Frailty and tertiary lymphoid structures in immunosenescence: Emerging associations, mechanistic clues and future perspectives
Zeyang Lin
1,2
,
Jiatong Chen
1
,
Sihui Liu
3
,
Shaodi Wen
2,*

,
Liangjie Zheng
1,*
*Correspondence to:
Shaodi Wen, Department of Oncology, Jiangsu Cancer Hospital & The Affiliated Cancer Hospital of Nanjing Medical University & Jiangsu Institute of Cancer Research, Nanjing 210009, Jiangsu, China.
E-mail: wenshaodi@njmu.edu.cn
Liangjie Zheng, Department of Anaesthesiology, Shantou Central Hospital, Shantou 515031, Guangdong, China. E-mail: 842709740@163.com
Liangjie Zheng, Department of Anaesthesiology, Shantou Central Hospital, Shantou 515031, Guangdong, China. E-mail: 842709740@163.com
Ageing Cancer Res Treat. 2026;3:202606. 10.70401/acrt.2026.0024
Received: February 02, 2026Accepted: June 09, 2026Published: June 09, 2026
Abstract
Frailty, a geriatric syndrome marked by multisystem functional decline and heightened vulnerability, threatens older adults’ health span. Immunosenescence and chronic low-grade inflammation are increasingly recognized as core drivers, with age-related impairments in cellular, humoral, and innate immunity disrupting immune homeostasis. Tertiary lymphoid structures (TLS), ectopic immune aggregates that orchestrate local immune responses, have emerged as candidate regulators in chronic disease, yet their role in frailty remains largely unexplored. Here, we propose a conceptual framework for frailty pathogenesis in which immunosenescence and chronic low-grade inflammation (inflammaging) may act as upstream biological drivers associated with systemic lymphatic dysfunction, potentially contributing to the structural and functional dysregulation of TLS, a candidate intermediate tissue-level histological link. TLS dysfunction may subsequently contribute to multi-organ functional decline and the progression of frailty. We synthesize current evidence on the dynamic characteristics of TLS in aging, discuss TLS as candidate biomarkers and putative therapeutic targets for frailty, and highlight intervention strategies including molecular modulation, lymphatic function enhancement, immunotherapy, and lifestyle modifications. This tissue-level perspective highlights TLS as a potential histological link connecting immunosenescence, lymphatic dysfunction, and frailty, offering novel avenues for geriatric precision medicine and the development of immune-targeted interventions to delay frailty progression.
Keywords
Frailty, immune aging, tertiary lymphoid structure, chronic inflammation, immune regulation
1. Introduction
Frailty is a prevalent geriatric syndrome characterized by a progressive decline in physiological reserves and reduced resilience to internal and external stressors, leading to increased vulnerability to adverse health outcomes[1]. With the rapid aging of the global population, frailty has emerged as a major determinant of health span and quality of life in older adults. Epidemiological studies estimate that approximately 10-15% of community-dwelling older adults are frail, while more than 30% are in a pre-frail state, with comparable prevalence observed across Western and Asian populations[2,3]. Clinically, frailty is strongly associated with increased risks of falls, infections, hospitalization, and mortality, highlighting its growing clinical and public health significance[4-6].
With the increasing understanding of the mechanisms underlying frailty, accumulating evidence indicates that immunosenescence and chronic low-grade inflammation (inflammaging) constitute key biological foundations for the onset and progression of frailty[7-10]. In the context of immunosenescence, the immune system gradually loses its capacity for regulation and repair, thereby contributing to the development of frailty. The immune system enters a continuous physiological aging trajectory from early life, among which thymic involution represents the earliest and irreversible hallmark[11]. Following puberty, the thymus undergoes rapid atrophy and progressive adipose replacement, leading to a marked decline in the production of naïve T cells[12]. Concurrently, peripheral lymphoid organs such as lymph nodes and the spleen exhibit irreversible structural and functional alterations, including follicular disorganization, reduction of high endothelial venules (HEVs), and decreased immune cell diversity[13]. Collectively, thymic involution, impaired immune cell renewal, and remodeling of immune regulatory networks constitute the core features of immunosenescence, resulting in a progressively diminished capacity to mount effective immune responses against novel antigens. Meanwhile, older individuals commonly exhibit persistent, low-grade elevation of pro-inflammatory mediators, giving rise to a state referred to as inflammaging[14]. Immunosenescence and chronic inflammation reinforce each other and are widely regarded as central biological drivers of frailty development and progression, potentially influencing the formation, maturation, and functional stability of tertiary lymphoid structures (TLS)[15,16]. Therefore, elucidating the mechanisms of immune aging is essential for a comprehensive understanding of frailty pathogenesis.
In this context, TLS have gained increasing attention as tissue-level regulators of local immune responses. TLS are ectopic immune aggregates that form in non-lymphoid tissues under conditions of chronic inflammation or persistent antigenic stimulation and share structural and functional similarities with secondary lymphoid organs (SLOs). By supporting localized immune cell recruitment, antigen presentation, and immune regulation, TLS play important roles in chronic inflammatory diseases, infections, and cancer[17,18].
Aging and frailty are accompanied by structural and functional deterioration of the lymphatic and immune systems[19,20], including impaired lymphatic drainage[21], altered immune cell trafficking[22], and disrupted stromal–immune interactions[20]. However, despite growing interest in TLS biology, their potential involvement in immunosenescence-associated frailty remains poorly understood. Cancer and frailty frequently coexist in older adults and share common biological drivers, including immunosenescence, chronic inflammation, and lymphoid tissue remodeling. Frailty, characterized by multi-organ functional decline and increased vulnerability, is a prominent risk factor for poor outcomes in cancer patients, including compromised response to treatment, increased postoperative complications, and higher mortality rates[23]. Similarly, frailty is driven by a combination of age-related immune dysfunction and chronic inflammation (often referred to as inflammaging), which undermines the body’s ability to recover from stressors, including infections, surgical procedures, and cancer treatment[8].
TLS, initially recognized in the context of cancer, are increasingly being investigated as potential contributors to local immune remodeling and age-related immune dysfunction associated with frailty[24]. In cancer, TLS provide crucial support for anti-tumor immunity by fostering the organization of local immune responses, antigen presentation, and immune cell activation[25]. These structures are formed under conditions of chronic inflammation, often driven by tumor progression or treatment-related inflammation. However, similar TLS or TLS-like structures have also been observed in organs affected by frailty, such as the lung, brain, and skeletal muscle, where they may contribute to ongoing inflammatory processes and tissue remodeling, a parallel that highlights TLS as a conserved immune feature across cancer and aging, albeit with distinct functional consequences[26].
Thus, cancer-associated TLS offer a valuable mechanistic model for understanding the role of TLS in frailty. Both conditions are characterized by an immune microenvironment that shifts from a protective immune response to a chronic, unresolved inflammatory state, driven by dysfunctional TLS. While TLS may initially function to orchestrate immune responses, their persistence in aging and frailty-related tissues may exacerbate chronic inflammation, further accelerating frailty progression[27]. This shared immune landscape highlights the potential of TLS as a candidate biomarker and therapeutic target for frailty, with implications for improving immune regulation and clinical outcomes in frail cancer patients[28].
