Decoding the clonal origins of mitochondrial pathology
Navdeep S. Chandel
1,2,5,*
,
Yogesh Goyal
3,4,5,*

*Correspondence to:
Navdeep S. Chandel, Division of Pulmonary and Critical Care, Department of Medicine, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
E-mail: nav@northwestern.edu
Yogesh Goyal, Department of Cell and Developmental Biology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA. E-mail: yogesh.goyal@northwestern.edu
Yogesh Goyal, Department of Cell and Developmental Biology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA. E-mail: yogesh.goyal@northwestern.edu
EXO. 2026;1:202612. 10.70401/EXO.2026.0016
Received: March 06, 2026Accepted: July 06, 2026Published: July 06, 2026
Abstract
Metabolic stress driven by mitochondrial dysfunction underlies a wide range of human diseases, yet the same defect can be detrimental to some cells while sparing their neighbors. We argue that this paradox reflects lineage mosaicism. Tissues are built from diverse clonal lineages whose differences remain hidden until mitochondrial dysfunction unmasks them. Rather than failing uniformly, cells diverge, engaging distinct stress programs shaped by developmental history and local context. By applying lineage-resolved approaches to mitochondrial dysfunction, we can move beyond average cellular behavior to understand when, where, and why individual cells adapt, persist, or fail.
Keywords
Lineage tracing, mitochondrial dysfunction, clonal mosaicism, metabolic stress response
1. Introduction
Metabolic stress underlies a wide range of an enormous spectrum of human diseases, ranging from inborn mitochondrial disorders to neurodegeneration, cancer, and the deterioration associated with aging[1,2]. A perplexing feature of these conditions is that the same metabolic perturbation can produce very different outcomes across tissues or even among adjacent cells within the same organ. Mitochondrial gene mutations underscore this paradox: the same molecular lesion can incapacitate neurons, partially weaken skeletal muscle, and leave liver function relatively intact[3-5]. Explanations invoking differences in metabolic demand or energetic buffering provide only partial insight. What is missing is an appreciation of the mosaicism of cell lineages that already exists within any organ, a hidden architecture that shapes how cells interpret and respond to metabolic stress. We propose that mitochondrial disease heterogeneity emerges from the interaction between mitochondrial defects, developmental lineage history, local tissue context, and the timing of stress-response engagement. Lineage heterogeneity may help explain why a shared metabolic insult generates such divergent phenotypes across biological systems.
2. Lineage Mosaicism and Metabolic Disease
Mosaicism in biology is like a Georges Seurat painting. When one steps back, many tissues look uniform, but if you apply stress and look closer, you see they were always built from distinct, unequal cellular dots. Every tissue is composed of thousands of clones that differ subtly or substantially in their developmental histories, mitochondrial DNA (mtDNA) complement, proteostatic capacity, epigenetic state, and exposure to microenvironmental cues. Under homeostatic conditions, many of these differences remain phenotypically silent. However, when mitochondrial function becomes compromised through genetic mutation, metabolic or nutrient stress, or age-associated decline, these latent distinctions become unmasked[6]. A central source of this clonal divergence is mtDNA itself. Because mitochondrial genomes replicate independently of the cell cycle, a somatic mutation arising on a single molecule can drift or be selected to high heteroplasmy within a given lineage, generating a mosaic of clonally distinct mtDNA states across a tissue well before any overt dysfunction[7]. Consistent with this, a recent population-scale analysis found that the mtDNA mutations accumulating in human blood with age are largely neutral passengers rendered detectable by clonal expansion rather than generated by oxidative damage, underscoring that the mtDNA signal often reports on clonal structure as much as on mitochondrial injury itself[8]. Cells that once appeared equivalent reveal marked differences in their ability to withstand, adapt to, or succumb to mitochondrial stress. From this perspective, the tissue-specificity of metabolic disease may reflect not inherent organ-level vulnerability but the aggregate outcome of clonal trajectories that diverge in both tempo and direction when faced with metabolic challenge.
Understanding these trajectories requires accounting for the fact that mitochondrial dysfunction does not elicit a uniform cellular response. Rather than a simple failure of adenosine triphosphate (ATP) production, mitochondrial defects initiate diverse stress programs whose manifestations vary from cell to cell. Some activate the mitochondrial unfolded protein response, others shift their redox metabolism or alter nucleotide or amino acid supply pathways, and still others modulate mitophagy or mitochondrial biogenesis[9]. Prominent among these is the integrated stress response, through which mitochondrial dysfunction converges on the transcription factor activating transcription factor 4 (ATF4) to reprogram amino acid and nucleotide metabolism, and which is coupled to the redox-sensitive regulator nuclear factor erythroid 2-related factor 2 (NRF2) to mount antioxidant defenses against the oxidative stress produced by a compromised electron transport chain[10,11]. Notably, these same stress and antioxidant programs are engaged not only by intrinsic genetic lesions but also by environmental insults such as hypoxia, nutrient limitation, and xenobiotic exposure, so differences in how clones activate them can shape how environmental stress is translated into mitochondrial pathology. These programs are influenced by developmental timing; neonatal tissues, for instance, transition abruptly from glycolytic to oxidative metabolism, revealing vulnerabilities that remain hidden during fetal growth[12]. Moreover, mitochondrial defects provoke wide-ranging retrograde signals, including alterations in metabolites, reactive oxygen species (ROS), cofactors, and redox states that remodel transcriptional and epigenetic landscapes in cell-type-specific ways[13-15]. The same mutation in mtDNA replication or translation machinery, extensively described in mitochondrial disease research, therefore produces divergent outcomes depending on when and where it arises[16,17]. Pathology cannot be reduced to biochemical insufficiency alone; it emerges from an interplay between stress-response dynamics and lineage-intrinsic properties.
3. Design Principles for Lineage-Resolved Metabolic Studies
Lineage-resolved studies of metabolic disease can be guided by a set of experimental design principles. First, distinguishing predetermined vulnerability from stress-induced divergence requires lineage information that precedes overt pathology. Second, resolving when fate decisions occur demands temporal recording rather than endpoint sampling. Third, separating lineage-intrinsic effects from microenvironmental influence requires comparing clonally related cells across anatomical contexts. Fourth, correlation must be paired with perturbation to establish necessity and sufficiency of candidate pathways. Together, these principles define a framework for moving from pathology to mechanism (for related discussions of conceptual frameworks and experimental design in lineage tracing, see the studies[18-20]).
