hepatocytes activates the KEAP1-NRF2 pathway and contributes to hepatocarcinogenesis. Redox Biol. 2026;90:104046.[DOI]
-
46. Álvarez-Garcia V, Tawil Y, Wise HM, Leslie NR. Mechanisms of PTEN loss in cancer: It’s all about diversity. Semin Cancer Biol. 2019;59:66-79.[DOI]
-
47. Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, et al. PTEN: Multiple functions in human malignant tumors. Front Oncol. 2015;5:24.[DOI]
-
48. Zhao Z, Cui T, Wei F, Zhou Z, Sun Y, Gao C, et al. Wnt/β-Catenin signaling pathway in hepatocellular carcinoma: Pathogenic role and therapeutic target. Front Oncol. 2024;14:1367364.[DOI]
-
53. Wilson CH, Dorstyn L, Kumar S. Fat, sex and caspase-2. Cell Death Dis. 2016;7(3):e2125.[DOI]
-
54. Machado MV, Michelotti GA, Jewell ML, Pereira TA, Xie G, Premont RT, et al. Caspase-2 promotes obesity, the metabolic syndrome and nonalcoholic fatty liver disease. Cell Death Dis. 2016;7(2):e2096.[DOI]
-
56. Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet. 2022;400(10360):1345-1362.[DOI]
-
57. Lopez-Pascual A, Cusachs M, Arechederra M, Berasain C, Herrero C, Ávila MA, et al. Caspase-2 in liver disease and hepatocellular carcinoma. Explor Dig Dis. 2022;1(2):80-96.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Dorstyn L, Kumar S. The caspase-2 paradox in liver polyploidy and cancer risk. EXO. 2026;1:202609. https://doi.org/10.70401/EXO.2026.0006
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Polyploidy: A Double-Edged Sword in Liver Biology
- 3. Polyploidy, Inflammation and Liver Disease
- 4. Caspase-2 in Liver Disease and Therapeutic Targeting
- 5. Conclusion and Future Perspectives
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Dorstyn L, Kumar S. The caspase-2 paradox in liver polyploidy and cancer risk. EXO. 2026;1:202609. https://doi.org/10.70401/EXO.2026.0006
copy
Share Link
copy