Such age-related changes may influence the formation, maturation, and functional stability of TLS, potentially shifting their role from effective immune coordination toward persistent, unresolved inflammation[16,29]. Elucidating how immunosenescence and lymphatic dysfunction affect TLS may therefore provide novel insights into the immune mechanisms of frailty[30].
In this review, we propose a conceptual framework in which immunosenescence and inflammaging may act as upstream biological processes associated with progressive lymphatic dysfunction, potentially contributing to structural and functional alterations of TLS, a candidate intermediate tissue-level immune niche.
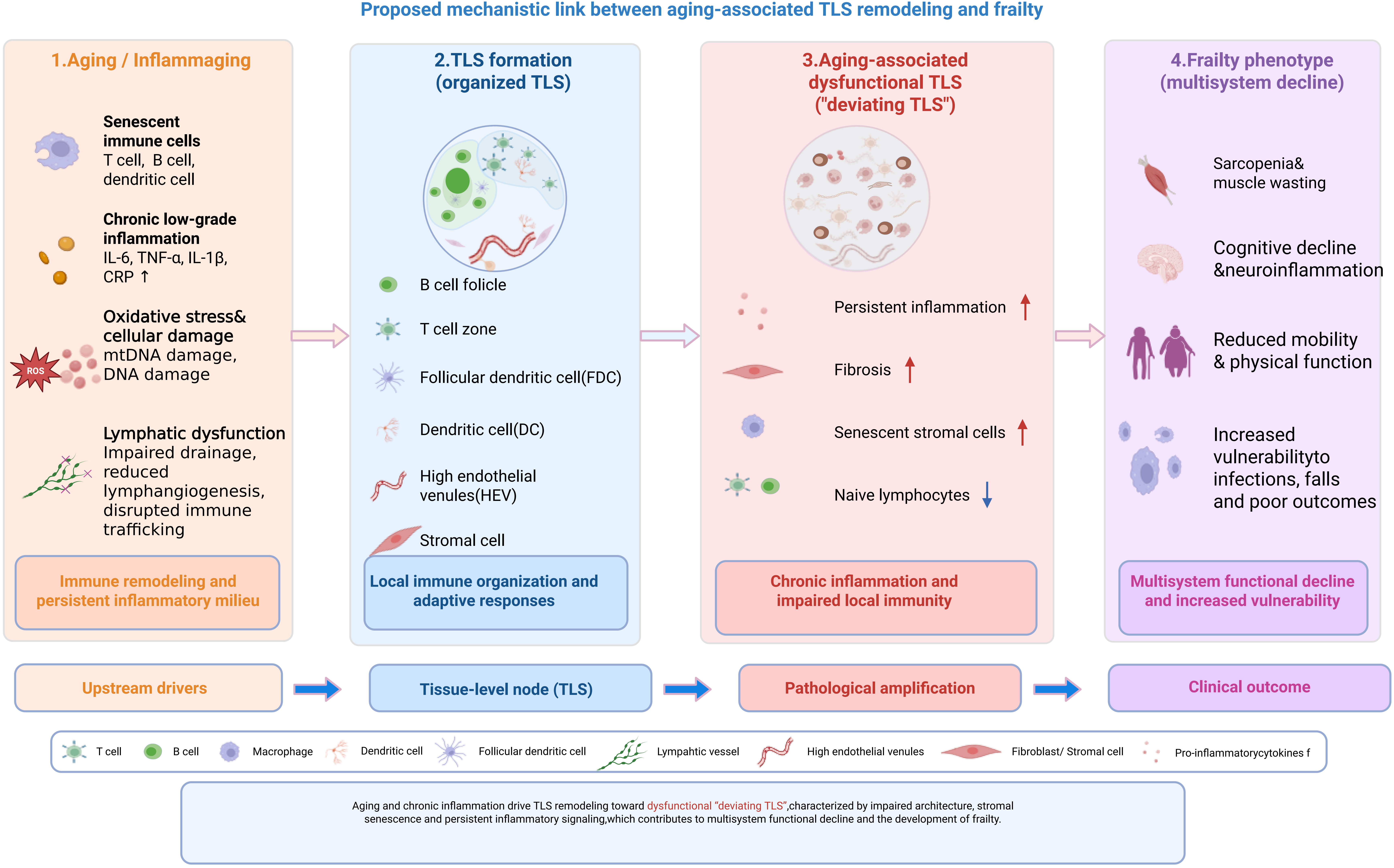
TLS alterations may further contribute to multi-organ functional decline and frailty progression as a downstream clinical phenotype. Within this conceptual framework, we synthesize current knowledge on frailty, immunosenescence, and TLS biology, examine emerging evidence linking age-related TLS remodeling with frailty-associated immune dysfunction[31-33], and discuss the exploratory translational relevance of TLS as potential biomarkers of age-associated immune remodeling and as putative targets for future frailty-related intervention research, largely extrapolated from chronic inflammatory and oncology-associated TLS studies[34]. A schematic overview of this core mechanistic framework is summarized in Figure 1.
{kind=link}
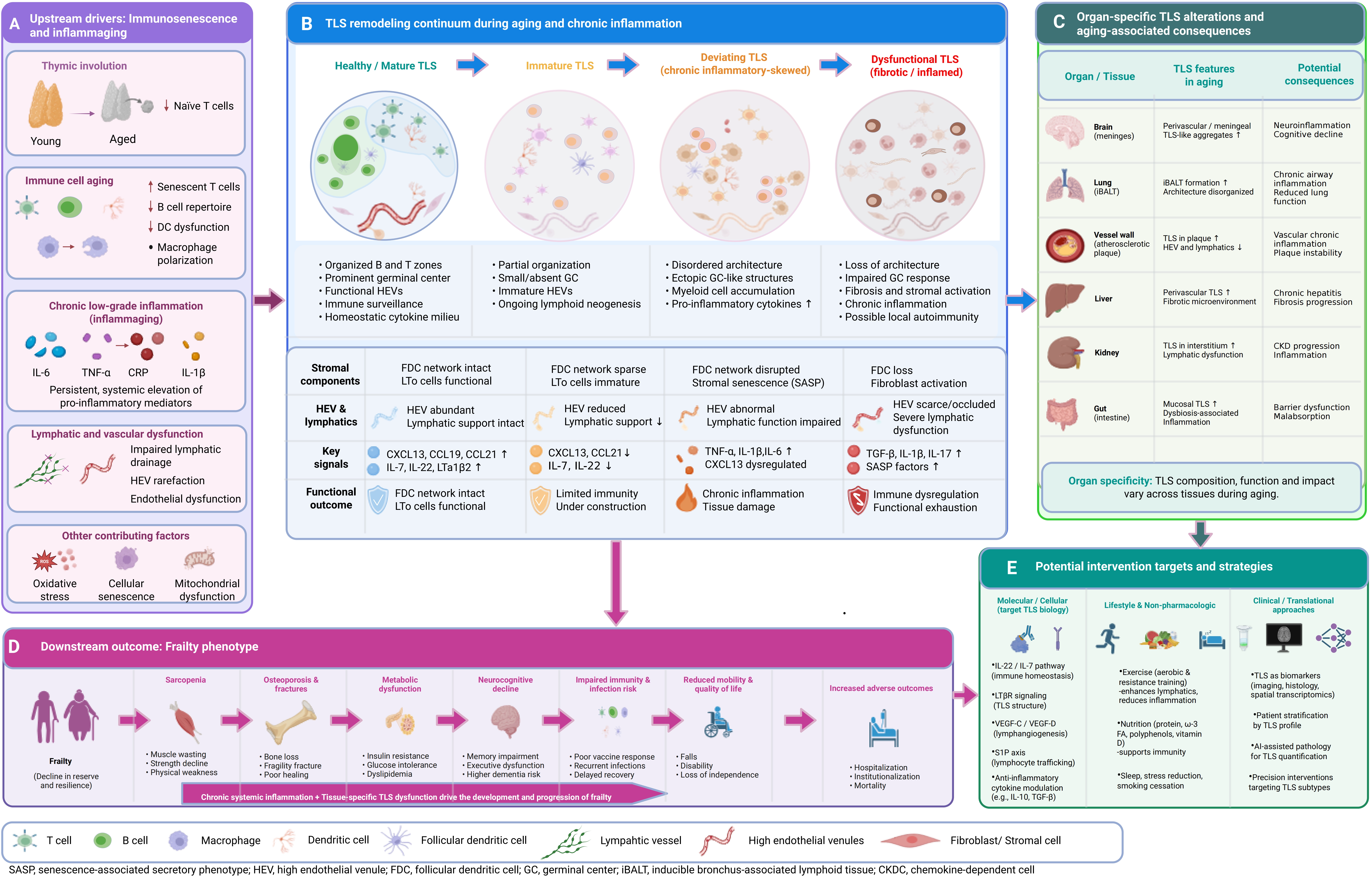
