Protein post-translational modifications in aging and cancer: Mechanisms and translational implications
Rou Zhang
1,#
,
Yunhua Fu
1,#
,
Huimin Chen
1,#
,
Jia Yu
1,#
,
Yingjie Li
1,#
,
Meng Hu
1
,
Litong Nie
2

,
Lunzhi Dai
1,*
*Correspondence to:
Lunzhi Dai, National Clinical Research Center for Geriatrics, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China.
E-mail: lunzhi.dai@scu.edu.cn
Ageing Cancer Res Treat. 2026;3:202619. 10.70401/acrt.2026.0023
Received: April 27, 2026Accepted: June 05, 2026Published: June 05, 2026
Abstract
Aging is a key risk factor for cancer, with complex, context-dependent processes influencing both tumor initiation and progression. While certain aging-associated processes restrain cellular proliferation, others drive tumor initiation and progression, revealing a context-dependent duality in the role of aging in cancer. Increasing evidence indicates that many of these transitions occur not through genomic alterations, but through reprogramming of protein post-translational modifications (PTMs). Phosphorylation, ubiquitination, acetylation, methylation and an expanding repertoire of acylations collectively regulate protein activity, stability, localization and interactions, translating aging-associated stresses, including metabolic imbalance, microenvironmental remodeling and accumulated damage, into functional cellular outcomes. In this review, we examine how PTMs bridge aging and cancer through three interconnected axes: integration of stress signals, encoding of metabolic states, and modulation of the tissue microenvironment. We discuss how major PTM systems coordinate core cellular processes, such as checkpoint control, proteostasis, chromatin organization and cell–environment communication, and how, through dynamic and combinatorial actions, they establish distinct regulatory states that determine whether tissues sustain homeostasis, progress to dysfunction, or undergo malignant transformation. An in-depth understanding of how PTMs define these states provides valuable insights into disease stratification and offers new avenues for therapeutic interventions.
Keywords
Protein modifications, post-translational modifications, cancer, aging, biomarkers, therapeutic targets, PTM crosstalk, aging and cancer interplay
1. Introduction
Aging and cancer are linked through a complex and context-dependent relationship in which some processes are shared, some are antagonistic and others shift in meaning over time. Several aging-associated hallmarks, including genomic instability, epigenetic alterations, chronic inflammation and dysbiosis, overlap significantly with the canonical features of tumorigenesis and may therefore serve as common drivers linking aging to cancer[1,2]. In contrast, other aging-related processes, particularly telomere attrition and stem cell exhaustion, limit cellular proliferative capacity and thus act as primary barriers against malignant transformation[3]. However, not all aging-associated processes can be clearly assigned to either category. Processes such as disabled macroautophagy and cellular senescence often occupy an intermediate state, as they may suppress malignant transformation at early stages but later promote tumor initiation, progression, and therapeutic resistance when chronic persistence in damaged tissues occurs[4]. This interplay among common drivers, antagonistic constraints and context-dependent dynamics helps explain why aging significantly increases cancer risk, yet not all aging tissues develop into malignant tumors[3].
This complexity cannot be understood adequately at the level of gene expression alone, because many decisive changes arise at the level of protein regulation. Aging tissues are exposed to persistent metabolic stress[5], oxidative injury[6], organellar dysfunction[7], immune perturbations[8] and microenvironmental remodeling[9] long before overt histological transformation appears. These disturbances reshape protein behavior through biochemical modifications. Consequently, protein post-translational modifications (PTMs) represent a particularly critical layer of molecular information, enabling aging-related stresses to be translated into functional consequences[10]. PTMs regulate nearly every dimension of protein behavior, including enzymatic activity, conformational switching, intracellular trafficking, phase separation, interaction partner selection, degradation and subcellular compartmentalization[11].
Multiple lines of evidence support a central role for PTMs in both aging and cancer. Many key aging phenotypes are shaped by modification-dependent pathways, including phosphorylation-regulated damage sensing and mitophagy[11], acetylation-dependent chromatin accessibility[12], ubiquitin-mediated proteostasis[13], glycosylation-dependent barrier control[14] and cysteine oxidation-linked stress responses[6]. Many of these same PTM-regulated systems are subsequently reused by cancer cells, where they are redirected to support survival, metabolic adaptability, immune evasion and therapeutic resistance[13]. Furthermore, PTMs rarely function as isolated events, their significance often depends on crosstalk between different modification systems that together determine whether a protein is activated or restrained, stabilized or degraded, retained or redistributed[15]. This interplay makes PTMs especially valuable for understanding why the same aging-associated perturbations may either suppress malignant evolution or create conditions that facilitate tumor emergence[10,16].
Aging and cancer are closely connected, but the mechanisms linking them remain difficult to resolve at the protein regulatory level. Here, we summarize how PTMs bridge the gap between aging and cancer from three dimensions: the integration of stress signals, encoding of metabolic states, and modulation of the tissue microenvironment. Then, we discuss major PTM systems, including phosphorylation, acetylation, ubiquitination, glycosylation, lactylation, methylation and redox modifications, emphasizing their roles in cellular senescence, stem cell dysfunction, proteostatic decline, microenvironmental change and malignant adaptation. Moreover, we discuss how a PTM-based perspective may support the development of new biomarkers and therapeutic strategies for precision medicine.
2. PTMs Linking Aging and Cancer
PTMs are particularly well suited to explain why the same aging-associated perturbations may either restrain malignant transformation or create conditions that facilitate tumorigenesis. Unlike genomic alterations, which often impose relatively stable constraints on cellular behavior, many PTMs are rapid, reversible and strongly context-dependent[17]. By regulating enzymatic activity[18], protein stability[19], and subcellular localization[20], PTMs translate oxidative injury[6], metabolic imbalance[21], and microenvironmental remodeling[22] into functional cellular outcomes.
This regulatory flexibility is especially important in aging tissues, where molecular stress accumulates long before overt histological transformation appears[21]. PTM remodeling does not usually arise from isolated modification events. Instead, it reflects interconnected regulatory networks that determine whether tissue perturbations are repaired, buffered, converted into chronic dysfunction or redirected toward malignant adaptation[23]. In this review, we use three interconnected dimensions to organize this process: the integration of stress signals, the encoding of metabolic states and the regulation of microenvironmental remodeling (Figure 1).
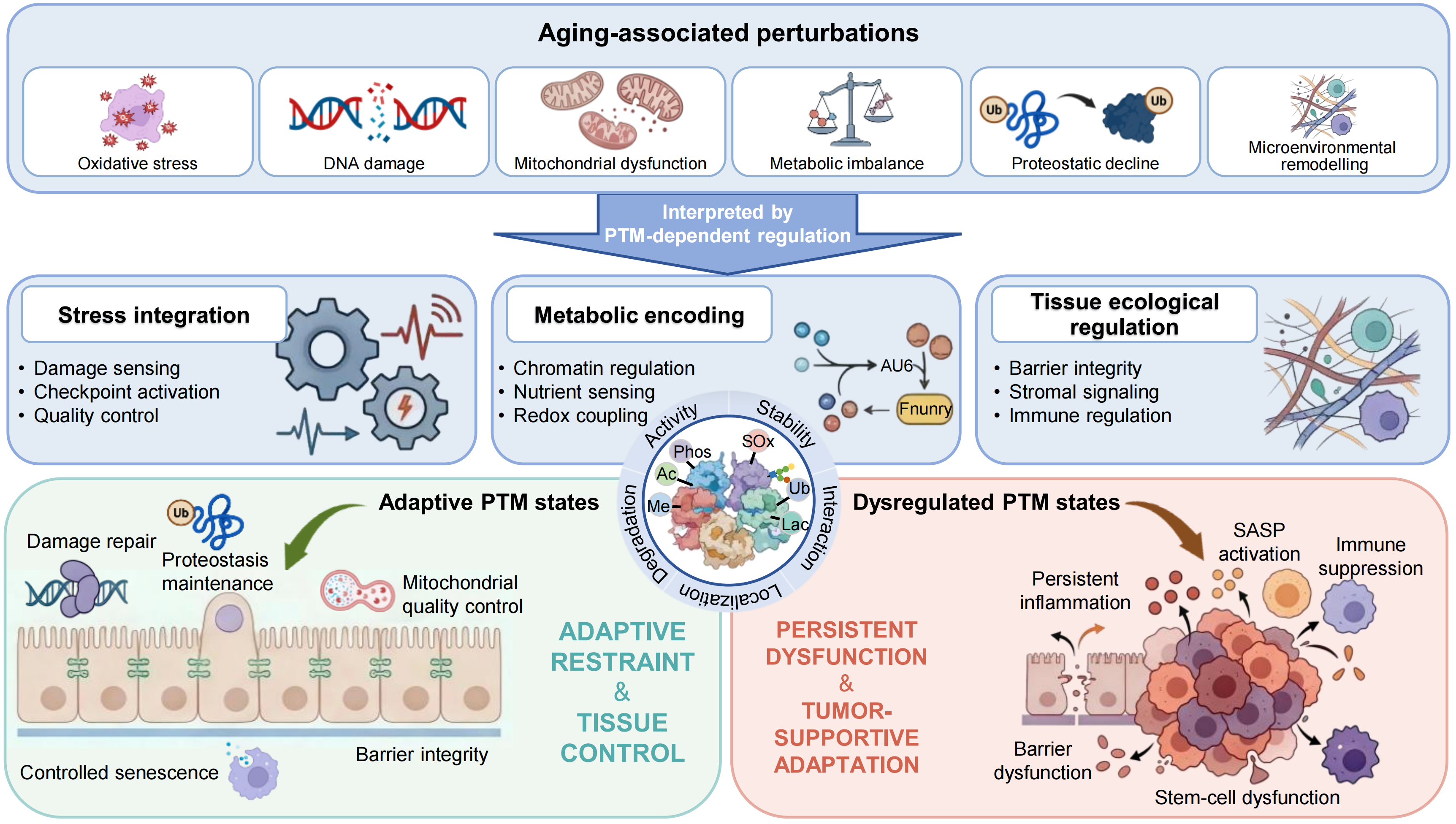{kind=link}
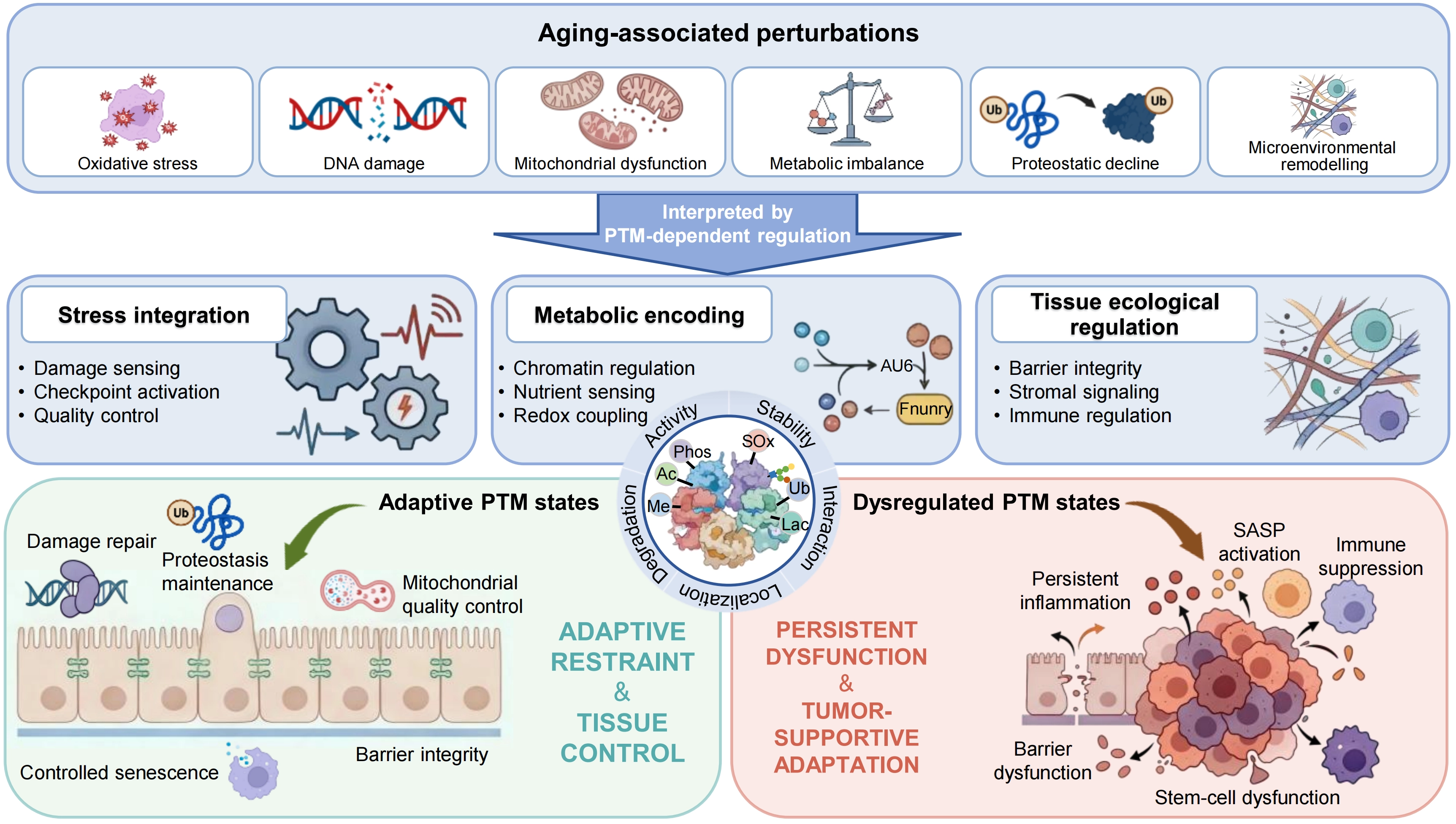
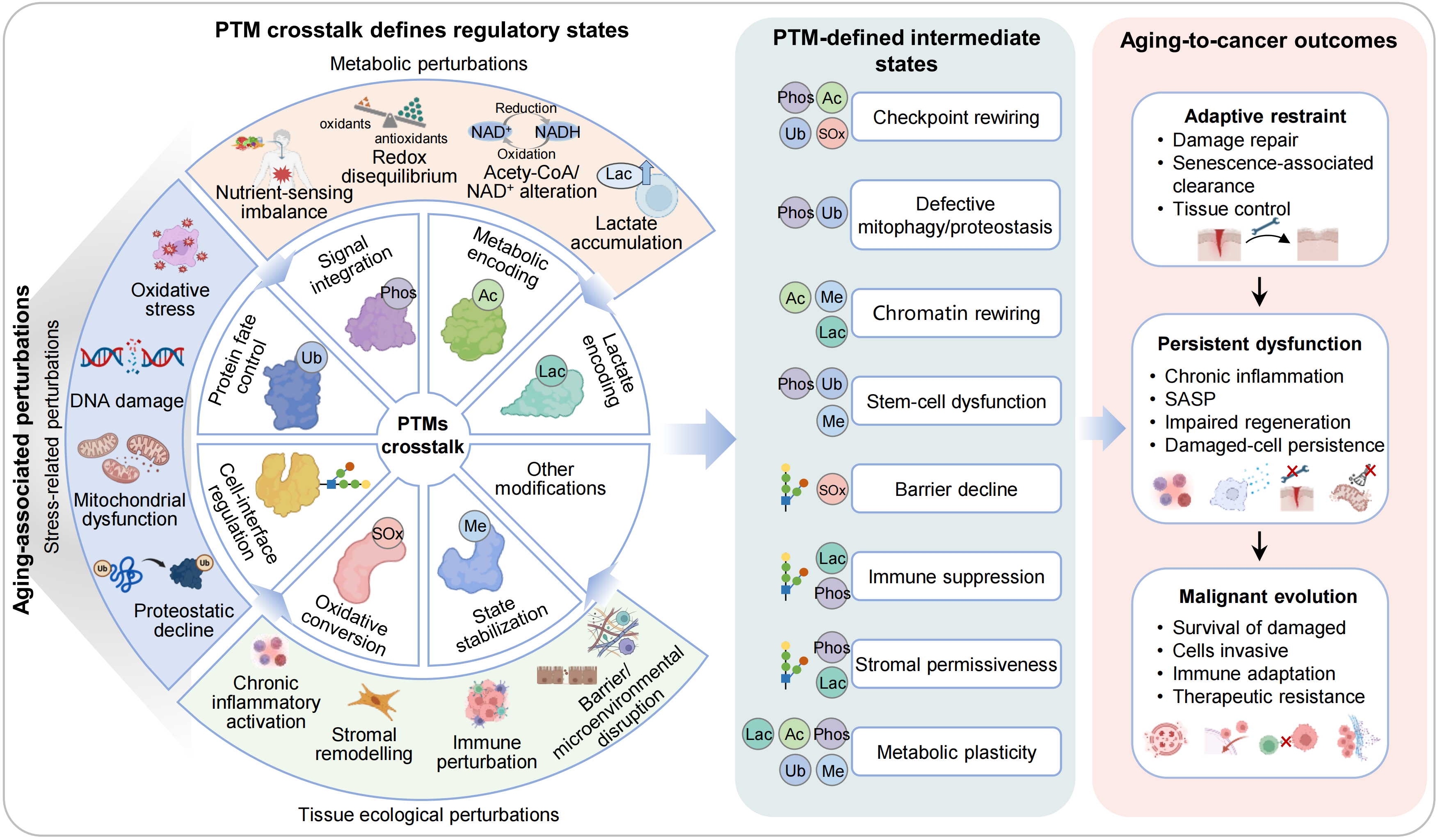
