Oral stem cell aging: From mechanisms to therapeutic rejuvenation
*Correspondence to:
Songtao Shi, Hospital of Stomatology, Guanghua School of Stomatology, South China Center of Craniofacial Stem Cell Research, Guangdong Provincial Key Laboratory of Stomatology, Sun Yat-sen University, Guangzhou 510055, Guangdong, China.
E-mail: shisongtao@mail.sysu.edu.cn
Ageing Cancer Res Treat. 2026;3:202618. 10.70401/acrt.2026.0022
Received: April 23, 2026Accepted: May 29, 2026Published: May 29, 2026
Abstract
Oral stem cells (OSCs) represent a group of mesenchymal stromal/stem cell-like populations localized within dental and craniofacial tissues, where they play vital roles in maintaining tissue integrity and supporting regeneration. Although OSCs possess robust self-renewal capacity and multilineage differentiation potential, they progressively develop senescent phenotypes in response to organismal aging, prolonged in vitro culture, and adverse microenvironmental stimuli. The senescent state of OSCs is characterized by diminished proliferation and differentiation, genomic instability, mitochondrial impairment, and the establishment of a senescence-associated secretory phenotype. Mechanistically, OSC senescence arises from a series of interacting processes, including DNA damage, metabolic imbalance, epigenetic reprogramming, and persistent inflammatory signaling. These alterations drive stem cell functional decline and compromise the surrounding regenerative niche. Consequently, OSC aging is closely associated with multiple craniofacial disorders, including periodontal tissue breakdown, defective dentin-pulp regeneration, alveolar bone resorption, and delayed mucosal healing. To reverse these age-related changes, various rejuvenation approaches have been explored, including epigenetic modulation, metabolic intervention, senescence-targeting therapies, extracellular vesicle-mediated strategies, and biomaterial-based niche engineering. Nonetheless, translational challenges remain, particularly in cellular heterogeneity, donor-related variability, and age-dependent functional changes in extracellular vesicles (EVs). Future efforts are expected to focus on developing targeted and clinically translatable strategies to rejuvenate OSCs and enhance regenerative outcomes.
Keywords
Oral stem cells, cellular senescence, inflammaging, senotherapy, extracellular vesicles
1. Introduction
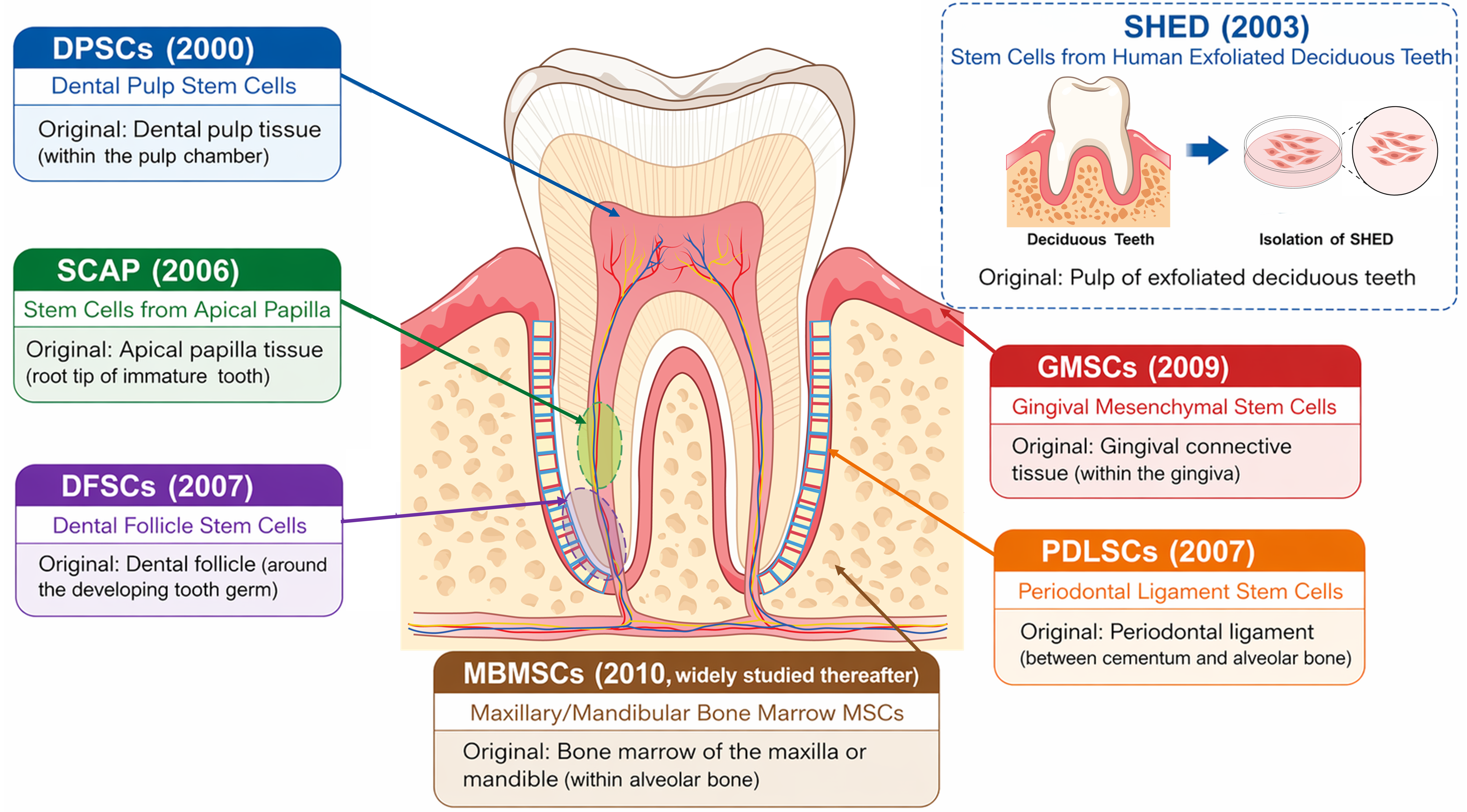
Oral stem cells (OSCs) constitute a diverse group of mesenchymal stromal/stem cell (MSC)-like populations residing in dental and craniofacial tissues. Research on OSCs emerged in 2000 when Gronthos and Shi et al. isolated dental pulp stem cells (DPSCs) from adult pulp tissue, demonstrating their clonogenic and odontogenic potential[1] (Figure 1). This discovery was soon followed by the identification of stem cells from human exfoliated deciduous teeth (SHED) in 2003[2], followed by subsequently periodontal ligament stem cells (PDLSCs) in 2004[3], dental follicle stem cells (DFSCs) in 2005[4], stem cells from the apical papilla (SCAP) in 2006[5], and gingival mesenchymal stem cells (GMSCs) in 2009[6]. These milestones collectively established OSCs as an easily accessible and highly regenerative stem cell population with broad applications in dental pulp regeneration, periodontal reconstruction, and craniofacial tissue engineering. Specifically, OSCs are known for their remarkable self-renewal ability, multilineage differentiation potential, and distinctive immunomodulatory properties[7].
{kind=link}
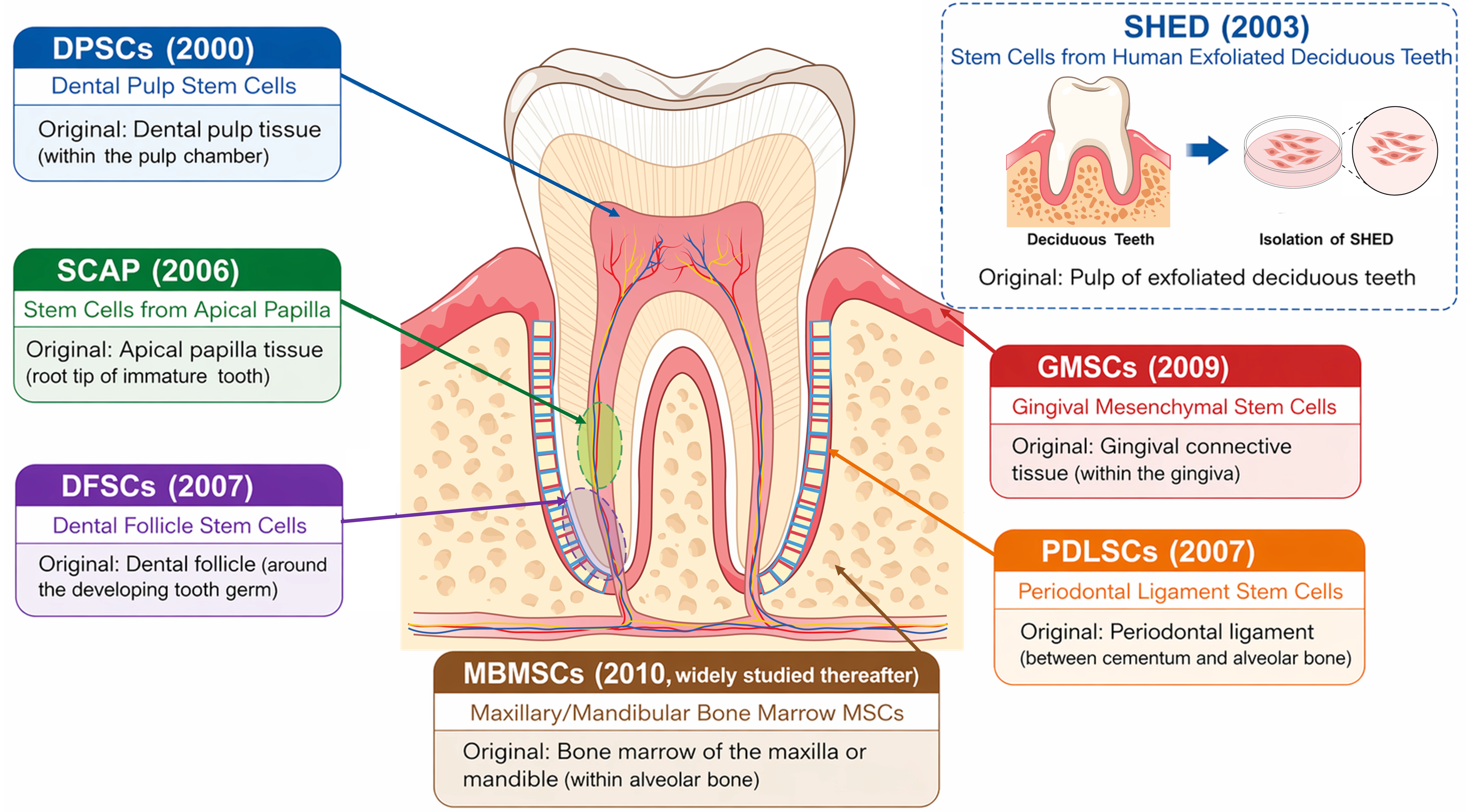
Figure 1. Anatomical distribution and discovery timeline of major OSCs population. Schematic illustration showing the major OSCs and their anatomical locations within the tooth and surrounding tissues. DPSCs (2000) arise from the dental pulp; SCAP (2006) from the apical papilla; DFSCs (2005) from the dental follicle; PDLSCs (2004) from the periodontal ligament; GMSCs (2009) from gingival connective tissue; and MBMSCs (2010s) from alveolar bone marrow. An inset depicts SHED (2003), isolated from exfoliated deciduous teeth. OSCs: oral stem cells; DPSCs: dental pulp stem cells; SCAP: stem cells from the apical papilla; DFSCs: dental follicle stem cells; PDLSCs: periodontal ligament stem cells; GMSCs: gingival mesenchymal stem cells; MBMSCs: maxillary/mandibular bone marrow-derived MSCs; SHED: stem cells from human exfoliated deciduous teeth.