4. From Cancer to Metabolism: Why Lineage History Matters
Single-cell lineage tracing has already demonstrated the success of these ideas, but to date has largely been confined to cancer biology. In cancer, lineage tracing has been useful for providing detailed understanding of tumor initiation, metastasis, and why therapies fail (covered in the work[21]). What is striking, though, is how little these ideas and technologies have been extended to primary mitochondrial and metabolic diseases. Notably, lineage barcoding studies in cancer have repeatedly implicated metabolic reprogramming in subsets of clonally related cells as determinants of cell fate under therapeutic stress. Using Watermelon, a single-cell lineage barcoding tool, one study showed that cycling persister cells arise from lineages with distinct metabolic programs, including elevated fatty acid oxidation and antioxidant defenses, across multiple cancers[22]. Similarly, lineage-resolved analyses in BRAF-mutant melanoma and endocrine-treated ER+ breast cancer found that therapy-resistant clones show increased reliance on mitochondrial respiration, implicating oxidative phosphorylation as a shared metabolic vulnerability[23,24]. Together, these studies demonstrate that clonal history, even within what appear to be homogeneous cell types, reveals metabolic determinants of survival that cell state profiling alone cannot detect (see also[21,25-27] for overview of experimental and computational frameworks addressing non-genetic variability in cancer). Several factors likely explain why this has not yet extended to primary mitochondrial disease: these disorders largely affect postmitotic tissues such as brain, muscle, and heart, where low cell turnover limits clonal expansion and complicates barcode-based tracing. In addition, they have been studied primarily through their biochemical and genetic basis rather than their clonal architecture. Only the recent advent of endogenous mtDNA- and epimutation-based tracing (Table 1) has made it feasible to reconstruct clonal histories in unmanipulated human tissue. We argue that applying lineage tracing technologies to diseases in which mitochondrial dysfunction is the primary lesion, rather than a secondary response, could yield equally deep insights.
Table 1. Lineage tracing and cellular recording technologies.
| Technology class | Specific methods | Principle | What it captures | Limitations | Key references |
| Static barcodes | ClonTracer, CloneSifter, CaTCH, ClonMapper, Watermelon, SPLINTR, Polylox, FateMap, LARRY, CellTags | Heritable DNA tags mark progenitors: barcode inherited by all descendants | Clonal identity; endpoint relationships | Endpoints only; no temporal resolution | [22,28,30-40,43,44] |
| CRISPR-based evolving recorders | GESTALT, scGESTALT, ScarTrace, CARLIN, macsGESTALT, DARLIN, iTracer, MEMOIR | Progressive Cas9-induced mutations accumulate over divisions | Lineage tree structure; branch-point timing | Mutation saturation limits recording depth | [46-50,52-54] |
| Endogenous mtDNA barcodes | mtscATAC-seq, MAESTER, ReDeeM, SCI-LITE, EMBLEM | Natural accumulation of mtDNA variants as endogenous barcodes | Clonal relationships in native human tissues | Lower barcode diversity; heteroplasmy complicates clonal assignment | [56-61] |
| Endogenous epimutation barcodes | EPI-clone, MethylTree, scTAM-seq | Clonally heritable somatic CpG methylation changes serve as natural lineage markers | Clonal relationships in native human tissues; compatible with simultaneous cell-state profiling | Requires computational separation of clonal vs. cell-type-driven methylation; limited phylogenetic depth | [63-66] |
| DNA signal recorders | TRACECAMERA, DNA Typewriter, ENGRAM | Engineered constructs convert transient signal activity into permanent genomic edits using base/prime editing | Molecular history of specific signals; temporal order of exposures | Requires pathway-specific reporter configuration; temporal resolution depends on promoter and editing kinetics | [67-70] |
| Imaging-based protein recorders | GEMINI, CytoTape/XRI | Computationally designed protein assemblies grow intracellularly, encoding signaling history as fluorescent patterns | Transcriptional and signaling dynamics with temporal resolution; spatially readable in intact tissue | Requires transgenic expression; not sequencing-compatible; limited multiplexing relative to DNA recorders | [62,71] |
| Cell-cell interaction recorders | LIPSTIC, uLIPSTIC | Sortase-mediated enzymatic labeling transfers substrate between contacting cells | History of cell-cell contacts; identity of interaction partners | Requires transgenic system; captures contact not signaling outcome | [72,73] |
SPLINTR: single-cell profiling and lineage tracing; LARRY: lineage and RNA recovery; CARLIN: CRISPR array repair lineage tracing; DARLIN: dual-acting recording of lineage information; EPI: epigenetic; scTAM-seq: single-cell targeted analysis of the methylome by sequencing; TRACE: temporal recording in arrays by CRISPR expansion.
5. Technologies for Reconstructing Cellular History
The cancer examples above only partially represent the rapidly expanding set of lineage tracing methods. More generally, dissecting lineage-specific responses requires technologies that can track individual clones and their fates. Over the past decade, a diverse and increasingly capable set of approaches has taken shape to make lineage-resolved analyses possible in complex tissues (summarized in Table 1, also see the study[20]). Rather than a single technique, lineage tracing encompasses methods that tackle different aspects of cellular history: ancestry, timing, and experience. Some focus on ancestry by assigning heritable identifiers to progenitor cells, others on timing by recording when lineage branches occur, and still others on experience by capturing the molecular states or interactions cells encounter along their trajectories. When combined with single-cell profiling, these approaches have shown, in our work and that of others, that clonally related cells carry functional information not apparent from molecular snapshots alone. In the context of mitochondrial dysfunction, lineage tracing enables analyses of failure, adaptation, and divergence under metabolic stress.
At the level of lineage and timing, static barcoding approaches, including lentiviral integration and recombinase-based systems, allow one to leave a permanent ‘name tag’ on progenitor cells. This enables retrospective reconstruction of clonal relationships across tissues[22,28-44]. A major axis of innovation in these systems is barcode design, with different implementations trading off barcode length (for example, ~8 bp in CellTags[39] versus ~100 bp in FateMap[30]), clonal diversity, and readout modality, including variants optimized for in situ visualization. Static barcoding strategies also enable creative experimental designs that are difficult to realize otherwise. For example, barcoded populations can be split into matched “twin” cohorts[30], akin to the landmark replica-plating experiments of Lederberg and Lederberg[45], with one exposed to metabolic stress and the other left untreated, allowing direct tests of whether lineage-specific vulnerability is pre-specified or emerges during the stress response.
Evolving CRISPR-based recorders extend lineage tracing into the time dimension by allowing cells to accumulate edits across successive divisions, creating a molecular record of their history as development unfolds. Systems such as the original GESTALT[46,47] and its derivatives encode lineage history in arrays of Cas9 target sites that progressively mutate, enabling reconstruction of lineage trees with branch-point resolution. Editing rate, target architecture, and mutation saturation together determine the tradeoff between temporal resolution and recording depth, typically spanning tens of cell divisions. Alternative designs, including scGESTALT[48], ScarTrace[49], iTracer[50], LINNAEUS[51], MEMOIR[52], and CARLIN/DARLIN[53,54] explore different positions in this tradeoff space, emphasizing robustness, readout modality, or dynamic range.
