Anchoring virtual cells on gene programs: From biological networks to patient digital twins
Tianhao Wang
1,2

,
Haoran Li
1,2
,
Jie Liao
1,2,*
*Correspondence to:
Jie Liao, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, 310058, China.
E-mail: liaojie@zju.edu.cn
EXO. 2026;1:202610. 10.70401/EXO.2026.0013
Received: February 27, 2026Accepted: June 11, 2026Published: June 11, 2026
Abstract
Predicting developmental trajectories and disease progression is a central goal of virtual biology. While conventional biological network models struggle with noise and computational complexity, black-box deep learning obscures mechanistic interpretability. In this Perspective, we introduce “gene programs” (GPs) as the essential intermediate layer to close this gap. Extracted from massive single-cell datasets, GPs provide relatively robust and interpretable modules that capture coherent biological functions. Supported by recent multi-modal and large-scale perturbation advances, GPs may reflect underlying cross-layer biophysical structures. We propose a framework where next-generation virtual cells encode GPs as latent variables in neural networks. By incorporating spatial multicellular contexts and multimodal GP extraction, this GP-centric framework will reduce data noise and allow direct perturbation-to-phenotype mapping, enabling high-fidelity simulations of complex tissue dynamics. Anchoring virtual models on GPs paves the way for highly personalized “Patient Digital Twins”, empowering predictive in silico clinical simulations and advancing mechanism-driven precision medicine.
Keywords
Virtual cells, gene programs, single-cell multi-omics, spatial multi-omics, precision medicine
1. Introduction
Organismal development and pathogenesis emerge from complex genetic and cellular interactions. Understanding the developmental trajectory of multicellular organisms and predicting pathogenesis are pivotal goals of virtual cell modeling. Over the past two decades, several approaches to virtual cells have been proposed. A conventional approach is modeling the target biological variables with ordinary differential equations[1], which is computationally intractable when the model becomes large. Another strategy is to use interpretable rule-based models that update cell states and predict cell behaviors according to predefined rules, with internal cellular states encoded as functions of environmental signals. However, these methods generally struggle with complex phenotypes involving nonlinear interactions, undefined cell types, unknown behaviors, spatial organization, temporal dynamics, and tissue-level characteristics in clinical data[2]. In contrast, systems biology provides a holistic framework by viewing cells as systems of interacting genes and proteins[3]. Signaling pathways and gene regulatory networks (GRNs) are inferred from gene expression profiles of perturbed cells and used as low-rank approximations of cellular states. Such models have provided valuable insights into transcriptional responses to genetic perturbations or mutations. However, several fundamental limitations make these simulations inadequate. In complex systems, perturbations often have context-dependent, non-additive effects, while conventional models are often static, based on local linear approximations, and do not consider spatial or cell-type heterogeneity[4]. GRNs have achieved some success in holistic modeling but are often limited to being a complex and abstract representation without clear system boundaries[5]. Recently, researchers have started to train virtual cell models by feeding vast amounts of single-cell RNA-seq data into deep generative models or agents. Although these models have shown great value in predicting cellular responses, their black-box nature makes it difficult to explicitly explain the mapping of genes to phenotypic effects and sometimes leads to uncontrolled model behaviors[6]. After all, the detailed principles bridging individual genes and the highly organized biological structures remain poorly understood. Although efforts have been made to organize gene sets to functional modules or “pathways”, these concepts remain nebulous owing to the lack of rigorous defining criteria and validation approaches[7]. Therefore, it is necessary to find a new anchor point for the next-generation virtual models.
2. Gene Programs as a Robust Interpretable Layer for Virtual Modeling
The advent of single-cell genomics has enabled the extraction of “gene programs” (GPs). Originating in the 1960s, this concept shares its roots with GRNs, positing that biomolecules operate modularly[8]. The discovery of GPs, driven primarily by component analysis or non-negative matrix factorization, allows covariant gene expression patterns to naturally emerge from large datasets[9]. These programs are highly interpretable and functionally specific representations of cellular systems. For example, Ling et al. identified a neuron-astrocyte concerted program that is associated with reduced schizophrenia risk and physiologically downregulated in older individuals[10]. Echoing Barabási’s view of diseases as breakdowns of biological modules, this program is enriched for known schizophrenia risk genes, which suggests that aberrant program dynamics might drive pathogenesis[11]. Furthermore, Gavish et al. found that diverse tumor categories shared common GPs that form clusters with defined biological functions[12]. These consistent gene sets, which they called meta-programs (MPs), are de-noised forms of GPs and may reflect fundamental states of cells. In developmental biology, Farrell et al. discovered that statistically inferred GPs form tree-like structures that mirror the cell differentiation trajectories during embryogenesis[13]. Transcription factors (TFs) of GPs can be inferred using SCENIC[12,14], indicating that GPs are coherent functional modules emerging from noisy biological networks and establishing GPs as robust frameworks for genetic research[14,15].