Cellular senescence is a fundamental biological process characterized by stable cell cycle arrest and widespread alterations in cellular function[8]. Senescence can be triggered by diverse stimuli and plays essential roles in tissue remodeling, physiological aging, and related pathologies[9]. Stem cells are particularly susceptible to aging, as it directly impairs their self-renewal capacity and disrupts tissue homeostasis[10]. In this context, OSCs undergo senescence during organismal aging, prolonged in vitro expansion, or exposure to pathological microenvironments[11]. Functionally, senescent OSCs exhibit reduced proliferation and clonogenicity, impaired differentiation potential, genomic instability, mitochondrial dysfunction, and activation of the senescence-associated secretory phenotype (SASP)[12]. These alternations weaken their intrinsic regenerative potential and perturb the surrounding niche, contributing to impaired dental pulp regeneration, periodontal degeneration, alveolar bone loss, and delayed oral mucosal healing[13].
Unlike conventional mesenchymal stem cells, OSCs undergo a unique aging trajectory shaped by lifelong microbial exposure and mechanical stress in the oral environment. These factors drive metabolic, epigenetic, and inflammatory alterations that progressively impair regenerative function. Therefore, future therapies should shift from broad anti-aging interventions toward precision rejuvenation strategies that restore OSC function while preserving lineage identity. This review discusses the mechanisms underlying OSC aging and emerging therapeutic opportunities for craniofacial regeneration.
2. Types of OSCs and Their Biological Characteristics
OSCs comprise a heterogeneous group of stem cell populations that can be broadly categorized into dental-derived and non-dental-derived stem cells based on their tissue origin. Dental-derived stem cells include DPSCs, SHED, SCAP, and DFSCs, whereas non-dental OSCs encompass PDLSCs, GMSCs, and maxillary/mandibular bone marrow-derived MSCs (MBMSCs)[14]. Diverse OSCs are readily accessible and exhibit high proliferative capacity, multilineage differentiation potential, and distinctive neurovascular and immunomodulatory properties[15].
DPSCs, the first identified OSC population, exhibit typical mesenchymal features, including clonogenicity, multilineage differentiation potential, and robust proliferative capacity[1]. They can differentiate into odontoblast-like cells and generate reparative dentin-like structures in vivo[16]. DPSCs can express mesenchymal markers, such as vascular cell adhesion molecule-1 (VCAM-1), CD146, alpha-smooth muscle actin (α-SMA), and bone-related markers such as alkaline phosphatase (ALP), osteopontin (OPN), and bone sialoprotein (BSP). SHED, isolated from exfoliated deciduous teeth, displays a more immature phenotype, characterized by the expression of mesenchymal markers (e.g., STRO-1, CD146), embryonic stem cell markers, and tumor-related antigens[2,17]. Compared with DPSCs, SHED exhibit higher proliferation rates and enhanced osteogenic potential, suggesting a greater regenerative capacity[2,18].
SCAPs are derived from the apical papilla of immature permanent teeth and exhibit strong proliferative capacity and odontogenic potential comparable to DPSCs[5]. SCAPs express multiple bone-/dentin-related markers together with the MSC marker STRO-1. Notably, SCAPs also show high expression of several neural markers, including Nestin, neuronal nuclei antigen (NeuN) and glial fibrillary acidic protein (GFAP), similar to DPSCs and SHED[19]. In organ cultures, SCAPs display a proliferation rate 2-3 times higher than that of DPSCs, while expressing lower levels of dentin sialoprotein (DSP), transforming growth factor β receptor II (TGFβRII), fms-like tyrosine kinase 1 (Flt-1), fibroblast growth factor receptor 1 (FGFR-1/Flg), and CD146. These differences likely reflect the lower vascularity and cellularity of the apical papilla, together with the fact that SCAP function as primary rather than reparative odontoblasts[20]. Owing to their intimate involvement in root development, DPSCs, SHED, and SCAPs serve as ideal seed cells for functional dental pulp regeneration, demonstrating superior potential in reconstructing neurovascularized pulp-like tissues and promoting continued root maturation[21].
DFSCs are neural crest-derived stem cells in the dental follicle tissue of tooth germs[22]. As direct precursor cells of the periodontium, DFSCs develop into the periodontal ligament, cementum, and alveolar bone proper during the later stages of tooth development. DFSCs exhibit a typical fibroblast-like morphology and express undifferentiated markers such as Notch receptor 1 (Notch-1), Nestin, and STRO-1, as well as osteogenic proteins, including osteocalcin (OCN) and BSP[4,23]. Although DFSCs show strong osteogenic and cementogenic potential in vitro, in vivo transplantation typically results in fibrous or rigid structures rather than organized mineralized tissues[24].
PDLSCs are a novel population of adult stem cell isolated from periodontal ligament tissues with high-level mesenchymal markers such as STRO-1 and CD146. PDLSCs feature a remarkable proliferative capacity in vitro comparable to DPSCs[3,25], and possess a unique osteogenic/cementogenic differentiation ability, with expression of the tendon-related transcription factor scleraxis. These potentials enable PDLSCs to form collagen fibers resembling Sharpey’s fibers. In vivo transplantation further demonstrates their ability to regenerate cementum/periodontal ligament-like structures[3], underscoring their therapeutic potential for periodontal diseases and highlighting the profound influence of the tissue-specific microenvironment on MSC developmental potency.
GMSCs are derived from human gingival tissues and are characterized by the expression of octamer-binding transcription factor 4 (OCT-4), stage-specific embryonic antigen-4 (SSEA-4), human telomerase reverse transcriptase (hTERT), and STRO-1. Notably, they can be stably expanded in vitro over multiple passages without loss of their early phenotypic characteristics[6]. In addition to multipotent differentiation capacity, GMSCs also possess potent immunomodulatory and anti-inflammatory functions through modulating the phenotype and activation of a variety of innate and adaptive immune cells both in vitro and in vivo[26]. Derived from maxillary/mandibular bone marrow, MBMSCs exhibit distinct site-specific characteristics because of their neural crest origin and intramembranous ossification pathway, distinguishing them from mesoderm-derived long bone MSCs[14,27]. Biologically, they possess superior proliferative capacity, delayed onset of senescence and enhanced osteogenic differentiation potential, characterized by higher alkaline phosphatase activity and calcium accumulation[28]. In contrast, MBMSCs display significantly lower adipogenic commitment and terminal differentiation due to the suppressed expression of key transcription factors such as zinc finger protein 423 (Zfp423), peroxisome proliferator-activated receptor γ (PPARγ), and CCAAT/enhancer-binding proteinδ (C/EBPδ)[29]. Furthermore, their unique immunomodulatory interplay with T-lymphocytes underscores their specialized efficacy for regenerating orofacial defects[30].
Drawing from their unique ectomesenchymal origin, OSCs represent a specialized subpopulation that differs significantly from classical MSCs. While OSCs maintain core mesenchymal hallmarks, such as fibroblast-like morphology, robust colony-forming units, and multilineage plasticity, they are developmentally distinct, as most originate from neural crest cells and peripheral nerve-associated glia. This distinct lineage endows them with specific neuro-vascular profiles, including the expression of glial proteins and α-SMA, alongside a superior capacity for angiogenic and neurogenic differentiation[15].
3. Characteristics and Molecular Mechanisms of OSC Aging
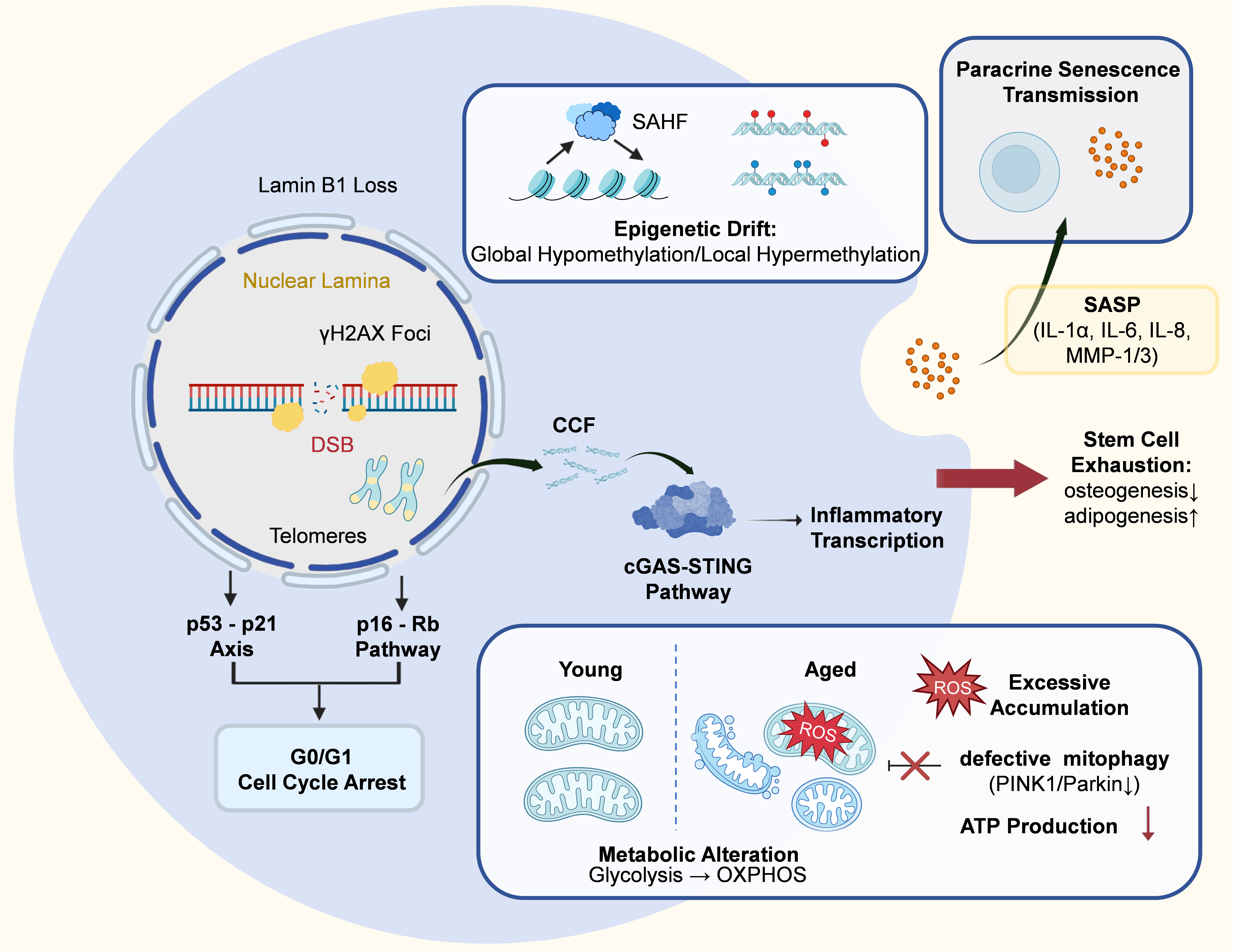
Unlike mesoderm-derived MSCs, many OSCs originate from cranial neural crest cells and retain distinct developmental and epigenetic features[31]. Together with lifelong exposure to microbial, inflammatory, and mechanical stress in the oral cavity, these characteristics may contribute to a unique aging trajectory[7]. However, direct mechanistic evidence linking neural crest origin to senescence remains limited and requires further investigation. Despite these unique upstream influences, their effects ultimately converge on several conserved molecular pathways that drive senescence progression. At the molecular level, the senescent decline is typically driven by multiple interconnected factors, such as genomic instability, mitochondrial dysfunction, epigenetic alterations, and microenvironmental shifts (Figure 2)[32].