Both static barcodes and evolving CRISPR recorders, however, rely on genetic engineering, which largely confines their use to animal models, organoids, and cell culture. An important advance toward overcoming this limitation comes from the use of naturally occurring somatic mutations in mtDNA as endogenous lineage markers. As mtDNA accumulates mutations that are inherited through mitochondrial segregation, methods such as mtscATAC-seq[55], MAESTER[56], ReDeeM[57], SCI-LITE[58], and others[59-61] can recover clonal structure directly from unmanipulated human tissues. Compared to engineered systems, mtDNA-based tracing trades experimental control over mutation rate and target architecture for immediate applicability in human samples, with the additional complication that heteroplasmy and limited barcode diversity must be computationally modeled. Nevertheless, this approach is particularly well suited for studying clonal dynamics in human development, aging, and mitochondrial disease. More recently, clonally heritable somatic epimutations at CpG sites have emerged as a complementary endogenous barcode, offering higher variant density per cell and applicability across tissues without genetic manipulation[62-66].
Beyond passive reconstruction of ancestry, a complementary class of technologies enables the capture of the molecular events that cells experience along their trajectories. DNA-based signal recorders such as TRACE[67], CAMERA[68], DNA Typewriter[69], and ENGRAM[70] convert transient pathway activity into stable genomic marks, typically by coupling signal-responsive promoters to base or prime editors that irreversibly modify predefined targets. Imaging-based protein assembly recorders such as GEMINI[71] and CytoTape[62] offer a complementary modality, encoding similar dynamics as fluorescent patterns readable in situ. Experimentally, stress-responsive promoters can be wired to these writers so that engagement of defined mitochondrial stress programs triggers recording in vivo, allowing the temporal sequence of exposures, such as oxidative stress, hypoxia, or proteotoxic signaling, to be written into the genome as a within-cell molecular history. In principle, such designs could be extended to mitochondrial-specific variables such as transient ROS or pH shifts.
So far, most lineage and molecular recording strategies capture what happens inside individual cells. They report external influences only indirectly, through the downstream effects those signals leave on cellular states. A distinct but equally important dimension of cellular experience is interaction history, which has recently become accessible through cell-cell contact recorders such as LIPSTIC[72] and uLIPSTIC[73]. These methods directly label physical contacts between cells in situ, enabling retrospective reconstruction of interaction networks within tissues. Together, these approaches make it possible to reconstruct not only where cells come from and when they diverge, but also which cellular environments and neighbors they have encountered.
Spatial profiling technologies[74,75] extend this logic by placing recorded histories back into tissue context, where metabolic gradients, including oxygen, nutrients, lactate, and signaling molecules, vary across tissues at micron scale. For example, cells near capillaries may experience different redox shifts than those deeper within a tissue, leading to distinct stress histories encoded in their genomes. Recovering lineage or DNA recorder states by spatial transcriptomics would allow these histories to be interpreted in their native microenvironmental context, enabling reconstruction of metabolic ‘maps’ that provide mechanistic explanations for why disease hotspots emerge in specific tissue regions. Such integration can be achieved computationally, by mapping lineage-resolved single-cell data onto spatial transcriptomic references (for example, TemSOMap[76] and others[77]), or experimentally, through approaches that directly couple lineage barcodes or molecular recorders to spatial transcriptomic readouts in situ (for example, SpaceBar[78], SPACE-seq[79], or Space-TREX[80]).
Metabolic zonation in certain tissues offers an opportunity for lineage-resolved methods to provide granularity to within-organ heterogeneity. For example, the liver has gradients in oxygen and nutrients that generate periportal, midzonal, and pericentral regions with distinct metabolic functions[81,82]. A mitochondrial defect may therefore have different consequences depending on the zone in which a clone resides. Lineage tracing could reveal whether clonally related hepatocytes found across nutrient gradients share a common origin that constrains stress responses across zones, or whether local metabolic conditions override lineage history. Unrelated clones within a single zone could test the opposite case, asking whether a common microenvironment drives convergence toward similar adaptive or pathological states. Comparable logic applies to organs such as the kidney, where specialized cell types are arrayed across steep corticomedullary gradients of oxygen and solute concentration[83]. Importantly, coupling lineage information with spatial transcriptomics and metabolic profiling would help determine whether regional disease patterns reflect clonal history, zonated tissue architecture, or both.
6. Lineage-Resolved Questions across Mitochondrial Disease
Lineage-resolved approaches offer a way to compare how related mitochondrial lesions generate divergent tissue pathologies. Static lineage barcoding combined with single-cell profiling in matched mouse models could distinguish whether vulnerable tissues harbor intrinsically distinct clonal populations, or whether a common progenitor pool diverges differently depending on the specific nucleotide metabolism defect encountered. Lineage tracing also provides a framework for defining when cells commit to failure versus compensation. Clones that appear equivalent under homeostasis may diverge sharply under mitochondrial stress. Evolving CRISPR-based lineage recorders could identify the developmental or disease-stage window at which neuronal lineages branch toward degeneration versus resilience and determine whether this divergence precedes or follows activation of stress programs such as the mitochondrial unfolded protein response. Lineage-resolved analyses also enable questions that bulk or static measurements cannot address, including how heteroplasmy[17] evolves under stress. Tracking mtDNA variants at clonal resolution can, for example, distinguish selective expansion from altered mitochondrial biogenesis and stochastic drift. Finally, lineage analysis may help resolve long-standing clinical puzzles that remain opaque to endpoint pathology. Some infants with severe mitochondrial disease survive an early life-threatening phase and partially recover, while others succumb[4]. This pattern suggests that apparent recovery does not reflect uniform correction of the underlying defect but instead arises from lineage-specific survival or compensation, system-wide metabolic adaptation, or stress effects that accumulate and only manifest at later developmental stages, distinctions that could be resolved by tracking clonal persistence and fate over time.
In postmitotic tissues, fate is defined differently than in proliferative systems. In proliferative tissues, clonal expansion, depletion, and state transitions are useful readouts of clonal behavior. By contrast, in neurons and other long-lived cells, mitochondrial stress may instead produce prolonged states in which cells remain alive but lose their function. A change in transcriptional trajectory is not a commitment to an irreversible fate decision. For example, a neuron experiencing respiratory chain impairment could activate stress programs, dysregulate anabolic functions, alter its interactions with glia or immune cells, and persist for an extended period before degenerating. Fate in this context should link lineage history to endpoints such as survival, degeneration markers, regulated cell death, senescence-like states, tissue-specific function, and direct metabolic measurements. These measurements are needed to separate cells that are adapting to mitochondrial stress from cells already committed to metabolic collapse. Because that distinction depends on when and in what order stress programs were engaged rather than on a single endpoint state, signaling recorders that write pathway activity into the genome over time (Table 1) would be especially valuable, allowing a neuron’s stress-response history to be reconstructed and matched against its eventual fate.