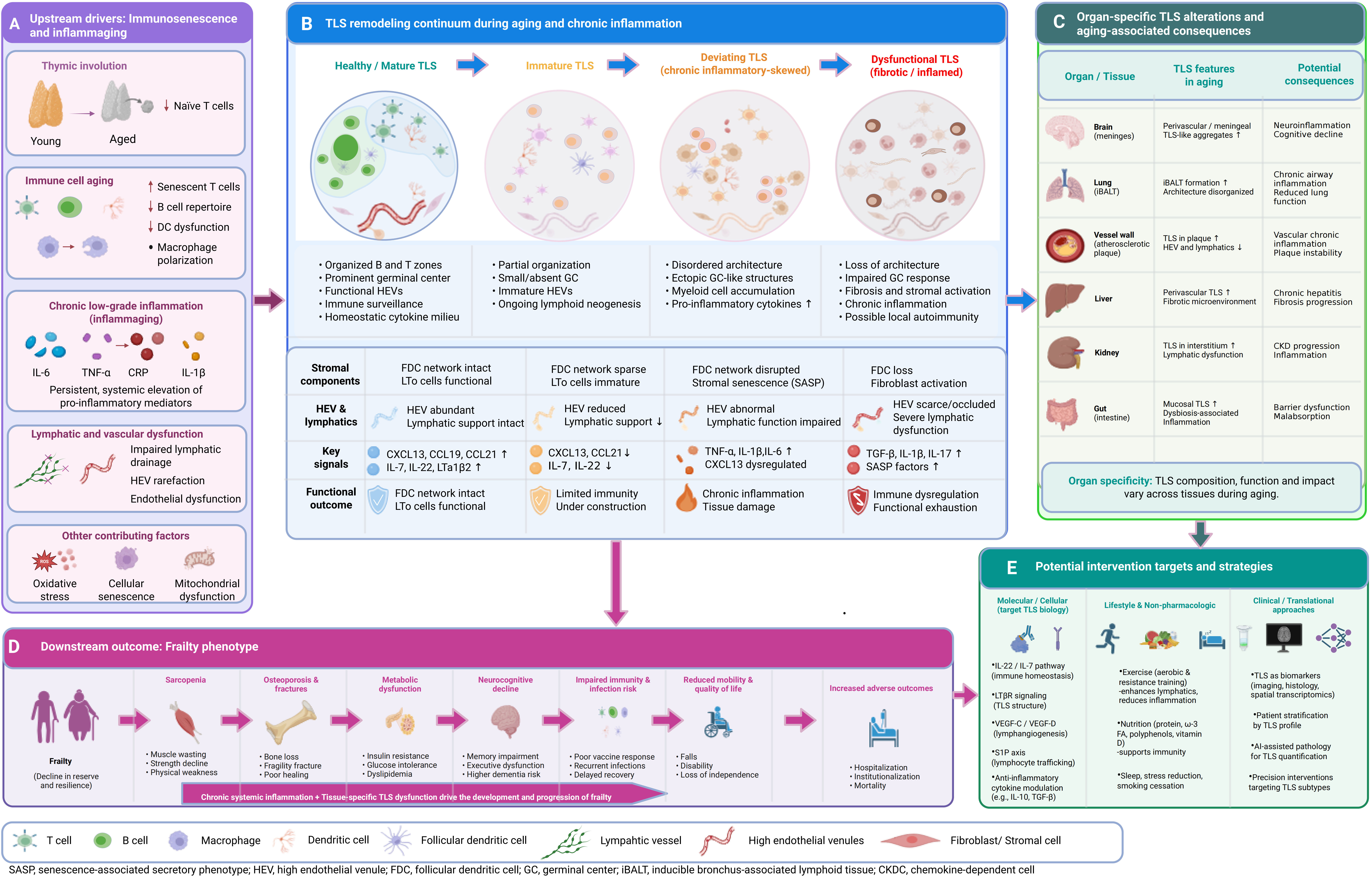
Figure 1. Schematic overview of the core mechanistic framework linking immunosenescence, TLS dysfunction, and frailty progression. Upstream immunosenescence and chronic low-grade inflammation (inflammaging) drive systemic lymphatic dysfunction, which further leads to the transition from healthy/mature TLS to age-related deviating TLS (dysfunctional TLS). Mature TLS are characterized by organized T/B cell zones, functional GCs, and effective immune regulation. In contrast, deviating TLS exhibit architectural disorganization, loss of T/B cell compartmentalization, impaired GC reactions, and sustained inflammatory signaling. TLS dysfunction reinforces a chronic immune-dysregulated milieu, exacerbating multi-organ functional decline and ultimately promoting frailty progression. Key intervention targets that can modulate TLS formation and function are indicated, including LTβR, CXCL13/CCL19, VEGF-C/VEGFR-3, and IL-22/STAT3. Created in BioRender. Lin, Z. (2026) https://BioRender.com/5p1wn9z. TLS: tertiary lymphoid structures; GCs: germinal centers.
Of note, aging and circadian rhythm disturbances can modulate tumor immunity and tissue inflammation through immune cell function and metabolic reprogramming, which may indirectly influence TLS formation and frailty progression[35]. Aging globally impairs cellular homeostasis, including proteostasis, mitochondrial function, and stress resistance, which collectively shape immune cell fitness and TLS structural integrity[36].
