The lymphatic endothelial-immune dialogue in cancer and immunotherapy
Diede Houbaert
1,2

,
Kathryn A Jacobs
1,2
,
Patrizia Agostinis
1,2,*
*Correspondence to:
Patrizia Agostinis ,Cell Death Research and Therapy Laboratory, Center for Cancer Biology, VIB, Leuven 3000, Belgium; Department of Cellular and Molecular Medicine, KU Leuven, Leuven 3000, Belgium.
E-mail: patrizia.agostinis@kuleuven.be
EXO. 2026;1:202604. 10.70401/EXO.2026.0007
Received: January 30, 2026Accepted: April 21, 2026Published: April 22, 2026
Abstract
Lymphatic endothelial cells (LECs) and the lymphatic vasculature have evolved from being viewed as passive conduits for fluid drainage and metastatic dissemination to active, dynamic regulators of inflammation and tumor immunity. In solid tumors, both tumor-associated and lymph node (LN)–resident LECs engage in complex interactions with their environment to orchestrate immune processes, including antigen transport and presentation to T cells, leukocyte recruitment and trafficking via chemokine gradients, and local immune modulation through the expression of co-inhibitory ligands such as programmed death-ligand 1 (PD-L1). These multifaceted roles enable LECs to either amplify effector responses or induce tolerance, profoundly influencing the efficacy of cancer immunotherapies depending on their activation state, tissue context, and molecular programming. This minireview synthesizes and discusses recent advances in tumor lymphangiogenesis, the role of LECs and their intensive crosstalk with the immune compartments, in the coordination of anti-tumor immune responses, with particular focus on LEC-autophagy as a lipid metabolic checkpoint controlling lymph node T cell egress, and its far-reaching implications for optimizing immunotherapy outcomes in solid tumors.
Keywords
Lymphatic endothelial cells, lymph node, anti-tumor immunity, autophagy, metabolism, immunotherapy
1. Lymphangiogenesis in Homeostasis
In mammals, the circulatory system comprises a vast vessel network essential for preserving tissue function and overall organism homeostasis. This vascular architecture is organized into two closely interconnected systems: the cardiovascular and the lymphatic systems. Blood vessels deliver oxygen and nutrients to cells throughout the body, whereas lymphatic vessels maintain tissue fluid homeostasis, facilitate dietary lipid absorption, and support immune surveillance[1-3].
Unlike blood capillaries, lymphatic capillaries are discontinuous, single-layered, thin-walled vessels (30-80 µm) that form a one-way network aligned with their draining function[2,4]. Initial lymphatic vessels, connected by ‘button-like’ junctions, trap newly formed lymph, which then flows to pre-collecting and collecting vessels, through lymph nodes (LNs), and back to the blood via the thoracic duct; these primary valves also permit dendritic cell (DC) trafficking to LNs, where DCs act as antigen-presenting cells (APCs)[1,5,6]. In mouse embryos, Sox18 activates Prospero Homeobox 1 (PROX1), which, together with the venous nuclear transcription factor chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII), drives segmental lymphatic endothelial cell (LEC) differentiation in the cardinal vein[7,8]. During this process LECs upregulate the expression of vascular endothelial growth factor receptor 3 (VEGFR3), the main receptor for the lymphangiogenic growth factor vascular endothelial growth factor C (VEGF-C), as well as lymphatic vessel hyaluronan receptor 1 (LYVE1), which facilitates DC migration, and podoplanin, a membrane glycoprotein that promotes DC infiltration via chemokine (C-C motif) ligand 21 (CCL21)[9,10]. Secretion of VEGF-C promotes dorsolateral sprouting, migration and the survival of nascent LECs, leading to the formation of lymph sacs[2].
PROX1 is indispensable for LEC identity, as loss of Prox1 prevents endothelial cells from expressing Vegfr3 and Lyve1, whereas ectopic PROX1 in blood endothelial cells (BECs) induces lymphatic programs and suppresses blood endothelial cell features[10-12]. Coexpression of PROX1, VEGFR3, and LYVE1, is therefore considered a hallmark of final LEC determination[10]. Later lymphatic maturation, including valve formation, is regulated by Neurogenic locus Notch Homolog protein 1 and Forkhead box C2 signaling[13-15].
Lymphangiogenesis, essential during embryogenesis, becomes largely inactive after birth, but can be reactivated during tissue repair and inflammation. In wound healing, macrophages and granulocytes release (lymph)angiogenic factors that stimulate new blood and lymphatic vessel growth. Because newly formed vessels are leaky, newly formed lymphatics help drain excess interstitial fluid and support immune cell transport. When lymphatic function is impaired, immune dysfunction and fluid accumulation may lead to lymphedema[16].
2. Lymphatic Endothelial Cells of the Lymph Node and Lymphocyte Trafficking
LNs are specialized secondary lymphoid organs within the lymphatic system, optimized for coordinating adaptive immune responses through compartmentalized interactions among antigens, antigen-presenting cells, and naïve lymphocytes. Although leukocytes constitute ~95% of LN cellularity, non-hematopoietic stromal populations, including BECs, LECs, and fibroblast reticular cells, play indispensable roles in structural organization, leukocyte positioning, and microenvironmental homeostasis[17,18].
Lymphocyte trafficking in LNs relies on tightly regulated entry from blood or lymph and subsequent exit back to circulation, with naïve cells continually recirculating to sustain immune surveillance. Naïve T cells rapidly scan for antigen-presenting cells and typically remain for only a few hours, whereas B cells can persist longer in LNs before leaving. LN trafficking comprises two main phases: entry into the node and egress into efferent lymph[19].