Unlike conventional GRNs, which suffer from high noise, ambiguous boundaries, and computational difficulties[8], GPs are robust to data heterogeneity and can be easily used, once established, for downstream tasks. For example, Zhang et al. showed that established MPs can be used as robust anchors for highly heterogeneous data integration[16]. Puram et al. found GPs directly correlated with tumor metastasis and cancer stages[17]. Other uses of established GPs include cell state scoring (for consistent cell type identification), and identifying new cellular activities[18,19]. These findings establish GPs as reusable and robust representations of biological systems. Crucially, Ota et al. demonstrated that GPs sometimes act as the “intermediate layer” translating genotype to phenotype[20]. Individual traits, such as mean corpuscular hemoglobin, are found to be bidirectionally regulated by programs related to hemoglobin synthesis, cell cycle, and autophagy across various cell types. This implies that GPs may function as the intermediate layer bridging the genome and cellular phenotypes and can potentially be used as computational nodes in virtual cell computation. In this context, we use “computational nodes” to refer to computable model units that represent GP activities and can receive perturbation inputs and update downstream cellular states or phenotypic outputs. Depending on the modeling architecture, such nodes may be implemented as interpretable latent dimensions, graph nodes, or constrained neural modules. For example, linearly decoded variational autoencoders restrict the decoder to a linear configuration, where GPs serve as interpretable variables within the latent space[21]. Similarly, but further extended to spatial modeling, the tool INSPIRE uses graph neural networks to find consistent GPs to integrate non-adjacent spatial transcriptomics (ST) data[22], where GPs are modeled as global basis vectors jointly optimized with the network. By mathematically grounding GPs as either latent variables or structured factors, these approaches provide a blueprint for embedding biological knowledge into computational models, offering capabilities to predict cell states in response to genetic perturbations or environmental signals. Specifically, genetic perturbations can be encoded as vectors over genes, such as one-hot encoding, and linearly projected onto the GP space using gene-to-GP membership weights. Pathway-level perturbations can be encoded similarly as vectors over the corresponding genes. In the simplest linear approximation, a loss-of-function or over-expression perturbation can be modeled as a negative or positive change in the perturbed gene value, which is then projected to GP-level changes according to gene-to-GP membership weights. The updated GP states can then be used to predict the cell responses. In a neural network implementation, perturbations may also be first represented as gene embeddings or functional gene signature representations derived from Perturb-seq or drug-response datasets[23,24]. Subsequently, perturbation effects can be propagated through GP regulatory relationships, spatial cell-cell interaction (CCI) graphs, or through learned state-update functions in different architectures. Here, propagation refers to transmitting the perturbation effects from direct molecular targets to downstream GPs, cell states, or spatial neighborhoods. For example, graph-enhanced gene activation and repression simulator (GEARS) uses graph neural networks trained on Perturb-seq datasets to predict single- or multi-gene perturbation outcomes, where the perturbation effects are propagated through a gene co-expression graph and a pathway-based gene knowledge graph[23]. Similar perturbation encoding and propagation strategies could be adapted to a GP-centered framework.
3. Beyond Single Cells: Multi-Modal and Spatial Dimensions
Although GPs are primarily inferred from gene expression data and are sometimes viewed as alternatives to network modules or pathways, multi-omics technologies have provided evidence that certain GPs are linked to cross-layer functional modules with identifiable biophysical underpinnings[25]. TFs, their binding motifs, and their experimentally verified regulons have been recurrently discovered by multi-omics sequencing and factor analysis[26,27]. This is especially significant in simple systems such as E. coli, where GPs inferred by component analysis can be directly mapped to known regulons[28]. Furthermore, existing evidence suggests that certain GPs, such as the one discovered by Puram et al., are downstream effects of specific ligand-receptor interactions[29]. Regarding more remote molecular layers, such as metabolites and 3D chromatin structure, while there are some studies proposing their correlations with GPs within specific biosynthetic pathways and chromatin loops[30,31], the precise and direct relationships among these structures remain an active area of investigation. These disparate molecular layers also complement the inherent noise and the temporal instability of transcriptomic state by providing modality-specific programs and cross-modality validation of top-weighted genes of GPs, serving as the cornerstone of a multi-level virtual model[32].