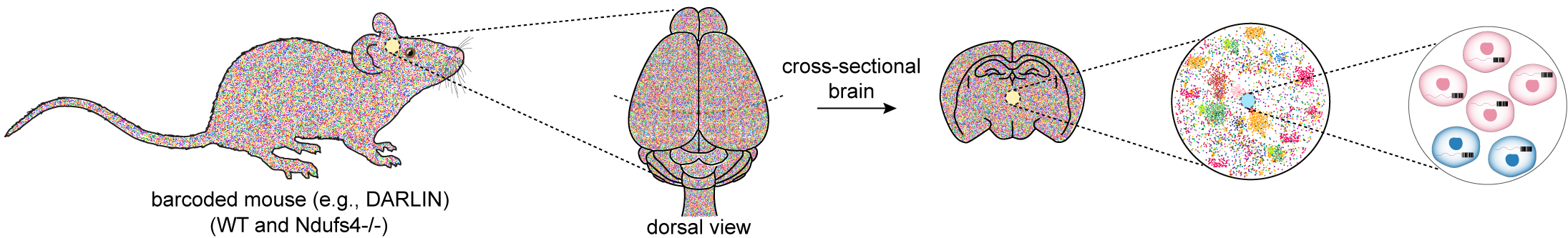
A vivid example of how these principles manifest in vivo comes from the global loss of NDUFS4, a nuclear-encoded subunit essential for the final assembly of mitochondrial complex I[84,85]. In humans, NDUFS4 mutations cause Leigh syndrome with profound degeneration of brainstem and basal ganglia circuits, whereas Ndufs4-null mice display a distinct pattern of regional vulnerability and pronounced neuroinflammation, with relative sparing of many peripheral tissues[86]. Such species- and tissue-specific divergence cannot be explained by metabolic demand alone. Instead, it likely reflects lineage-intrinsic differences in how clones tolerate partial complex I assembly and the resulting redox, metabolic, and inflammatory stress. Developmental timing likely amplifies these discrepancies: the prolonged maturation of human neural circuits exposes certain lineages to extended windows of vulnerability, while the more rapid postnatal maturation in mice drives distinct clonal trajectories under the same mitochondrial lesion. The NDUFS4 system therefore illustrates how a single, uniform molecular defect gives rise to heterogeneous lineage-specific stress responses whose aggregate behavior produces sharply different pathological landscapes across tissues and species.
Viewed through a lineage-resolved lens, the NDUFS4 system becomes a test case for dissecting how clonal histories constrain cellular responses to a shared mitochondrial defect (Figure 1). The question this raises is not simply which cells are lost, but how these histories shape the space of possible responses to a common insult.
{kind=link}
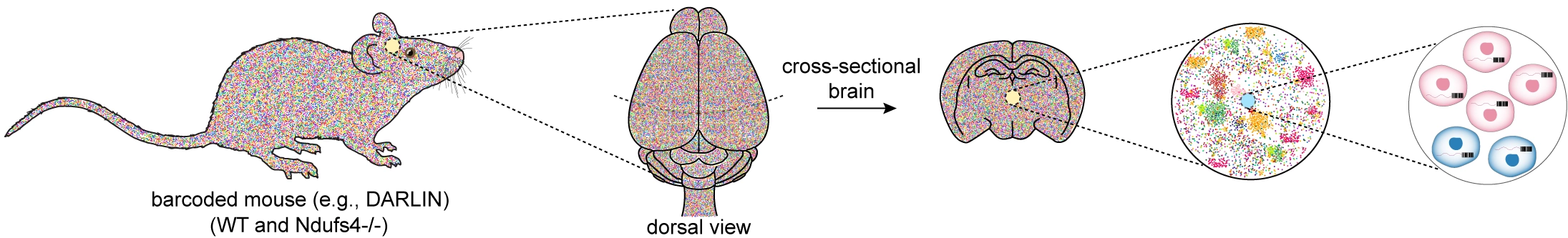
Figure 1. Schematic of a mouse model to track clonal evolution in Ndufs4-/- relative to WT. WT: wild-type; DARLIN: dual-acting recording of lineage information.
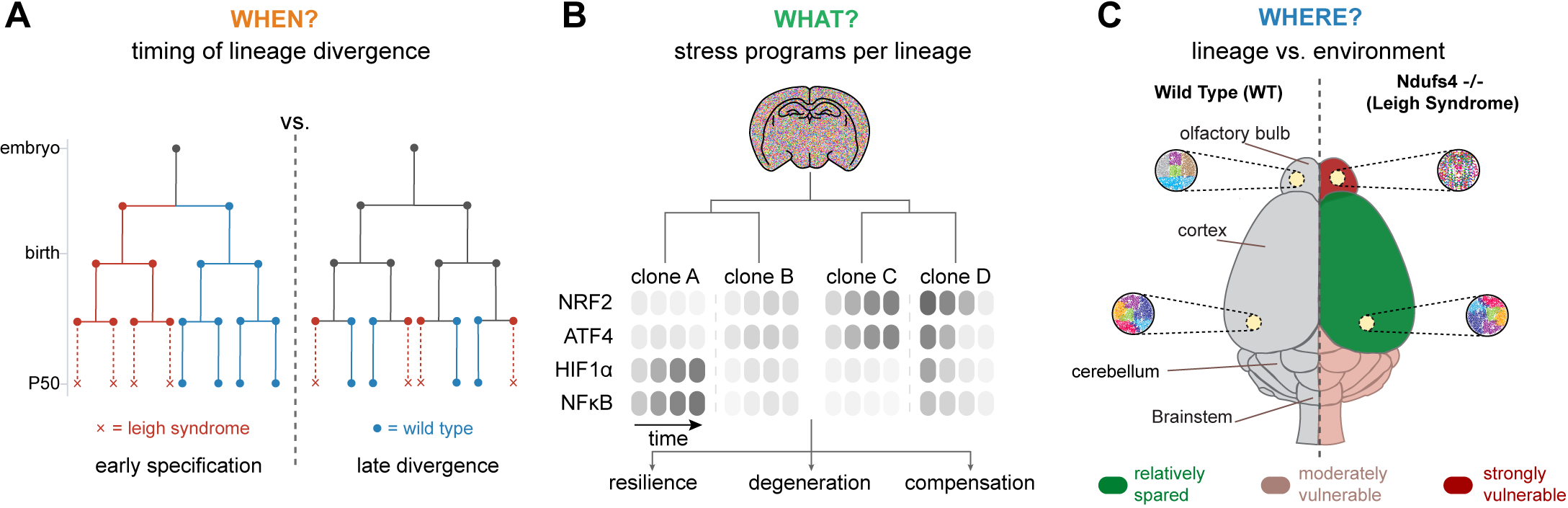
One unresolved issue is whether vulnerable and resilient lineages are specified early, or whether most cells initially follow similar trajectories and only diverge after prolonged stress exposure (Figure 2A). This distinction bears directly on whether selective vulnerability reflects early developmental or epigenetic priming, or instead emerges from time-integral threshold effects in redox buffering, proteostasis, or inflammatory signaling. This can be tested directly by combining evolving lineage recorders with time-resolved sampling in Ndufs4-null tissue: early separation would appear as deep branch points whose descendant clones are already fate-biased, whereas late separation would produce largely shared lineage structure until shortly before overt pathology, with fate divergence emerging only at terminal branches.