2. Definition and Clinical Features of Frailty
2.1 Definition of frailty
Frailty describes a multidimensional clinical syndrome resulting from the cumulative decline of multiple physiological systems, leading to diminished functional reserve and increased vulnerability to stressors. Unlike single-organ diseases, frailty reflects a state in which biological aging exceeds chronological aging, manifesting as reduced resilience, delayed recovery, and heightened susceptibility to adverse health outcomes. This systemic nature distinguishes frailty as a dynamic and potentially reversible condition rather than an inevitable consequence of aging.
Currently, the commonly used clinical frailty assessment models can be classified into two major frameworks: the Fried Phenotype Model and the frailty index (FI) Model. The Fried model primarily evaluates physiological phenotypic decline, whereas the FI focuses on the accumulation of multi-system deficits (Table 1).
Table 1. Comparison of frailty assessment models[37].
| Feature | Fried Phenotype Model[1] | Frailty Index Model (Rockwood)[38] |
| Theoretical Basis | Physiological phenotypic decline | Accumulation of multi-system deficits |
| Assessment Domains | Physical performance, activity, energy metabolism | Diseases, symptoms, functional impairments, laboratory parameters |
| Representative Indicators | Weight loss, grip strength, gait speed, fatigue | Deficit proportion (FI) |
| Variable Type | Categorical (frail/pre-frail/robust) | Continuous (0-1) |
| Advantages | Simple, easy to use, clinically intuitive | Comprehensive, systematic, quantifiable |
| Limitations | Subjective, overlooks cognitive and social factors | Computationally complex, heavily dependent on chosen indicators |
| Primary Applications | Clinical screening, phenotypic studies | Cohort follow-up, risk prediction |
FI: frailty index.
The Fried Phenotype Model was proposed by Fried et al. in 2001 and defines frailty based on five objective criteria: unintentional weight loss, decreased grip strength, fatigue, slowed gait speed, and reduced physical activity[1]. Individuals meeting three or more criteria are classified as “frail”, while those meeting one or two criteria are considered “pre-frail”. This model emphasizes declines in physical function and strength, is simple to use, and has been widely applied in community-based screening of older adults. However, it primarily reflects physical function deterioration and does not comprehensively capture cognitive, psychological, or social support dimensions.
The FI, proposed by Rockwood and Mitnitski, quantifies the accumulation of deficits across multiple health domains, including diseases, symptoms, laboratory abnormalities, and functional impairments, providing a more comprehensive reflection of the physiological complexity and systemic nature of frailty[38]. The FI is calculated by counting the number of health deficits present in an individual and dividing by the total number of potential deficits: FI = number of health deficits present/total number of deficit items. Generally, an FI ≥ 0.25 is considered “frail”, 0.10-0.25 as “pre-frail”, and FI < 0.10 as “robust” or “healthy”. Unlike the categorical Fried Phenotype Model, the FI is a continuous variable that better reflects gradation and multidimensionality of frailty and is suitable for longitudinal studies and health outcome prediction[38].
Overall, the Fried model emphasizes identification of physiological phenotypes focusing on external manifestations such as muscle strength, weight, and physical performance changes[1], whereas the Rockwood model reflects quantitative accumulation of multi-system deficits and is more suitable for predicting long-term outcomes such as mortality, readmission, and functional decline. Together, these two models complement each other, forming the theoretical framework of modern frailty research and providing practical assessment tools for geriatric medicine, cardiology, and immunology[37].
2.2 Clinical characteristics of frailty
The clinical characteristics of frailty extend well beyond physical decline and are closely associated with dysregulation of immune, inflammatory, endocrine, and metabolic systems, underscoring its systemic, multi-organ nature. The major clinical features of frailty can be summarized as follows.
2.2.1 Decline in physical strength and motor function
Reduced physical strength and impaired exercise capacity are hallmark features of frailty. Sarcopenia is widely recognized as one of the structural foundations of frailty. The European Working Group on Sarcopenia in Older People defines sarcopenia by the concurrent presence of reduced muscle mass, decreased muscle strength, and impaired physical performance, all of which constitute core components of frailty[39]. Loss of skeletal muscle mass results in diminished energy metabolism, slowed gait speed, and an increased risk of falls. In addition, decreased bone mineral density and osteoporosis frequently coexist in frail individuals. Khosla and Riggs demonstrated that reduced bone remodeling activity, closely associated with estrogen and testosterone deficiency, represents a key mechanism underlying frailty-related skeletal fragility[40].
2.2.2 Chronic low-grade inflammatory state
Chronic low-grade inflammation is a central biological hallmark of frailty. Franceschi and Campisi introduced the concept of “inflammaging”, describing aging as a state of persistent, low-level immune activation characterized by elevated inflammatory mediators in the absence of overt infection[7]. Systematic reviews and meta-analyses have consistently shown that elevated serum levels of C-reactive protein (CRP), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) are significantly associated with an increased risk of frailty[10]. This chronic inflammatory milieu not only impairs muscle and endothelial function but also promotes cellular senescence and oxidative stress through activation of the nuclear factor-kappa B (NF-κB) signaling pathway, thereby establishing a self-perpetuating cycle of inflammation and tissue degeneration.
2.2.3 Endocrine and metabolic dysfunction
Frailty is also closely associated with declines in anabolic hormone levels. In the InCHIANTI cohort study, Maggio et al. reported that reduced levels of testosterone, dehydroepiandrosterone, and insulin-like growth factor-1 (IGF-1) were significantly correlated with frailty severity in older men[41]. Similar associations have been observed in women, in whom deficiencies in anabolic hormones, including estrogen and IGF-1, accelerate muscle atrophy and disrupt metabolic homeostasis[42]. Hormonal decline leads to reduced protein synthesis, increased skeletal muscle apoptosis, and impaired metabolic responsiveness, ultimately weakening physiological resilience. In addition, insulin resistance and mitochondrial dysfunction contribute to the metabolic pathology of frailty by reducing energy production efficiency and enhancing oxidative stress.
2.2.4 Oxidative stress and cellular damage
Oxidative stress represents a critical molecular mechanism in the progression of frailty. Evidence indicates that frail individuals exhibit elevated serum levels of reactive oxygen species and lipid peroxidation products, accompanied by reduced activity of antioxidant enzymes such as glutathione peroxidase[43]. Accumulated oxidative stress damages cellular membranes and mitochondrial structures and promotes cellular senescence through DNA damage and protein carbonylation. Accumulated oxidative stress is not only a key molecular event in frailty, but also a core driver of cellular senescence and systemic aging. Oxidative stress-induced DNA damage and protein carbonylation can directly trigger cellular senescence, and the senescence-associated secretory phenotype (SASP) further amplifies oxidative stress and chronic low-grade inflammation, forming a self-perpetuating vicious cycle that accelerates aging and frailty progression[44]. Ji reported that moderate physical activity improves redox homeostasis in older adults, whereas excessive oxidative stress exacerbates muscle wasting and functional decline[45]. Accordingly, oxidative damage is considered a key link connecting metabolic dysregulation with tissue degeneration during frailty. Recent studies have further clarified the crosstalk between oxidative stress, cellular senescence, and age-related functional decline, supporting its central role in frailty pathogenesis.