Beyond cellular programs, the manifestation of biological complexity resides in the spatial architecture of tissues and intercellular communication. The integration of ST with GP inference has unveiled how GPs coordinate across spatial niches to form “multicellular programs” or “tissue programs”. For instance, Miller et al. discovered that meaningful GPs can be directly inferred from ST data by component analysis, and some of these GPs share a similar functional interpretation with single-cell GPs[15]. After that, by mapping single-cell GPs to spatial datasets, they found that an immunosuppressive myeloid program within the tumor microenvironment is colocalized with tumor mesenchymal programs and physically entangled with angiogenesis or hypoxia niches defined by spatial GPs, serving as a critical prognostic factor for glioblastoma patients. Shi et al. discovered multicellular programs and demonstrated their strong correlation with aging and tumor progression by ST and in vivo perturbation data[33]. Furthermore, spatial CCI analyses on ST datasets have begun to move beyond pairwise ligand-receptor interactions by incorporating sender-receiver gene co-expression and pathway-level contexts[34-36], which motivates a GP-centered integration in which GP activities are analyzed together with spatial CCI networks to link niche-level phenotypes with candidate communication routes. These examples hint that spatial GPs may be associated with CCI, cell migration, and disease progression in processes like tumor invasion and drug resistance. Consequently, modeling virtual cells in microenvironments similar to in vivo states requires incorporating spatial context into GP extraction (Figure 1).