Most naïve T cells and B cells enter LNs via specialized lymphocyte-attracting blood vessels termed high endothelial venules (HEVs), with a smaller fraction arriving through afferent lymphatics. Entry through HEVs involves L-selectin–mediated interaction with HEV-specific coating named peripheral node addressin, which allows tethering and rolling, followed by chemokine-induced integrin activation, firm adhesion, and transendothelial migration[19-21]. CCL21 on HEVs binds C-C chemokine receptor type 7 (CCR7) on lymphocytes, enhancing lymphocyte function-associated antigen 1 (LFA-1) affinity for intercellular adhesion molecule 1/2 (ICAM-1/2) and enabling parenchymal entry. Within LNs, CCR7 ligands (CCL19/CCL21) guide CCR7+ T cells to the T cell zone, whereas C-X-C motif chemokine ligand 13 (CXCL13) directs CXCR5+ B cells into follicles. All leukocytes, including lymphocytes and dendritic cells, can also access LNs via afferent lymphatics, where LECs attract and guide tissue-activated CCR7-expressing T cells and DCs through the production of CCL19 and CCL21. Consistently, CCR7 deficiency severely impairs this route and compromises immune homeostasis[19,22,23].
In contrast, lymphocyte LN exit via efferent lymphatics is primarily controlled by a sphingosine-1-phosphate (S1P) gradient, which is low inside LNs due to degradative enzymes and high in lymph and blood. Sphingosine-1-phosphate receptor 1 (S1PR1) on T and B cells senses this gradient and drives migration from parenchyma into cortical and medullary sinuses, enabling recirculation; loss of S1PR1 or its desensitization permits T cell entry but blocks efficient egress[24,25]. CCR7 and S1PR1 signaling thus oppose each other: CCR7 promotes retention in the T cell zone, whereas S1PR1 favors movement toward S1P-rich sinuses and exit[25]. Perturbing Gαi-coupled chemokine receptors can alleviate egress defects in S1PR1-deficient cells, indicating that reduced retention can partially compensate for impaired S1P sensing[26]. During circulation, naïve lymphocytes cyclically adjust surface expression: high CCR7 and low S1PR1 favor HEV entry and LN retention, while re-expression of S1PR1 and attenuation of CCR7 signaling at later stages allow cells to follow the S1P gradient out of the node and back into the lymph (Figure 1).
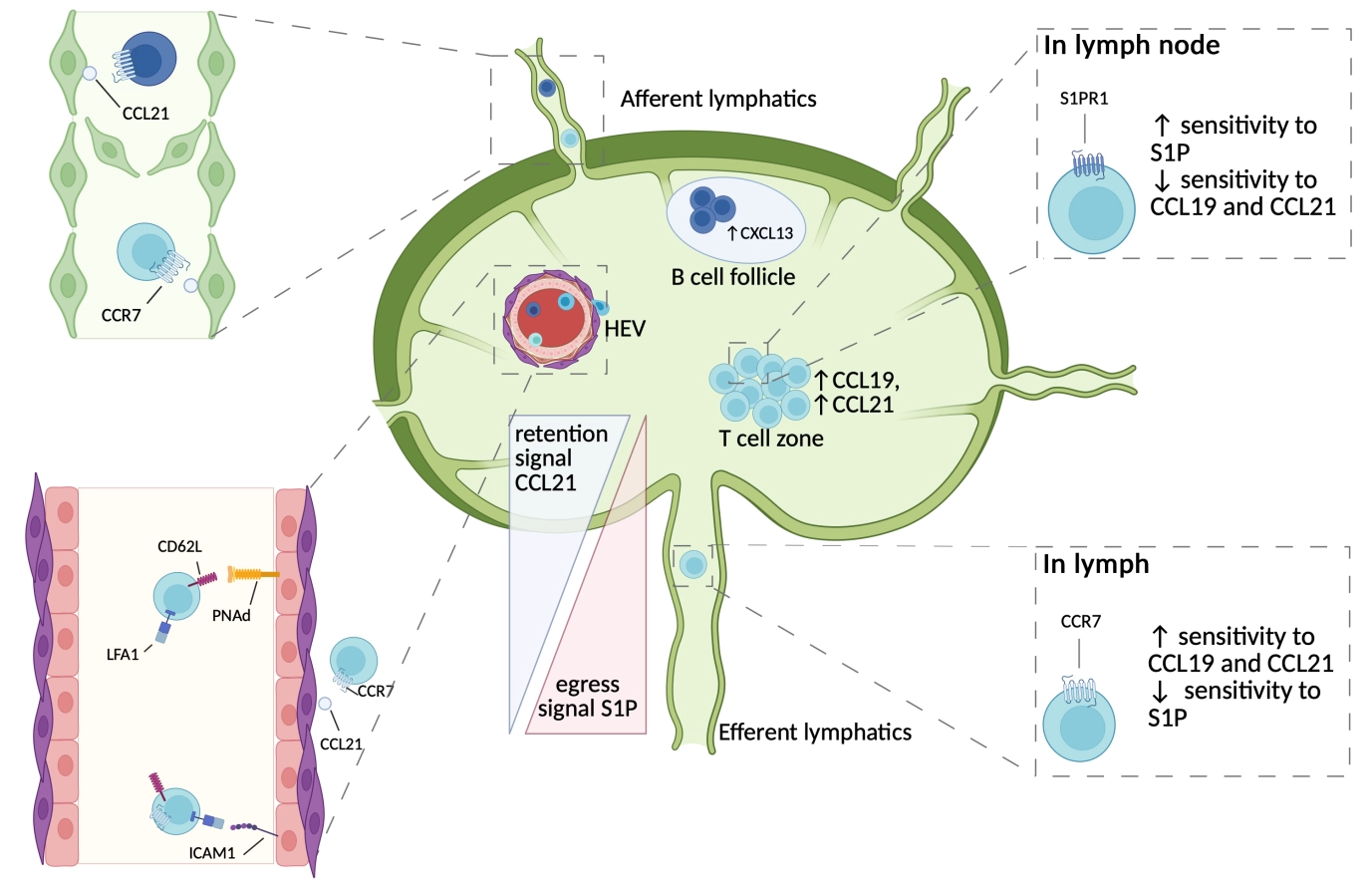
Figure 1. T cell trafficking in the lymph node. Representative scheme of major trafficking mechanisms for B and T lymphocytes enter the LNs. Lymphocyte entry occurs via afferent lymphatic vessels and HEVs in a CCR7-dependent manner. HEVs constitutively express homing molecules (PNAd) allowing the tethering, adhesion and arrest of lymphocytes, followed by their transmigration. Once in the LNs, B cells and T cells will migrate to B cell follicles and T cell zones, guided by CXCL13 or CCL19/21 signaling, respectively. T cells are guided out of the LN by the increasing concentration of S1P egress signal in efferent lymphatic vessels, in combination with a lower CCL21 retention signal. Recirculation is ensured by the transient desensitization of CCR7 or S1PR1, after exposure to CCL21 or S1P, respectively. Other trafficking molecules[1,6] exist but are not depicted for the sake of clarity. LNs: lymph nodes; HEVs: high endothelial venules; CCR7: C-C chemokine receptor type 7; CXCL13: C-X-C motif chemokine ligand 13; CCR7: C-C chemokine receptor type 7; S1PR1: sphingosine-1-phosphate receptor 1; CCL21: chemokine (C-C motif) ligand 21; S1P: sphingosine-1-phosphate; PNAd: peripheral node addressin; CCL19/21: chemokine (C-C motif) ligand 19/21.