Figure 1. PTM-dependent integration of aging-associated perturbations into adaptive and maladaptive tissue states. Aging-associated perturbations, including oxidative stress, DNA damage, mitochondrial dysfunction, metabolic imbalance, proteostatic decline and microenvironmental remodeling, are integrated by PTM-dependent regulation. Through this layer, PTMs coordinate three principal functional dimensions: stress integration, metabolic encoding and tissue ecological regulation, encompassing processes such as damage sensing, checkpoint activation, chromatin regulation, redox coupling, barrier integrity, stromal signaling and immune regulation. Under balanced conditions, PTM-defined states support adaptive tissue functions, including damage repair, proteostasis maintenance, mitochondrial quality control, senescent cell clearance and barrier integrity, thereby maintaining tissue restraint and homeostasis. With aging, however, dysregulated PTM states drive a transition towards persistent dysfunction, characterized by persistent inflammation, SASP activation, immune suppression, barrier dysfunction and stem-cell dysfunction, ultimately promoting tumor-supportive adaptation. PTM: post-translational modification; SASP: senescence-associated secretory phenotype.
2.1 Stress integration by PTMs
Aging tissues are exposed to persistent oxidative injury[24], DNA damage[25], impaired proteostasis[26], mitochondrial dysfunction and chronic inflammation[27]. PTMs are central to these responses because they determine whether stress signals are repaired, buffered, converted into senescence or propagated as chronic dysfunction. Rather than serving only as biochemical indicators of stress, PTMs regulate the duration, intensity and outcome of stress responses[28].
Some examples support this principle. For example, DNA-PK-mediated phosphorylation of STAT6 at Ser807 helps preserve DNA repair capacity and limits macrophage senescence[29], whereas stress-induced O-GlcNAcylation of GATA4 can reinforce senescence-associated inflammatory programs[30]. In tumor cells undergoing therapy-induced senescence, increased PD-L1 glycosylation stabilizes PD-L1 and reduces immune clearance, showing how a persistent stress state can be converted into immune evasion[31]. These examples highlight a key feature of PTM regulation: the biological outcome of stress depends not only on the presence of damage, but also on how PTM networks interpret and resolve that damage.
Thus, the relevance of PTMs at the aging-cancer interface lies in stress resolution. Under physiological conditions, PTM-dependent mechanisms support repair[32], mitophagy[33], proteostasis[34] and inflammatory control[35]. When these mechanisms remain chronically shifted, damaged proteins, dysfunctional organelles and senescent cells become easier to maintain. This persistent stress-retaining state can weaken tissue resilience and create conditions that favor malignant transition[31].
2.2 Metabolic-state encoding by PTMs
A second major axis linking aging and cancer is metabolism. Aging is accompanied by changes in nutrient sensing[36], mitochondrial function[37], redox balance[38], biosynthetic capacity[39] and energy allocation[31]. Cancer cells also depend on metabolic plasticity, but in the tumor context this plasticity is redirected toward survival, proliferation, immune evasion and therapeutic tolerance[40]. PTMs provide a direct molecular interface between these metabolic states and protein function.
This interface exists because many PTMs depend on metabolically sensitive substrates or cofactors. NAD+ availability influences sirtuin-dependent deacetylation[37], acetyl-CoA shapes acetylation[41], lactate can support lactylation[42] and redox state alters cysteine-centered modifications[38]. Through these links, metabolic conditions are translated into changes in chromatin accessibility, enzyme activity, protein stability, signaling thresholds and cell-state control[40].
PTMs therefore do more than reflect metabolic remodeling. They help determine whether metabolic fluctuation remains adaptive or becomes embedded into persistent tissue dysfunction. In aging tissues, chronic metabolic imbalance can progressively reduce repair capacity, alter inflammatory tone and weaken regenerative competence[43]. In tumors, the same PTM-sensitive metabolic axes can be redirected to sustain glycolytic adaptation, ferroptosis resistance, immune escape and therapeutic tolerance[44]. Metabolism is therefore not only a source of energy and biomass, but also a regulatory input that PTMs convert into cellular state.
2.3 Microenvironmental remodeling by PTMs
The consequences of aging are not restricted to intracellular dysfunction, but also extend to the tissue microenvironment[45]. Over time, tissue architecture, cellular composition, extracellular matrix organization and immune communication undergo progressive remodeling[45]. PTMs are central to this dimension because they regulate cell-cell adhesion[46], extracellular matrix interactions[47], cytokine signaling[48], barrier integrity[49] and immune recognition[50].
Microenvironmental remodeling is therefore not merely a passive consequence of cellular decline. PTM-dependent changes in junctional stability, secretory behavior, stromal signaling and immune communication actively reshape how damaged, senescent or metabolically stressed cells interact with their surroundings. In barrier tissues, such remodeling can weaken compartmentalization and mucosal defense[51]. In stromal-rich tissues, it can amplify fibroblast activation, inflammatory signaling and immune suppression[52]. These changes alter not only individual cell behavior, but also the ecological conditions under which abnormal cells are either eliminated or retained.
PTMs therefore regulate both cell-intrinsic state and tissue-level ecology. By controlling barrier function, stromal signaling and immune environment, they help determine whether aging tissues continue to restrain abnormal cells or progressively create conditions compatible with malignant persistence[53].
2.4 The PTM-regulated aging-to-cancer transition
PTMs link aging and cancer by coordinating three interdependent dimensions: stress responsiveness[54], metabolic organization[55], and microenvironmental remodeling[56]. These dimensions do not operate independently. Persistent stress reshapes metabolic priorities[54]; metabolic dysfunction alters inflammatory and repair programs[57]; and microenvironmental remodeling feeds back on both stress signaling and resource use[58]. PTMs provide the regulatory layer through which these disturbances are either resolved or stabilized into persistent pathological states.
This perspective helps explain why aging markedly increases cancer risk but does not inevitably lead to malignancy. Many PTM-dependent responses are initially protective because they preserve proteostasis[59], promote repair[60], and limit the propagation of damage[61]. However, when PTM remodeling becomes persistent or dysregulated, the same regulatory systems can reinforce senescence-associated dysfunction[54], weaken immune control[31], and create tissue states that favor tumor emergence[56]. The biological significance of a given PTM therefore depends on timing, substrate identity, cellular source, and tissue context[23].
We next examine how each major PTM system shapes this continuum through a distinct regulatory logic. Phosphorylation is emphasized as a rapid stress-integrating system; acetylation as a mechanism for metabolic-state encoding; ubiquitination as a regulator of selective protein fate; glycosylation as a mediator of extracellular organization and recognition; lactylation as a marker and driver of chronic lactate-rich adaptation; methylation as a mechanism of transcriptional memory; and redox-associated modifications as chemical embedding of oxidative stress. Representative targets and biological contexts are summarized in Table 1.