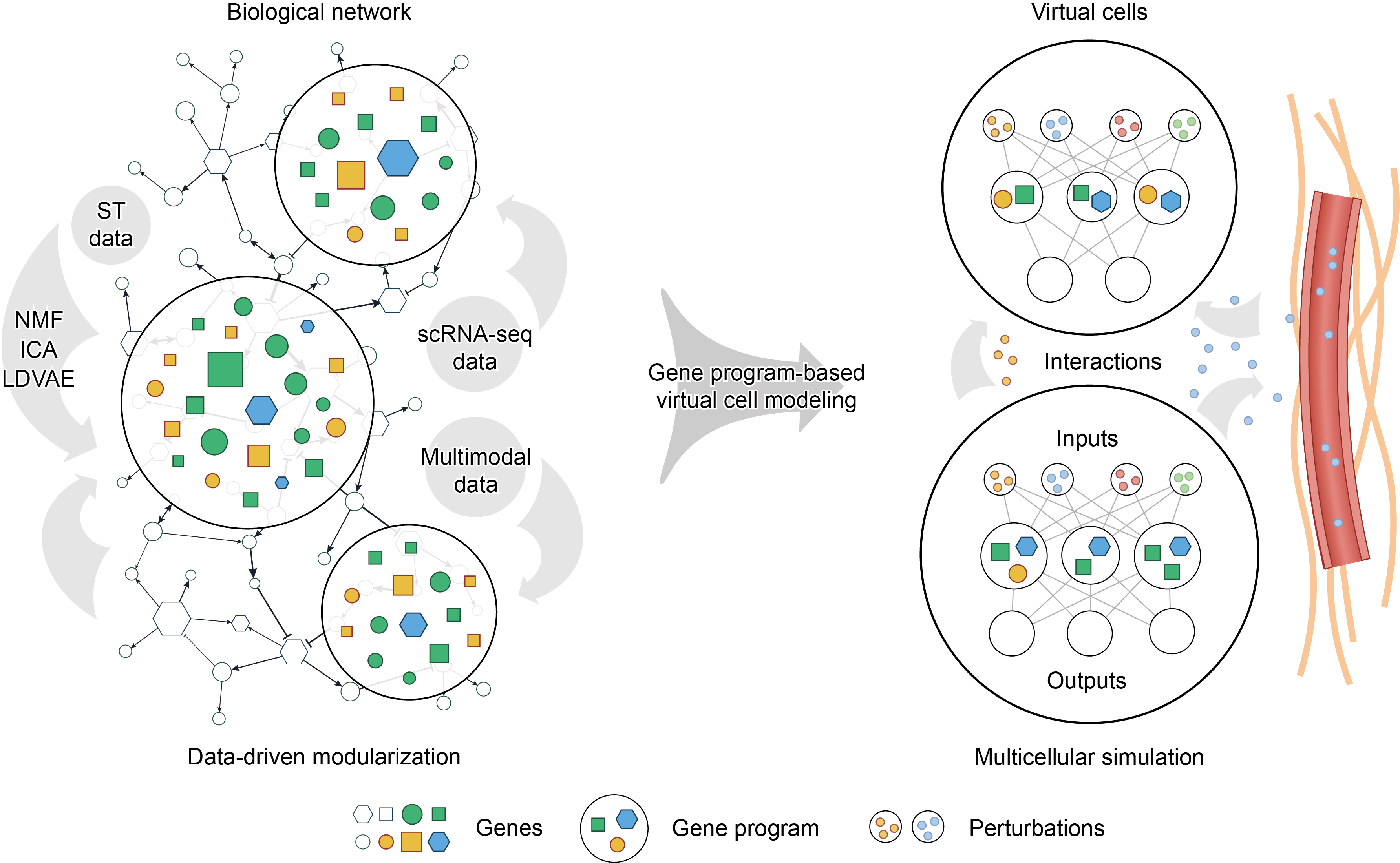{kind=link}
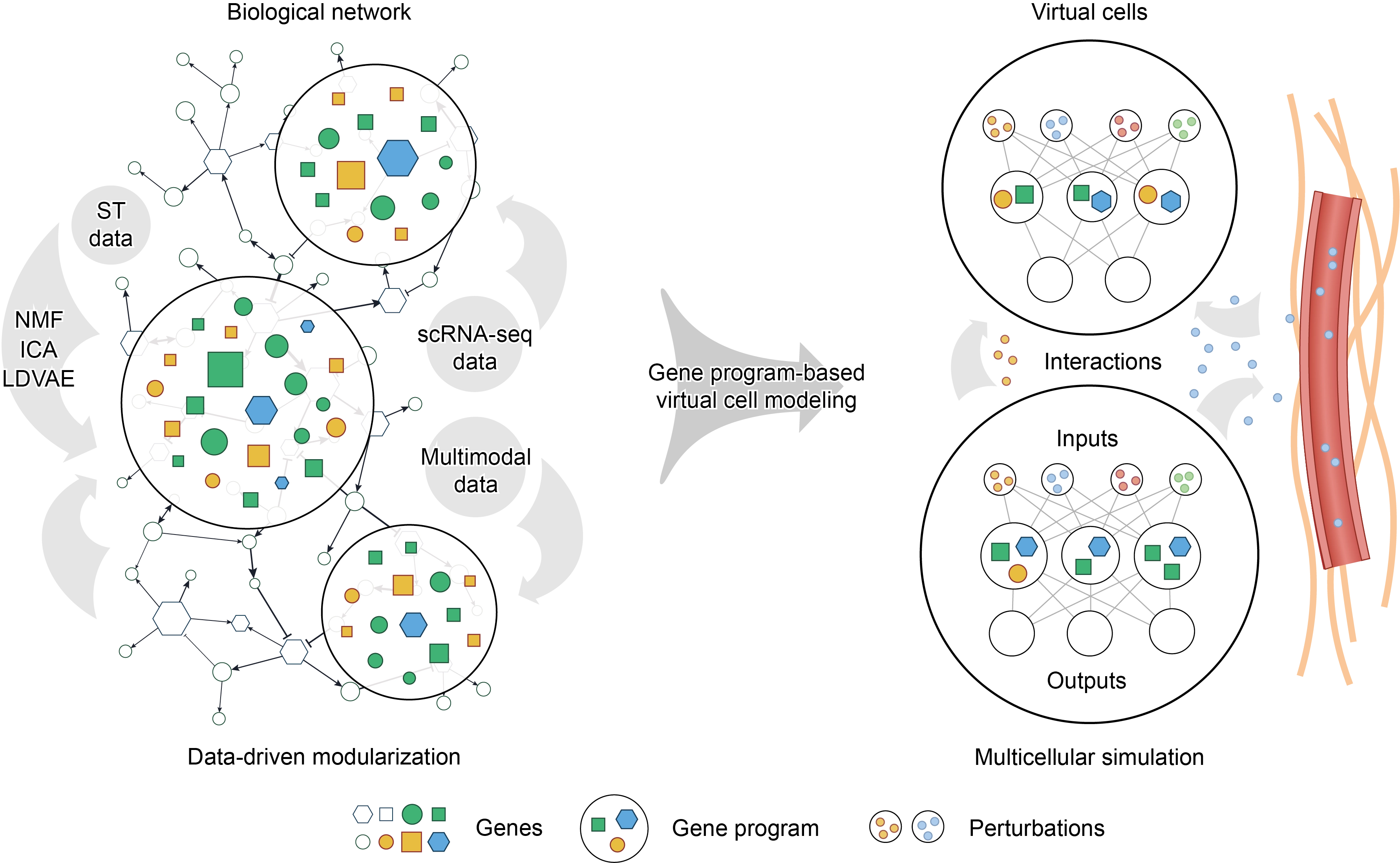
Figure 1. Schematic overview of gene program-based virtual cell modeling. Left, noisy biological networks are modularized from single-cell multi-omics data to yield robust and interpretable GPs, thereby reducing computational complexity while preserving biologically meaningful structure. Representative methods for GP extraction include NMF, ICA, and LDVAE. Right, GPs are encoded as explicit computational nodes, such as predefined latent variables in deep learning models. This framework accepts inputs from intrinsic or extrinsic perturbations and update cell states. Within this framework, intrinsic or extrinsic perturbations can be encoded as model inputs to update GP activities and downstream cell states. This enables interpretable genotype-to-phenotype mapping or in silico prediction of perturbation responses. By extending the framework to multiple interacting virtual cells and spatial cell-cell communication, it may also support simulation of tissue microenvironments in a manner closer to in vivo contexts. GPs: gene programs; NMF: non-negative matrix factorization; ICA: independent component analysis; LDVAE: linearly decoded variational autoencoder; ST: spatial transcriptomics.