Precise segregation of immune cells within LNs is critical for their role in protective immunity, and LECs of the lymphatic vasculature actively orchestrate immune cell movement and spatial organization throughout the nodal microenvironment. In the LN, LECs function as bona fide APCs, presenting antigens on major histocompatibility complex class I (MHC-I) and major histocompatibility complex class II (MHC-II) in the absence of co-stimulatory molecules, therefore inducing CD4 and CD8 T cell tolerance through mechanisms also involving the expression of programmed death-ligand 1 (PD-L1)[27,28].
Recent single-cell RNA sequencing has identified five major Prox1+ lymph node LEC subsets with distinct transcriptional profiles and spatial localization, consistent with discrete and specialized functions. In mouse LNs, these include valve LECs, ceiling LECs (cLECs), floor LECs (fLECs), and two medullary subsets marked by pentraxin 3 (Ptx3) or Marco. cLECs and fLECs reside in the subcapsular sinus (SCS) that forms the outer lymphatic lining, with cLECs facing the capsule and fLECs oriented toward the parenchyma and paracortex, while medullary LECs occupy deeper medullary sinuses. cLECs express high levels of atypical chemokine receptor 4 (Ackr4), which shapes CCL21 gradients and supports the recruitment of CCR7+ cells, in particular DCs, and cluster of differentiation 36 (CD36), enabling low-density lipoprotein uptake. fLECs preferentially express inflammatory chemokines such as CCL20 and ELR+ CXCL family members, aligning with a role in immune cell trafficking. SCS LECs also express mucosal addressin cell adhesion molecule 1 (MADCAM1) and integrin αIIb (ITGA2B/CD41) for lymphocyte adhesion[27,29]. Medullary LECs present self-antigens alongside PD-L1 to delete alloreactive CD8+ T cells, with interferon signaling driving PD-L1 induction[28]. Among the medullary subsets, Marco+ LECs express Cd209, implicated in neutrophil interactions, whereas Ptx3+ LECs show elevated CCL21 and sphingosine kinase 1 (SPHK1), suggesting they are implicated in lymphocyte positioning and egress. Human datasets revealed similar LEC subtypes (cLECs, fLECs, valve LECs, and medullary LECs) with additional bridge LECs and collecting vessel LEC subclusters. Bridge LECs have an intermediate bone morphogenetic protein 2 (BMP2) expression between cLECs and fLECs and likely act as transition cells between the subtypes. Collecting vessel LECs expressed high levels of microfibril-associated protein 4 (MFAP4) and were closely associated with valve LECs[30,31]. A study of human LECs from breast cancer draining and distant LNs, further confirmed the major LEC subtypes in humans with higher resolution. The study additionally identified a metastasis-induced LEC subcluster, characterized by expression of transcription factors HEY1 and SRY-box transcription factor 4 (SOX4) as well as the chemokine CXCL1 and immunosuppressive molecule CD200. These LECs were described as capillary-like and mostly found in metastatic LNs in close contact with cancer cells[32]. Despite this emerging atlas, functional characterization of LN LEC subsets remains incomplete and represents a promising area for future work. Understanding the diversity of homeostatic LECs is essential to appreciating the changes that occur during pathological lymphangiogenesis, and to developing novel therapies to restore LEC homeostasis and function.
3. Lymphangiogenesis in Inflammation
Lymphangiogenesis is tightly linked to inflammation, where extensive remodeling of lymphatic vessels occurs and can either promote or limit immune responses. Lymphatics drain APCs to LNs to initiate immunity but also remove edema and inflammatory cytokines from inflamed tissues[16,33]. In bacterial infection, lipopolysaccharide and inflammatory cytokines induce vascular endothelial growth factor C (VEGF-C) via nuclear factor kappa B (NF-κB) signaling in macrophages and granulocytes[34,35]. In asthma and airway infection, lymphangiogenesis is required to reduce bronchial lymphedema and airflow obstruction[34], whereas in chronic inflammatory diseases such as psoriasis and rheumatoid arthritis, high VEGF-C levels are found in affected joints[16,35].
In organ transplantation, inflammatory cells in the graft, including macrophages, secrete high VEGF-C, inducing lymphatic vessel growth and enhancing APC mobilization through increased lymph flow and CCL21 secretion, which activates CCR7+ T cells and drives acute rejection[36,37]. Blockade of lymphangiogenesis in this context has been shown to inhibit acute graft rejection after organ transplantations[36,37]. While the rationale for blockade of lymphangiogenesis in chronic inflammation is clear, the situation in cancer is substantially more complex.
4. Lymphangiogenesis in Cancer And Antitumor Immunity: A Double-Edged Sword
A functional lymphatic network is essential for effective antigen transport, DC migration, and T cell priming. In line with this, in mice lacking dermal lymphatic capillaries, intradermal melanomas exhibit severely reduced leukocyte infiltration, impaired DC trafficking and antigen presentation in tumor draining LNs (TdLNs), and failure of endogenous adaptive anti-tumor responses[38,39]. Likewise, local ablation of peritumoral lymphatics leads to accumulation of immunosuppressive myeloid cells and Tregs and promotes a suppressive microenvironment[40]. These studies thus indicate that functional lymphatic drainage between the tumor and TdLNs is essential for the coordination of effective anti-tumor immune responses.