Table 1. Representative PTM targets and regulatory functions in aging and cancer.
| PTM class | Target_protein/site | Aging/cancer context | Functions |
| Phosphorylation | STAT6_S807[29] | Macrophage senescence | Limits macrophage senescence |
| p53_S15[66] | Helicobacter pylori induced atrophic gastritis | Links DNA damage response, checkpoint activation and senescence-associated inflammatory injury | |
| Parkin_S65[68] | Mitochondrial stress in aging tissues | Initiates phosphorylation-dependent mitophagy signaling | |
| AMPK_Y172[54] | PAGln-associated cellular senescence | Connects metabolic stress with mitochondrial dysfunction and senescence signaling | |
| DRP1_S616[54] | PAGln-associated cellular senescence | Promotes mitochondrial fragmentation under sustained metabolic stress | |
| AKT_S473[71] | Gastric inflammation and tumorigenesis | Reflects activation of pro-survival signaling in an inflammatory tumorigenic context | |
| ERK1/2_T202/Y204[71] | Gastric inflammation and tumorigenesis | Reflects MAPK pathway activation during inflammatory gastric tumorigenesis | |
| SRC_Y416[72] | Interleukin 6 induced colorectal cancer EMT | Supports migration, invasion and stress-tolerance signaling | |
| FAK_Y397[72] | Interleukin 6 induced colorectal cancer EMT | Supports focal adhesion signaling and migration | |
| Acetylation | H3K27ac[91] | Early tumorigenesis and enhancer remodeling | Marks active enhancers and stabilizes growth-associated transcription |
| AFP_K194/K211/K242[94] | Hepatocellular carcinoma | Increases AFP stability and promotes malignant progression | |
| IDH1_K224[95] | Colorectal cancer and liver metastasis | Regulates IDH1 acetylation state and metabolic tumor suppression | |
| α-Tubulin_K40[96] | Paclitaxel resistant lung cancer | Enhances microtubule acetylation and stabilizes MCL1-associated survival | |
| GAC_K311[97] | Lung cancer metabolism | Regulates glutaminase activity through acetylation and ubiquitin crosstalk | |
| Ubiquitination | NOTCH1[111] | Intestinal epithelial renewal and aging-related homeostasis | FBXW7-mediated turnover restrains abnormal renewal signaling |
| p53[110] | Malignant selection | Modulates checkpoint restraint and damaged cell survival | |
| β-catenin_K508[112] | Colorectal cancer progression and ferroptosis | SPOP-dependent polyubiquitination promotes β-catenin degradation | |
| YAP_K90[113] | Hippo pathway signaling and tumorigenesis | PARK2 mediated K48-linked ubiquitination promotes YAP degradation and restrains tumor progression | |
| KYNU_K96/K163[114] | Gastric cancer redox adaptation | Controls redox-dependent ubiquitin linkage switching and supports tumor survival | |
| Glycosylation | GATA4_S406[30] | Cellular senescence | Stabilizes GATA4 and reinforces SASP-associated inflammatory programs |
| PD-L1_N192/N200/N219[127] | Tumor immune evasion | Stabilizes PD-L1 and weakens T cell clearance | |
| SPOP_S58/S59[112] | Colorectal cancer progression and ferroptosis | Modulates SPOP stability and β-catenin degradation | |
| Lactylation | H3K18la[42] | Immune evasive lung cancer microenvironment | Potentiates transcriptional programs linked to immune escape |
| NOL6_K54[118] | Colorectal cancer progression | Stabilizes NOL6 and supports MYC-dependent malignant programs | |
| H4K12la[184] | Colorectal cancer stem cell chemoresistance | Upregulates GCLC expression and suppresses ferroptosis | |
| Methylation | H3K4me3[147] | Spermatid development and leukemia transcriptional control | Regulates broad active chromatin domains and gene expression programs |
| GPX4_R152me2s[155] | Cancer ferroptosis resistance | Stabilizes GPX4 and suppresses ferroptosis | |
| LSH_R309me1[158] | Cancer stem-like properties | Crosstalk with LSH Ser503 phosphorylation drives stem-like behavior | |
| Redox modification | Cytoskeletal proteins[6] | Tissue aging | Alters protein interactions and epithelial maintenance |
| SUMOylation | YAP1_K97/K280[75] | Gastric cancer chemoresistance and malignant persistence | Promotes YAP1 SUMOylation, stability and nuclear retention |
| Palmitoylation | CLDN4_C104/C107[183] | Lenvatinib-resistant hepatocellular carcinoma | Sustains CLDN4 lipid raft anchoring and promotes hepatic-to-biliary lineage transition |
| Acylation | OPTN_K108[173] | Diabetic retinopathy | SIRT5-mediated desuccinylation protects against autophagic flux blockade |
PTM: post-translational modification; AMPK: AMP-activated protein kinase; MAPK: mitogen-activated protein kinase; EMT: epithelial–mesenchymal transition; LSH: lymphoid-specific helicase; AFP: alpha-fetoprotein; MCL1: myeloid cell leukemia 1; PD-L1: programmed death-ligand 1; SPOP: speckle-type POZ protein; NOL6: nucleolar protein 6; GCLC: glutamate-cysteine ligase catalyzing subunit.
3. Major PTM Systems Across the Aging-Cancer Continuum
3.1 Phosphorylation
Among major PTM systems, phosphorylation is most prominently involved in the rapid integration of stress signals[62]. Because kinases and phosphatases respond quickly to DNA damage, inflammatory stimulation, mitochondrial dysfunction, metabolic perturbation and microenvironmental cues, phosphorylation often represents the earliest regulatory layer through which aging-associated disturbances are converted into cellular decisions. In young or resilient tissues, phosphorylation supports checkpoint control, organelle surveillance, adaptive repair and restoration of homeostasis[63]. During aging, however, chronic or incompletely resolved stress can progressively alter signaling duration, threshold sensitivity, and pathway resolution[64]. As a result, phosphorylation networks that initially preserve tissue integrity may become redirected toward senescence persistence, stress tolerance, weakened growth restraint, and malignant adaptation (Figure 2).
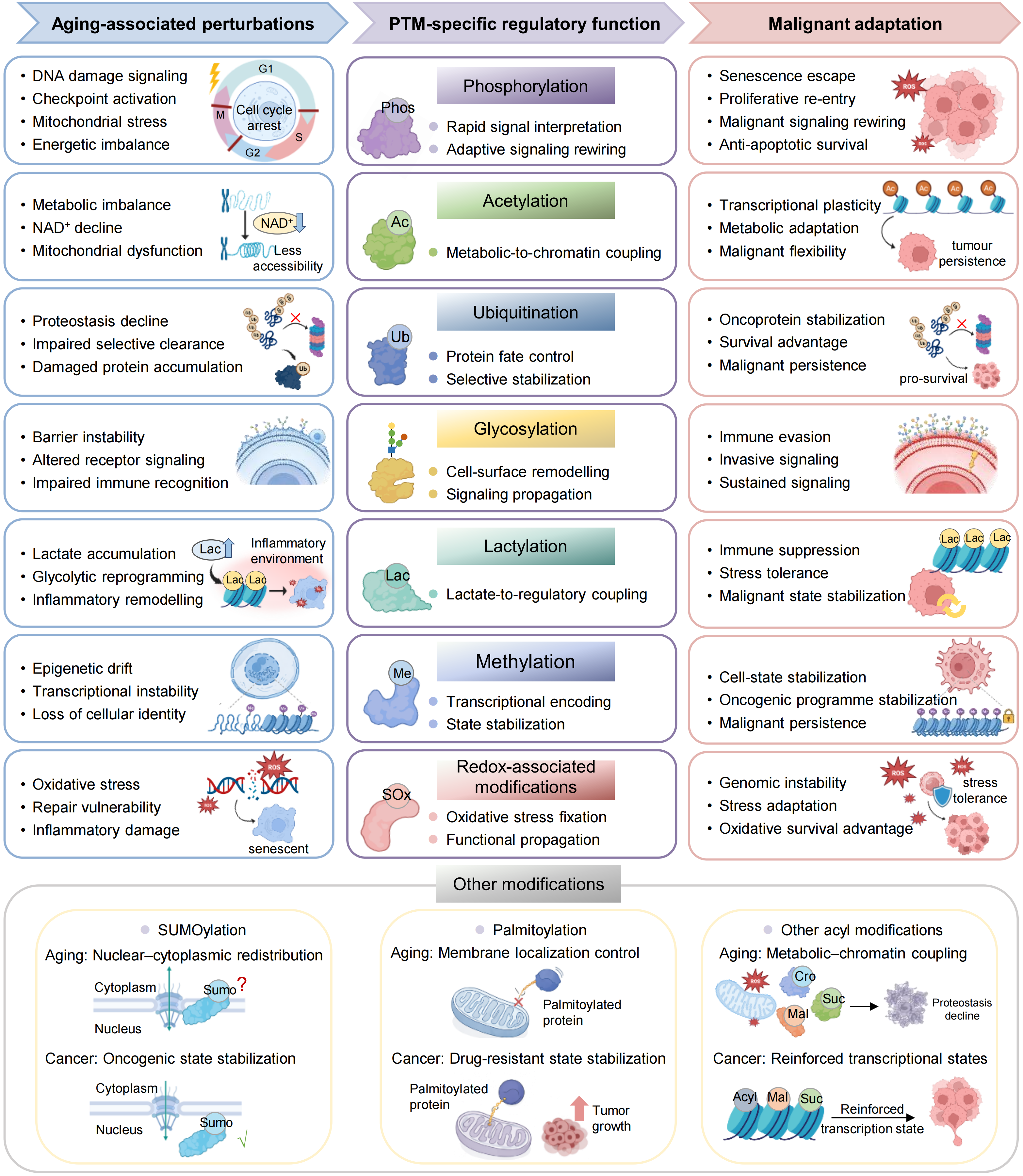{kind=link}
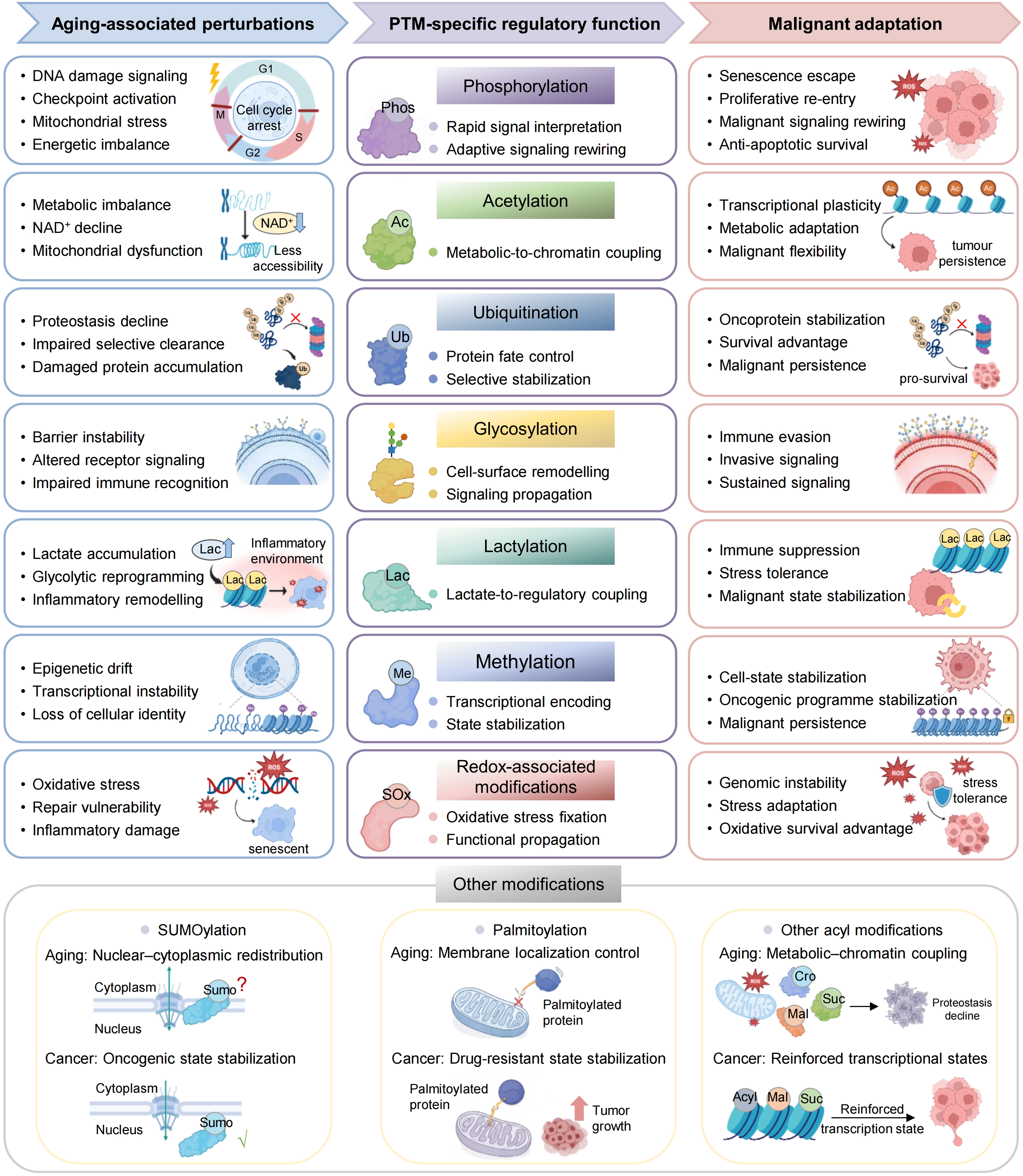
Figure 2. PTM-specific regulatory functions linking aging-associated states to malignant adaptation. Major PTM systems translate aging associated states into distinct regulatory functions that support malignant adaptation. Phosphorylation integrates stress and signaling cues to enable adaptive rewiring, whereas acetylation couples metabolic state to chromatin regulation and transcriptional plasticity. Ubiquitination controls protein stability and selective stabilization, and glycosylation reshapes cell-surface organization and signaling. Lactylation links glycolytic metabolism to regulatory and inflammatory states, whereas methylation stabilizes transcriptional programmes and cell identity. Redox-associated modifications propagate oxidative stress into functional states in signaling and genome stability. Additional PTMs, including SUMOylation, palmitoylation and other acyl modifications, further refine subcellular organization, membrane signaling and chromatin regulation. Together, these PTM systems convert aging-associated perturbations into regulatory states that enable survival, plasticity and progression in cancer. PTM: post-translational modification.