4. Next-Generation Virtual Cells and Patient Digital Twins
While pioneering studies are already leveraging GPs alongside large-scale single-cell datasets such as Tahoe-100M to construct virtual cells[37], primary bottlenecks include a scarcity of experimentally validated programs, inherent noise in perturbation profiles, and the limitations of current model architectures in encoding GPs as explicit variables[20]. Integrating a broader array of programs as prior knowledge, along with their long-term temporal dynamics and multi-modal features, will establish a comprehensive toolkit and enable simulations of multicellularity, multi-perturbation responses, and continuous cell dynamics[38,39]. Furthermore, how to assess the simulation quality is another critical issue that should be addressed. Technically, all criteria should be based on high-quality ground truth data, such as spatially resolved single-cell transcriptomics data for benchmarking multicellular simulations[40], multi-gene perturbation experiments for prediction of multi-perturbation responses[41], and temporally resolved transcriptomics for continuous cell dynamics modeling[42]. Using these datasets, researchers can conduct multi-level assessment against the ground truth, such as the dynamics of GPs, marker genes associated with specific clinical outcomes, and cell migration, and compare model performance across different datasets and in multi-center validation. A useful model should be robust in real-world scenarios and require minimal retuning between scenarios.
The ultimate clinical promise of virtual cell technology lies in the construction of highly personalized “Patient Digital Twins”. Clinically derived multi-modal biopsy data are inherently noisy and heavily confounded by batch effects[43]. In this context, GPs, serving as robust biological representations, emerge as the ideal framework to align macroscopic clinical phenotypes with microscopic molecular states. By leveraging GPs to extract features from an individual’s multi-omics profile, generalized virtual organ models can be rapidly fine-tuned into digital twins that accurately recapitulate the patient-specific pathophysiological states[44]. This empowers clinicians to execute counterfactual simulations in silico prior to physical intervention, predictively evaluating the efficacies and toxicities of therapeutics. Thus, anchoring virtual biology on the foundation of GPs represents a practical route. We anticipate that a broad range of fields such as systems medicine, genetics, and oncology will be further propelled by this framework, moving towards mechanism-driven, highly individualized precision health.
Authors contribution
Wang T: Conceptualization, formal analysis, writing-original draft, writing-review & editing.
Li H, Liao J: Formal analysis, writing-review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82522092).
Copyright
© The Author(s) 2026.
References
-
1. Rachel T, Brombacher E, Wöhrle S, Groß O, Kreutz C. Dynamic modelling of signalling pathways when ordinary differential equations are not feasible. Bioinformatics. 2024;40(12):btae683.[DOI]
-
2. Johnson JAI, Bergman DR, Rocha HL, Zhou DL, Cramer E, McLean IC, et al. Human interpretable grammar encodes multicellular systems biology models to democratize virtual cell laboratories. Cell. 2025;188(17):4711-4733.e37.[DOI]
-
3. Fischer DS, Villanueva MA, Winter PS, Shalek AK. Adapting systems biology to address the complexity of human disease in the single-cell era. Nat Rev Genet. 2025;26(8):514-531.[DOI]
-
4. Yao D, Binan L, Bezney J, Simonton B, Freedman J, Frangieh CJ, et al. Scalable genetic screening for regulatory circuits using compressed Perturb-seq. Nat Biotechnol. 2024;42(8):1282-1295.[DOI]
-
7. Milano M, Agapito G, Cannataro M. Challenges and limitations of biological network analysis. BioTech. 2022;11(3):24.[DOI]
-
9. Johnson JAI, Tsang AP, Mitchell JT, Zhou DL, Bowden J, Davis-Marcisak E, et al. Inferring cellular and molecular processes in single-cell data with non-negative matrix factorization using Python, R and GenePattern Notebook implementations of CoGAPS. Nat Protoc. 2023;18(12):3690-3731.