2.2.5 Neurological and psychological manifestations
Frailty is not limited to physiological decline but is frequently accompanied by cognitive and emotional impairments. Buchman et al. demonstrated that frailty is an independent predictor of Alzheimer’s disease and cognitive decline, potentially mediated by chronic inflammation, impaired neuronal energy metabolism, and cerebrovascular dysfunction[46]. Depressive symptoms are also highly prevalent among frail individuals, with a bidirectional relationship likely driven by hyperactivation of the hypothalamic–pituitary–adrenal axis, elevated inflammatory mediators, and mitochondrial dysfunction[47]. Cognitive impairment and depression further exacerbate functional dependence and reduce adherence to exercise and nutritional interventions, thereby accelerating frailty progression.
Overall, frailty is a complex, multisystem syndrome characterized by dynamic interactions among chronic low-grade inflammation, hormonal decline, oxidative stress, sarcopenia, and cognitive–emotional disturbances. These factors collectively form a biological cascade linking metabolic dysregulation, inflammatory activation, and progressive functional decline. Such multisystem involvement underscores frailty as a global failure of physiological integration rather than isolated organ dysfunction. Understanding these clinical features provides the necessary framework for exploring immunological mechanisms of frailty and their relationship with immune aging and TLS dysfunction, which are discussed in subsequent sections.
3. Relationship Between Immunosenescence and Frailty
Immunosenescence refers to the progressive, age-associated remodeling of the immune system, characterized by reduced immune responsiveness, impaired immune regulation, and persistent low-grade inflammation. Rather than representing uniform decline in immune activity, immunosenescence is marked by the paradoxical coexistence of weakened adaptive immune responses and sustained inflammatory signaling. This immune imbalance is increasingly recognized as a central mechanism underlying frailty and its associated vulnerability to stressors[8,14].
Decline in cellular immunity is a hallmark of immunosenescence. Age-related thymic involution markedly reduces naïve T cells output, resulting in a peripheral T-cell pool dominated by memory T cells and reduced capacity to respond to novel antigens[11,48]. Although CD8+ T cells may increase numerically with age, their function is compromised, with reduced cytotoxicity, lower cytokine production, clonal expansions, and decreased T-cell receptor diversity. Dysregulation of CD4+ helper T-cell subsets, including altered Th1/Th2 balance and increased proportion of regulatory T cells (Tregs), further suppresses effective immune responses[49,50]. Studies have shown that T cells from frail individuals exhibit shortened telomeres, reduced proliferative capacity, and impaired metabolic activity, collectively contributing to functional immune aging.
Humoral immunity is also adversely affected in frailty. B-cell development and differentiation are impaired, leading to reduced numbers of mature B cells and diminished antibody production, which results in blunted antigen responses and decreased vaccine efficacy[51,52]. Older individuals exhibit reduced plasma cell generation, lower antibody affinity, and shorter maintenance of immunological memory, increasing susceptibility to recurrent infections. Abnormal distributions of IgM and IgG subclasses have been reported in frail individuals, indicating disrupted regulation of humoral immune networks. Elevated expression of inhibitory receptors on B cells, such as PD-1 and FcRL4, is associated with higher activation thresholds and persistent inflammatory states.
Frailty is further accompanied by widespread dysfunction of the innate immune system. Macrophages in older individuals show reduced phagocytic capacity and impaired chemotaxis; dendritic cell (DC) numbers and antigen-presenting function decline; and although natural killer cells may increase numerically, their cytotoxic activity is significantly diminished[53]. Chronic exposure of monocytes to pro-inflammatory signals sustains a state of low-grade inflammation, often termed inflammaging. This condition is characterized by persistently elevated IL-6, TNF-α, and CRP levels, promoting oxidative stress, mitochondrial dysfunction, and tissue injury, thereby accelerating frailty progression. Cumulative dysfunction across immune cell populations leads to systemic immune decline and contributes to frailty.
Impaired immune tolerance is another critical aspect of immune dysregulation in frailty. In younger individuals, central and peripheral tolerance mechanisms maintain immune homeostasis and prevent aberrant activation. Aging diminishes thymic negative selection efficiency and alters peripheral Treg function, weakening immune tolerance and permitting accumulation of autoreactive cells[54]. Chronic, low-grade inflammation that develops with aging, often termed inflammaging, is characterized by persistent elevation of pro-inflammatory mediators and is strongly associated with frailty and other age-related pathologies[9]. This chronic inflammatory milieu impairs tissue repair and further disrupts lymphoid architecture and immune cell communication.
Overall, immune alterations in frailty are characterized by paradoxical coexistence of weakened immune responses and persistent inflammation. While older individuals exhibit inadequate responses to external pathogens, endogenous inflammatory processes persist. This imbalance increases susceptibility to infection, reduces vaccine efficacy, impairs tissue repair, and contributes to multisystem functional decline[10]. Immune dysfunction is therefore not only a defining feature of frailty but also a core pathogenic mechanism. Further studies on frailty-associated immune alterations, particularly interactions among immune cells, the lymphatic system, and TLS, will be essential for elucidating the link between immunosenescence and systemic frailty.
4. Tertiary Lymphoid Structures and Their Functions
Within the conceptual framework proposed in this review, immunosenescence/inflammaging→lymphatic dysfunction→TLS remodeling→multi-organ functional decline associated with frailty, the lymphatic system and TLS may represent an important intermediate tissue-level interface linking systemic immune aging with end-organ functional impairment. The lymphatic system is a key component for maintaining immune homeostasis and tissue fluid balance, consisting primarily of primary lymphoid organs (bone marrow and thymus), SLOs (lymph nodes, spleen, and mucosa-associated lymphoid tissue), and lymphatic networks[55]. Its main functions include mediating generation, differentiation, migration, and antigen recognition of immune cells. With aging, the structure and function of the lymphatic system gradually decline, manifested as thymic atrophy, reduced lymphocyte production, impaired lymphatic drainage, and decreased immune cell activity. These age-related lymphatic degenerative changes constitute the critical intermediate link between upstream immunosenescence/inflammaging and downstream TLS dysregulation, laying the essential histological basis for frailty pathogenesis.
Within the lymphatic system, primary lymphoid organs are responsible for immune cell development and maturation. Bone marrow is the source of hematopoietic stem cells, generating and releasing B cells, myeloid cells, and other immune components. The thymus serves as the site for T cell development, selection, and establishment of immune tolerance. As individuals age, the thymus is progressively replaced by adipose tissue, and its epithelial network atrophies, resulting in decreased production of naïve T cells, reduced T cell diversity, and impaired immune tolerance. This process, termed thymic involution, represents one of the earliest hallmarks of immunosenescence[11].
SLOs, such as lymph nodes and the spleen, serve as central sites for antigen recognition and initiation of immune responses. DCs in lymph nodes capture exogenous antigens, subsequently activating naïve T cells and inducing humoral or cellular immune responses. The spleen primarily clears senescent red blood cells and circulating antigens, maintaining peripheral immune homeostasis. Aging leads to decreased numbers of lymph node stromal cells, cortical atrophy of lymph nodes, blurred follicular architecture, and weakened demarcation between B cell and T cell zones, all indicative of immune function decline[56,57].
Concurrently, reduced expression of chemokines such as CXCL13 and CCL21 further impairs immune cell homing and local immune response capacity, resulting in delayed T cell priming and weakened germinal centers (GCs) reactions[58]. Collectively, these structural and molecular changes form an important histological basis for immunosenescence. These systemic lymphoid degenerative changes directly shape the formation, maturation, and functional fate of TLS, which are systematically elaborated below.