3.1.1 Phosphorylation as a stress-integrating system
Phosphorylation does not merely indicate that a stress pathway has been activated. Its broader significance lies in determining how different forms of stress are interpreted and coordinated[65]. For example, chronic inflammatory injury engages phosphorylation-centered NF-κB and p53 signaling, linking immune activation with damage sensing and checkpoint control[66]. Similarly, phosphorylation-dependent activation of the PINK1-Parkin axis determines whether damaged mitochondria are selectively removed or retained[67]. Efficient activation of this response limits mitochondrial stress and prevents prolonged reactive oxygen species (ROS) accumulation, whereas insufficient activation allows damaged mitochondria, oxidative stress and inflammatory signaling to persist[68].
Metabolic stress is processed through a similar logic. AMP-activated protein kinase-centered phosphorylation networks couple nutrient availability, mitochondrial dynamics and epithelial maintenance, thereby connecting energetic imbalance with adaptive remodeling[54]. These pathways can initially support repair and metabolic adjustment, but repeated or unresolved stress may convert them into sustained signaling states that promote mitochondrial fragmentation, impaired tissue renewal and senescence-associated phenotypes[54]. Extracellular cues from cytokines, growth factors and stromal cells further reshape phosphorylation networks controlling survival, migration and stress tolerance[52]. Phosphorylation therefore acts as a central stress-integrating system, translating inflammatory, metabolic, organellar and microenvironmental disturbances into decisions about repair, arrest, adaptation or persistence.
3.1.2 Aging-associated phospho-network rewiring
The key change during aging is not simply stronger phosphorylation signaling, but a gradual loss of signaling resolution[69]. When phosphorylation-dependent stress responses remain chronically active or improperly reset, damaged tissues become less able to return to homeostasis[69]. Persistent phospho-network imbalance can maintain senescence-associated reprogramming, reduce repair capacity, impair mitochondrial quality control and weaken tissue resilience[68]. These changes do not by themselves constitute malignant transformation, but they generate a tissue state in which damaged or metabolically stressed cells are more likely to persist.
Malignant transition begins when phosphorylation-dependent systems that normally contain damage gradually lose their restraining capacity[70]. In chronically injured tissues, persistent AKT-, ERK-, SRC- or FAK-associated signaling can weaken checkpoint control and bias cells toward survival, migration and proliferative re-entry[71,72]. In the liver, aging-associated phosphoproteome remodeling overlaps with signaling patterns observed in metabolic liver disease and hepatocellular carcinoma, suggesting that malignant potential can emerge before overt tumor formation at the level of phosphorylation networks[10]. Similarly, growth factor-rich or inflammatory microenvironments can reshape epithelial phospho-signaling, making energetic or inflammatory stress less effective at maintaining growth restraint[52].
Once malignant transition has occurred, phosphorylation networks are further repurposed to maintain tumor states[73]. Pathways that originally support transient stress adaptation are reused to sustain metabolic flexibility, invasive behavior, immune tolerance and survival under unfavorable tissue conditions[10,52]. Thus, the defining feature of malignant progression is not merely increased kinase activity, but the conversion of conditional phospho-signaling into a more stable regulatory architecture that continuously supports tumor persistence.
3.1.3 Crosstalk with other PTM systems
The biological consequences of phosphorylation are rarely determined by phosphorylation alone. Across the transition from aging to cancer, phosphorylation outputs are shaped by other PTM systems that influence substrate stability, localization, chromatin accessibility, metabolic state and signaling duration (Figure 3)[74]. Crosstalk with ubiquitination provides a clear example: phosphorylation can create substrate states that permit ubiquitin-dependent degradation, thereby converting transient signaling decisions into more durable changes in protein abundance[10]. In liver cancer-related signaling, phosphorylation-dependent regulation of FBP1 can facilitate its ubiquitin-mediated turnover, remove a metabolic restraint and allow oncogenic signaling to persist[10].
{kind=link}
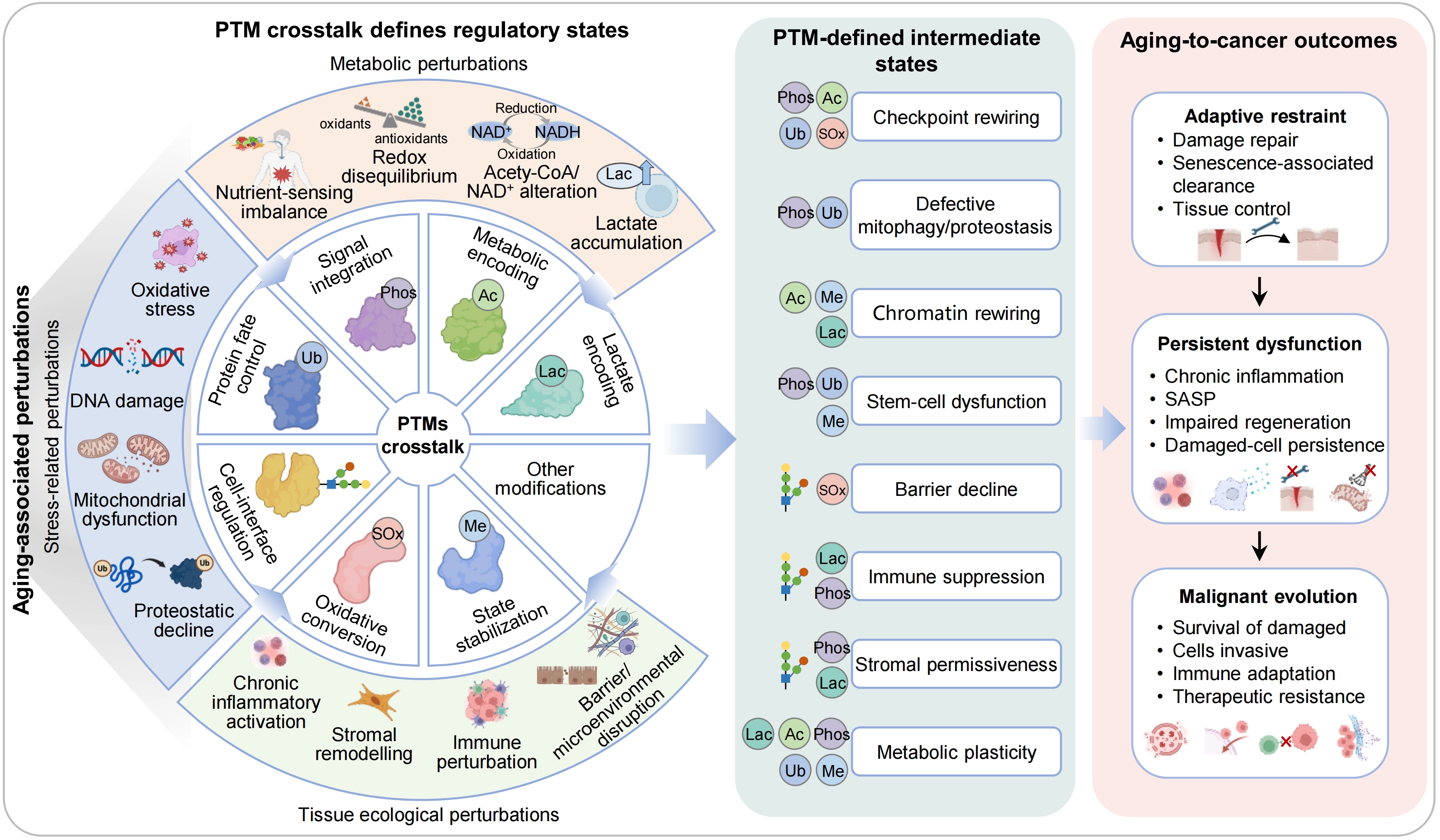
Figure 3. PTM crosstalk organizes regulatory states linking aging to cancer. Aging-associated perturbations arising from stress, metabolic imbalance and tissue ecological disruption are integrated by a network of interacting PTMs. Through crosstalk, PTMs act combinatorially to define regulatory states, including checkpoint rewiring, defective mitophagy and proteostasis, chromatin rewiring, stem-cell dysfunction, barrier decline, immune suppression, stromal permissiveness and metabolic plasticity. These PTM-defined intermediate states determine whether tissues maintain adaptive restraint, progress to persistent dysfunction or undergo malignant evolution, characterized by survival of damaged cells, invasion, immune adaptation and therapeutic resistance. PTM: post-translational modification.
Other PTMs can redirect phosphorylation outcomes by altering protein localization, structural accessibility or signaling persistence[75]. Acetylation and lactylation can stabilize metabolically induced signaling states, whereas glycosylation can reshape receptor organization and extracellular signal interpretation[76]. This context dependence is especially important in aging tissues, where metabolic imbalance, proteostatic decline and microenvironmental remodeling continuously reshape the broader PTM landscape[77]. Phosphorylation should therefore be viewed not as an isolated signaling switch, but as part of an integrated PTM network that determines whether stress-responsive signaling remains adaptive or becomes stabilized into a tumor-promoting state.
3.2 Acetylation
Among major PTM systems, acetylation is especially important for translating metabolic state into durable cellular programs[78]. Because acetylation depends on acetyl-CoA availability and is reversed by NAD+-dependent or Zn2+-dependent deacetylases, it provides a direct molecular interface between nutrient supply, mitochondrial function, redox balance and protein regulation[79,80]. Unlike phosphorylation, which rapidly transmits stress signals, acetylation is particularly suited to stabilizing metabolically induced changes in chromatin accessibility, transcriptional competence and non-histone protein function (Figure 2)[81,82]. Across the transition from aging to cancer, this property allows transient metabolic fluctuations to become progressively embedded into longer-lasting cellular states[81].
3.2.1 Acetylation as a metabolic-encoding system
Acetylation links metabolism to gene regulation by coupling substrate availability to chromatin structure and transcriptional output[81]. Acetyl-CoA-dependent histone acetylation can increase local chromatin accessibility and enable enhancer activation, whereas NAD+-dependent deacetylation by sirtuins connects cellular energy status to mitochondrial function, stress resistance and transcriptional control[81,83]. In this way, metabolic conditions are not only reflected in acetylation patterns but are also converted into regulatory programs that influence cell identity, repair capacity and tissue function[84,85].
This mechanism is especially relevant during aging, when NAD+ decline, mitochondrial dysfunction, altered acetyl-CoA handling and chronic inflammatory stress progressively disturb the balance between acetylation and deacetylation[86]. Reduced sirtuin activity can increase acetylation of mitochondrial and nuclear proteins, weakening metabolic flexibility and stress tolerance[87]. At the chromatin level, altered histone acetylation can reduce the ability of cells to mount adaptive transcriptional responses[88]. Acetylation therefore helps explain how metabolic aging becomes linked to impaired repair, reduced regenerative competence and persistent tissue dysfunction[89,90].
3.2.2 Acetylation-state remodeling in aging and cancer
During malignant transition, acetylation can convert metabolic instability into more stable oncogenic transcriptional states[81,82]. Increased acetyl-CoA availability, altered activity of acetyltransferases and deacetylases, and redistribution of enhancer acetylation can strengthen growth-associated gene programs[91,92]. In several tumors, elevated H3K27ac or H4 acetylation marks active enhancers and super-enhancers that support proliferation, stem-like features and survival[91]. These changes do not merely accompany malignant transformation; they help stabilize transcriptional programs that make damaged or metabolically stressed cells more capable of persistence and expansion[92].
Once tumors are established, acetylation further supports malignant plasticity through both histone and non-histone substrates[81,91]. Histone acetylation maintains accessible chromatin regions required for repeated activation of growth, stress-response and lineage plasticity programs[93]. Non-histone acetylation can regulate the stability, activity or localization of proteins involved in apoptosis, metabolism and invasion, such as alpha-fetoprotein (AFP), IDH1, MCL1 or glutaminase C in different tumor settings[94-97]. Together, these mechanisms allow tumor cells to remain adaptable without losing the core regulatory architecture required for continued malignant persistence.
3.2.3 Crosstalk with other PTM systems
The effects of acetylation are strongly shaped by crosstalk with other PTM systems (Figure 3)[98,99]. This is particularly important because acetylation exists within a broader network of metabolically linked acyl modifications[79]. Under conditions of metabolic rewiring, the key issue is often not simply whether acetylation increases or decreases, but which acyl state gains regulatory dominance over chromatin accessibility, protein stability and transcriptional output[99,100].
Ubiquitination can determine whether acetylated proteins are retained or degraded, whereas phosphorylation can influence the activity of acetyltransferases, deacetylases and chromatin-associated regulators[85,98]. Other acyl modifications, such as crotonylation or lactylation, may compete with or redirect acetylation-dependent regulation under altered metabolic conditions[99,101]. Through this crosstalk, acetylation helps convert metabolic imbalance into more persistent regulatory states[101]. In aging tissues, this can reinforce dysfunctional adaptation; in tumors, it can stabilize malignant transcriptional and metabolic programs[99,101].
3.3 Ubiquitination
Ubiquitination is the PTM system most directly responsible for determining selective protein fate[102]. Rather than primarily controlling rapid signaling responses, ubiquitination governs which proteins are removed, retained or stabilized over time, thereby shaping the proteostatic and regulatory landscape of aging tissues[102]. Across the transition from aging to cancer, altered ubiquitin control progressively changes which damaged proteins, organelles and cellular programs are efficiently eliminated and which are allowed to persist (Figure 2)[103].