{kind=link}
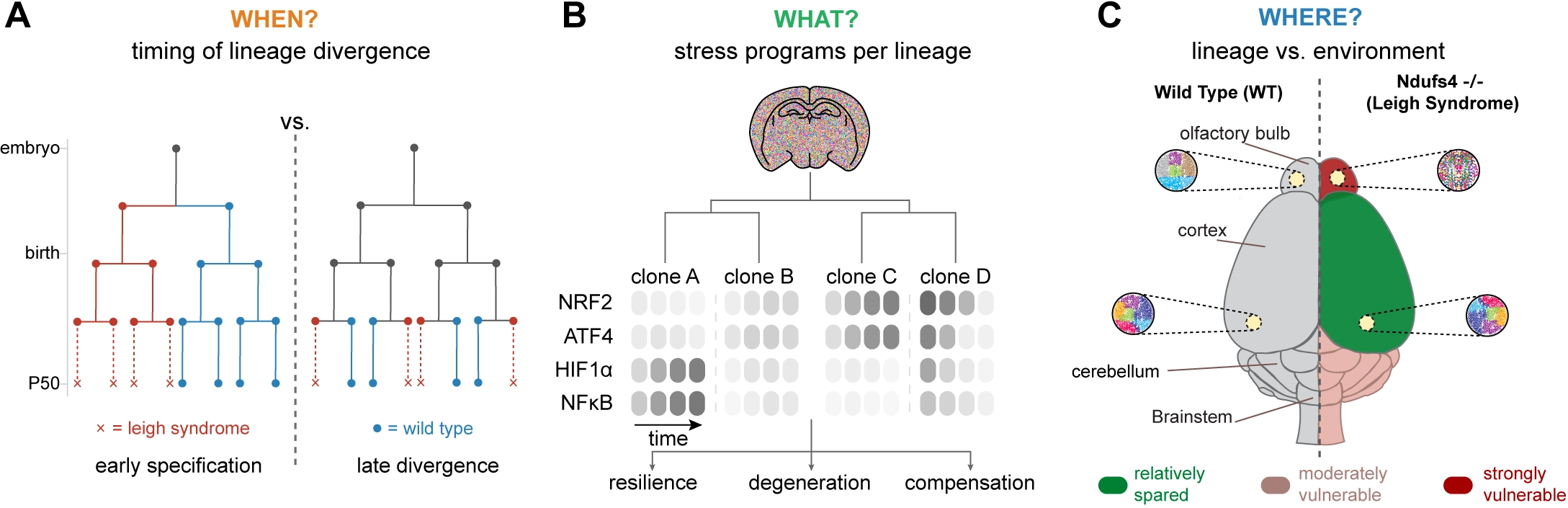
Figure 2. Schematics to capture (A) the timing, (B) regulatory programs, and (C) spatial consequences on clonal evolution in Ndufs4-/-pathology. NRF2: nuclear factor erythroid 2-related factor 2; ATF4: activating transcription factor 4; HIF: hypoxia-inducible factor; NFκB: nuclear factor kappa B.
A key question is what truly distinguishes lineages at the onset of divergence. Even if fate bias becomes apparent only at later stages, it is likely shaped earlier by differences in a cell’s cumulative stress-response history, which can ‘latently’ precondition downstream decisions (Figure 2B). Mitochondrial dysfunction activates multiple stress programs[13]. DNA signal recorders responsive to canonical regulators such as NRF2, ATF4, or HIF1α could be used to track which lineages engage these defenses early and which fail to do so. This can be achieved by combining an inducible, high-diversity lineage recording system (for example, a DARLIN-type Cas9 recorder in mice) with conditional Ndufs4 loss and viral delivery of an orthogonal DNA-based signal recorder, enabling simultaneous reconstruction of lineage relationships and intracellular stress-response histories within the same cells. One can then ask whether lineages that later persist versus contract differ in the identity, combination, or temporal ordering of stress programs they engage before fate differences become apparent.
A third issue concerns the relative contributions of lineage-intrinsic properties versus local tissue environment in shaping vulnerability (Figure 2C). In NDUFS4 deficiency, regional pathology is highly stereotyped, suggesting that fate is imposed primarily by extrinsic cues such as inflammatory milieu, vascularization, circuit activity, or interactions with glial and immune cells[87]. Lineage tracing provides a direct way to disentangle these effects by comparing clonally related cells across different regions with unrelated cells within the same region. If lineage-intrinsic factors dominate, descendants of the same clone should exhibit correlated molecular trajectories even in distinct anatomical contexts; if environment dominates, cells within a given region should converge in fate regardless of clonal origin. Cell-cell interaction recorders such as uLIPSTIC offer a further layer of resolution by directly capturing physical contacts between neurons, glia, and infiltrating immune cells, enabling tests of whether specific interaction histories correlate with lineage-specific vulnerability.
Finally, these questions can be extended to human disease using endogenous lineage tracing based on somatic mtDNA mutations. Because mtDNA variants accumulate and segregate with mitochondrial inheritance, they serve as natural lineage markers in unmanipulated human tissues. Applied to postmortem samples from Leigh syndrome and related mitochondrial disorders, mtDNA-based barcoding can reveal whether clonally related cells exhibit coordinated vulnerability or depletion across affected regions, providing a lineage-resolved view of tissue-selective degeneration in humans.
7. From Lineage Correlation to Causal Inference in Mitochondrial Dysfunction-Dependent Pathology
Lineage-resolved analyses in models of mitochondrial dysfunction such as those caused by NDUFS4 mutations are useful for revealing relationships between lineage history, cellular stress responses, and fate, but on their own they stop short of establishing causality. To move beyond correlation, the key step is to perturb candidate pathways and ask whether lineage trajectories change. Pooled perturbation approaches[88-95] such as Perturb-seq and CROP-seq fit naturally into this framework, because they allow genetic or metabolic perturbations to be introduced at scale while preserving lineage relationships, making it possible to follow the fate of perturbed clones over time. For instance, deleting NRF2 in barcoded neurons would directly test whether antioxidant capacity is required for lineage persistence under mitochondrial dysfunction. By comparing which clones persist and which are depleted, one can distinguish pathways that actively shape vulnerability from those that merely track with lineage fate, using loss-of-function perturbations to test necessity and gain-of-function perturbations to test sufficiency. When combined with evolving lineage recorders or molecular signal recorders, these perturbations can further be placed in temporal context, revealing when along a lineage trajectory a given pathway exerts its effect. Finally, incorporating spatially resolved methods allows these causal relationships to be interpreted within intact tissue architecture, whether through imaging-based approaches like perturb-FISH[94,96] and Perturb-Multi[97] that offer single-cell spatial resolution, or sequencing-based strategies such as Perturb-DBiT[93] that trade cellular precision for transcriptome-wide coverage. Beyond mechanistic insight, such experiments could nominate therapeutic targets: pathways whose activation stabilizes vulnerable lineages become candidates for pharmacological enhancement.