{kind=link}
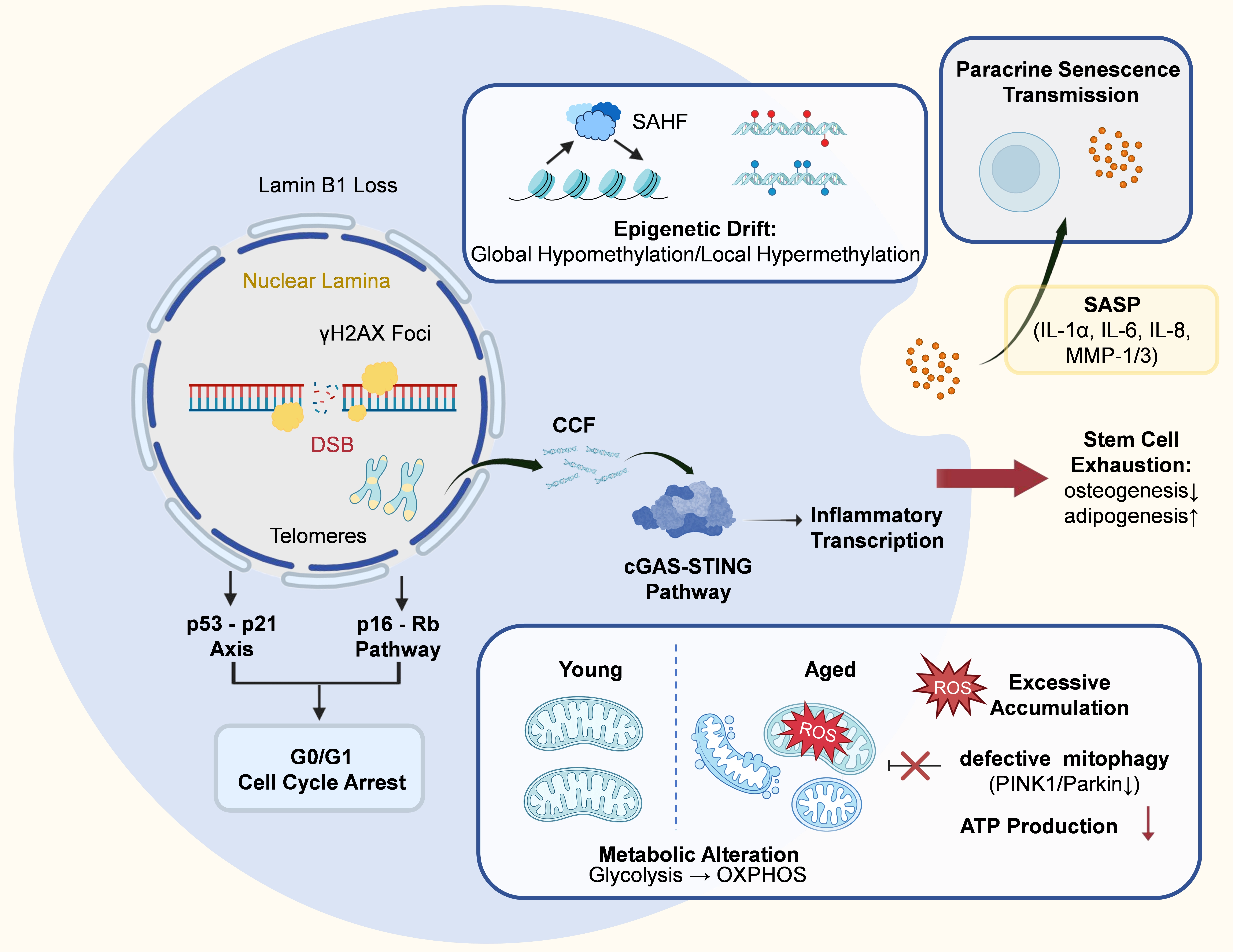
Figure 2. Molecular landscape and underlying mechanisms of OSC senescence. Schematic illustration of the key molecular and functional alterations in senescent OSCs. Senescent OSCs exhibit nuclear and genomic instability, characterized by persistent DNA damage (e.g., double-strand breaks and γH2AX accumulation), telomere attrition, and loss of nuclear lamina integrity (e.g., Lamin B1 depletion). These alterations activate the p53-p21 and p16-Rb pathways, ultimately leading to irreversible G0/G1 cell cycle arrest. Epigenetic remodeling further reinforces senescence, including global epigenetic drift and the formation of SAHF, resulting in stable transcriptional reprogramming. The leakage of CCF triggers the cGAS-STING pathway, driving inflammatory transcription. In parallel, mitochondrial dysfunction develops, characterized by metabolic inflexibility (a shift from glycolysis toward oxidative phosphorylation), impaired mitophagy (PINK1/Parkin downregulation), excessive ROS accumulation, and reduced ATP production. Collectively, these molecular alterations drive stem cell exhaustion, manifested as reduced osteogenic differentiation and enhanced adipogenic bias. In addition, senescent OSCs secrete a range of SASP factors (e.g., IL-1α, IL-6, IL-8, MMP-1, and MMP-3), which disrupt the local microenvironment and propagate senescence to neighboring cells via paracrine signaling. OSC: oral stem cell; γH2AX: phosphorylated histone H2AX; SAHF: senescence-associated heterochromatin foci; CCF: cytoplasmic chromatin fragments; ROS: reactive oxygen species; SASP: senescence-associated secretory phenotype; DSB: DNA double-strand break; cGAS-STING: cyclic GMP-AMP–stimulator of interferon genes; OXPHOS: oxidative phosphorylation.
Importantly, activation of these molecular pathways does not necessarily lead to a uniform senescence outcome, and accumulating evidence suggests substantial heterogeneity in the progression and reversibility of stem cell aging. Cellular senescence has traditionally been defined as an irreversible cell-cycle arrest state. However, emerging evidence suggests that this definition may oversimplify the biological heterogeneity of stem cell aging. In OSCs, certain early dysfunctions, including mitochondrial metabolic imbalance, epigenetic alterations, and SASP-associated inflammatory signaling, may remain partially reversible through pharmacological or bioengineering interventions. In contrast, advanced senescence characterized by severe genomic instability, telomere erosion, and stem cell pool depletion may represent a more stable and difficult-to-reverse state. Therefore, OSC aging may be better viewed as a dynamic continuum ranging from reversible functional decline to terminal regenerative exhaustion.
3.1 Genomic instability and DNA damage
Genomic instability and DNA damage constitute fundamental molecular mechanisms of OSC aging[33]. Both physiological metabolism and external pathological stressors, including oxidative stress, ionizing radiation, and chronic periodontal inflammation, constantly generate DNA lesions[34]. While young OSCs deploy robust DNA damage repair (DDR) to maintain genomic integrity, senescent OSCs exhibit markedly reduced DDR efficiency and accuracy[35].
Molecularly, senescent OSCs exhibit significantly elevated levels of phosphorylated histone H2AX (γH2AX)[36], an early and sensitive marker for DNA double-strand breaks, in the nuclei of aged DPSCs and PDLSCs. The accumulation of γH2AX indicates marked defects in major DNA repair pathways, including homologous recombination and non-homologous end joining[37,38]. The defects of persistent DNA repair ultimately manifest as large-scale genomic alterations, consistent with scRNA-seq findings that substantial chromosomal instability and subclonal aneuploidy are present in senescent OSCs[39].
Telomere attrition represents a classic mechanism of stem cell aging, in which insufficient telomerase activity leads to progressive shortening of telomeric repeats and subsequent activation of DDR signaling that enforces tumor protein (p53)-cyclin-dependent kinase inhibitor 1A (p21)-mediated cell-cycle arrest[40-43]. However, contemporary evidence suggests that telomere erosion in OSCs more often reflects cumulative oxidative stress rather than serving as the primary driver of natural senescence[44]. Unrepaired nuclear DNA break fragments leak out into the cytoplasm as cytoplasmic chromatin fragments (CCF), which are abnormally recognized and activated by the cyclic GMP-AMP-stimulator of interferon genes (cGAS-STING) pathway, thereby driving the transcription of inflammatory factors[45]. Thus, telomere dysfunction is one component of a broader genomic instability landscape.
3.2 Mitochondrial dysfunction and metabolic alterations
Mitochondrial dysfunction and metabolic alterations constitute another primary hallmark of OSC aging[46]. Senescent OSCs exhibit profound metabolic inflexibility, characterized by a shift from glycolysis toward oxidative phosphorylation (OXPHOS) despite a diminished respiratory capacity[47]. Consequently, ATP production declines, while reactive oxygen species (ROS) accumulate excessively. Concomitantly, aging OSCs display fragmented mitochondria with disrupted cristae organization[48].
A central mechanism underlying mitochondrial dysfunction is the progressive deterioration of the electron transport chain (ETC). Mitochondrial DNA (mtDNA), located in close proximity to ROS-generating complexes, is highly susceptible to oxidative damage[10]. Accumulated mtDNA mutations compromise the synthesis of essential ETC components, further increasing ROS production and establishing a self-reinforcing cycle of ROS accumulation, mtDNA instability, and OXPHOS deficiency[49]. This vicious cycle is exacerbated by defective PTEN-induced kinase 1 (PINK1)/Parkin-mediated mitophagy, leading to the persistence of dysfunctional mitochondria[50,51]. Additionally, loss of mitochondrial deacetylase sirtuin 3 (SIRT3) suppresses antioxidant defenses, thereby amplifying oxidative stress and metabolic dysfunction[52]. Collectively, these defects converge to produce a sustained mitochondrial collapse in aged OSCs.
Importantly, emerging evidence indicates that ROS exerts a dual role in stem cell biology. While excessive ROS accumulation drives mitochondrial damage and senescence, physiological ROS levels function as essential signaling molecules that regulate stemness and activate pathways such as hypoxia-inducible factor-1 alpha (HIF-1α) signaling[53,54]. This context-dependent role of ROS highlights a critical therapeutic challenge: rejuvenation strategies should aim to selectively mitigate pathological oxidative stress while preserving physiological redox signaling.
3.3 Epigenetic alterations
Epigenetic alterations represent a central driver of transcriptional reprogramming in senescent OSCs, arising from coordinated disruptions in both DNA methylation and histone-modifying systems[55]. Senescent cells exhibit a characteristic pattern of “epigenetic drift,” in which global DNA hypomethylation coexists with focal promoter hypermethylation[56]. Mechanistically, aging markedly suppresses the core DNA methyltransferases (DNMT), including DNMT1, DNMT3A, and DNMT3B, thereby contributing to the progressive loss of genome-wide methylation stability[57]. Paradoxically, despite overall hypomethylation, selective promoters, including those governing stemness maintenance, undergo hypermethylation, resulting in transcriptional silencing and diminished regenerative competence[8].
A rapidly expanding research frontier emphasizes the metabolic-epigenetic coupling underlying these methylation defects. Senescent DPSCs downregulate the serine synthesis enzymes phosphoserine aminotransferase 1 (PSAT1)/phosphoglycerate dehydrogenase (PHGDH) expression, which sharply reduces intracellular S-adenosylmethionine (SAM) availability. Because SAM is the universal methyl donor, its depletion preferentially induces hypomethylation at specific genomic loci such as the cyclin-dependent kinase inhibitor 2A (p16) promoter, enabling aberrant transcriptional activation of senescence programs[58]. These findings highlight that metabolic rewiring does not globally suppress methylation but rather redefines methylation specificity in a locus-dependent manner, a process that remains mechanistically unresolved.
In parallel, senescent OSCs accumulate repressive histone marks, particularly histone H3 lysine 27 trimethylation (H3K27me3), further constraining chromatin accessibility and silencing pro-regenerative genes[59]. Loss of nuclear lamina integrity, especially Lamin B1 depletion, promotes large-scale chromatin reorganization and the formation of senescence-associated heterochromatin foci (SAHF). This structural remodeling reinforces the transcriptional repression of developmental and stemness networks while simultaneously enabling the inappropriate activation of pro-senescence genes[33]. Together, these multilayered epigenetic alterations converge to reprogram the chromatin landscape of senescent OSCs, locking cells into a stable, self-perpetuating state of regenerative exhaustion.
3.4 Stem cell exhaustion
Stem cell exhaustion represents a major functional consequence of cumulative senescence mechanisms and contributes directly to impaired craniofacial tissue regeneration in aged individuals[60]. Senescent OSCs typically display enlarged morphology, diminished proliferation, and markedly reduced colony-forming ability, features that collectively reflect reduced cellular fitness[61].