However, within the tumor microenvironment (TME), cancer and stromal immune cells produce VEGF-C and vascular endothelial growth factor D (VEGF-D) to induce expansion of pre-existing lymphatics, increasing contact surfaces for cancer–LEC interactions. These lymphangiogenic tumors often display a more immunosuppressive TME with higher infiltration of inhibitory immune populations that dampen cytotoxic T cell function[38,41]. VEGF-C additionally drives lymphangiogenesis in TdLNs, expanding the lymphatic network before cancer cell arrival and establishing a lymphovascular niche or premetastatic LN that facilitates metastatic seeding. LEC-derived CCL21, induced by VEGF-C, can attract both CCR7+ immune cells and CCR7+ cancer cells, further promoting LN colonization[4,39]. In line with this, tumoral lymphatic density also correlates with metastasis and poor prognosis in several cancers, and regional LN metastasis is an early and clinically important marker of cancer dissemination[42]. Moreover, the lymphatic compartment provides a permissive milieu for disseminating cancer cells by lowering oxidative stress during LN homing and reducing susceptibility to ferroptosis, an iron- and lipid peroxide–driven necrotic cell death[43]. Hence, blockade of the VEGF-C/VEGFR3 axis is an attractive anti-metastatic target[1,4,44]. Additionally, a recent study revealed that intratumoral interferon gamma (IFN-γ) blocks lymphangiogenesis, as well as metastasis via metabolic reprogramming of LECs. Altering mitochondrial respiration maintained LECs in a quiescent state, preventing phenotype switch to a tip-like state, which reduced proliferation and prometastatic function[45] (Figure 2).
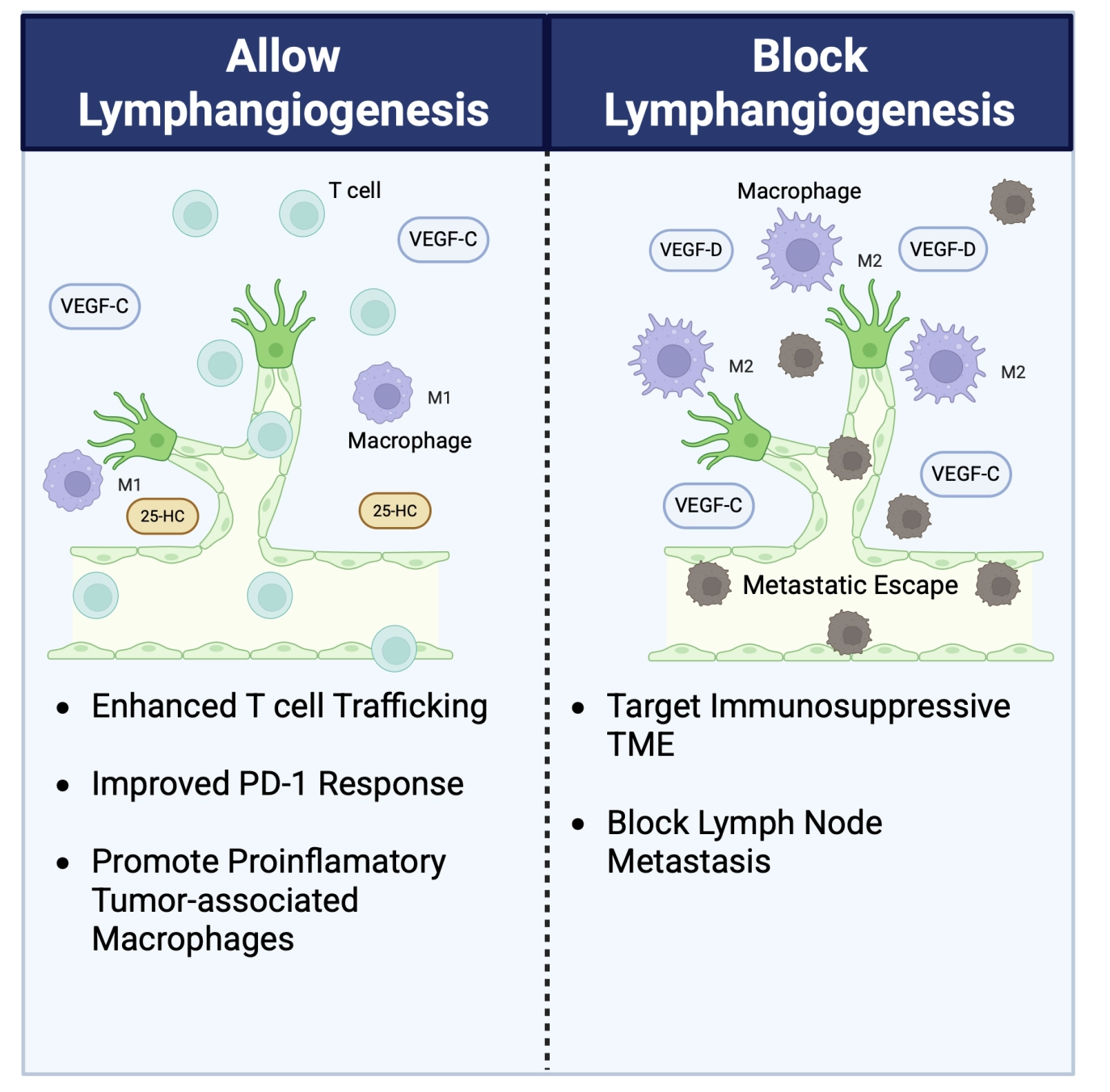
Figure 2. Pros and cons of anti- lymphangiogenic therapy. In support of allowing lymphangiogenic growth, VEGF-C driven lymphangiogenesis can improve T cell trafficking in mouse models of melanoma and improve responses to anti-PD1 therapy. Additionally, lymphangiogenic LECs can release 25-HC to promote proinflammatory M1 polarization. In support of blocking lymphangiogenesis, a tumor milieu with high VEGF-C and VEGF-D can promote an immunosuppressive TME with anti-inflammatory M2 macrophages. Moreover, this lymphangiogenesis can promote cancer cell metastasis to the lymph nodes. VEGF-C: vascular endothelial growth factor C; TME: tumor microenvironmen; LECs: lymphatic endothelial cells; 25-HC: 25-hydroxycholesterol; VEGF-D: vascular endothelial growth factor D.