Lineage-based methods serve as discovery tools. We suggest that a practical guide would be to use lineage tracing, molecular recording, and spatial profiling to discover programs linked to vulnerability or resilience, then perturb those programs to test necessity, followed by appropriate metabolic measurements that explain how the perturbation changes cell physiology. For example, lineage studies might indicate the necessity of increased antioxidant capacity, a change in fuel choices, respiratory-chain compensation, or inflammatory signaling. Each possibility would point to different assays that measure ROS levels, mitochondrial membrane potential, glucose or fatty acid oxidation, ATP reserve, and spatial metabolomics. A further challenge is that the relevant measurements unfold over very different time scales and are often spatially compartmentalized within tissues. Mitochondrial depolarization, ROS production, redox imbalance, and changes in NADH/NAD+ can appear within minutes to hours. Transcriptional and inflammatory states may persist much longer, while lineage and molecular recording systems often integrate cellular history over days to weeks. This complicates interpretation: a recorded stress signal may reflect the metabolic event that pushed a lineage toward decline, but it may also represent a later adaptive or compensatory response to an earlier mitochondrial defect. Perturbation experiments are therefore essential, but they introduce their own complications, because altering NRF2, ATF4, lipid metabolism, mitophagy, or inflammatory signaling can itself reshape the metabolic state being measured. Lineage-based discovery is therefore most informative when recording is coupled to time-resolved perturbations, metabolic measurements matched to the time scale of the process under study, and later functional readouts of cell fate.
Much of this will depend on building new computational frameworks. Most existing perturbation datasets, in vitro and in vivo, are endpoint snapshots[98], so they cannot easily separate stress responses that drive clonal collapse from those that simply follow it. Adding clonal and lineage information to these datasets and accompanying computational efforts[99-102] is one way to recover when these programs act. In the NDUFS4 setting, this might mean asking whether NRF2, ATF4, inflammatory, or fuel-use programs are activated before vulnerable lineages decline, or only after the cells are already committed to dysfunction. The point is not that computation replaces mechanism, but that it can help build testable models of how mitochondrial stress plays out across lineages.
8. Outlook
Mitochondrial disease has long been framed in terms of metabolic dysregulation and tissue-level vulnerability, yet growing evidence suggests that lineage history plays a central role in shaping cellular responses to metabolic stress. By integrating lineage tracing, molecular recording, and pooled perturbations, it becomes possible to move beyond endpoint pathology and ask when fate bias emerges, how stress histories differ across clones, and which pathways causally determine resilience or failure. Lineage-resolved approaches may also have translational value. By defining which clonal populations are vulnerable, and which pathways distinguish adaptation from failure, they could yield readouts that are potentially more informative than bulk tissue measurements. Such lineage-informed biomarkers might help identify tissues at risk before overt pathology, stratify patients by the clonal architecture underlying their disease course, and guide when and where intervention is most likely to succeed. This need is acute given how few validated biomarkers exist for primary mitochondrial disease, largely limited to circulating factors such as FGF21 and GDF15. Applied across model systems and human organoids that mimic tissues in vitro, lineage-resolved approaches offer a unifying framework for linking uniform molecular lesions to heterogeneous outcomes, reframing mitochondrial disease as a problem of clonal dynamics rather than average cellular behavior.
Acknowledgements
We thank P. Pholraksa for assistance with figure design. During the preparation of this work, the authors used Claude (Anthropic) to improve the wording and readability of the text. The core ideas, arguments, and conclusions are the authors’ own. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Authors contribution
Chandel NS, Goyal Y: Conceptualization, writing-original draft, writing-review & editing.
Conflicts of interest
Yogesh Goyal receives consultancy from Schmidt Sciences and Gilead Sciences. The other author declares no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
None.
Copyright
© The Author(s) 2026.
References
-
1. DeBerardinis RJ, Keshari KR. Metabolic analysis as a driver for discovery, diagnosis, and therapy. Cell. 2022;185(15):2678-2689.[DOI]
-
3. Suomalainen A, Nunnari J. Mitochondria at the crossroads of health and disease. Cell. 2024;187(11):2601-2627.[DOI]
-
4. Suomalainen A, Battersby BJ. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol. 2018;19(2):77-92.[DOI]
-
5. Burr SP, Chinnery PF. Origins of tissue and cell-type specificity in mitochondrial DNA (mtDNA) disease. Hum Mol Genet. 2024;33(R1):R3-R11.[DOI]
-
6. Ng YS, Bindoff LA, Gorman GS, Klopstock T, Kornblum C, Mancuso M, et al. Mitochondrial disease in adults: Recent advances and future promise. Lancet Neurol. 2021;20(7):573-584.[DOI]
-
8. Gupta R, Durham TJ, Chau G, Kanai M, Uddin MM, Lu W, et al. Mechanism of age-related accumulation of mtDNA mutations in human blood. Nature. 2026.[DOI]
-
10. Quirós PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol. 2016;17(4):213-226.[DOI]
-
11. Kreß JKC, Jessen C, Hufnagel A, Schmitz W, da Silva TNX, dos Santos AF, et al. The integrated stress response effector ATF4 is an obligatory metabolic activator of NRF2. Cell Rep. 2023;42(7):112724.[DOI]
-
12. Tai S, Kimura W. Transition from fetal to postnatal state in the heart: Crosstalk between metabolism and regeneration. Dev Growth Differ. 2024;66(9):[DOI]
-
13. Chakrabarty RP, Chandel NS. Beyond ATP, new roles of mitochondria. Biochemist. 2022;44(4):2-8.[DOI]