Beyond these general defects, aging profoundly disrupts lineage fidelity. Senescent OSCs exhibit impaired osteogenic commitment accompanied by a pronounced shift toward adipogenic differentiation. This adipogenic bias alters niche composition and exacerbates alveolar bone resorption in the elderly[11]. In addition, stem cell exhaustion is often associated with a progressive depletion of the stem cell pool, driven by impaired self-renewal and increased senescence-associated cell loss, further limiting long-term regenerative capacity. Mechanistically, persistent activation of cell-cycle arrest pathways drives exhaustion[62]. Diverse stressors converge on the p53/p21 and p16/retinoblastoma protein (Rb) axes, where elevated cyclin-dependent kinase inhibitors permanently lock OSCs into irreversible G0/G1 arrest[7,33].
Recent scRNA-seq analyses further indicate that senescent OSCs are not a uniform population[63]. P16+ and p21+ subsets display distinct transcriptional signatures and divergent SASP profiles, suggesting that stem cell exhaustion emerges from heterogeneous senescent subpopulations rather than a single terminal state[64,65]. This heterogeneity underscores the need to refine therapeutic interventions toward selectively targeting pathological senescent subsets.
3.5 Inflammaging and SASP
A defining pathological feature of senescent OSCs is the development of a hyperactive SASP, which contributes to both local tissue degeneration and systemic inflammaging[7]. Senescent OSCs secrete high levels of pro-inflammatory cytokines, including IL-1α, IL-6, and IL-8, as well as matrix metalloproteinases such as MMP-1 and MMP-3. Chronic exposure to SASP progressively compromises niche integrity, promotes extracellular matrix degradation, and establishes a persistent pro-inflammatory microenvironment that propagates tissue dysfunction[61].
Beyond general inflammatory effects, SASP directly impairs the regenerative functions of OSCs. Pro-inflammatory cytokines, particularly IL-6 and IL-1β, suppress osteogenic and odontogenic differentiation by inhibiting key transcription factors such as runt-related transcription factor 2 (Runx2) and reducing ALP activity[66]. In parallel, SASP reshapes the immune microenvironment by recruiting and activating immune cells, including macrophages, thereby sustaining chronic inflammation and further amplifying tissue damage[67].
SASP expression is predominantly regulated through nuclear factor kappa B (NF-κB)–dependent transcriptional programs. Functionally, SASP exerts strong paracrine activity: cytokines including IL-1α and IL-8 can induce “secondary senescence” in neighboring cells, thereby amplifying senescence burden across the tissue[61]. This paracrine amplification not only expands the senescent cell burden but also accelerates stem cell functional decline.
Notably, SASP exhibits a context-dependent dual role. While prolonged SASP signaling accelerates aging and tissue degeneration, transient SASP activation during acute injury facilitates wound healing by recruiting immune cells and promoting tissue remodeling[68]. These opposing roles highlight the importance of developing strategies that modulate, rather than globally inhibit, SASP activity.
3.6 Interconnected regulatory network of OSC senescence
Rather than functioning as isolated events, the molecular hallmarks of OSC senescence are increasingly recognized as a highly interconnected regulatory network that collectively drives progressive regenerative decline. Persistent DNA damage serves as an early initiating event by activating DNA repair pathways, which can lead to excessive consumption of intracellular nicotinamide adenine dinucleotide (NAD+). NAD+ depletion subsequently impairs sirtuin activity and mitochondrial homeostasis, resulting in reduced oxidative phosphorylation efficiency, defective mitophagy, and progressive mitochondrial dysfunction[7,33]. In turn, dysfunctional mitochondria generate excessive ROS, which further exacerbate nuclear and mitochondrial DNA damage, thereby forming a self-amplifying DNA damage-mitochondrial stress loop[48].
In parallel, metabolic rewiring directly influences epigenetic stability by altering the availability of key metabolites that serve as substrates or cofactors for chromatin-modifying enzymes. For example, reduced levels of SAM, α-ketoglutarate, and NAD+ may disrupt DNA methylation, histone acetylation, and chromatin accessibility, thereby accelerating epigenetic drift[58]. These epigenetic alterations further reinforce senescence-associated transcriptional programs, including activation of inflammatory pathways such as cGAS-STING signaling. Consequently, senescent OSCs establish a robust SASP, which propagates inflammatory signaling to neighboring cells and induces secondary senescence through paracrine mechanisms.
Collectively, these processes form a self-reinforcing feed-forward loop in which genomic instability, mitochondrial dysfunction, metabolic imbalance, epigenetic remodeling, and inflammatory signaling continuously amplify one another. This integrated model may better explain why OSC aging often progresses from reversible functional decline to stable regenerative exhaustion, while also highlighting multiple potential intervention nodes for therapeutic rejuvenation.
Beyond canonical hallmarks such as DNA damage and mitochondrial dysfunction, we propose that OSC aging may involve a unique metabolism–epigenetics–microenvironment coupling mechanism. Due to their neural crest-derived developmental origin and lifelong exposure to the oral environment, OSCs continuously encounter microbial stimulation, inflammatory stress, and mechanical loading[31]. These external stressors may reshape intracellular metabolic states, and further influence chromatin remodeling and accelerate epigenetic drift. This integrated model may partially explain why OSC aging exhibits both common stem cell aging hallmarks and tissue-specific features distinct from other mesenchymal stem cell populations.
4. Functional Consequences of OSC Aging
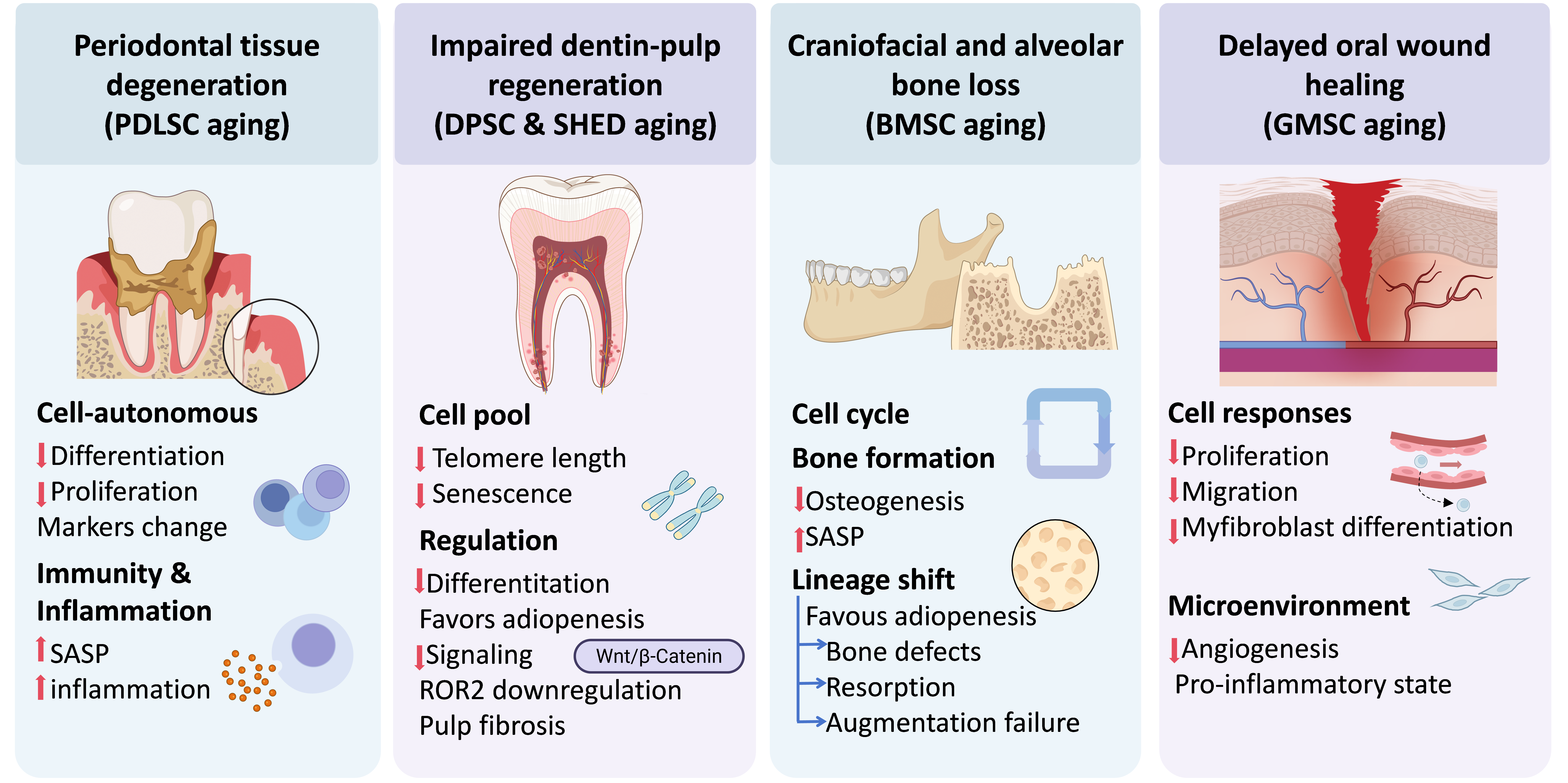
OSC aging is a fundamental driver of the decline in regenerative capacity, immune homeostasis, and structural integrity within the craniofacial region. Rather than a simple reduction in cell number, OSC aging leads to a profound shift in the local microenvironment from a pro-regenerative state to a chronic inflammatory microenvironment[24]. Oral tissue dysfunctions caused by the aging of OSCs include periodontal tissue degeneration, impaired dentin-pulp regeneration, craniofacial and alveolar bone loss, and delayed oral wound healing (Figure 3).