As antigen-presenting stromal cells, LECs integrate MHC-I/II presentation with PD-L1 and other checkpoints to shape T cell functions through multiple regulatory axes. VEGF-C–driven lymphangiogenesis in melanoma expands LN LECs that cross-present tumor antigens on MHC-I and promote CD8+ T cell tolerance rather than productive immunity[46,47]. MHC-II–restricted antigen presentation by tumor LECs programs regulatory T cells (Tregs) toward a more suppressive phenotype and constrains effector CD4+ and CD8+ T cell accumulation, and genetic ablation of LEC MHC-II markedly slows tumor growth and increases effector tumor infiltrating lymphocytes while reducing Tregs[48,49]. Intratumoral LECs upregulate PD-L1 in response to IFN-γ from tumor antigen-experienced CD8+ T cells, thereby limiting their effector activity[49,50], and control the egress of tumor-specific CD8+ T cells, constraining tumor control[38,41,51,52]. Furthermore, lymphatic-specific PD-L1 selectively deletes tumor-specific CD8+ central memory cells in TdLNs, limiting the size and quality of the anti-tumor T cell pool and diminishing the efficacy of adoptive T cell therapy[28].
However, VEGF-C–dependent lymphatics and VEGF-C–driven lymphangiogenesis can also potentiate anti-tumor immunity and immunotherapy by favoring immune cell trafficking under the right conditions[41,48,53]. In lymphangiogenic melanoma models, tumor cell–derived VEGF-C enhances T cell and DC trafficking via CCL21–CCR7, and improves responses to PD-1 blockade and other immunotherapies[41]. This dynamic view further emphasizes that LECs’ tolerogenic function can be reprogrammed by the strength of the immune response itself and the cancer cell-LEC interactions (Figure 2).
Beyond regulating adaptive immunity, LECs also reprogram the myeloid compartments through immunomodulatory and metabolic cues. In lymphangiogenic melanoma, tumor LECs upregulate cholesterol 25-hydroxylase (Ch25h) to secrete 25-hydroxycholesterol (25-HC), which blocks PPARγ in macrophages and monocytes, preventing their immunosuppressive polarization while promoting a proinflammatory phenotype that enhances effector T cell activity. In human melanoma, LEC Ch25h expression aligns with lymphatic signatures, proinflammatory macrophage infiltration, improved survival, and better immunotherapy responses[54] (Figure 2).
These data add a metabolic layer to LEC-mediated immune regulation and suggest that LEC-derived lipids can tune the balance between suppressive and supportive myeloid cells within the tumor microenvironment. Many mechanistic aspects of cancer-driven lymphangiogenesis remain to be solved, and further work is needed to exploit tumor-associated lymphatic remodeling for therapeutic benefit.
5. Autophagy in Lymphatic Endothelial Cells: A Key Rheostat of Lymphocyte Dynamics and Immunotherapy Responses
Within LNs, LECs orchestrate lymphocyte dynamics (Figure 1). Our recent studies identify a critical role of autophagy, the major lysosomal degradation and recycling pathway[55], in LEC identity, lipid metabolism[56], and systemic control of lymphocyte dynamics[57]. In mice lacking Atg5 in LECs (ATG5LEC-KO), naïve T cells, B cells, and NK cells fail to egress from LNs and spleen, causing systemic lymphopenia and pointing to a disrupted S1P–S1PR1 axis that normally guides lymphocyte exit. Single-cell RNA-seq of TdLNs showed that cLECs along the SCS roof critically depend on autophagy to sustain their transcriptional identity, as Atg5 deletion downregulated hallmark cLEC genes (Ackr4, Ackr3, CD36) while upregulating Lyve1 and Ptx3, markers of paracortical and medullary LECs[29,57,58]. Given the essential role of Ackr4 in maintaining intranodal CCL21 gradients, these autophagy-dependent adaptations of LEC identity perturb lymphocyte guidance, even though bulk LN entry via HEVs appears largely preserved[59]. Functionally, loss of LEC-autophagy alters sphingolipid metabolism and increases S1P secretion within LNs, reshaping NK cell positioning from T cell zones to medullary regions, and preventing the egress of naïve and tumor-experienced T cells from the LNs. Of note, in tumor-bearing mice, LEC-specific deletion of Atg5 reduces peritumoral lymphangiogenesis, dampens the influx of immunosuppressive cells, and delays tumor growth, yet paradoxically renders anti-PD-1 and anti-CTLA4 immunotherapy treatment ineffective by limiting intratumoral T cell recruitment and local reactivation[59] (Figure 3).