-
14. Poor TA, Chandel NS. SnapShot: Mitochondrial signaling. Mol Cell. 2023;83(6):1012.[DOI]
-
15. Picard M, Shirihai OS. Mitochondrial signal transduction. Cell Metab. 2022;34(11):1620-1653.[DOI]
-
16. Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491(7424):374-383.[DOI]
-
17. Gupta R, Kanai M, Durham TJ, Tsuo K, McCoy JG, Kotrys AV, et al. Nuclear genetic control of mtDNA copy number and heteroplasmy in humans. Nature. 2023;620(7975):839-848.[DOI]
-
18. Sankaran VG, Weissman JS, Zon LI. Cellular barcoding to decipher clonal dynamics in disease. Science. 2022;378(6616):eabm5874.[DOI]
-
21. Sun H, Kumari N, Melzer ME, Backman V, Goyal Y. Navigating nongenetic plasticity in cancer drug resistance. Annu Rev Cancer Biol. 2025;9:245-266.[DOI]
-
26. Marine JC, Dawson SJ, Dawson MA. Non-genetic mechanisms of therapeutic resistance in cancer. Nat Rev Cancer. 2020;20(12):743-756.[DOI]
-
44. Guernet A, Mungamuri SK, Cartier D, Sachidanandam R, Jayaprakash A, Adriouch S, et al. Crispr-barcoding for intratumor genetic heterogeneity modeling and functional analysis of oncogenic driver mutations. Mol cell. 2016;63(3):526-538.[DOI]
-
46. McKenna A, Findlay GM, Gagnon JA, Horwitz MS, Schier AF, Shendure J. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science. 2016;353(6298):aaf7907.[DOI]
-
47. Simeonov KP, Byrns CN, Clark ML, Norgard RJ, Martin B, Stanger BZ, et al. Single-cell lineage tracing of metastatic cancer reveals selection of hybrid EMT states. Cancer Cell. 2021;39(8):1150-1162.[DOI]
-
48. Raj B, Wagner DE, McKenna A, Pandey S, Klein AM, Shendure J, et al. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat Biotechnol. 2018;36(5):442-450.[DOI]
-
49. Alemany A, Florescu M, Baron CS, Peterson-Maduro J, van Oudenaarden A. Whole-organism clone tracing using single-cell sequencing. Nature. 2018;556(7699):108-112.[DOI]
-
50. He Z, Maynard A, Jain A, Gerber T, Petri R, Lin HC, et al. Lineage recording in human cerebral organoids. Nat Methods. 2022;19(1):90-99.[DOI]
-
51. Spanjaard B, Hu B, Mitic N, Olivares-Chauvet P, Janjuha S, Ninov N, et al. Simultaneous lineage tracing and cell-type identification using CRISPR–Cas9-induced genetic scars. Nat Biotechnol. 2018;36(5):469-473.[DOI]
-
52. Frieda KL, Linton JM, Hormoz S, Choi J, Chow KK, Singer ZS, et al. Synthetic recording and in situ readout of lineage information in single cells. Nature. 2017;541(7635):107-111.[DOI]
-
53. Bowling S, Sritharan D, Osorio FG, Nguyen M, Cheung P, Rodriguez-Fraticelli A, et al. An engineered CRISPR-Cas9 mouse line for simultaneous readout of lineage histories and gene expression profiles in single cells. Cell. 2020;181(7):1693-1694.[DOI]
-
54. Li L, Bowling S, McGeary SE, Yu Q, Lemke B, Alcedo K, et al. A mouse model with high clonal barcode diversity for joint lineage, transcriptomic, and epigenomic profiling in single cells. Cell. 2023;186(23):5183-5199.[DOI]
-
55. Lareau CA, Liu V, Muus C, Praktiknjo SD, Nitsch L, Kautz P, et al. Mitochondrial single-cell ATAC-seq for high-throughput multi-omic detection of mitochondrial genotypes and chromatin accessibility. Nat Protoc. 2023;18(5):1416-1440.[DOI]
-
56. Miller TE, Lareau CA, Verga JA, DePasquale EAK, Liu V, Ssozi D, et al. Mitochondrial variant enrichment from high-throughput single-cell RNA sequencing resolves clonal populations. Nat Biotechnol. 2022;40(7):1030-1034.[DOI]
-
57. Weng C, Yu F, Yang D, Poeschla M, Liggett LA, Jones MG, et al. Deciphering cell states and genealogies of human haematopoiesis. Nature. 2024;627(8003):389-398.[DOI]
-
58. Kotrys AV, Durham TJ, Guo XA, Vantaku VR, Parangi S, Mootha VK. Single-cell analysis reveals context-dependent, cell-level selection of mtDNA. Nature. 2024;629(8011):458-466.[DOI]
-
59. Xu J, Nuno K, Litzenburger UM, Qi Y, Corces MR, Majeti R, et al. Single-cell lineage tracing by endogenous mutations enriched in transposase accessible mitochondrial DNA. eLife. 2019;8:e45105.[DOI]
-
60. Ludwig LS, Lareau CA, Ulirsch JC, Christian E, Muus C, Li LH, et al. Lineage tracing in humans enabled by mitochondrial mutations and single-cell genomics. Cell. 2019;176(6):1325-1339.[DOI]
-
61. Lareau CA, Ludwig LS, Muus C, Gohil SH, Zhao T, Chiang Z, et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat Biotechnol. 2021;39(4):451-461.[DOI]
-
62. Zheng L, Shi D, Yan Y, Zhou B, Lim J, Hou Y, et al. Scalable and multiplexed recorders of gene regulation dynamics across weeks. Nature. 2026;652(8111):1038-1048.[DOI]
-
63. Chen M, Fu R, Chen Y, Li L, Wang SW. High-resolution, noninvasive single-cell lineage tracing in mice and humans based on DNA methylation epimutations. Nat Methods. 2025;22(3):488-498.[DOI]
-
64. Bianchi A, Scherer M, Zaurin R, Quililan K, Velten L, Beekman R. scTAM-seq enables targeted high-confidence analysis of DNA methylation in single cells. Genome Biol. 2022;23:229.[DOI]
-
65. Scherer M, Singh I, Braun MM, Szu-Tu C, Sanchez Sanchez P, Lindenhofer D, et al. Clonal tracing with somatic epimutations reveals dynamics of blood ageing. Nature. 2025;643(8071):478-487.[DOI]
-
66. Gabbutt C, Schenck RO, Weisenberger DJ, Kimberley C, Berner A, Househam J, et al. Fluctuating methylation clocks for cell lineage tracing at high temporal resolution in human tissues. Nat Biotechnol. 2022;40(5):720-730.[DOI]
-
67. Sheth RU, Yim SS, Wu FL, Wang HH. Multiplex recording of cellular events over time on CRISPR biological tape. Science. 2017;358(6369):1457-1461.[DOI]
-
68. Tang W, Liu DR. Rewritable multi-event analog recording in bacterial and mammalian cells. Science. 2018;360(6385):eaap8992.[DOI]
-
69. Choi J, Chen W, Minkina A, Chardon FM, Suiter CC, Regalado SG, et al. A time-resolved, multi-symbol molecular recorder via sequential genome editing. Nature. 2022;608(7921):98-107.[DOI]
-
70. Chen W, Choi J, Li X, Nathans JF, Martin B, Yang W, et al. Symbolic recording of signalling and cis-regulatory element activity to DNA. Nature. 2024;632(8027):1073-1081.[DOI]
-
71. Yan Y, Lu J, Li Z, Zhao Z, Shay TF, Wang S, et al. Genetically encoded assembly recorder temporally resolves cellular history. Nature. 2026;652(8111):1049-1059.[DOI]
-