{kind=link}
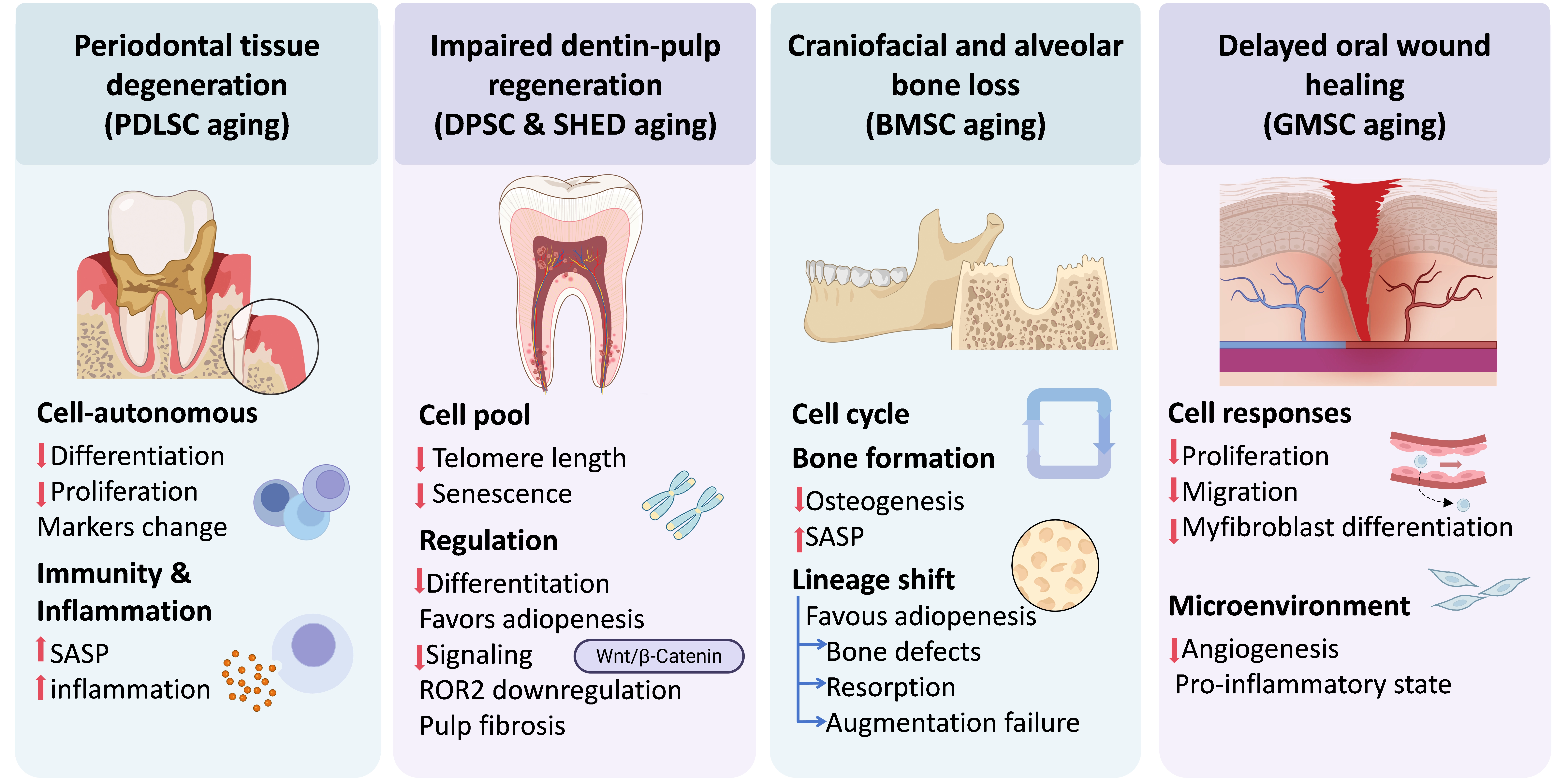
Figure 3. Functional consequences of OSC aging: drivers of craniofacial degenerative pathologies. Schematic illustrates how OSC senescence orchestrates a transition from a pro-regenerative microenvironment to one dominated by chronic inflammation and impaired tissue repair. PDLSC aging manifests as diminished osteogenic differentiation and immune dysregulation, driving the pathogenesis of periodontitis. In DPSCs and SHED, telomere attrition and ROR2-mediated signaling shifts promote a pro-adipogenic lineage skewing and pulp fibrosis. MBMSC senescence triggers cell cycle arrest and reduced osteogenic commitment, leading to alveolar bone resorption and failure of clinical augmentation. Concurrently, GMSC aging impairs myofibroblastic differentiation and angiogenesis through a pro-inflammatory secretory phenotype, resulting in delayed oral wound healing. Ultimately, these integrated cell-intrinsic deficits and microenvironmental shifts culminate in the progressive loss of craniofacial structural integrity. OSC: oral stem cell; PDLSC: periodontal ligament stem cell; DPSCs: dental pulp stem cell; SHED: stem cells from human exfoliated deciduous teeth; MBMSC: maxillary/mandibular bone marrow-derived MSCs; GMSC: gingival mesenchymal stem cell; BMSC: bone marrow-derived MSC; ROR: receptor tyrosine kinase-like orphan receptors.
4.1 Periodontal tissue degeneration: PDLSC ageing
Aging leads to profound functional impairment of PDLSCs and further contributes to periodontal tissue degeneration through two interrelated mechanisms. Aged PDLSCs exhibit impaired osteogenic/cementogenic differentiation capacity[69], with reduced ALP activity, diminished mineralized nodule formation, and downregulation of key osteogenic transcription factors such as Runx2, ALP, and collagen type I alpha 1 chain (COL1A1)[70]. These cellular defects are accompanied by reduced proliferative capacity, decreased expression of stem cell markers STRO-1 and CD146, and impaired regenerative potential in vivo, as evidenced by the failure to form cementum/periodontal ligament-like structures in ectopic transplantation models[71].
Moreover, aging compromises the immunomodulatory functions of PDLSCs, manifesting as a reduced capacity to suppress peripheral blood mononuclear cell (PBMC) proliferation and a diminished ability to maintain long-term immune tolerance. Impaired immunomodulation and senolytic phenotype likely contribute to a chronic inflammatory microenvironment within the periodontium[69]. Collectively, age-related alterations encompass both cell-autonomous defects in regenerative capacity and dysregulation of immune homeostasis. Therefore, PDLSCs aging underscores the pivotal role of PDLSC aging in the pathogenesis of periodontitis and the compromised efficacy of regenerative therapies in elderly populations.
4.2 Impaired dentin-pulp regeneration: DPSC and SHED ageing
Aging significantly impairs the proliferative capacity of DPSCs and SHED. Advanced donor age or extensive in vitro expansion leads to senescence-associated functional decline, such as impaired dentin-pulp regeneration, decreased dentin formation, and pulp fibrosis. The exhaustion of the functional stem cell pool, driven by critical telomere shortening and cell cycle arrest[72], ensures that an insufficient number of progenitor cells are available for mobilization upon injury, fundamentally causing impaired dentin-pulp regeneration[73,74].
The aging process compromises the multilineage differentiation potential of DPSCs and SHEDs, directly causing a decrease in dentin regeneration. Senescent cells exhibit a markedly reduced capacity for osteogenic and odontogenic differentiation, evidenced by fewer mineralization nodules and downregulated functional markers like ALP and OCN[72,75]. Furthermore, aging induces a "lineage shift" where cells favor adipogenesis over hard tissue formation[74]. A decline in Wnt/β-Catenin signaling is also specifically linked to the loss of neurogenic differentiation capacity in aged dental stem cells, further impairing the complex neuro-vascular regeneration of the pulp[76].
Deep molecular shifts drive the pathological progression of aging. The downregulation of receptor tyrosine kinase-like orphan receptor 2 (ROR2) acts as a central trigger, promoting senescence through the MSX2/NSUN2/p21 regulatory axis[77] or by inhibiting the STK4-FOXO1/SMS1 axis[74], which disrupts sphingomyelin biosynthesis and alters free fatty acid compositions[78]. These metabolic changes, combined with SASP characterized by increased secretion of inflammatory cytokines like IL-6, further deteriorate the dental pulp microenvironment. This chronic inflammatory state inhibits functional regeneration and promotes the over-proliferation of fibroblasts and abnormal collagen deposition, ultimately resulting in pulp fibrosis[75,76].
4.3 Craniofacial and alveolar bone loss: Bone marrow-derived MSC (BMSC) ageing
The senescence of BMSCs is a pivotal factor in the decline of bone regenerative capacity, particularly in the maxillofacial region. This region is characterized by a unique neural crest-derived embryonic origin. With advancing age or chronic inflammation, MBMSCs undergo cell cycle arrest and a marked reduction in osteogenic differentiation potential. This functional impairment leads to the downregulation of osteogenic genes and the emergence of SASP, which further exacerbates the deterioration of the local bone microenvironment[79,80].
Under pathological conditions, senescent MBMSCs exhibit a lineage commitment shift, favoring adipogenesis over osteogenesis, which results in maxillary and alveolar bone defects[81]. This senescence-mediated decline in bone formation is a primary cause of clinical alveolar ridge resorption and the failure of bone augmentation procedures[11,80].
4.4 Delayed oral wound healing: GMSC ageing
Aging of GMSCs and fibroblasts is a primary driver of delayed oral wound healing. Research indicates that aging significantly impairs cell proliferation, migration, and myofibroblastic differentiation, leading to observed healing delays in aged rat models. This impairment is closely linked to decreased angiogenesis, mainly because lower levels of key factors such as vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) in the serum of aged individuals hinder vascularization and granulation tissue maturation at the wound site[82,83]. Regarding immune regulation, aging shifts the local environment toward a pro-inflammatory state characterized by increased expression of monocyte chemoattractant protein-1 (MCP-1) and tumor necrosis factor (TNF)[82,84]. Although GMSCs can retain some age-independent immunomodulatory activity in systemic models, their diminished osteogenic potential and the accumulation of senescence-associated factors in aged serum ultimately compromise the regenerative capacity of gingival tissues[84].
OSC aging serves as a fundamental driver of the progressive decline in regenerative capacity, disruption of immune homeostasis, and deterioration of structural integrity within the craniofacial region. Rather than simply reducing stem cell numbers, this process fundamentally shifts the local microenvironment from a pro-regenerative state toward a chronic inflammatory state and further impaired tissue repair. Specifically, OSC aging contributes to functional deterioration through four interrelated pathways. Aging of PDLSCs leads to diminished osteogenic/cementogenic differentiation, impaired immunomodulatory functions, and the development of a SASP, thereby driving periodontal tissue degeneration. Senescence of DPSCs and SHED results in reduced proliferative and differentiation capacities, lineage skewing toward adipogenesis, and molecular alterations such as telomere shortening, collectively compromising dentin-pulp regeneration and promoting pulp fibrosis. Aging of BMSCs causes cell cycle arrest, reduced osteogenic potential, and a shift toward adipogenic differentiation, directly contributing to craniofacial and alveolar bone loss. Finally, aging of GMSCs impairs proliferation, migration, and myofibroblastic differentiation. When combined with a pro-inflammatory microenvironment and reduced angiogenesis, defects caused by GMSCs aging lead to delayed oral wound healing. Collectively, these findings highlight that OSC aging acts through a combination of cell-intrinsic regenerative deficits and microenvironmental immune dysregulation, positioning it as a central pathological mechanism in age-related degenerative diseases of the oral tissues.
5. Strategies to Rejuvenate Aging OSCs
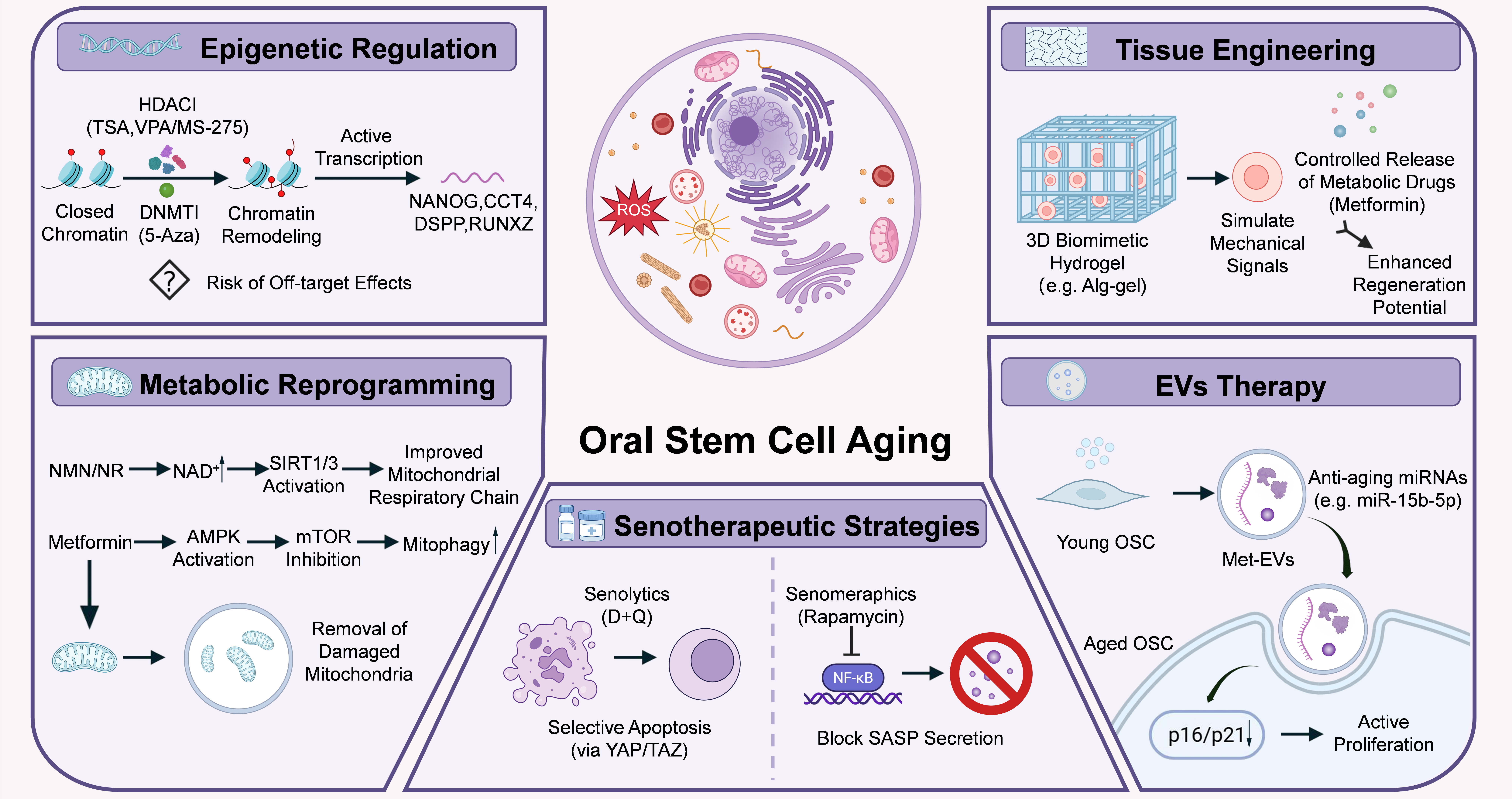
Rejuvenating senescent OSCs has emerged as a central objective in oral regenerative medicine[7]. Current therapeutic strategies collectively aim to reverse intrinsic cellular defects, eliminate detrimental senescent populations, and reconstruct a pro-regenerative microenvironment, thereby restoring tissue homeostasis at multiple levels (Figure 4)[13,85,86].