3.3.1 Ubiquitination as a protein-fate selection system
Under physiological conditions, ubiquitination supports tissue homeostasis by directing damaged proteins for proteasomal degradation, regulating organellar quality control, and controlling the abundance of key signaling proteins[104,105]. This selectivity is especially important in long-lived or repeatedly stressed tissues, where timely removal of abnormal proteins and dysfunctional mitochondria prevents proteotoxic burden, oxidative stress, and chronic inflammatory activation[105].
During aging, ubiquitin-dependent clearance becomes progressively less efficient and less precise[106]. Damaged proteins and organelles that would normally be removed begin to persist, whereas some regulatory proteins escape normal turnover and remain aberrantly stabilized[107]. Impaired proteasome activity, altered E3 ligase or deubiquitinase function, and defective mitophagy all contribute to this shift[103]. As a result, proteotoxic stress, mitochondrial dysfunction, and senescence-associated signaling become increasingly difficult to resolve[108].
The biological significance of ubiquitination therefore lies not only in degradation itself, but also in determining which cellular states are permitted to persist[103]. When ubiquitin-dependent filtering becomes impaired, aging tissues lose part of their ability to eliminate damaged or maladapted components, creating a background in which stress-retaining and tumor-permissive states become easier to maintain[108].
3.3.2 Ubiquitin-network rewiring in aging and cancer
During malignant transition, altered ubiquitin control weakens the selective elimination of damaged, stress-adapted, or proliferative cellular states[109]. Proteins that normally restrain abnormal expansion may be destabilized, whereas survival-promoting regulators can escape turnover[110]. In this way, ubiquitination contributes to the shift from aging-associated dysfunction to malignant selection by changing the balance between restraint and persistence[110].
This logic is evident in pathways controlling stem-cell renewal, checkpoint activity, and tumor-suppressive signaling. For example, FBXW7-mediated ubiquitination of NOTCH1 helps restrain inappropriate epithelial expansion, whereas loss or weakening of such control can support tumor-promoting renewal[111]. Similarly, altered ubiquitination of p53, β-catenin, YAP, or apoptosis-associated regulators can determine whether damaged cells remain constrained or acquire increased survival and proliferative capacity[110,112,113]. The key point is not that ubiquitination simply promotes or suppresses cancer, but that it selectively edits the protein landscape on which malignant selection acts.
Once tumors are established, ubiquitination is further redirected to support malignant advantage[114]. Tumor cells can degrade pro-apoptotic proteins, stabilize metabolic enzymes, preserve transcriptional regulators, or modulate immune-related proteins[115]. These changes allow malignant cells to maintain survival, metabolic flexibility, invasive behavior, and immune evasion[114,115]. Thus, ubiquitination functions as a protein-state selection system that determines which regulatory programs remain transient and which become durably retained during tumor evolution.
3.3.3 Crosstalk with other PTM systems
The outcome of ubiquitination depends strongly on crosstalk with other PTM systems[116]. Ubiquitin-dependent fate decisions require substrate recognition, accessibility, and linkage specificity, all of which can be altered by phosphorylation, acetylation, glycosylation, lactylation, or methylation[116]. Other PTMs can therefore determine whether proteins become available for degradation, escape turnover, or undergo selective stabilization under stress (Figure 3)[116].
Phosphorylation often acts upstream by creating degradation-permissive substrate states[117]. In other contexts, O-GlcNAcylation, acetylation, or lactylation can protect proteins from ubiquitin-dependent clearance and prolong the function of oncogenic or stress-adaptive regulators[118]. Through these interactions, ubiquitination becomes more than a terminal degradation signal[116]. It serves as a decision layer that integrates competing PTM inputs and converts them into durable changes in protein abundance, signaling persistence, and cellular state[116].
In this sense, ubiquitination links aging and cancer by controlling selective persistence[119]. During aging, impaired ubiquitin regulation allows damaged components and dysfunctional programs to accumulate[108]. During cancer evolution, the same protein-fate machinery can be repurposed to preserve proteins that sustain malignant adaptation[114].
3.4 Glycosylation
Glycosylation is uniquely positioned at the interface between cells and their surrounding environment[120]. By modifying membrane receptors, secreted proteins, extracellular matrix-associated proteins and selected intracellular substrates, glycosylation regulates receptor responsiveness, cell-cell adhesion, barrier integrity, immune recognition and extracellular communication (Figure 2)[120-122]. Across the transition from aging to cancer, glycosylation progressively alters how cells are presented to neighboring cells, stromal compartments and immune systems, thereby reshaping the extracellular conditions that restrain or support malignant persistence[120].
3.4.1 Glycosylation as an extracellular-recognition system
Unlike intracellular PTMs that mainly regulate signaling or chromatin state, glycosylation directly shapes the surface architecture through which tissues interact with their environment[120]. N-glycosylation and O-glycosylation influence receptor folding, ligand binding, secretory protein stability, and barrier-associated structures[121], whereas O-GlcNAcylation links nutrient state to intracellular stress responses and transcriptional regulation[123]. Together, these modifications help maintain tissue boundaries, extracellular communication, and recognition systems[120,122].
During aging, glycosylation patterns are progressively remodeled rather than simply reduced[124]. Barrier-associated glycans, receptor-linked glycan structures, and secretory glycoprotein states can lose precision, weakening tissue compartmentalization, immune regulation, and environmental responsiveness[120,122,124]. These changes illustrate how glycosylation remodeling can convert aging-associated tissue disruption into altered extracellular recognition and communication[122].
3.4.2 Glycan remodeling and PTM crosstalk
During malignant transition and progression, glycosylation is frequently redirected to stabilize tumor-environment interactions[125]. Altered glycosylation can increase the stability or activity of cell-surface proteins, strengthen receptor signaling, remodel extracellular protease function, and reduce immune recognition[126]. For example, glycosylation of PD-L1 stabilizes its surface expression and weakens T-cell-mediated clearance, providing a direct link between glycan remodeling and tumor immune evasion[127].
Intracellular O-GlcNAcylation adds another layer by stabilizing selected proteins involved in DNA repair, transcriptional control, and metabolic adaptation[128,129]. Glycosylation also interacts extensively with other PTMs (Figure 3). For example, O-GlcNAcylation can influence phosphorylation, glycan structures can protect proteins from ubiquitin-dependent degradation, and surface glycans can reorganize receptor clustering and ligand responsiveness[116,130]. Glycosylation therefore links aging and cancer by controlling extracellular organization, immune recognition, and tissue-level communication[120].
3.5 Lactylation
Lactylation is positioned at the interface between chronic metabolic stress and malignant persistence[101]. Unlike broader acetylation-dependent regulation, lactylation specifically reflects sustained lactate accumulation arising from glycolytic remodeling, mitochondrial dysfunction, and inflammatory tissue environments (Figure 2)[131,132]. Its importance lies not only in indicating increased glycolysis, but also in allowing lactate-rich states to acquire longer-lasting regulatory effects on chromatin organization, protein stability, and cellular adaptation[101,131].
3.5.1 Lactylation in lactate-rich tissue states
Lactylation usually does not represent the earliest response to metabolic disturbance[133]. Instead, it tends to emerge when glycolytic stress, mitochondrial dysfunction, or inflammatory remodeling become chronically sustained[133]. In aging tissues, reduced mitochondrial efficiency, altered redox balance, and persistent inflammation can increase reliance on glycolytic compensation, creating biochemical conditions that favor lactate accumulation and lactylation[134].
This process is especially relevant in tissue environments shaped by stromal activation and chronic inflammation[135,136]. Glycolytic fibroblasts, inflammatory cells, and metabolically stressed epithelial cells can generate lactate-rich niches, allowing lactate to function not only as a metabolic by-product but also as a regulatory substrate[135,136]. Through histone and non-histone lactylation, prolonged metabolic imbalance can become incorporated into transcriptional regulation, protein function, and local cell-cell communication[101].
3.5.2 Lactate-driven malignant persistence
During malignant transition, lactylation can convert persistent metabolic stress into more stable oncogenic regulation[137]. In lactate-rich environments, histone lactylation can activate transcriptional programs that support proliferation, stress tolerance, inflammatory adaptation, and immune suppression[138]. Cancer-associated fibroblasts and other glycolytic stromal populations can further amplify this process by supplying lactate to tumor cells, thereby linking microenvironmental metabolism to malignant cell-state remodeling[135,138].
Once malignant states are established, lactylation can reinforce tumor persistence by maintaining communication between tumor metabolism and the surrounding microenvironment[139]. In this setting, lactylation links glycolytic remodeling not only to intracellular transcriptional adaptation, but also to stromal communication, immune modulation, and resistance to cellular stress[139,140]. Thus, lactylation links aging and cancer by encoding chronic lactate-rich adaptation that can be exploited during tumor evolution[141,142].
Lactylation also operates within broader PTM networks (Figure 3)[118]. Lactylation in lactate-rich states can intersect with acetylation and other acyl modifications in response to shifts in glycolysis, lactate accumulation, and broader acylation metabolism[101,143], reinforce phosphorylation-dependent inflammatory or survival signaling[138], and protect selected proteins from ubiquitin-dependent turnover[118]. Thus, lactylation should be viewed not only as a lactate-derived mark, but also as part of a metabolic PTM network that stabilizes chronic adaptation in aging and cancer.
3.6 Methylation
Methylation is especially suited to stabilize long-lasting regulatory states[144]. Whereas phosphorylation rapidly transmits stress signals and acetylation links metabolic state to chromatin accessibility, methylation more often contributes to transcriptional memory, lineage identity, and persistence of stress-adapted cellular programs (Figure 2)[144,145]. Across the transition from aging to cancer, methylation helps determine whether aging-associated disturbances remain reversible or become fixed into durable dysfunctional or malignant states[146].
3.6.1 Methylation remodeling during aging
Aging is accompanied by progressive changes in methylation-dependent involving both histone and non-histone substrates[144,147]. Histone methylation influences chromatin organization, transcriptional competence, and lineage-specific gene expression, whereas protein methylation can alter protein interactions, stability, localization, and signaling output[144,147]. Together, these mechanisms provide a regulatory memory that helps cells preserve identity and respond to stress[148].
During aging, this memory system can become distorted[149]. Altered methylation states may weaken transcriptional programs required for repair, differentiation, and tissue maintenance, while stabilizing inflammatory or stress-adapted states[150]. Unlike transient signaling events, methylation-dependent changes can persist after the initial stress has subsided, making aging-associated dysfunction more difficult to reverse[149].
3.6.2 Methylation-dependent stabilization of aging and malignant states
During malignant transition, methylation can convert unstable regulatory changes into more fixed oncogenic states[151]. At the chromatin level, altered histone methylation can silence tumor-suppressive programs, maintain stem-like transcriptional states, or reinforce lineage plasticity[152,153]. At the protein level, methylation of transcription factors, signaling mediators, or RNA-binding proteins can stabilize oncogenic interactions and support survival under stress[154,155].
The effects of methylation are also shaped by crosstalk with other PTM systems (Figure 3)[156,157]. Histone methylation interacts with acetylation to determine whether chromatin regions remain repressed, poised, or active[147,157], whereas protein methylation can influence phosphorylation-dependent signaling[158], ubiquitin-mediated degradation[159] or transcriptional complex assembly[160]. Through these interactions, methylation helps determine whether regulatory states remain flexible or become fixed[157].
3.7 Redox-associated modifications
Redox-associated modifications represent the PTM layer most directly linked to oxidative imbalance[161]. Unlike phosphorylation, which rapidly transmits regulatory signals, redox modifications arise when oxidative conditions alter the chemical reactivity of specific amino acid residues, especially cysteine[161,162]. Their importance at the aging-cancer interface lies not only in reflecting elevated ROS, but also in embedding oxidative stress into protein function, repair capacity, inflammatory signaling, and metabolic control (Figure 2)[163].
3.7.1 Redox modifications in oxidative-stress remodeling
Aging tissues are exposed to persistent oxidative stress arising from mitochondrial dysfunction, inflammatory activation, and declining antioxidant capacity[161,164]. Redox-associated modifications provide a chemical mechanism through which this oxidative burden is written into specific proteins[161]. Cysteine residues are particularly important because their thiol groups are highly sensitive to oxidation and can undergo reversible or irreversible modifications depending on the local redox environment[161].
Site-resolved redoxomic studies show that oxidative modifications are not randomly distributed across the proteome[9]. Instead, they often affect functionally important residues in metabolic enzymes, cytoskeletal proteins, stress-response regulators, and epithelial maintenance factors[6]. As these modifications accumulate, they can alter enzyme activity, protein stability, interaction networks, and subcellular localization[6]. In aging tissues, such changes may impair metabolic flexibility, weaken barrier integrity, and reduce the capacity to restore homeostasis after injury[6].