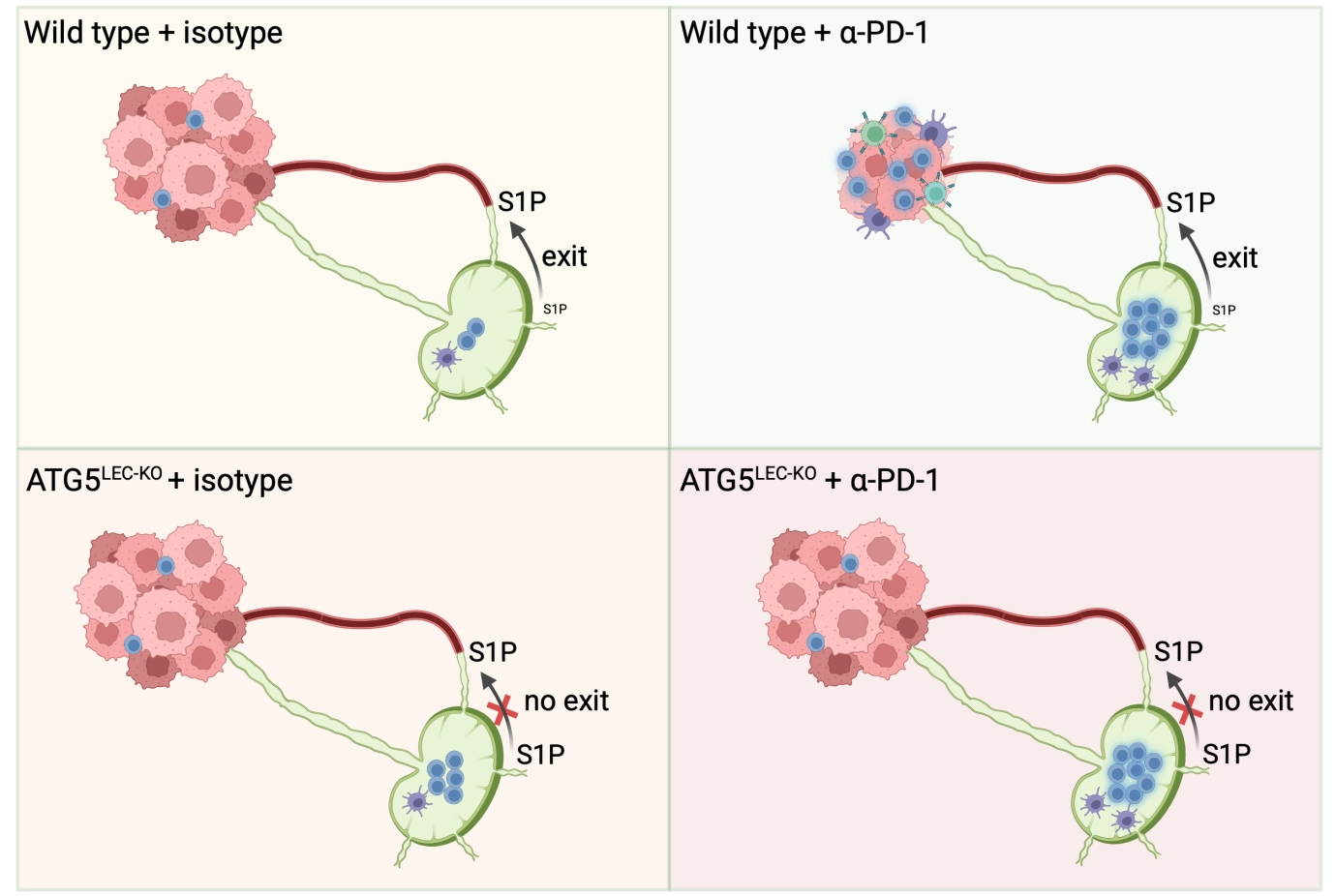
Figure 3. Working model of LEC autophagy’s impact on ICB efficacy. In wild-type tumors, ICB enhances lymphocyte activation in the LN, and the subsequent trafficking of tumor-antigen-experienced T cells within the tumor parenchyma allows tumor burden control. In ATG5LEC-KO mice, the egress of naïve and activated T lymphocytes is prevented due to an imbalance in the S1P gradient within the LN. In these mice, preventing the dynamics of lymphocyte trafficking, blunts the efficacy of ICB-mediated antitumor responses. LEC: lymphatic endothelial cell; ICB: immune checkpoint blockade; LN: lymph node; ATG5LEC-KO: autophagy related gene 5; S1P: sphingosine-1-phosphate.
Consistent with this, pharmacologic manipulation of S1P signaling with agents such as FTY720, which sequester T cells in secondary lymphoid organs, similarly negates immune checkpoint blockade (ICB) efficacy when administered during the priming phase, underscoring that effective lymphocyte egress from TdLNs is a prerequisite for durable anti-tumor responses[19,41,60-62]. Notably, in a model of collagen-induced arthritis, where Atg5 is deleted in LECs, differential Th17 egress is promoted due to their high S1PR1 expression[63]. These findings emphasize that genetic or chemical rewiring of intranodal S1P landscapes can have context-dependent effects on T cell subsets.
Together, these data support a model in which LEC-autophagy integrates lipid metabolic and inflammatory signals to define subset identity, promote lymphangiogenesis, and control S1P-mediated lymphocyte trafficking in a tissue- and context-dependent fashion, with implications for autoimmunity, inflammation, and immunotherapy-driven anti-tumor immunity. Interestingly, a separate work in melanoma-bearing mice has shown that the route and kinetics of lymphatic antigen delivery into TdLNs compartmentalize antigen access to different APC populations and niches, thereby controlling the balance between “stem-like” and terminally differentiated cytotoxic CD8+ T cell pools[64]. Lymph node LECs thus centrally govern both the quantity (via autophagy-tuned egress/retention) and quality (differentiation fate) of T cell responses critical for T cell dynamics and checkpoint blockade efficacy.
6. Concluding Remarks
Tumor lymphangiogenesis critically shapes the local and peripheral tumor microenvironment, yet its impact on cancer dissemination, immunosurveillance, and response to immunotherapy remains context-dependent and difficult to fit into a single paradigm. Recent literature increasingly supports LEC reprogramming rather than blockade as the mechanism to improve patient outcomes under the right circumstances. In support of this, recent work shows that, when spatially and temporally controlled, inducing lymphangiogenesis at defined sites can enhance lymphatic transport, T cell priming in the draining lymph nodes, and the efficacy of cancer vaccines and immune checkpoint blockade. Intriguingly, a similar conceptual work in glioma has shown that boosting meningeal lymphatic drainage with VEGF-C results in enhancement of CD8+ T cell priming in deep cervical LNs, which strongly improves anti–PD1 efficacy[65]. Single-cell RNAseq studies further reveal that LECs can adopt different phenotypes linked to distinct functions that are highly tissue- and context-specific. Recent data further underscore that metabolic plasticity, and in particular lipid metabolism, shapes LEC interactions with immune cells, thereby either amplifying or restraining anti-tumor responses. Collectively, these findings reposition LECs and lymphatic networks as central, programmable nodes in the cancer immune ecosystem. Rather than globally ablating lymphatic vessels, which may impair beneficial priming, particularly under immunotherapy, a more effective strategy is to preserve or induce functional lymphangiogenesis where it supports immunity. This could be achieved by selectively reprogramming LEC-intrinsic pathways (e.g., PD-L1, MHC II/Treg interactions, IFN-γ–driven metabolic programs, autophagy/S1P signaling, Ch25h/25-HC) to convert lymphatics from tolerogenic, pro-metastatic conduits into facilitators of durable anti-tumor responses. Future studies will be crucial to define when and where lymphangiogenesis and LEC reprogramming most effectively control inflammatory and immune responses, and synergize with immunotherapies, to rationally design proper lymphatic-targeted interventions in patients.