4.1 Formation and functions of TLS
To ensure conceptual clarity, we provide unified criteria for TLS-related structures:
Typical TLS: Ectopic immune aggregates with clear T/B cell compartmentalization, follicular dendritic cell (FDC) networks, HEVs, and functional GCs.
TLS-like structures: Immune aggregates with partial structural organization but lacking complete compartmentalization or GCs.
Lymphoid aggregates: Loose immune cell infiltration without structural organization or vascular/chemokine support.
Under chronic inflammation or persistent immune stimulation, TLS can be induced and formed in non-lymphoid tissues[18,59]. Also known as ectopic lymphoid tissue, TLS are immune aggregates that spontaneously form in local inflammatory microenvironments. Structurally, they are similar to SLOs but are not congenitally present[60,61]. TLS comprise B cell zones, T cell zones, DCs, stromal cells, HEVs, as well as FDC networks and GCs in their mature state. They support local antigen presentation, lymphocyte recruitment and activation, and the integration of humoral and cellular immunity.
TLS can be classified as mature or immature. Mature TLS possess complete compartmentalization and GCs, whereas immature TLS generally have a loose structure and limited functions[62]. Recent studies further subdivide immature TLS into Conforming TLS, which are close to mature and have potential functional capacity, and deviating TLS, which exhibit developmental deviation and functional impairment due to microenvironmental suppression. This classification emphasizes TLS structural and functional heterogeneity[63].
TLS formation is typically triggered by chronic inflammation or persistent immune stimulation. Pro-inflammatory cytokines (e.g., IL-1β, IL-6, TNF-α) induce chemokine expression (e.g., CXCL13, CCL19, CCL21), recruiting lymphoid tissue inducer cells or functionally similar cells, as well as naïve B/T cells and DCs. Stromal and endothelial cells are then activated via lymphotoxin α/β–lymphotoxin β receptor signaling, promoting HEV formation and adhesion molecule upregulation, enabling lymphocyte infiltration and aggregation to form rudimentary TLS. Through stromal/extracellular matrix remodeling, FDC network establishment, and GC development, TLS can mature; however, microenvironmental factors may arrest TLS in the deviating TLS state[64]. The quantity, maturity, and spatial distribution of TLS also dynamically change in response to treatments, demonstrating their high plasticity[65].
4.1.1 Immune cell recruitment and activation
DCs are professional antigen-presenting cells that bridge innate and adaptive immune responses; after antigen capture, they migrate to draining lymph nodes and localize in T-cell-rich areas to initiate adaptive responses by presenting antigens to T cells[66]. Homeostatic chemokines such as CCL19, CCL21, CXCL13, and CXCL12 regulate lymphocyte and DC trafficking, directing them into lymphoid tissues and contributing to organization of T- and B-cell zones and structured immune aggregates[67]. Ectopic expression of homeostatic chemokines induces lymphoid neogenesis with organized lymphocyte and DC infiltration, resembling the architecture of SLOs[68].
Fibroblasts, especially cancer-associated fibroblasts (CAFs), act as lymphoid tissue organizers in TLS formation and maintenance. CAFs actively secrete immune chemokines and signals, participating in TLS spatial organization and immune regulation[69]. B cells activate CAFs via LTβR, forming a positive feedback loop that promotes TLS maturation[69,70]. In solid tumors, specific CAF subsets (CXCL13+ or CCL19+) serve as TLS structural cells, promoting TLS neogenesis and maturation[69,71,72]. Mature TLS correlate positively with immune checkpoint inhibitor efficacy, highlighting the CAF–TLS axis in tumor immunity[17].
4.1.2 The role of TLS in tumors (brief summary)
TLS have been most extensively characterized in cancer, where mature TLS with organized T/B cell compartments and functional GCs act as critical anti-tumor immune hubs[17]. This well-established cancer TLS paradigm provides a valuable mechanistic reference for understanding age-related TLS, although their functional orientations differ significantly. Cancer-associated TLS primarily orchestrate protective anti-tumor immunity, whereas age-related TLS in frailty predominantly sustain chronic local inflammation and tissue damage. Given the review’s core focus on immunosenescence and frailty pathogenesis, detailed discussions of cancer-specific TLS biology are streamlined to avoid redundancy.
4.1.3 The supporting role of lymphatic and vascular pathways
Lymphatic vessels and HEVs support TLS formation and maintenance.
First, lymphatic vessels surrounding TLS provide essential conduits for immune cell trafficking. Ruddle noted that lymphatic structures within or adjacent to TLS closely resemble those of SLOs, such as lymph nodes, and are capable of draining antigen-bearing cells (e.g., DCs) or lymphocytes from peripheral tissues into regional lymph nodes or the systemic circulation[73]. These lymphatic channels maintain routes for cellular entry into and egress from TLS, allowing local immune responses to be sustained within TLS while simultaneously communicating with systemic immunity.
Second, lymphatic vessels and associated lymphoid structures contribute to the regulation and transport of tissue fluid and antigen–cell mixtures. The inflammatory microenvironments in which TLS arise are often characterized by extensive cellular infiltration, altered vascular permeability, and tissue edema. The presence of lymphatic vessels facilitates the clearance of interstitial fluid, prevents excessive edema accumulation, and enables the drainage of soluble antigens, cellular debris, and immune cells into the lymphatic system or bloodstream, thereby preserving local microenvironmental homeostasis[73].
HEVs represent another critical vascular component of TLS. These specialized blood vessels are lined by plump, cuboidal endothelial cells and express high levels of adhesion molecules, including PNAd, ICAM-1, and VCAM-1, enabling efficient recruitment of circulating lymphocytes. Through HEVs, naïve and memory T and B cells are selectively transported from the bloodstream into tissues, thereby establishing stable immune cell aggregates within TLS. Concurrently, HEVs cooperate with FDC networks and stromal cells to support B- and T-cell compartmentalization and GC formation. HEV density positively correlates with anti-tumor immune responses and favorable clinical outcomes. Thus, blood vessels (particularly HEVs) together with lymphatic vessels, not only provide structural support but also constitute indispensable determinants of TLS functional maturation, immune cell trafficking, and localized immune activity.
4.2 The role of TLS in immunosenescence
With aging and age-related chronic disease burden, the immune system undergoes progressive immunosenescence and functional dysfunction, which is particularly prominent in the context of frailty. Aging globally impairs cellular homeostasis, inducing senescence of stromal cells, endothelial cells, and immune cells, which is the fundamental cellular basis of TLS structural and functional degeneration[36]. In this process, structural and functional degenerative changes of the lymphatic system are key underpinnings of frailty pathogenesis, and collectively determine the formation, maturation and functional fate of TLS[73-75].
4.2.1 Degenerative changes in lymphoid structures
In aged or immunosenescent states, lymph nodes undergo histological remodeling, with reduced stromal cells, disorganized follicles, and impaired lymphatic function. Chemokines (CCL19, CCL21, IL-7) are downregulated, lymphatic transport efficiency decreases, and compartment boundaries blur[74]. Accumulated cellular senescence in stromal cells disrupts the lymphoid-stromal microenvironment, impairing immune cell localization and signaling[76].