The key point is that redox-associated modifications convert oxidative stress from a diffuse biochemical condition into discrete protein-level changes[163]. When these changes remain reversible and properly regulated, they can support adaptive stress responses[161]. When oxidative stress persists, however, redox remodeling can become embedded into tissue dysfunction, making repair failure, inflammatory persistence, and cellular vulnerability more difficult to reverse[164].
3.7.2 Redox-associated repair failure and malignant transition
Redox-associated modifications contribute to malignant transition when oxidative stress begins to compromise genome maintenance, inflammatory control, and cell-fate regulation[163]. Persistent oxidation can alter proteins involved in DNA repair, mitochondrial homeostasis, and stress signaling, thereby weakening the systems that normally prevent damaged cells from persisting[163,165]. In this setting, oxidative stress is not only a source of molecular damage but also a driver of altered protein function[163].
This process is particularly relevant in chronically injured tissues, where repeated oxidative stress can impair repair proteins and promote inflammatory signaling[165]. Redox-dependent disruption of DNA repair capacity may increase genomic instability, while concurrent activation of inflammatory pathways can create a tissue environment that favors survival of damaged cells[165]. These changes do not directly define malignancy, but they lower the threshold for malignant transition by allowing oxidative damage, inflammatory signaling, and defective repair to reinforce one another[165].
Redox-associated modifications therefore provide an important bridge between aging-associated oxidative injury and cancer risk[163]. They help explain how persistent ROS can move beyond nonspecific damage and become converted into specific molecular alterations that impair repair, sustain inflammation, and create a more permissive environment for malignant selection[165].
3.7.3 Redox adaptation and PTM crosstalk
Once tumors are established, redox-associated modifications can be exploited to support malignant adaptation[166]. Tumor cells often experience high oxidative pressure due to rapid proliferation, metabolic rewiring, hypoxia, and inflammatory microenvironments[166]. Rather than simply being damaged by ROS, malignant cells can remodel redox-sensitive proteins and antioxidant systems to maintain survival under oxidative stress.
This adaptation affects multiple aspects of tumor biology. Redox modifications can regulate metabolic enzymes, signaling proteins, transcriptional regulators, and cell-death pathways, thereby supporting metabolic flexibility, stress tolerance, and resistance to therapy[167]. In some contexts, tumor cells use altered redox states to avoid ferroptosis or apoptosis; in others, redox-sensitive signaling promotes inflammatory or immune-modulatory programs that support tumor persistence[167].
Redox-associated modifications therefore link aging and cancer by embedding oxidative stress into protein function[161,163]. During aging, this embedding contributes to impaired repair, barrier decline, and inflammatory persistence[163]. During tumor evolution, the same redox-modified protein landscape can be exploited to sustain stress tolerance, metabolic adaptation, and malignant progression[167].
Redox-associated modifications also intersect with other PTM systems (Figure 3)[163]. Oxidative changes can alter kinase and phosphatase activity, influence ubiquitin-dependent degradation, and reshape the availability of metabolic cofactors that support acetylation or lactylation[168,169]. In this way, redox remodeling can amplify or redirect broader PTM networks, allowing oxidative stress to become embedded not only in individual proteins but also in regulatory systems that sustain aging-associated dysfunction and tumor adaptation.
3.8 Other modifications
Several additional PTMs further broaden the regulatory landscape linking aging and cancer[75,170]. Their importance lies less in the depth of current evidence than in the mechanisms they highlight[170]. SUMOylation illustrates how changes in protein localization and stability can modify the outcome of other PTMs (Figure 2)[75]. For example, SUMOylation can oppose phosphorylation-dependent restraint and promote nuclear retention of oncogenic regulators such as YAP1, thereby shifting transient signaling states toward more persistent malignant programs[75]. Palmitoylation adds another layer by controlling membrane association, organellar positioning, and the stability of signaling or metabolic complexes[171]. In tumor settings, palmitoylation can suppress ubiquitin-dependent turnover of selected substrates and stabilize metabolic or membrane-associated proteins that support malignant progression[171].
Other acyl modifications, including crotonylation, succinylation, malonylation, and glutarylation, should currently be viewed as components of a broader metabolically sensitive acylation landscape rather than fully independent aging-cancer pathways (Figure 2)[170]. They link acyl-CoA availability, mitochondrial function, and chromatin or protein regulation[172], and may become especially relevant in tissues characterized by oxidative stress or metabolic decline[173,174]. These emerging modifications therefore serve as supplementary but informative examples of how aging-associated stress and metabolic imbalance can be translated into protein localization, membrane organization, and acylation-state remodeling[75].
4. Translational Implications
The translational value of PTM biology lies in its ability to describe functional disease states rather than static molecular categories[175]. At the aging-cancer interface, PTMs can indicate whether stress responses remain protective, whether metabolic imbalance has become stabilized, whether microenvironmental remodeling has created a tumor-permissive niche, and whether malignant cells have acquired therapy-resistant dependencies[176]. However, this potential must be interpreted with caution. Although PTM profiling has generated many candidate biomarkers and therapeutic targets, only a limited number of PTM-defined biomarkers or PTM-directed interventions have reached routine clinical use[177]. The central challenge is therefore to distinguish PTM changes that are merely associated with disease from those that are analytically robust, mechanistically necessary, and clinically actionable (Figure 4)[178].
{kind=link}
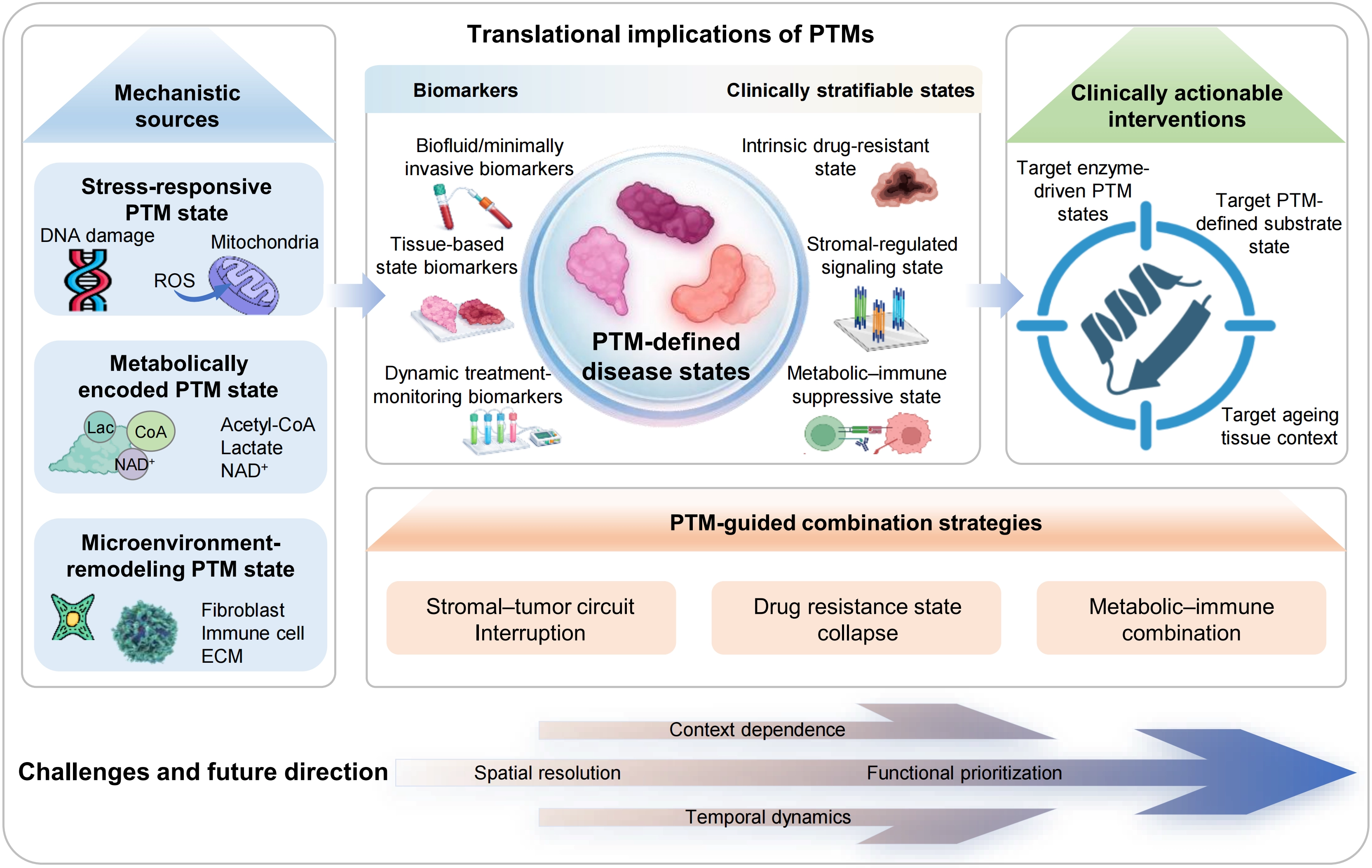
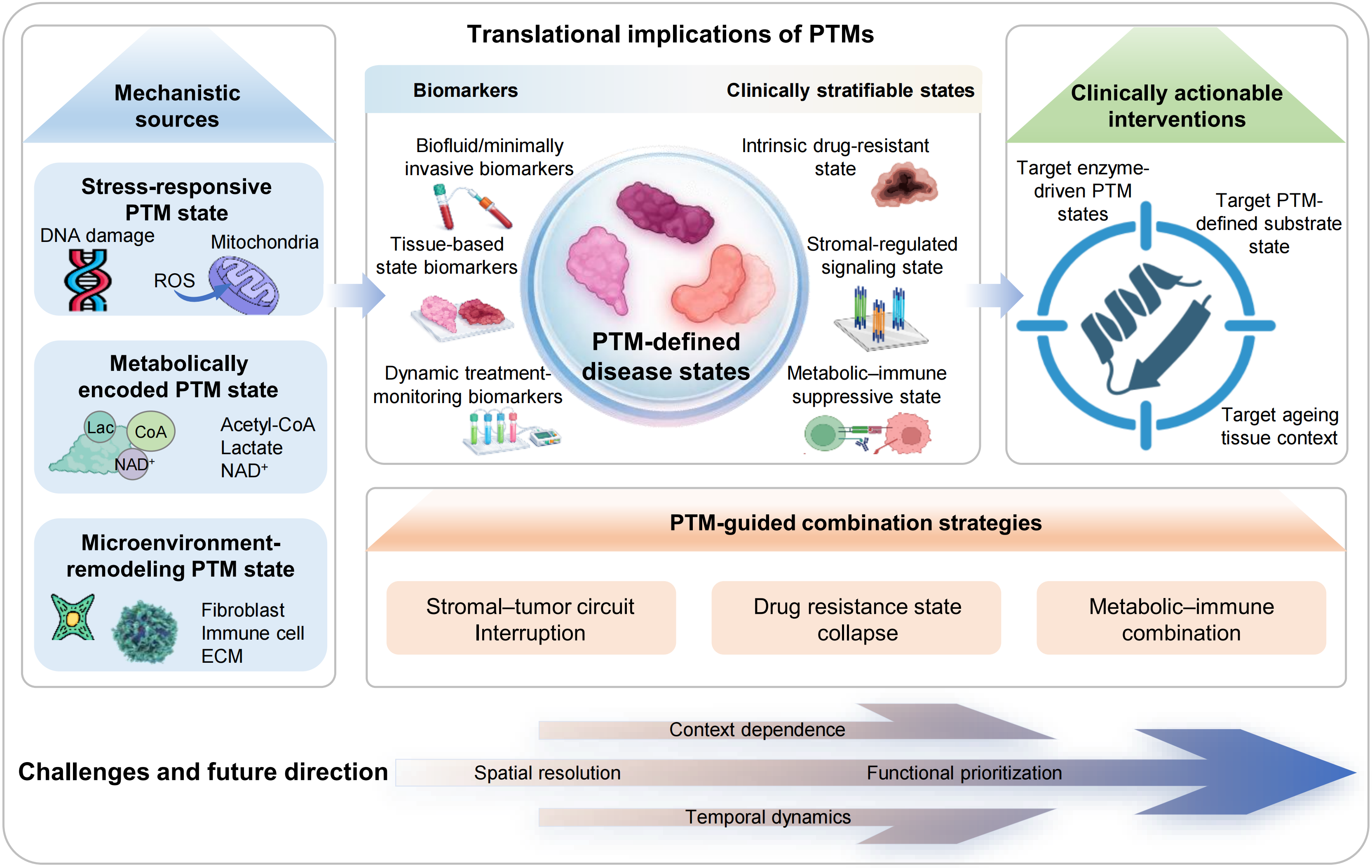
Figure 4. Translational framework of PTM-defined disease states. PTMs integrate stress, metabolic and microenvironmental inputs to generate distinct regulatory states. These PTM-defined states can be captured by biofluid or minimally invasive, tissue-based and dynamic monitoring biomarkers, enabling clinical stratification into intrinsic drug-resistant, stromal-regulated signaling and metabolic–immune suppressive states. This framework supports clinically actionable interventions targeting enzyme-driven PTM states, substrate states and aging tissue context, and informs PTM-guided combination strategies, including stromal–tumor circuit interruption, drug-resistance state collapse and metabolic–immune combination. Key challenges include context dependence, spatial resolution, functional prioritization and temporal dynamics. PTM: post-translational modification.