{kind=link}
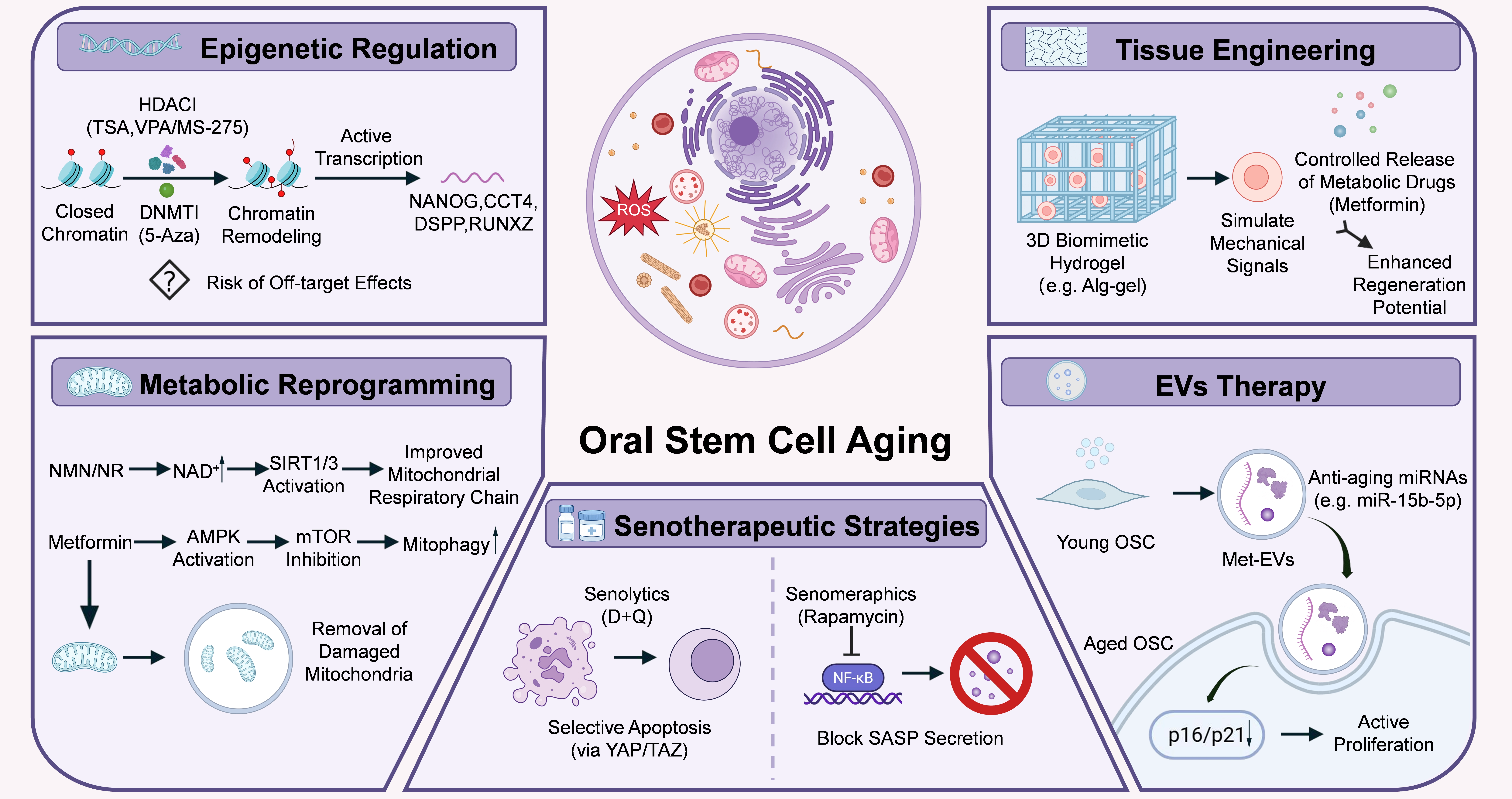
Figure 4. Contemporary rejuvenation strategies for aged OSCs. Schematic illustration showing five major therapeutic interventions targeting cellular senescence to restore the regenerative capacity of OSCs. Epigenetic regulation utilizes HDACi and DNMTi to promote chromatin remodeling and active transcription of key genes (NANOG, OCT4, DSPP, RUNX2); metabolic reprogramming employs NMN/NR to enhance the mitochondrial respiratory chain via NAD+/SIRT1/3 signaling, alongside Metformin-induced AMPK activation and mitophagy for the removal of damaged mitochondria; senotherapeutic strategies apply senolytics (D + Q) to induce selective apoptosis via the YAP/TAZ pathway, and senomorphics (Rapamycin) to block NF-κB and inhibit SASP secretion; EVs therapy leverages young OSC-derived EVs or metformin-preconditioned PDLSCs to produce EVs (met-EVs) to deliver anti-aging miRNAs (e.g., miR-15b-5p) for transcriptome remodeling and downregulating p16/p21-mediated proliferation; and biomaterials and tissue engineering utilize 3D biomimetic hydrogels (e.g., Alg-Gel) to simulate young niche mechanical signals coupled with the controlled release of metabolic drugs (Metformin) to achieve enhanced regeneration potential. OSCs: oral stem cells; HDACi: histone deacetylase inhibitors; DNMTi: DNA methyltransferases inhibitors; DSPP: dentin sialophosphoprotein; NMN: nicotinamide mononucleotide; NR: nicotinamide riboside; NAD+: nicotinamide adenine dinucleotide; NF-κB : nuclear factor kappa B; SASP: senescence-associated secretory phenotype; EVs: extracellular vesicles; PDLSCs: periodontal ligament stem cells; NANOG: homeobox protein NANOG; OCT4: octamer-binding transcription factor 4; RUNX2: runt-related transcription factor 2; SIRT1/3: sirtuin 1/3; AMPK: AMP-activated protein kinase.
5.1 Epigenetic regulation
Epigenetic regulation represents a fundamental strategy to reverse senescence-associated transcriptional repression and improve stem cell identity. Histone deacetylase (HDAC) inhibitors, including trichostatin A (TSA), valproic acid (VPA), and entinostat (MS-275), prevent histone deacetylation, thereby loosening chromatin structure and enhancing DNA accessibility[55,87]. Optimal TSA concentrations significantly promote DPSC proliferation via the activation of JNK/c-Jun and Smad2/3 pathways. Moreover, TSA accelerates mineralized nodule formation and elevates the expression of the dentin marker dentin sialophosphoprotein (DSPP)[88]. Similarly, MS-275 and VPA effectively restore the odontogenic and osteogenic differentiation capacity of senescent DPSCs[89]. In parallel, DNMT inhibitors, such as 5-azacytidine, effectively reverse abnormal hypermethylation, thereby reactivating silenced stemness-associated genes. Consequently, the intervention restores the osteogenic differentiation capacity of PDLSCs[90].
However, current epigenetic interventions remain largely non-specific. Global epigenetic alteration inevitably triggers widespread off-target effects, significantly elevating the risks of genomic instability and tumorigenesis[90]. Therefore, emerging approaches such as locus-specific epigenetic editing (e.g., CRISPR/dCas9-based systems) may provide more precise and safer rejuvenation strategies in the future.
5.2 Metabolic reprogramming
Metabolic reprogramming aims to restore bioenergetic balance and mitochondrial function, both of which are essential for maintaining stem cell self-renewal and differentiation capacity[7,91]. Supplementation with nicotinamide adenine dinucleotide (NAD+) precursors, including nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR), effectively restores sirtuin family activity, improves mitochondrial respiratory function and alleviate oxidative stress[52,91]. In DPSCs, activated SIRT1, and SIRT1-mediated histone H3 lysine 36 acetylation (H3K36ac) deacetylation directly delay cellular senescence. Additionally, activation of other sirtuin family members has also been shown to enhance mitochondrial function and promote osteogenic regeneration[7].
In addition to NAD+-sirtuin signaling, other energy-sensing pathways also play critical roles in metabolic rejuvenation. Metformin, a well-established metabolic modulator, specifically activates AMPK while concurrently inhibiting the mTOR pathway. This dual regulation promotes autophagy and mitophagy, thereby facilitating the clearance of damaged mitochondria and reducing ROS accumulation[58,92]. Ongoing clinical trials evaluate the systemic efficacy of metformin. However, the long-term safety and potential toxicity of metabolic modulators remain highly controversial[58]. Consequently, developing intermittent dosing strategies represents a critical research priority[91].
5.3 Senolytic and senomorphic therapies
Senotherapeutic strategies aim to mitigate the detrimental effects of cellular senescence either by eliminating senescent cells or by modulating their secretory phenotype[93].
Senolytics selectively induce apoptosis in senescent cells, thereby reducing senescent cell burden[9]. Intermittent administration of dasatinib and quercetin (D + Q) has been shown to alleviate senescence accumulation in periodontal tissues[94]. Additionally, D + Q treatment directly preserves stemness and enhances osteogenic differentiation in PDLSCs via the activation of the YAP/TAZ pathway[95,96]. In contrast, senomorphics do not eliminate senescent cells but instead suppress the SASP. Agents such as rapamycin inhibit key inflammatory pathways, including NF-κB and JAK/STAT signaling, thereby reducing the secretion of pro-inflammatory cytokines such as IL-6 and IL-8 and limiting the paracrine propagation of senescence[97].
Despite their therapeutic potential, both approaches present notable limitations. Excessive clearance of senescent cells may impair physiological processes such as tissue repair and remodeling, whereas senomorphic therapies typically require sustained administration, which may increase the risk of systemic immunosuppression. Therefore, the development of tissue-specific and precisely controlled delivery strategies remains a critical priority for clinical translation.
5.4 Extracellular vesicles (EVs)
EVs have emerged as a promising cell-free therapeutic strategy that recapitulates the paracrine functions of stem cells, offering a viable alternative to conventional cell transplantation. EV-based therapies exhibit low immunogenicity and high tissue penetration, and have demonstrated substantial potential in rejuvenating senescent OSCs[98].
EVs derived from young stem cells are enriched in bioactive cargos, particularly microRNAs such as miR-15b-5p and miR-290a-5p. Following internalization by senescent cells, these microRNAs suppress the expression of senescence-associated genes, including p16 and p21, thereby promoting transcriptomic reprogramming and restoring proliferative capacity[99]. Beyond native EVs, engineered or preconditioned EVs further enhance therapeutic efficacy. For example, metformin-preconditioned PDLSCs produce EVs (met-EVs) with increased yield and enhanced bioactivity. Upon uptake, met-EVs modulate cellular metabolism by promoting OXPHOS and mitophagy while reducing excessive glycolytic reliance. Consequently, Met-EV treatment restores osteogenic differentiation under inflammatory conditions and attenuates alveolar bone loss in vivo[100].