72. Pasqual G, Chudnovskiy A, Tas JMJ, Agudelo M, Schweitzer LD, Cui A, et al. Monitoring T cell–dendritic cell interactions in vivo by intercellular enzymatic labelling. Nature. 2018;553(7689):496-500.[DOI]
-
73. Nakandakari-Higa S, Walker S, Canesso MCC, van der Heide V, Chudnovskiy A, Kim DY, et al. Universal recording of immune cell interactions in vivo. Nature. 2024;627(8003):399-406.[DOI]
-
74. Liu L, Chen A, Li Y, Mulder J, Heyn H, Xu X. Spatiotemporal omics for biology and medicine. Cell. 2024;187(17):4488-4519.[DOI]
-
75. Moses L, Pachter L. Museum of spatial transcriptomics. Nat Methods. 2022;19(5):534-546.[DOI]
-
76. Pan X, Danies-Lopez A, Zhang X. Mapping lineage-resolved scRNA-seq data with spatial transcriptomics using TemSOMap. bioRxiv [Preprint]. 2024.[DOI]
-
77. Xue Y, Su J, Chao Y, Liu L, Lin X, Xiang Y, et al. Single-cell mitochondrial lineage tracing decodes fate decision and spatial clonal architecture in human hematopoietic organoids. Adv Sci. 2026;13(17):e18084.[DOI]
-
78. Kinsler G, Fagan C, Li H, Kaster J, Dunne M, Vander Velde RJ, et al. SpaceBar enables single-cell-resolution clone tracing with imaging-based spatial transcriptomics. Nat Methods. 2026;23(2):328-333.[DOI]
-
79. Jia Y, Sun D, Weir JA, Liu X, Russell AJC, Kim Y, et al. SPACE-seq integrates spatial transcriptomics and lineage tracing in native tissues. Cell Stem Cell. 2026.[DOI]
-
80. Ratz M, von Berlin L, Larsson L, Martin M, Westholm JO, La Manno G, et al. Clonal relations in the mouse brain revealed by single-cell and spatial transcriptomics. Nat Neurosci. 2022;25(3):285-294.[DOI]
-
81. Ben-Moshe S, Itzkovitz S. Spatial heterogeneity in the mammalian liver. Nat Rev Gastroenterol Hepatol. 2019;16(7):395-410.[DOI]
-
82. Chembazhi UV, Bangru S, Dutta RK, Das D, Peiffer B, Natua S, et al. Dysregulated RNA splicing impairs regeneration in alcohol-associated liver disease. Nat Commun. 2025;16:8049.[DOI]
-
83. Arnoux G, Serre J, Verissimo T, Tihy M, Davidson SM, Placier S, et al. Integrated spatial and functional metabolic profiling identified the thick ascending limb as a mitochondrial vulnerability hub in acute kidney injury. Kidney Int. 2025;108(5):866-882.[DOI]
-
84. Quintana A, Zanella S, Koch H, Kruse SE, Lee D, Ramirez JM, et al. Fatal breathing dysfunction in a mouse model of Leigh syndrome. J Clin Invest. 2012;122(7):2359-2368.[DOI]
-
85. Quintana A, Kruse SE, Kapur RP, Sanz E, Palmiter RD. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc Natl Acad Sci U S A. 2010;107(24):10996-11001.[DOI]
-
86. van de Wal MAE, Adjobo-Hermans MJW, Keijer J, Schirris TJJ, Homberg JR, Wieckowski MR, et al. Ndufs4 knockout mouse models of Leigh syndrome: Pathophysiology and intervention. Brain. 2022;145(1):45-63.[DOI]
-
87. Hanaford AR, Khanna A, Truong V, James K, Chen Y, Mulholland M, et al. Peripheral macrophages drive CNS disease in the Ndufs4(-/-) model of Leigh syndrome. Brain Pathol. 2023;33(6):e13192.[DOI]
-
88. Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell. 2016;167(7):1867-1882.[DOI]
-
89. Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods. 2017;14(3):297-301.[DOI]
-
90. Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, et al. Perturb-seq: Dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell. 2016;167(7):1853-1866.[DOI]
-
91. Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell. 2016;167(7):1883-1896.[DOI]
-
92. Bock C, Datlinger P, Chardon F, Coelho MA, Dong MB, Lawson KA, et al. High-content CRISPR screening. Nat Rev Methods Primers. 2022;2(1):8.[DOI]
-
93. Baysoy A, Tian X, Zhang F, Renauer P, Bai Z, Shi H, et al. Spatially resolved in vivo CRISPR screen sequencing via perturb-DBiT. bioRxiv [Preprint]. 2024.[DOI]
-
94. Binan L, Jiang A, Danquah SA, Valakh V, Simonton B, Bezney J, et al. Simultaneous CRISPR screening and spatial transcriptomics reveal intracellular, intercellular, and functional transcriptional circuits. Cell. 2025;188(8):2141-2158.[DOI]
-
95. Binan L. Investigating spatial gene circuits and gene-phenotype mechanisms with Perturb-FISH. Nat Rev Genet. 2025;26(8):507-508.[DOI]
-
96. Feldman D, Singh A, Schmid-Burgk JL, Carlson RJ, Mezger A, Garrity AJ, et al. Optical pooled screens in human cells. Cell. 2019;179(3):787-799.[DOI]
-
97. Saunders RA, Allen WE, Pan X, Sandhu J, Lu J, Lau TK, et al. Perturb-Multimodal: A platform for pooled genetic screens with imaging and sequencing in intact mammalian tissue. Cell. 2025;188(17):4790-4809.[DOI]
-
98. Bunne C, Roohani Y, Rosen Y, Gupta A, Zhang X, Roed M, et al. How to build the virtual cell with artificial intelligence: Priorities and opportunities. Cell. 2024;187(25):7045-7063.[DOI]
-
99. Kuznets-Speck B, Schwartz L, Sun H, Melzer ME, Kumari N, Haley B, et al. Fluctuation structure predicts genome-wide perturbation outcomes. bioRxiv [Preprint]. 2025.[DOI]
-
100. Roohani Y, Huang K, Leskovec J. Predicting transcriptional outcomes of novel multigene perturbations with GEARS. Nat Biotechnol. 2024;42(6):927-935.[DOI]
-
101. Wei Z, Wang Y, Gao Y, Wang S, Li P, Si D, et al. Benchmarking algorithms for generalizable single-cell perturbation response prediction. Nat Methods. 2026;23(2):451-464.[DOI]
-
102. Theodoris CV, Xiao L, Chopra A, Chaffin MD, Al Sayed ZR, Hill MC, et al. Transfer learning enables predictions in network biology. Nature. 2023;618(7965):616-624.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Chandel NS, Goyal Y. Decoding the clonal origins of mitochondrial pathology. EXO. 2026;1:202612. https://doi.org/10.70401/EXO.2026.0016
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Lineage Mosaicism and Metabolic Disease
- 3. Design Principles for Lineage-Resolved Metabolic Studies
- 4. From Cancer to Metabolism: Why Lineage History Matters
- 5. Technologies for Reconstructing Cellular History
- 6. Lineage-Resolved Questions across Mitochondrial Disease
- 7. From Lineage Correlation to Causal Inference in Mitochondrial Dysfunction-Dependent Pathology
- 8. Outlook
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Chandel NS, Goyal Y. Decoding the clonal origins of mitochondrial pathology. EXO. 2026;1:202612. https://doi.org/10.70401/EXO.2026.0016
copy
Share Link
copy