4.1 PTM-defined biomarkers
PTM-based biomarkers are attractive because they can report biological activity more directly than total protein abundance[176]. A phosphorylated receptor, a glycosylated immune checkpoint, or an acetylated chromatin state may indicate whether a pathway is functionally engaged, whether a tumor is adapting to treatment, or whether a tissue has entered a persistent dysfunctional state[179]. This is particularly relevant in aging-associated disease, where progression is gradual and often shaped by stress adaptation, metabolic remodeling, and tissue-level ecological change[176].
Clinically implemented PTM-based biomarkers remain limited. A recent study highlighted only a small number of modified proteoforms that have reached clinical use, including HbA1c, β2-transferrin, phosphorylated tau proteoforms, and AFP-L3[180]. Among these, AFP-L3, often clinically reported as AFP-L3%, is a fucosylated AFP fraction used in hepatocellular carcinoma risk assessment and is the most directly relevant example for oncology[181]. Phosphorylated tau proteoforms illustrate the clinical utility of PTM based biomarkers in aging related disease[182]. This limited list underscores that most PTM readouts in aging and cancer remain investigational rather than routine clinical tools.
Several investigational examples illustrate the promise and current limits of this field[183,184]. Dynamic epidermal growth factor receptor (EGFR) phosphorylation can provide pharmacodynamic information about EGFR-TKI engagement, while altered phospho-signaling states may help identify aggressive or therapy-resistant tumor programs[185]. Other investigational examples include CLDN4 palmitoylation in lenvatinib resistance in hepatocellular carcinoma and histone lactylation in chemoresistance in colorectal cancer[52]. These examples are valuable because they connect PTM states to specific disease behaviors[176]. Yet they also highlight why PTM biomarkers require rigorous validation: clinical utility depends not only on detecting a modification, but on proving that it is reproducible, disease-specific, temporally informative, and able to guide management[177,178].
4.2 PTM-directed therapeutics
PTM biology has already influenced cancer therapy, but most successful examples target PTM-regulating enzymes or systems rather than individual modified residues[186]. Kinase inhibitors represent the most mature PTM-directed therapeutic class[187]. Proteasome inhibitors provide another established example by targeting ubiquitin-dependent protein turnover[188], and histone deacetylase inhibitors demonstrate that chromatin-modifying enzymes can be therapeutically exploited in selected malignancies[189]. These successes show that PTM-regulating machinery can be druggable when disease dependence is strong and pharmacological windows are acceptable.
However, enzyme-level success does not mean that PTM-based therapy is straightforward[190]. Many PTM enzymes act on broad substrate repertoires, and the same enzyme may support protective adaptation in one context while promoting tumor persistence in another[186]. Broad inhibition can therefore produce toxicity, compensatory signaling, or loss of beneficial stress responses[191]. This issue is especially relevant in aging tissues, where PTM networks often maintain residual repair capacity even while contributing to chronic dysfunction[16].
A more precise strategy is to target disease-relevant modified protein states or PTM-dependent interactions. Examples such as CLDN4 palmitoylation in lenvatinib resistance or H4K12 lactylation in oxaliplatin resistance illustrate how a specific modification-defined circuit can create a therapeutic vulnerability[183]. However, such approaches remain difficult because residue-specific targeting, modified-proteoform detection, and substrate-selective intervention are technically challenging[192]. In many cases, therapeutic actionability depends less on the PTM class itself than on the specific relationship among the modifying enzyme, the substrate, and the disease context[183,184].
PTM-directed therapy should therefore be viewed at three levels: targeting the machinery that installs, removes, or recognizes a modification; disrupting a modified protein state that drives disease behavior; and modifying the tissue context that repeatedly recreates the pathological PTM state[16]. This distinction is especially important for aging-associated cancer, where the aged microenvironment may continue to regenerate the same stress, metabolic, and inflammatory inputs even after a tumor-intrinsic PTM dependency is inhibited[16].
4.3 PTM-guided combination strategies
PTM states can help rationalize combination therapy because malignant cells often preserve disease states through compensatory PTM crosstalk[193]. Blocking one kinase pathway may be bypassed by altered ubiquitination, acetylation, or metabolic acylation, and inhibiting a chromatin regulator may fail if the metabolic condition that supports the chromatin state remains unchanged[193]. The value of PTM-guided combination therapy is therefore not simply to add more inhibitors, but to identify the regulatory dependencies that allow tumor cells to maintain persistence under treatment pressure[193]. Combination strategies may be most effective when they pair pathway inhibition with disruption of the PTM mechanism that sustains resistance, such as modified protein stabilization, chromatin-state maintenance, altered membrane organization, or ferroptosis suppression[193].
4.4 Barriers to clinical translation
Several barriers explain why PTM-based clinical translation remains limited. For biomarkers, the first challenge is analytical robustness[192]. Many PTMs occur at low stoichiometry, are dynamically regulated, and require enrichment or site-specific detection[192]. Measurement can be affected by sample handling, pre-analytical degradation, antibody specificity, peptide localization uncertainty, enrichment bias, and platform variability[192]. These issues make it difficult to establish standardized thresholds across laboratories and cohorts[194].
The second challenge is biological interpretation[191]. The same PTM class may have opposite meanings depending on substrate identity, cell type, disease stage, and tissue context[191]. Bulk PTM measurements often obscure whether a signal arises from tumor cells, stromal cells, senescent fibroblasts, immune populations, or biofluid-derived material[195]. This is a major limitation for minimally invasive markers: saliva-, plasma- or serum-based PTM readouts are attractive, but their clinical value depends on knowing which tissue process they reflect[196].
For therapeutics, the main challenge is specificity. PTM writers, erasers, and readers usually regulate many substrates, and disease may depend on only a subset of their activities[186]. A broadly acting inhibitor may suppress a tumor-promoting PTM state while also impairing adaptive repair or immune function[190,191]. In addition, PTM networks are highly compensatory. Blocking one modification may redirect signaling through another PTM system, allowing the disease state to persist[193]. Therapies directed at a specific modified proteoform or PTM-dependent protein interaction could improve selectivity, but such strategies are still technically demanding.
Emerging technologies may help overcome these limitations[194,197]. Low-input PTMomics, single-cell or cell-type-resolved proteomics, spatial proteomics, imaging mass spectrometry, and improved modified-proteoform detection can help define where a PTM state arises, which cell population carries it, and how it changes during disease progression or treatment[195]. Longitudinal sampling and minimally invasive monitoring may further clarify when PTM states become clinically informative. Future progress will depend less on expanding PTM catalogues and more on identifying PTM states that are mechanistically necessary, temporally ordered, and actionable in a defined tissue context[178,192].
5. Perspective
Aging and cancer are connected by shared disturbances, protective constraints, and context-dependent transitions. PTMs provide a protein-level regulatory layer through which these relationships can be understood. Rather than acting as isolated biochemical marks, PTMs translate stress, metabolism, and tissue ecology into functional cellular states[23]. Whether these states remain adaptive, become chronically sustained, or are repurposed for malignant persistence depends on the timing, substrate, cellular source, and tissue context of each modification[198].
A key message of this review is that different PTM systems contribute to the aging-cancer continuum through distinct but interconnected regulatory roles[199]. Phosphorylation integrates stress signals; acetylation encodes metabolic state; ubiquitination controls selective protein fate; glycosylation shapes extracellular recognition; lactylation reflects chronic lactate-rich adaptation; methylation stabilizes regulatory memory; and redox-associated modifications embed oxidative stress into protein function[199]. These distinctions help move the field beyond cataloguing PTM changes toward understanding how PTM networks determine disease-relevant cellular states[177].
Future studies should focus on identifying PTM states that are causal, temporally ordered, spatially localized, and clinically actionable[198]. The most informative readouts are unlikely to be broad changes in a modification class, such as increased phosphorylation or elevated lactylation[177]. Instead, they will probably involve defined modified proteoforms, modification-dependent protein interactions, or PTM signatures linked to specific cell types and disease stages[200]. In this sense, PTM biology offers more than a molecular explanation for the aging-cancer connection; it provides a framework for defining disease states by the regulatory processes that sustain them[199].
Authors contribution
Dai L: Project administration, writing-review & editing.
Zhang R, Fu Y, Chen H, Yu J, Li Y, Hu M: Writing-original draft, formal analysis.
Nie L: Conceptualization, writing-review & editing.
Conflicts of interest
Lunzhi Dai is an Editorial Board Member of Ageing and Cancer Research & Treatment. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 325B2061), Science and Technology Project of Sichuan Province (Grant No. 2026NSFSC0924), and China Postdoctoral Science Foundation (Grant No. 2024M762204).
Copyright
© The Author(s) 2026.
References
-
1. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell. 2023;186(2):243-278.[DOI]
-
5. Palmer AK, Jensen MD. Metabolic changes in aging humans: Current evidence and therapeutic strategies. J Clin Invest. 2022;132(16):e158451.[DOI]
-
7. Sarver DC, Saqib M, Chen F, Wong GW. Mitochondrial respiration atlas reveals differential changes in mitochondrial function across sex and age. eLife. 2024;13:RP96926.[DOI]
-
8. Hu M, Zhu C, Sun R, Xu Z, Gong Y, Liu Y, et al. Low-input deep learning platform for citrullinated peptide identification, autoantigen discovery and rheumatoid arthritis treatment stratification. Nat Biomed Eng. 2026.[DOI]
-
10. Gu L, Zhu Y, Nandi SP, Lee M, Watari K, Bareng B, et al. FBP1 controls liver cancer evolution from senescent MASH hepatocytes. Nature. 2025;637(8045):461-469.[DOI]
-
17. Freed DM, Bessman NJ, Kiyatkin A, Salazar-Cavazos E, Byrne PO, Moore JO, et al. EGFR ligands differentially stabilize receptor dimers to specify signaling kinetics. Cell. 2017;171(3):683-695.[DOI]
-
21. Wang X, Luo Y, He S, Lu Y, Gong Y, Gao L, et al. Age-, sex- and proximal–distal-resolved multi-omics identifies regulators of intestinal aging in non-human primates. Nat Aging. 2024;4(3):414-433.[DOI]
-
24. Nie B, Gan W, Shi F, Hu GX, Chen LG, Hayakawa H, et al. Age-dependent accumulation of 8-oxoguanine in the DNA and RNA in various rat tissues. Oxid Med Cell Longev. 2013;2013:303181.[DOI]
-
28. Chibaya L, Karim B, Zhang H, Jones SN. Mdm2 phosphorylation by Akt regulates the p53 response to oxidative stress to promote cell proliferation and tumorigenesis. Proc Natl Acad Sci U S A. 2021;118(4):e2003193118.[DOI]