-
14. Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: Single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083-1086.[DOI]
-
15. Miller TE, El Farran CA, Couturier CP, Chen Z, D’Antonio JP, Verga J, et al. Programs, origins and immunomodulatory functions of myeloid cells in glioma. Nature. 2025;640(8060):1072-1082.[DOI]
-
19. Kinker GS, Greenwald AC, Tal R, Orlova Z, Cuoco MS, McFarland JM, et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet. 2020;52(11):1208-1218.[DOI]
-
22. Zhao J, Zhang X, Wang G, Lin Y, Liu T, Chang RB, et al. Interpretable, flexible and spatially aware integration of multiple spatial transcriptomics datasets from diverse sources. Nat Genet. 2026;58(5):1138-1150.[DOI]
-
23. Roohani Y, Huang K, Leskovec J. Predicting transcriptional outcomes of novel multigene perturbations with GEARS. Nat Biotechnol. 2024;42(6):927-935.[DOI]
-
24. Chen H, King FJ, Zhou B, Wang Y, Canedy CJ, Hayashi J, et al. Drug target prediction through deep learning functional representation of gene signatures. Nat Commun. 2024;15:1853.[DOI]
-
26. Wu J, Huang B, Chen H, Yin Q, Liu Y, Xiang Y, et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature. 2016;534(7609):652-657.[DOI]
-
29. Browaeys R, Saelens W, Saeys Y. NicheNet: Modeling intercellular communication by linking ligands to target genes. Nat Meth. 2020;17(2):159-162.[DOI]
-
32. Liu T, Huang T, Ding T, Wu H, Humphrey P, Perincheri S, et al. Leveraging multi-modal foundation models for analysing spatial multi-omic and histopathology data. Nat Biomed Eng. 2026;1-18.[DOI]
-
35. Zhu J, Wang Y, Chang WY, Malewska A, Napolitano F, Gahan JC, et al. Mapping cellular interactions from spatially resolved transcriptomics data. Nat Methods. 2024;21(10):1830-1842.[DOI]
-
36. Zhu J, Dai H, Chen L. Revealing cell-cell communication pathways with their spatially coupled gene programs. Brief Bioinform. 2024;25(3):bbae202.[DOI]
-
37. Swinderman JT, Tung PY, Winters A, Goudy L, Wilson CM, Bounds LR, et al. Scalable probe-based single-cell transcriptional profiling for virtual cell perturbation mapping and synthetic biology phenotyping. BioRxiv [Preprint]. 2026.[DOI]
-
40. Russell AJC, Weir JA, Nadaf NM, Shabet M, Kumar V, Kambhampati S, et al. Slide-tags enables single-nucleus barcoding for multimodal spatial genomics. Nature. 2024;625(7993):101-109.[DOI]
-
43. Sadée C, Testa S, Barba T, Hartmann K, Schuessler M, Thieme A, et al. Medical digital twins: Enabling precision medicine and medical artificial intelligence. Lancet Digit Heal. 2025;7(7):100864.[DOI]
-
44. Wei R, Wang B, Yan B, Yang Y, Liu W, Ding J, et al. From equations to agents: The artificial intelligence virtual cell reshaping precision oncology. Inno Oncol. 2026;1(1):100002.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Wang T, Li H, Liao J. Anchoring virtual cells on gene programs: From biological networks to patient digital twins. EXO. 2026;1:202610. https://doi.org/10.70401/EXO.2026.0013
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Gene Programs as a Robust Interpretable Layer for Virtual Modeling
- 3. Beyond Single Cells: Multi-Modal and Spatial Dimensions
- 4. Next-Generation Virtual Cells and Patient Digital Twins
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Wang T, Li H, Liao J. Anchoring virtual cells on gene programs: From biological networks to patient digital twins. EXO. 2026;1:202610. https://doi.org/10.70401/EXO.2026.0013
copy
Share Link
copy