Despite these promising findings, the clinical translation of EV-based therapies remains challenging. Key obstacles include the lack of standardized isolation and characterization protocols, as well as their relatively short half-life within the complex oral microenvironment[101]. Therefore, improving EV stability, scalability, and targeted delivery represents a critical direction for future research. Despite these promising findings, several major challenges remain before clinical translation can be achieved. For example, EV populations are inherently heterogeneous, and their biological activity can vary substantially depending on donor age, cell source, culture conditions, and isolation methods[102]. The heterogeneity complicates quality control and may lead to inconsistent therapeutic outcomes.
5.5 Biomaterial and tissue engineering
Biomaterial-based strategies aim to reconstruct a pro-regenerative stem cell niche and provide both structural and biochemical support for OSC rejuvenation. By mimicking key features of the native microenvironment, engineered biomaterials can partially reverse age-associated niche deterioration. Engineered three-dimensional hydrogels, such as nano-phosphate-loaded alginate-gelatin composites, provide tunable mechanical stiffness and excellent biocompatibility. The scaffolds closely mimic the physical properties of the native extracellular matrix in dental pulp or periodontal tissues, thereby enhancing DPSC adhesion and osteogenic differentiation[102].
Beyond structural support, biomaterials can serve as versatile delivery platforms for bioactive molecules. For example, incorporating metabolic modulators such as metformin into thermosensitive hydrogel microspheres enables controlled and sustained drug release. This approach not only reduces local oxidative stress but also provides continuous three-dimensional structural support for senescent OSCs, ultimately improving regenerative outcomes in periodontal tissues[102].
Despite these advances, current biomaterials remain largely static and fail to fully replicate the dynamic nature of native stem cell niches. Future efforts should focus on developing adaptive biomaterials with microenvironment-sensing capability and spatiotemporal control of therapeutic delivery to enable precise stem cell rejuvenation.
6. Future Perspective
Although significant progress has been made in elucidating the hallmarks, mechanisms, and functional consequences of OSC aging, several critical challenges remain before these insights can be translated into effective rejuvenation strategies and clinically applicable regenerative therapies.
6.1 OSC heterogeneity
A major frontier lies in resolving the substantial heterogeneity that characterizes OSC aging[103,104]. Accumulating evidence suggests that different OSC populations do not characterize in a uniform manner[105,106]. The rates of senescence, dominant molecular pathways, metabolic vulnerabilities, and inflammatory responsiveness may vary considerably depending on tissue origin, developmental lineage, microenvironmental exposure, and even donor-specific factors[107]. Current aging markers, such as SA-β-gal, γH2AX, p16/p21, mitochondrial ROS, or methylation drift, only capture limited aspects of this complexity. Future research will require integrating multi-omics profiling, lineage tracing, and single-cell technologies to construct high-resolution “aging atlases” of each OSC subtype, enabling precise identification of aging-driving nodes and the distinction between reversible dysfunction and irreversible senescence[108,109]. Understanding this cellular and molecular heterogeneity is essential for designing tailored rejuvenation approaches rather than assuming a one-size-fits-all anti-aging strategy[110].
6.2 Frontier therapeutic opportunities
While current rejuvenation strategies have demonstrated encouraging results, most interventions remain relatively broad and non-specific. Future therapeutic development may shift toward precision rejuvenation approaches. Emerging strategies such as transient partial reprogramming may reset epigenetic age without erasing cell identity, while CRISPR-based epigenetic editing could enable locus-specific rejuvenation with fewer off-target effects compared with conventional HDAC or DNMT inhibitors[112,113]. In addition, microbiome-responsive biomaterials, and senescence subtype-specific interventions guided by single-cell profiling may represent next-generation strategies for OSC rejuvenation[114,115]. These approaches may move the field from generalized anti-aging interventions toward personalized regenerative therapies.
Beyond currently investigated strategies in oral regenerative medicine, several emerging anti-aging interventions from broader aging research fields may provide promising translational opportunities for OSC rejuvenation. One notable example is partial epigenetic reprogramming, which has emerged as a breakthrough strategy for reversing age-associated cellular dysfunction. Transient expression of Shinya Yamanaka factors has been shown to reverse multiple hallmarks of aging without complete dedifferentiation[116]. In a landmark study published in Cell, Juan Carlos Izpisua Belmonte and colleagues demonstrated that cyclic partial reprogramming alleviated age-associated phenotypes and improved tissue regeneration in progeroid mice. Subsequent studies further showed that transient reprogramming could rejuvenate aged tissues by resetting epigenetic age, improving mitochondrial function, and restoring stem cell regenerative potential[117]. Despite its promise, this strategy has not yet been systematically explored in OSCs, where maintaining lineage identity and preventing tumorigenic risks remain major challenges.
Another emerging strategy involves clearance of senescent cells. Conventional senolytic drugs such as Dasatinib and Quercetin often suffer from limited specificity and potential off-target toxicity. Recently, hypobaric pressure-based approaches have been reported to promote senescent cell clearance through mechanical microenvironment remodeling. In preclinical studies, controlled hypobaric pressure was shown to reduce senescent cell burden and ameliorate senescence-associated pathologies[118]. Compared with traditional senolytics, this physical strategy may provide a more spatially controllable and less toxic alternative for selectively targeting senescent cells while preserving functional stem cell populations. Although not yet explored in oral stem cell aging, it may represent a promising next-generation approach for craniofacial regeneration.
6.3 Challenges of laboratory translation
An additional but underappreciated challenge lies in the influence of donor age. Increasing evidence indicates that MSCs derived from aged donors exhibit reduced proliferative capacity, impaired differentiation potential, and accelerated senescence during ex vivo expansion, which significantly compromises their therapeutic efficacy[111,112]. Moreover, prolonged in vitro culture itself induces replicative senescence and epigenetic drift, even in cells derived from young donors, suggesting that both intrinsic aging and culture-induced stress jointly determine OSC functional decline[113]. Beyond cellular aging, emerging studies highlight that EVs also undergo age-associated functional deterioration. EVs derived from aged MSCs display altered cargo profiles, including reduced pro-regenerative microRNAs and increased pro-inflammatory factors, which impair their ability to promote tissue repair and may even propagate senescence signals[114]. Importantly, replicative aging during cell expansion further modifies EV composition and bioactivity, indicating that EV-based therapies are not exempt from donor- and culture-dependent variability[115]. These findings underscore a critical knowledge gap that current rejuvenation strategies largely overlook how donor age and expansion-induced aging collectively shape both OSCs and their secretome. By addressing these issues, standardized criteria for donor selection, optimization of expansion protocols to minimize senescence, and rigorous characterization of EV quality across different aging states can be established.
Another critical direction is the translation of laboratory findings into clinically applicable therapies that can effectively restore craniofacial regenerative function in aged individuals[116]. Although numerous interventions, epigenetic regulators, metabolic reprogramming, senolytics/senomorphics, EVs, and biomaterial-based microenvironmental modulation, have demonstrated substantial rejuvenating potential in vitro or in animal models, several key obstacles limit their clinical advancement[104]. One challenge is ensuring long-term safety: rejuvenation therapies that involve epigenetic remodeling or metabolic enhancement carry potential risks of uncontrolled proliferation or tumorigenicity[117]. Another challenge lies in maintaining therapeutic efficacy within the highly inflammatory and structurally complex aging oral niche, where senescent immune cells, altered vascular supply, and degraded extracellular matrix can counteract stem cell–based regeneration[118]. Furthermore, the lack of standardized manufacturing protocols for OSCs and OSC-derived EVs remains a major barrier to clinical translation.
Future translational research should focus on developing targeted delivery strategies capable of reprogramming aged OSCs directly within their native microenvironment, optimizing biomaterial scaffolds that recreate youthful niche cues, and establishing GMP-level platforms for producing rejuvenated OSCs or their secretome[119]. Integrating senescence-modifying therapies with regenerative engineering, such as combining senolytics with EV-based microenvironment repair, or pairing metabolic enhancement with 3D biomimetic scaffolds, may offer synergistic benefits[120]. Ultimately, bridging the gap between mechanistic discoveries and clinically viable therapies will require multidisciplinary collaboration, long-term safety evaluation, and the establishment of aging-specific regenerative treatment frameworks. These efforts hold the promise of transforming OSC rejuvenation from an experimental concept into a realistic strategy to restore oral tissue homeostasis and promote craniofacial regeneration in aging populations.
Authors contribution
Ren Q, Liu X, Hu Y: Conceptualization, writing-original draft.
Shi S, Ou Q: Conceptualization, writing-review & editing.
Conflicts of interest
The authors declare no competing interests.
Ethical approval
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by grants from the National Key Research and Development Program of China (2021YFA1100600 to Shi S.), the Pearl River Talent Recruitment Program (2019ZT08Y485 and 2019JC01Y182 to Shi S.).
Copyright
© The Author(s) 2026.