-
29. Zhou Z, Li X, Wang Y, Liang L, Wang C, Sun Y, et al. DNA-PK-mediated phosphorylation of STAT6 establishes a non-canonical type 2 immunity axis to prevent macrophage senescence. Nat Commun. 2026;17:3123.[DOI]
-
32. Tian Y, Feng T, Zhang J, Meng Q, Zhan W, Tang M, et al. Histone H1 deamidation facilitates chromatin relaxation for DNA repair. Nature. 2025;641(8063):779-787.[DOI]
-
38. Mo HY, Wang RB, Ma MY, Zhang Y, Li XY, Wen WR, et al. MTHFD2-mediated redox homeostasis promotes gastric cancer progression under hypoxic conditions. Redox Rep. 2024;29:2345455.[DOI]
-
41. Chen G, Bao B, Cheng Y, Tian M, Song J, Zheng L, et al. Acetyl-CoA metabolism as a therapeutic target for cancer. Biomed Pharmacother. 2023;168:115741.[DOI]
-
43. Omrani O, Krepelova A, Rasa SMM, Sirvinskas D, Lu J, Annunziata F, et al. IFNγ-Stat1 axis drives aging-associated loss of intestinal tissue homeostasis and regeneration. Nat Commun. 2023;14:6109.[DOI]
-
45. Zabransky DJ, Chhabra Y, Fane ME, Kartalia E, Leatherman JM, Hüser L, et al. Fibroblasts in the aged pancreas drive pancreatic cancer progression. Cancer Res. 2024;84(8):1221-1236.[DOI]
-
46. Marchelletta RR, Krishnan M, Spalinger MR, Placone TW, Alvarez R, Sayoc-Becerra A, et al. T cell protein tyrosine phosphatase protects intestinal barrier function by restricting epithelial tight junction remodeling. J Clin Invest. 2021;131(17):e138230.[DOI]
-
48. O’Keefe ME, Kondolf HC, De Santis S, Pizarro TT, Abbott DW. Restraint of inflammasome-driven cytokine responses through the mRNA stability protein TTP. Cell Rep. 2025;44(3):115340.[DOI]
-
53. Skrupskelyte G, Rojo Arias JE, Ajith H, Dang Y, Rossetti D, Han S, et al. Precancerous niche remodelling dictates nascent tumour persistence. Nature. 2026;653(8113):242-253.[DOI]
-
57. Li X, Gong W, Wang H, Li T, Attri KS, Lewis RE, et al. O-GlcNAc transferase suppresses inflammation and necroptosis by targeting receptor-interacting serine/threonine-protein kinase 3. Immunity. 2019;50(3):576-590.[DOI]
-
58. Alicea GM, Patel P, Portuallo ME, Fane ME, Wei M, Chhabra Y, et al. Age-related increases in IGFBP2 increase melanoma cell invasion and lipid synthesis. Cancer Res Commun. 2024;4(8):1908-1918.[DOI]
-
60. Sampson LL, Davis AK, Grogg MW, Zheng Y. mTOR disruption causes intestinal epithelial cell defects and intestinal atrophy postinjury in mice. FASEB J. 2016;30(3):1263-1275.[DOI]
-
62. Wang Y, Wang P, Xu J. Phosphorylation: A fast switch for checkpoint signaling. Adv Exp Med Biol. 2020;1248:347-398.[DOI]
-
65. Houles T, Yoon SO, Roux PP. The expanding landscape of canonical and non-canonical protein phosphorylation. Trends Biochem Sci. 2024;49(11):986-999.[DOI]
-
67. Zhuang N, Li L, Chen S, Wang T. PINK1-dependent phosphorylation of PINK1 and Parkin is essential for mitochondrial quality control. Cell Death Dis. 2016;7(12):e2501.[DOI]
-
68. Yang R, Zhan Y, Deng Z, Zhang J, Chen S, Zhang Y, et al. Arylsulfatase K attenuates airway epithelial cell senescence in COPD by regulating parkin-mediated mitophagy. Redox Biol. 2025;86:103793.[DOI]
-
71. Fu K, Cheung AHK, Wong CC, Liu W, Zhou Y, Wang F, et al. Streptococcus anginosus promotes gastric inflammation, atrophy, and tumorigenesis in mice. Cell. 2024;187(4):882-896.[DOI]
-
72. Huang YH, Chen HK, Hsu YF, Chen HC, Chuang CH, Huang SW, et al. Src-FAK signaling mediates interleukin 6-induced HCT116 colorectal cancer epithelial–mesenchymal transition. Int J Mol Sci. 2023;24(7):6650.[DOI]
-
75. Gu Y, Xu T, Fang Y, Shao J, Hu T, Wu X, et al. CBX4 counteracts cellular senescence to desensitize gastric cancer cells to chemotherapy by inducing YAP1 SUMOylation. Drug Resist Updat. 2024;77:101136.[DOI]
-
80. Gong Y, Zhan H, Wei N, Liu M, Liu Y, Guan P, et al. Acetylation profiling by Iseq-Kac reveals insights into HSC aging and lineage decision. Nat Chem Biol. 2025;21(11):1675-1687.[DOI]
-
82. Murthy D, Attri KS, Shukla SK, Thakur R, Chaika NV, He C, et al. Cancer-associated fibroblast-derived acetate promotes pancreatic cancer development by altering polyamine metabolism via the ACSS2–SP1–SAT1 axis. Nat Cell Biol. 2024;26(4):613-627.[DOI]
-
84. Chen S, Yang H, Hu Z, Jin J, Xiong X, Zhang Z, et al. Deacetylation by SIRT6 increases the stability of GILZ to suppress NSCLC cell migration and invasion. Cell Signal. 2024;124:111414.[DOI]
-
94. Xue J, Cao Z, Cheng Y, Wang J, Liu Y, Yang R, et al. Acetylation of alpha-fetoprotein promotes hepatocellular carcinoma progression. Cancer Lett. 2020;471:12-26.[DOI]
-
95. Wang B, Ye Y, Yang X, Liu B, Wang Z, Chen S, et al. SIRT2-dependent IDH1 deacetylation inhibits colorectal cancer and liver metastases. EMBO Rep. 2020;21(4):e48183.[DOI]
-
105. Liao Y, Zhang W, Liu Y, Zhu C, Zou Z. The role of ubiquitination in health and disease. MedComm. 2024;5(10):e736.[DOI]
-
107. Xu M, Feng P, Yan J, Li L. Mitochondrial quality control: A pathophysiological mechanism and potential therapeutic target for chronic obstructive pulmonary disease. Front Pharmacol. 2025;15:1474310.[DOI]
-
108. Madrigal-Matute J, De Bruijn J, Van Kuijk K, Riascos-Bernal DF, Diaz A, Tasset I, et al. Protective role of chaperone-mediated autophagy against atherosclerosis. Proc Natl Acad Sci U S A. 2022;119(14):e2121133119.[DOI]
-
109. Wei J, Cao Z, Li Q, Li X, Wang Q, Zhang Y, et al. Nuclear ubiquitination permits Hippo–YAP signal for liver development and tumorigenesis. Nat Chem Biol. 2025;21(10):1565-1576.[DOI]
-
111. Kar R, Jha SK, Ojha S, Sharma A, Dholpuria S, Raju VSR, et al. The FBXW7-NOTCH interactome: A ubiquitin proteasomal system-induced crosstalk modulating oncogenic transformation in human tissues. Cancer Rep. 2021;4(4):e1369.[DOI]
-
120. He M, Zhou X, Wang X. Glycosylation: Mechanisms, biological functions and clinical implications. Sig Transduct Target Ther. 2024;9:194.[DOI]
-
123. Mody AC, Ramirez DH, Woo CM. Targeted protein O-GlcNAc reveals transcriptional functions for O-GlcNAc. Cell Chem Biol. 2025;32(12):1486-1502.[DOI]
-
127. Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632.[DOI]
-
129. Wang H, Sun J, Sun H, Wang Y, Lin B, Wu L, et al. The OGT–c-Myc–PDK2 axis rewires the TCA cycle and promotes colorectal tumor growth. Cell Death Differ. 2024;31(9):1157-1169.[DOI]
-
130. Zhao Q, Zhou S, Lou W, Qian H, Xu Z. Crosstalk between O-GlcNAcylation and phosphorylation in metabolism: Regulation and mechanism. Cell Death Differ. 2025;32(7):1181-1199.[DOI]
-
133. Chen LH, Wei JP, Li MD, Lu X, Ma YC, Wang Y, et al. AhR-mediated histone lactylation drives cellular senescence during benzo[a]pyrene-evoked chronic obstructive pulmonary disease. J Hazard Mater. 2025;495:139083.[DOI]
-
134. Li L, Dong J, Xu C, Wang S. Lactate drives senescence-resistant lineages in hepatocellular carcinoma via histone H2B lactylation of NDRG1. Cancer Lett. 2025;616:217567.[DOI]
-
137. Li A, Gong Z, Long Y, Li Y, Liu C, Lu X, et al. Lactylation of LSD1 is an acquired epigenetic vulnerability of BRAFi/MEKi-resistant melanoma. Dev Cell. 2025;60(14):1974-1990.[DOI]
-
139. Liang L, Zong Y, Huang J, Chen Y, Xu N, Yan P, et al. Lactylation stabilizes PD-L1 to promote tumor immune evasion and cell growth. Cell Death Dis. 2026;17:335.[DOI]
-
141. Wang X, Liu X, Xiao R, Fang Y, Zhou F, Gu M, et al. Histone lactylation dynamics: Unlocking the triad of metabolism, epigenetics, and immune regulation in metastatic cascade of pancreatic cancer. Cancer Lett. 2024;598:217117.[DOI]
-
143. Wang Y, Du G, Zhang J, Zhai N, Liu W, Cao G, et al. Mitochondrial PDHA1 acetylation orchestrates lactate-dependent epigenetic reprogramming to promote fibrosis via NUAK2. Cell Mol Life Sci. 2026;83:198.[DOI]
-
144. An D, Kim J, Moon B, Kim H, Nguyen H, Park S, et al. PRMT1-mediated methylation regulates MLL2 stability and gene expression. Nucleic Acids Res. 2025;53(4):gkae1227.[DOI]
-
151. Wang N, Pachai MR, Li D, Lee CJ, Warda S, Khudoynazarova MN, et al. Loss of Kmt2c or Kmt2d primes urothelium for tumorigenesis and redistributes KMT2A–menin to bivalent promoters. Nat Genet. 2025;57(1):165-179.[DOI]
-
152. Ge S, Zhang Q, Tian Y, Hao L, Duan J, Zhang B. Cell metabolic profiling of colorectal cancer via 1H NMR. Clin Chim Acta. 2020;510:291-297.[DOI]
-
154. Yang F, Fan G, Cao J, Zhao Q, Guo K, Liu M, et al. Cancer-specific bivalent promoters featuring low-level H3K27me3 signals favor active transcription and govern the cancer cell state transition. Acta Biochim Biophys Sin. 2026;58(5):989-1007.[DOI]
-
161. Meng J. An emerging role for cysteine-mediated redox signaling in aging. Redox Biol. 2025;86:103852.[DOI]
-
162. Yang X, Rocks JW, Jiang K, Walters AJ, Rai K, Liu J, et al. Engineering synthetic phosphorylation signaling networks in human cells. Science. 2025;387(6729):74-81.[DOI]
-
165. Li X, Huang Y, Zu D, Liu H, He H, Bao Q, et al. PMMA nanoplastics induce gastric epithelial cellular senescence and cGAS-STING-mediated inflammation via ROS overproduction and NHEJ suppression. Ecotoxicol Environ Saf. 2024;287:117284.[DOI]
-
174. Guo Y, Li J, Zhang K. Crotonylation modification and its role in diseases. Front Mol Biosci. 2024;11:1492212.[DOI]
-
179. Bruno PS, Arshad A, Gogu MR, Waterman N, Flack R, Dunn K, et al. Post-translational modifications of proteins orchestrate all hallmarks of cancer. Life. 2025;15(1):126.[DOI]
-
182. Barthélemy NR, Saef B, Li Y, Gordon BA, He Y, Horie K, et al. CSF tau phosphorylation occupancies at T217 and T205 represent improved biomarkers of amyloid and tau pathology in Alzheimer’s disease. Nat Aging. 2023;3(4):391-401.[DOI]
-
185. Zhang X, Maity T, Kashyap MK, Bansal M, Venugopalan A, Singh S, et al. Quantitative tyrosine phosphoproteomics of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor-treated lung adenocarcinoma cells reveals potential novel biomarkers of therapeutic response. Mol Cell Proteomics. 2017;16(5):891-910.
-
187. Planchard D, Jänne PA, Cheng Y, Yang JC, Yanagitani N, Kim SW, et al. Osimertinib with or without chemotherapy in EGFR-mutated advanced NSCLC. N Engl J Med. 2023;389(21):1935-1948.[DOI]
-
188. Sonneveld P, Dimopoulos MA, Boccadoro M, Quach H, Ho PJ, Beksac M, et al. Daratumumab, bortezomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2024;390(4):301-313.[DOI]
-
191. Hong Y, Fu Y, Long Q. Post-translational governance of NF-κB in cancer immunity: Mechanisms and therapeutic horizons. Front Immunol. 2025;16:1627084.[DOI]
-
192. Prus G, Satpathy S, Weinert BT, Narita T, Choudhary C. Global, site-resolved analysis of ubiquitylation occupancy and turnover rate reveals systems properties. Cell. 2024;187(11):2875-2892.[DOI]
-
193. Miao C, Huang Y, Zhang C, Wang X, Wang B, Zhou X, et al. Post-translational modifications in drug resistance. Drug Resist Updat. 2025;78:101173.[DOI]
-
194. Haines M, Thorup JR, Gohsman S, Ctortecka C, Newton C, Rohrer DC, et al. High-throughput proteomic and phosphoproteomic analysis of formalin-fixed paraffin-embedded tissues. Mol Cell Proteom. 2025;24(9):101044.[DOI]
-
196. Pickering C, Aiyetan P, Xu G, Mitchell A, Rice R, Najjar YG, et al. Plasma glycoproteomic biomarkers identify metastatic melanoma patients with reduced clinical benefit from immune checkpoint inhibitor therapy. Front Immunol. 2023;14:1187332.[DOI]
-
199. Zhang H, Yan Q, Jiang S, Hu D, Lu P, Li S, et al. Protein post-translational modifications and tumor immunity: A pan-cancer perspective. Phys Life Rev. 2025;55:142-209.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Zhang R, Fu Y, Chen H, Yu J, Li Y, Hu M, et al. Protein post-translational modifications in aging and cancer: Mechanisms and translational implications. Ageing Cancer Res Treat. 2026;3:202619. https://doi.org/10.70401/acrt.2026.0023
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. PTMs Linking Aging and Cancer
- 3. Major PTM Systems Across the Aging-Cancer Continuum
- 4. Translational Implications
- 5. Perspective
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Zhang R, Fu Y, Chen H, Yu J, Li Y, Hu M, et al. Protein post-translational modifications in aging and cancer: Mechanisms and translational implications. Ageing Cancer Res Treat. 2026;3:202619. https://doi.org/10.70401/acrt.2026.0023
copy
Share Link
copy