4.2.2 Impaired immune cell migration and responsiveness
Degeneration of lymphatic structures compromises immune cell migration and activation. Aged lymph nodes show indistinct T/B zones, fewer GCs, reduced naïve T cells, and excessive memory T cells, leading to attenuated responses to novel antigens[74,75]. This explains the delayed immune responses and slow recovery in frail individuals.
4.2.3 Chronic low-grade inflammation and lymphoid dysregulation
Chronic low-grade inflammation in frailty induces cellular senescence and stromal damage in lymphoid organs. SASP from senescent cells alters stromal signaling and chemokine distribution, exacerbating structural disorder and immune decline[76]. The lymphatic system is actively remodeled in the inflammatory microenvironment, driving a vicious cycle of immunosenescence.
During aging, the formation and functional regulation of TLS may be closely associated with immune system remodeling. Several studies have reported that in elderly individuals or frailty models, TLS or TLS-like often display incomplete architecture, aberrant cellular composition, and disorganized chemokine distribution, suggesting a potential role in the decline of immune function associated with frailty[33,77,78]. Recent evidence further indicates that the immunological functions of TLS are not static but are profoundly influenced by the host’s age and immune status. In the context of immunosenescence, TLS are more prone to sustained immune activation and amplification of inflammatory signaling, thereby contributing to the progression of age-related diseases[16]. TLS may therefore be regarded as a manifestation of “local reprogramming” of the immune system under frailty conditions, with alterations in their structure and function reflecting the pathological interplay between immunosenescence and chronic inflammation.
In summary, TLS exhibit functional duality. On the one hand, they can serve as local sites for antigen presentation and immune responses, facilitating pathogen clearance and antitumor immunity; on the other hand, the persistent presence of TLS may sustain chronic inflammation, promote autoimmune responses or tissue fibrosis, and disrupt immune tolerance, ultimately leading to tissue damage.
Notably, this functional duality of TLS is not uniformly expressed across different tissues; instead, it is shaped by organ-specific microenvironments and aligns with the functional decline of key organs in frailty[79]. Collectively, age-related TLS dysfunction represents a tissue-level manifestation of immunosenescence, characterized by structural instability, impaired maturation, and a pro-inflammatory functional shift. These age-related abnormalities exhibit significant organ specificity, shaped by distinct tissue microenvironments. Below, we systematically elaborate on the characteristics of age-related TLS dysfunction and their relevance to organ aging in core systems.
4.3 Organ-specific TLS in aging: Evidence beyond cancer-centered paradigms
While TLS have been extensively studied in the context of cancer, emerging evidence indicates that TLS or TLS-like immune aggregates also arise in multiple non-lymphoid organs during aging and chronic inflammation[33]. Importantly, many of these organs, particularly the heart, brain, lung, liver, kidney, and gastrointestinal tract, are central to age-related functional decline and frailty. However, compared with cancer-associated TLS, direct evidence linking TLS to organ-specific aging remains limited, fragmented, and highly context dependent. Nevertheless, observations across different organ systems suggest that TLS may represent a shared immunological feature of chronic tissue stress and immune remodeling during aging.
4.3.1 Cardiovascular system
TLS have been identified in the adventitia of large arteries and within atherosclerotic lesions, especially in aged individuals with chronic vascular inflammation[33]. Available evidence from aged vascular inflammatory models suggests that vascular TLS-like structures may exhibit distinct age-associated features, including predominantly immature or deviating TLS phenotypes, reduced GC formation, increased expression of the profibrotic chemokine CCL2, and impaired perivascular lymphatic drainage, potentially involving LTβR/CCL21-related signaling pathways. In addition, these TLS-like structures have been reported to display increased infiltration of pro-inflammatory macrophages, reduced follicular B-cell abundance, and less organized T/B-cell compartmentalization. Given that cardiovascular aging is characterized by reduced vascular compliance, impaired endothelial signaling, and increased inflammatory burden, these TLS abnormalities promote vascular stiffness and endothelial dysfunction, which are directly associated with age-related cardiovascular reserve decline.
4.3.2 Central nervous system
In the aging brain, TLS-like immune aggregates have been predominantly identified in the perivascular spaces of the leptomeninges, particularly in association with neurodegenerative and neuroinflammatory disorders[80]. These structures may exhibit a distinct age-related phenotype: dominated by innate immune cells (macrophages and microglia), lacking functional FDC networks and organized B cell follicles, and characterized by high expression of pro-inflammatory cytokines IL-6 and TNF-α via the IL6/STAT3 pathway. The meningeal lymphatic vessels adjacent to these aggregates show impaired drainage function in aged individuals, further exacerbating local inflammatory cell accumulation. TLS-like structures in the aging central nervous system may contribute to local neuroinflammatory remodeling and may be associated with impaired blood–brain barrier integrity and persistent inflammatory signaling, which have been linked to age-related cognitive decline.
4.3.3 Lung
The lung represents one of the most extensively studied sites of inducible TLS formation outside of tumors, particularly in the form of inducible bronchus-associated lymphoid tissue (iBALT) distributed around the bronchi and bronchioles, regulated by the NF-κB/IL-17 pathway[81]. Age-related accumulation of iBALT-like structures has been observed in chronic pulmonary inflammation and infectious conditions. Emerging evidence from aging-associated pulmonary inflammation models suggests that TLS/iBALT-like structures in the aging lung may display altered immune organization, including impaired HEV formation, reduced stromal CXCL13 expression, low GC activity, and shifts in lymphocyte composition characterized by relatively increased memory T cells and decreased naïve lymphocytes. In older adults, pulmonary aging is characterized by diminished immune surveillance, impaired tissue repair, and persistent low-grade inflammation. These TLS abnormalities reduce the local anti-infection immune capacity of the lung, which is closely associated with age-related respiratory dysfunction and increased infection susceptibility.
4.3.4 Liver
The liver is a highly immunologically active organ, and TLS-like structures have been reported in chronic liver diseases, including fibrosis and cirrhosis, conditions more prevalent with aging[79]. These immune aggregates are predominantly located in the portal areas adjacent to bile ducts and vascular structures, exhibiting a unique age-associated phenotype: high TGF-β expression, dominated by fibroblastic stromal cells, and classified as deviating TLS with prominent profibrotic properties, mediated by the TGF-β/Smad signaling pathway. Aging liver TLS show reduced numbers of cytotoxic T cells and increased infiltration of regulatory T cells, leading to impaired immune surveillance. Given that hepatic aging involves altered immune tolerance, reduced regenerative capacity, and increased inflammatory burden, these TLS alterations may contribute to hepatic fibrosis and metabolic dysregulation, which are core features of age-related liver dysfunction.
4.3.5 Kidney
TLS have been observed in aged kidneys and in chronic kidney disease, where they are mainly distributed in the renal interstitium, often in close proximity to fibrotic lesions and atrophic tubules, associated with interstitial inflammation and progressive fibrosis via the CCL21/CCR7 pathway[79]. Renal aging is characterized by declining glomerular filtration, vascular rarefaction, and chronic immune activation. TLS-like structures in the kidney are mostly immature and may contribute to persistent inflammatory microenvironments that accelerate functional decline. These changes are closely associated with progressive renal functional decline in aging.