Authors contribution
Houbaert D: Visualization, writing-original draft.
Jacobs KA: Visualization, writing-review & editing.
Agostinis P: Writing-original draft, writing-review & editing.
Conflicts of interest
The authors declare no competing interests.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
D.H. is the recipient of an FWO Doctoral Fellowship (FWO-Vlaanderen, 1155121N), Belgium. K.A.J. is the recipient of an FWO Postdoctoral Fellowship (FWO-Vlaanderen, 12Y4322N). P.A. is supported by the C1 KU Leuven Consortium InterAction (Grant No. C14/21/095), FWO grants (G0A3320N and G094922N), the EOS MetaNiche consortium (No. 40007532) and the iBOF/21/053 ATLANTIS consortium.
Copyright
© The Author(s) 2026.
References
-
1. Dieterich Lothar C, Carlotta T, Luca D, Michael D. Lymphatic vessels in cancer. Physiol Rev. 2022;102(4):1837-1879.[DOI]
-
2. Hu Z, Zhao X, Wu Z, Qu B, Yuan M, Xing Y, et al. Lymphatic vessel: Origin, heterogeneity, biological functions and therapeutic targets. Sig Transduct Target Ther. 2024;9:9.[DOI]
-
3. Pugsley MK, Tabrizchi R. The vascular system An overview of structure and function. J Pharmacol Toxicol Meth. 2000;44(2):333-340.[DOI]
-
4. Karakousi T, Mudianto T, Lund AW. Lymphatic vessels in the age of cancer immunotherapy. Nat Rev Cancer. 2024;24(6):363-381.[DOI]
-
5. Liao S, von der Weid PY. Inflammation-induced lymphangiogenesis and lymphatic dysfunction. Angiogenesis. 2014;17(2):325-334.[DOI]
-
6. Oliver G, Kipnis J, Randolph GJ, Harvey NL. The lymphatic vasculature in the 21st century: Novel functional roles in homeostasis and disease. Cell. 2020;182(2):270-296.[DOI]
-
7. François M, Caprini A, Hosking B, Orsenigo F, Wilhelm D, Browne C, et al. Sox18 induces development of the lymphatic vasculature in mice. Nature. 2008;456(7222):643-647.[DOI]
-
8. Srinivasan RS, Geng X, Yang Y, Wang Y, Mukatira S, Studer M, et al. The nuclear hormone receptor Coup-TFII is required for the initiation and early maintenance of Prox1 expression in lymphatic endothelial cells. Genes Dev. 2010;24(7):696-707.[DOI]
-
9. Schulte-Merker S, Sabine A, Petrova TV. Lymphatic vascular morphogenesis in development, physiology, and disease. J Cell Biol. 2011;193(4):607-618.[DOI]
-
10. Wigle JT, Oliver G. Prox1 function is required for the development of the murine lymphatic system. Cell. 1999;98(6):769-778.[DOI]
-
11. Petrova TV. Lymphatic endothelial reprogramming of vascular endothelial cells by the Prox-1 homeobox transcription factor. EMBO J. 2002;21(17):4593-4599.[DOI]
-
12. Hong YK, Harvey N, Noh YH, Schacht V, Hirakawa S, Detmar M, et al. Prox1 is a master control gene in the program specifying lymphatic endothelial cell fate. Dev Dyn. 2002;225(3):351-357.[DOI]
-
13. Petrova TV, Karpanen T, Norrmén C, Mellor R, Tamakoshi T, Finegold D, et al. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat Med. 2004;10(9):974-981.[DOI]
-
14. Murtomaki A, Uh MK, Kitajewski C, Zhao J, Nagasaki T, Shawber CJ, et al. Notch signaling functions in lymphatic valve formation. Development. 2014;141(12):2446-2451.[DOI]
-
15. Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. 2004;5(1):74-80.[DOI]
-
16. Schwager S, Detmar M. Inflammation and lymphatic function. Front Immunol. 2019;10:308.[DOI]
-
17. Onder L, Ludewig B. A fresh view on lymph node organogenesis. Trends Immunol. 2018;39(10):775-787.[DOI]
-
18. Grasso C, Pierie C, Mebius RE, van Baarsen LGM. Lymph node stromal cells: Subsets and functions in health and disease. Trends Immunol. 2021;42(10):920-936.[DOI]
-
19. Martens R, Permanyer M, Werth K, Yu K, Braun A, Halle O, et al. Efficient homing of T cells via afferent lymphatics requires mechanical arrest and integrin-supported chemokine guidance. Nat Commun. 2020;11:1114.[DOI]
-
20. Vella G, Guelfi S, Bergers G. High endothelial venules: A vascular perspective on tertiary lymphoid structures in cancer. Front Immunol. 2021;12:736670.[DOI]
-
21. Blanchard L, Girard JP. High endothelial venules (HEVs) in immunity, inflammation and cancer. Angiogenesis. 2021;24(4):719-753.[DOI]
-
22. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355-360.[DOI]
-
23. Davalos-Misslitz AM, Rieckenberg J, Willenzon S, Worbs T, Kremmer E, Bernhardt G, et al. Generalized multi-organ autoimmunity in CCR7-deficient mice. Eur J Immunol. 2007;37(3):613-622.[DOI]
-
24. Benechet AP, Menon M, Xu D, Samji T, Maher L, Murooka TT, et al. T cell-intrinsic S1PR1 regulates endogenous effector T-cell egress dynamics from lymph nodes during infection. Proc Natl Acad Sci U S A. 2016;113(8):2182-2187.[DOI]
-
25. Baeyens AAL, Schwab SR. Finding a way out: S1P signaling and immune cell migration. Annu Rev Immunol. 2020;38:759-784.[DOI]