References
-
2. Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG, et al. SHED: Stem cells from human exfoliated deciduous teeth. Proc Natl Acad Sci U S A. 2003;100(10):5807-5812.[DOI]
-
3. Seo BM, Miura M, Gronthos S, Bartold PM, Batouli S, Brahim J, et al. Investigation of multipotent postnatal stem cells from human periodontal ligament. Lancet. 2004;364(9429):149-155.[DOI]
-
5. Sonoyama W, Liu Y, Fang D, Yamaza T, Seo BM, Zhang C, et al. Mesenchymal stem cell-mediated functional tooth regeneration in swine. PLoS One. 2006;1(1):e79.[DOI]
-
8. Schüler SC, Gebert N, Ori A. Stem cell aging: The upcoming era of proteins and metabolites. Mech Ageing Dev. 2020;190:111288.[DOI]
-
9. Saliev T, Singh PB. Targeting senescence: A review of senolytics and senomorphics in anti-aging interventions. Biomolecules. 2025;15(6):860.[DOI]
-
10. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194-1217.[DOI]
-
14. Zong C, Zhao L, Huang C, Chen Y, Tian L. Isolation and culture of bone marrow mesenchymal stem cells from the human mandible. J Vis Exp. 2022;182:e63811.[DOI]
-
21. Sui B, Wu D, Xiang L, Fu Y, Kou X, Shi S. Dental pulp stem cells: From discovery to clinical application. J Endod. 2020;46(9):S46-S55.[DOI]
-
26. Kim D, Lee AE, Xu Q, Zhang Q, Le AD. Gingiva-derived mesenchymal stem cells: Potential application in tissue engineering and regenerative medicine - A comprehensive review. Front Immunol. 2021;12:667221.[DOI]
-
27. Yamaza T, Ren G, Akiyama K, Chen C, Shi Y, Shi S. Mouse mandible contains distinctive mesenchymal stem cells. J Dent Res. 2011;90(3):317-324.[DOI]
-
28. Akintoye SO, Lam T, Shi S, Brahim J, Collins MT, Robey PG. Skeletal site-specific characterization of orofacial and iliac crest human bone marrow stromal cells in same individuals. Bone. 2006;38(6):758-768.[DOI]
-
29. Miyata H, Ishii M, Suehiro F, Komabashiri N, Ikeda N, Sakurai T, et al. Elucidation of adipogenic differentiation regulatory mechanism in human maxillary/mandibular bone marrow-derived stem cells. Arch Oral Biol. 2023;146:105608.[DOI]
-
30. Lee DJ, Kwon J, Current L, Yoon K, Zalal R, Hu X, et al. Osteogenic potential of mesenchymal stem cells from rat mandible to regenerate critical sized calvarial defect. J Tissue Eng. 2019;10:2041731419830427.[DOI]
-
31. Zhang L, Pitcher LE, Yousefzadeh MJ, Niedernhofer LJ, Robbins PD, Zhu Y. Cellular senescence: A key therapeutic target in aging and diseases. J Clin Invest. 2022;132(15):e158450.[DOI]
-
32. Miwa S, Kashyap S, Chini E, von Zglinicki T. Mitochondrial dysfunction in cell senescence and aging. J Clin Invest. 2022;132(13):e158447.[DOI]
-
33. Kirichenko TV, Markina YV, Markin AM, Vasilyev VS, Hua H, Li D, et al. Functional features of senescent cells and implications for therapy. Int J Mol Sci. 2025;26(11):5390.[DOI]
-
37. Gong P, Guo Z, Wang S, Gao S, Cao Q. Histone phosphorylation in DNA damage response. Int J Mol Sci. 2025;26(6):2405.[DOI]
-
38. Merighi A, Gionchiglia N, Granato A, Lossi L. The phosphorylated form of the histone H2AX (γH2AX) in the brain from embryonic life to old age. Molecules. 2021;26(23):7198.[DOI]
-
41. Liu J, Wang L, Wang Z, Liu JP. Roles of telomere biology in cell senescence, replicative and chronological ageing. Cells. 2019;8(1):54.[DOI]
-
43. Lin J, Epel E. Stress and telomere shortening: Insights from cellular mechanisms. Ageing Res Rev. 2022;73:101507.[DOI]
-
44. Barnes RP, Fouquerel E, Opresko PL. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech Ageing Dev. 2019;177:37-45.[DOI]
-
46. Amorim JA, Coppotelli G, Rolo AP, Palmeira CM, Ross JM, Sinclair DA. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat Rev Endocrinol. 2022;18(4):243-258.[DOI]
-
47. Alam S, Aijaz M. Age-related declines in stem cell function: Molecular insights and future therapies. J Exp Clin Med. 2025;42(2):170-198.[DOI]
-
49. Somasundaram I, Jain SM, Blot-Chabaud M, Pathak S, Banerjee A, Rawat S, et al. Mitochondrial dysfunction and its association with age-related disorders. Front Physiol. 2024;15:1384966.[DOI]
-
50. Barazzuol L, Giamogante F, Brini M, Calì T. PINK1/parkin mediated mitophagy, Ca2+ signalling, and ER-mitochondria contacts in Parkinson’s disease. Int J Mol Sci. 2020;21(5):1772.[DOI]
-
51. Cairns G, Thumiah-Mootoo M, Burelle Y, Khacho M. Mitophagy: A new player in stem cell biology. Biology. 2020;9(12):481.[DOI]
-
52. Mu WC, Ohkubo R, Widjaja A, Chen D. The mitochondrial metabolic checkpoint in stem cell aging and rejuvenation. Mech Ageing Dev. 2020;188:111254.[DOI]
-
53. Viña J. The free radical theory of frailty: Mechanisms and opportunities for interventions to promote successful aging. Free Radic Biol Med. 2019;134:690-694.[DOI]
-
54. Feng X, Wei Z, Hu Y, Chen T, Yu B. Stem cell-based strategies for vascular aging: From mechanistic insights to clinical translation. Vessel Plus. 2025;9:27.[DOI]
-
55. Yu H, Feng T, Zhang C, Jiao Z, Fan W, Jiang R, et al. Epigenetic pharmacology in aging: From mechanisms to therapies for age-related disorders. Front Pharmacol. 2025;16:1699296.[DOI]
-
56. Soto-Palma C, Niedernhofer LJ, Faulk CD, Dong X. Epigenetics, DNA damage, and aging. J Clin Invest. 2022;132(16):e158446.[DOI]
-
57. Camacho P, Ribeiro E, Pereira B, Nascimento J, Caldeira Rosa P, Henriques J, et al. DNA methyltransferase expression (DNMT1, DNMT3a, and DNMT3b) as a potential biomarker in age-related macular degeneration. J Clin Med. 2025;14(2):559.[DOI]
-
59. Zhou X, Hong Y, Zhang H, Li X. Mesenchymal stem cell senescence and rejuvenation: Current status and challenges. Front Cell Dev Biol. 2020;8:364.[DOI]
-
60. Boyette L, Tuan R. Adult stem cells and diseases of aging. J Clin Med. 2014;3(1):88-134.[DOI]
-
61. Chou LY, Ho CT, Hung SC. Paracrine senescence of mesenchymal stromal cells involves inflammatory cytokines and the NF-κB pathway. Cells. 2022;11(20):3324.[DOI]
-
63. Idda ML, McClusky WG, Lodde V, Munk R, Abdelmohsen K, Rossi M, et al. Survey of senescent cell markers with age in human tissues. Aging. 2020;12(5):4052-4066.[DOI]
-
65. Saul D, Jurk D, Doolittle ML, et al. Distinct secretomes in p16-and p21-positive senescent cells across tissues. BioRxiv [Preprint]. 2023.[DOI]
-
66. Chen J, Wang Z, Du Y, Liu B, Chen Y, Chen G, et al. Injectable CRISPRa-microspheres for targeted A20 activation rescue age-related osteogenic impairment via senescence mitigation. Biomaterials. 2026;328:123830.[DOI]
-
67. Schloesser D. Senescent cells suppress macrophage-mediated corpse removal via upregulation of the CD47-QPCT/l axis. J Cell Biol. 2022;222(2):e202207097.[DOI]
-
68. Alqahtani S, Alqahtani T, Venkatesan K, Sivadasan D, Ahmed R, Sirag N, et al. SASP modulation for cellular rejuvenation and tissue homeostasis: Therapeutic strategies and molecular insights. Cells. 2025;14(8):608.[DOI]
-
69. Wu RX, Bi CS, Yu Y, Zhang LL, Chen FM. Age-related decline in the matrix contents and functional properties of human periodontal ligament stem cell sheets. Acta Biomater. 2015;22:70-82.[DOI]
-
70. Li X, Zhang B, Wang H, Zhao X, Zhang Z, Ding G, et al. The effect of aging on the biological and immunological characteristics of periodontal ligament stem cells. Stem Cell Res Ther. 2020;11(1):326.[DOI]
-
73. Wang GY, Liao L, Tian WD. Senolytic effects on dental pulp stem cell’s proliferation and differentiation during long-term expansion. Chin J Stomatol. 2024;59(5):444-453.[DOI]
-
78. Ivan A, Cristea MI, Telea A, Oprean C, Galuscan A, Tatu CA, et al. Stem cells derived from human exfoliated deciduous teeth functional assessment: Exploring the changes of free fatty acids composition during cultivation. Int J Mol Sci. 2023;24(24):17249.[DOI]
-
79. Lin H, Sohn J, Shen H, Langhans MT, Tuan RS. Bone marrow mesenchymal stem cells: Aging and tissue engineering applications to enhance bone healing. Biomaterials. 2019;203:96-110.[DOI]
-
83. Cáceres M, Oyarzun A, Smith PC. Defective wound-healing in aging gingival tissue. J Dent Res. 2014;93(7):691-697.[DOI]
-
85. Larijani B, Foroughi-Heravani N, Alaei S, Rezaei-Tavirani M, Alavi-Moghadam S, Payab M, et al. Opportunities and challenges in stem cell aging. Adv Exp Med Biol. 2021;1341:143-175.[DOI]
-
87. Lee EC, Kim YM, Lim HM, Ki GE, Seo YK. The histone deacetylase inhibitor (MS-275) promotes differentiation of human dental pulp stem cells into odontoblast-like cells independent of the MAPK signaling system. Int J Mol Sci. 2020;21(16):5771.[DOI]
-
89. Duncan HF, Smith AJ, Fleming GJP, Cooper PR. Histone deacetylase inhibitors epigenetically promote reparative events in primary dental pulp cells. Exp Cell Res. 2013;319(10):1534-1543.[DOI]
-
91. Delrue C, Speeckaert R, Speeckaert MM. Rewinding the clock: Emerging pharmacological strategies for human anti-aging therapy. Int J Mol Sci. 2025;26(19):9372.[DOI]
-
92. Khorraminejad-Shirazi M, Farahmandnia M, Kardeh B, Estedlal A, Kardeh S, Monabati A. Aging and stem cell therapy: AMPK as an applicable pharmacological target for rejuvenation of aged stem cells and achieving higher efficacy in stem cell therapy. Hematol Oncol Stem Cell Ther. 2018;11(4):189-194.
-
95. Hickson LJ, Prata LGL, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019;47:446-456.[DOI]
-
98. Yu L, Wen H, Liu C, Wang C, Yu H, Zhang K, et al. Embryonic stem cell-derived extracellular vesicles rejuvenate senescent cells and antagonize aging in mice. Bioact Mater. 2023;29:85-97.[DOI]
-
101. Ghodasara A, Raza A, Wolfram J, Salomon C, Popat A. Clinical translation of extracellular vesicles. Adv Healthc Mater. 2023;12(28):2301010.[DOI]
-
104. Alarcón-Apablaza J, Salazar LA, Loren P, Martínez-Cardozo C, Fuentes R. Effect of aging on the morphofunctional characteristics of oral cavity mesenchymal stromal cells: A scoping review. Biomedicines. 2025;13(11):2776.[DOI]
-
105. Kok ZY, Alaidaroos NYA, Alraies A, Colombo JS, Davies LC, Waddington RJ, et al. Dental pulp stem cell heterogeneity: Finding superior quality “needles” in a dental pulpal “haystack” for regenerative medicine-based applications. Stem Cells Int. 2022;2022:9127074.[DOI]
-
106. Krivanek J, Soldatov RA, Kastriti ME, Chontorotzea T, Herdina AN, Petersen J, et al. Dental cell type atlas reveals stem and differentiated cell types in mouse and human teeth. Nat Commun. 2020;11:4816.[DOI]
-
107. Maličev E, Jazbec K. An overview of mesenchymal stem cell heterogeneity and concentration. Pharmaceuticals. 2024;17(3):350.[DOI]
-
108. Uyar B, Palmer D, Kowald A, Murua Escobar H, Barrantes I, Möller S, et al. Single-cell analyses of aging, inflammation and senescence. Ageing Res Rev. 2020;64:101156.[DOI]
-
109. Li Y, Wang Q, Xuan Y, Zhao J, Li J, Tian Y, et al. Investigation of human aging at the single-cell level. Ageing Res Rev. 2024;101:102530.[DOI]
-
110. Česnik AB, Švajger U. The issue of heterogeneity of MSC-based advanced therapy medicinal products–a review. Front Cell Dev Biol. 2024;12:1400347.[DOI]
-
112. Balikov D, Crowder S, Lee J, Lee Y, Ko U, Kang ML, et al. Aging donor-derived human mesenchymal stem cells exhibit reduced reactive oxygen species loads and increased differentiation potential following serial expansion on a PEG-PCL copolymer substrate. Int J Mol Sci. 2018;19(2):359.[DOI]
-
113. Choudhery MS, Badowski M, Muise A, Pierce J, Harris DT. Donor age negatively impacts adipose tissue-derived mesenchymal stem cell expansion and differentiation. J Transl Med. 2014;12:8.[DOI]
-
115. Yin Y, Chen H, Wang Y, Zhang L, Wang X. Roles of extracellular vesicles in the aging microenvironment and age-related diseases. J Extracell Vesicle. 2021;10(12):e12154.[DOI]
-
117. Wong RSY, Tan EW, Goh BH. Mesenchymal stem cell-based therapies: Challenges and enhancement strategies. Cell Biochem Biophys. 2026;84(1):131-147.[DOI]
-
120. Wu H, Zhang Q, Zhu J, Wu L, Xiao Y, Yang X. Biomaterials targeting senescent cells for bone regeneration: State-of-the-art and future perspectives. Bioact Mater. 2025;54:686-714.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Ren Q, Liu X, Hu Y, Ou Q, Shi S. Oral stem cell aging: From mechanisms to therapeutic rejuvenation. Ageing Cancer Res Treat. 2026;3:202618. https://doi.org/10.70401/acrt.2026.0022
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Types of OSCs and Their Biological Characteristics
- 3. Characteristics and Molecular Mechanisms of OSC Aging
- 4. Functional Consequences of OSC Aging
- 5. Strategies to Rejuvenate Aging OSCs
- 6. Future Perspective
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Ren Q, Liu X, Hu Y, Ou Q, Shi S. Oral stem cell aging: From mechanisms to therapeutic rejuvenation. Ageing Cancer Res Treat. 2026;3:202618. https://doi.org/10.70401/acrt.2026.0022
copy
Share Link
copy