-
26. Pham THM, Okada T, Matloubian M, Lo CG, Cyster JG. S1P1 receptor signaling overrides retention mediated by Gαi-coupled receptors to promote T cell egress. Immunity. 2008;28(1):122-133.[DOI]
-
27. Rouhani SJ. Regulation of T-cell tolerance by lymphatic endothelial cells. J Clin Cell Immunol. 2014;5(4):1000242.[DOI]
-
28. Cousin N, Cap S, Dihr M, Tacconi C, Detmar M, Dieterich LC. Lymphatic PD-L1 expression restricts tumor-specific CD8+ T-cell responses. Cancer Res. 2021;81(15):4133-4144.[DOI]
-
29. Xiang M, Grosso RA, Takeda A, Pan J, Bekkhus T, Brulois K, et al. A single-cell transcriptional roadmap of the mouse and human lymph node lymphatic vasculature. Front Cardiovasc Med. 2020;7:52.[DOI]
-
30. Takeda A, Hollmén M, Dermadi D, Pan J, Brulois KF, Kaukonen R, et al. Single-cell survey of human lymphatics unveils marked endothelial cell heterogeneity and mechanisms of homing for neutrophils. Immunity. 2019;51(3):561-572.e5.[DOI]
-
31. Abe Y, Sakata-Yanagimoto M, Fujisawa M, Miyoshi H, Suehara Y, Hattori K, et al. A single-cell atlas of non-haematopoietic cells in human lymph nodes and lymphoma reveals a landscape of stromal remodelling. Nat Cell Biol. 2022;24(4):565-578.[DOI]
-
32. Eichin D, Lehotina D, Kauko A, Uenaka M, Leppänen M, Elima K, et al. Breast cancer remodels lymphatics in sentinel lymph nodes. Nat Commun. 2025;16:10056.[DOI]
-
33. Kang S, Lee SP, Kim KE, Kim HZ, Mémet S, Koh GY. Toll-like receptor 4 in lymphatic endothelial cells contributes to LPS-induced lymphangiogenesis by chemotactic recruitment of macrophages. Blood. 2009;113(11):2605-2613.[DOI]
-
34. Baluk P, Tammela T, Ator E, Lyubynska N, Achen MG, Hicklin DJ, et al. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest. 2005;115(2):247-257.[DOI]
-
35. Paavonen K, Mandelin J, Partanen T, Jussila L, Li TF, Ristimaki A, et al. Vascular endothelial growth factors C and D and their VEGFR-2 and 3 receptors in blood and lymphatic vessels in healthy and arthritic synovium. J Rheumatol. 2002;29(1):39-45.
-
36. Nykänen AI, Sandelin H, Krebs R, Keränen MAI, Tuuminen R, Kärpänen T, et al. Targeting lymphatic vessel activation and CCL21 production by vascular endothelial growth factor receptor-3 inhibition has novel immunomodulatory and antiarteriosclerotic effects in cardiac allografts. Circulation. 2010;121(12):1413-1422.[DOI]
-
37. Yin N, Zhang N, Xu J, Shi Q, Ding Y, Bromberg JS. Targeting lymphangiogenesis after islet transplantation prolongs islet allograft survival. Transplantation. 2011;92(1):25-30.[DOI]
-
38. Lund AW, Wagner M, Fankhauser M, Steinskog ES, Broggi MA, Spranger S, et al. Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J Clin Investig. 2016;126(9):3389-3402.[DOI]
-
39. Delclaux I, Ventre KS, Jones D, Lund AW. The tumor-draining lymph node as a reservoir for systemic immune surveillance. Trends Cancer. 2024;10(1):28-37.[DOI]
-
40. Kataru RP, Ly CL, Shin J, Park HJ, Baik JE, Rehal S, et al. Tumor lymphatic function regulates tumor inflammatory and immunosuppressive microenvironments. Cancer Immunol Res. 2019;7(8):1345-1358.[DOI]
-
41. Fankhauser M, Broggi MAS, Potin L, Bordry N, Jeanbart L, Lund AW, et al. Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci Transl Med. 2017;9(407):eaal4712.[DOI]
-
42. Commerford CD, Dieterich LC, He Y, Hell T, Montoya-Zegarra JA, Noerrelykke SF, et al. Mechanisms of tumor-induced lymphovascular niche formation in draining lymph nodes. Cell Rep. 2018;25(13):3554-3563.e4.[DOI]
-
43. Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature. 2020;585(7823):113-118.[DOI]
-
44. Lei PJ, Fraser C, Jones D, Ubellacker JM, Padera TP. Lymphatic system regulation of anti-cancer immunity and metastasis. Front Immunol. 2024;15:1449291.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Houbaert D, Jacobs KA, Agostinis P. The lymphatic endothelial-immune dialogue in cancer and immunotherapy. EXO. 2026;1:202604. https://doi.org/10.70401/EXO.2026.0007
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Lymphangiogenesis in Homeostasis
- 2. Lymphatic Endothelial Cells of the Lymph Node and Lymphocyte Trafficking
- 3. Lymphangiogenesis in Inflammation
- 4. Lymphangiogenesis in Cancer And Antitumor Immunity: A Double-Edged Sword
- 5. Autophagy in Lymphatic Endothelial Cells: A Key Rheostat of Lymphocyte Dynamics and Immunotherapy Responses
- 6. Concluding Remarks
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Houbaert D, Jacobs KA, Agostinis P. The lymphatic endothelial-immune dialogue in cancer and immunotherapy. EXO. 2026;1:202604. https://doi.org/10.70401/EXO.2026.0007
copy
Share Link
copy