4.3.6 Gastrointestinal tract
The gastrointestinal tract contains extensive mucosal immune networks, and TLS-like aggregates have been described in the context of chronic intestinal inflammation and age-associated dysbiosis, predominantly located in the lamina propria of the small and large intestinal mucosa, regulated by the TLR4/NF-κB pathway[79]. Aging intestinal TLS exhibit a distinct phenotype: disrupted T/B cell compartmentalization, driven by gut microbiota dysbiosis and increased bacterial translocation, and characterized by high TLR4/NF-κB pathway activation. The intestinal lymphatic vessels adjacent to these aggregates show impaired drainage function in aged individuals, further promoting systemic dissemination of inflammatory mediators. Aging-related changes in gut immunity include impaired barrier function, altered microbiota composition, and increased inflammatory tone. These TLS alterations may be associated with impaired intestinal barrier integrity and systemic low-grade inflammation, which is a key upstream driver of multi-organ functional decline in aging.
4.3.7 Integrative perspective
Collectively, although the majority of mechanistic insights into TLS biology originate from cancer research, accumulating evidence across multiple organ systems suggests that TLS may also emerge as part of the immune remodeling process during aging.
A cross-organ comparison reveals both common rules and distinct organ-specific characteristics of age-related TLS changes:
Common features: Most TLS observed in aging non-lymphoid organs are TLS-like structures or immature deviating TLS, rather than typical mature TLS with complete structure and function; all exhibit impaired GC formation, reduced chemokine expression, and disrupted vascular/lymphatic support.
Organ-specific phenotypes:
Cardiovascular and hepatic TLS are predominantly profibrotic deviating TLS;
Central nervous system TLS are innate immune-dominated TLS-like structures;
Pulmonary TLS are mainly iBALT with impaired HEV formation;
Gastrointestinal TLS are microbiota-driven mucosal aggregates.
A critical distinction between cancer-associated and aging-associated TLS lies in their functional orientation: cancer-related mature TLS primarily serve as anti-tumor immune hubs, while most TLS observed in aging non-lymphoid organs are TLS-like structures or immature deviating TLS that sustain chronic local inflammation and impair organ resilience. This functional divergence is driven by differences in microenvironmental cytokine profiles, cancer TLS are dominated by IL-22 and LTβR signaling, whereas aging TLS are characterized by elevated IL-6, TNF-α, and TGF-β[79]. Further organ-specific and longitudinal studies are required to clarify whether TLS actively drive age-related functional decline or serve as markers of immune dysregulation during aging.
Further organ-specific and longitudinal studies are required to clarify whether TLS actively drive age-related functional decline or serve as markers of immune dysregulation during aging. To systematically synthesize the current evidence base, stratify evidence strength, and highlight key limitations across different organ systems, we have compiled a comprehensive evidence summary table (Table 2). This table organizes all available studies by organ system, study model, evidence type, TLS phenotype, core mechanistic pathways, and translational limitations, enabling readers to clearly distinguish conclusions supported by moderate-strength indirect evidence from those based on preliminary or extrapolated findings.
Table 2. Summary of existing evidence linking TLS to aging and frailty.
| Organ/Tissue | Disease/Model Status | Evidence Type | TLS Phenotype | Association Type | Core Mechanisms | Evidence Confidence | Key Limitations | Ref. |
| Lung | Aged mouse model, chronic pulmonary inflammation | Animal model study | Immature TLS/iBALT-like structures | Experimental causal | Aging-induced impaired DC function reduces TLS/iBALT maturation efficiency; NF-κB/IL-17 pathway drives TLS formation | Moderate | Only verified in mouse models; lack of direct evidence from frail human lung tissues | [82,83] |
| Central Nervous System | Aged mouse model, neurodegenerative disorders | Animal model + human histological study | Putative TLS-like structures (no complete T/B compartmentalization) | Indirect correlational | Meningeal TLS-like structures drive chronic neuroinflammation via IL-6/STAT3 pathway, contributing to cognitive decline | Moderate | No direct verification in frail human cohorts | [84] |
| Cardiovascular System | Aged human atherosclerotic lesions, mouse atherosclerosis model | Human histological + animal model study | Immature/Deviating TLS | Indirect correlational | Vascular TLS in aging are dominated by profibrotic signaling via LTβR/CCL21 pathway | Moderate | Lack of direct evidence linking vascular TLS to frailty phenotype | [33] |
| Kidney | Aged mouse kidney, chronic kidney disease | Animal model + human histological study | Immature TLS-like structures | Indirect correlational | TLS in aged kidney drive interstitial inflammation and fibrosis via CCL21/CCR7 pathway | Moderate | No direct verification in frail populations | [85] |
| Liver | Aged human liver tissues, chronic liver fibrosis | Human histological study | Deviating TLS (profibrotic phenotype) | Indirect correlational | Aging-associated hepatic TLS are dominated by TGF-β/Smad profibrotic signaling | Moderate | Lack of longitudinal evidence linking hepatic TLS to frailty onset | [86] |
| Gastrointestinal Tract | Aged mouse model, gut dysbiosis | Animal model study | TLS-like immune aggregates | Mechanistic extrapolation | Gut dysbiosis in aging drives TLS formation via TLR4/NF-κB pathway | Low | Only verified in animal models; no direct evidence linking intestinal TLS to human frailty | [87] |
| Systemic Immune System | Aged mouse lymph node model, immunosenescence | Animal model study | TLS structural and functional degeneration | Experimental causal | Aging-induced stromal cell senescence, chemokine downregulation and HEV dysfunction impair TLS formation | Moderate | Lack of verification in human frail cohorts | [80] |
| Oncology-derived mechanistic reference | Multiple solid tumors, human cancer cohorts | Human clinical + preclinical study | Mature/immature TLS | Mechanistic extrapolation | Tumor-associated TLS provide mechanistic reference, with shared core regulatory pathways (LTβR, CXCL13/CCL19) | Moderate | Indirect extrapolation from oncology literature | [17,24] |
TLS: tertiary lymphoid structures; iBALT: inducible bronchus-associated lymphoid tissue; DC: dendritic cell; LTβR: lymphotoxin beta receptor; CCL: C-C motif chemokine ligand; CCR: C-C motif chemokine receptor; HEV: high endothelial venule; NF-κB: nuclear factor kappa B; TGF-β: transforming growth factor beta.
As summarized in Table 2, while moderate-strength indirect evidence supports an association between TLS dysfunction and age-related organ decline in the lung and brain, evidence for other organ systems remains preliminary. This evidence gap reinforces the need for cautious interpretation of the proposed mechanistic framework and highlights priority areas for future research.
5. The Role and Clinical Significance of TLS in Frailty
Building on the core mechanistic framework outlined in Figure 1, a comprehensive, multi-layered mechanistic framework detailing the full cascade of age-related TLS remodeling and its contribution to frailty pathogenesis is presented in Figure 2. This framework integrates upstream drivers of TLS dysfunction, the continuum of TLS structural and functional degeneration, organ-specific TLS phenotypes, downstream frailty manifestations, and potential intervention strategies, providing a holistic roadmap for the subsequent discussion.
{kind=link}
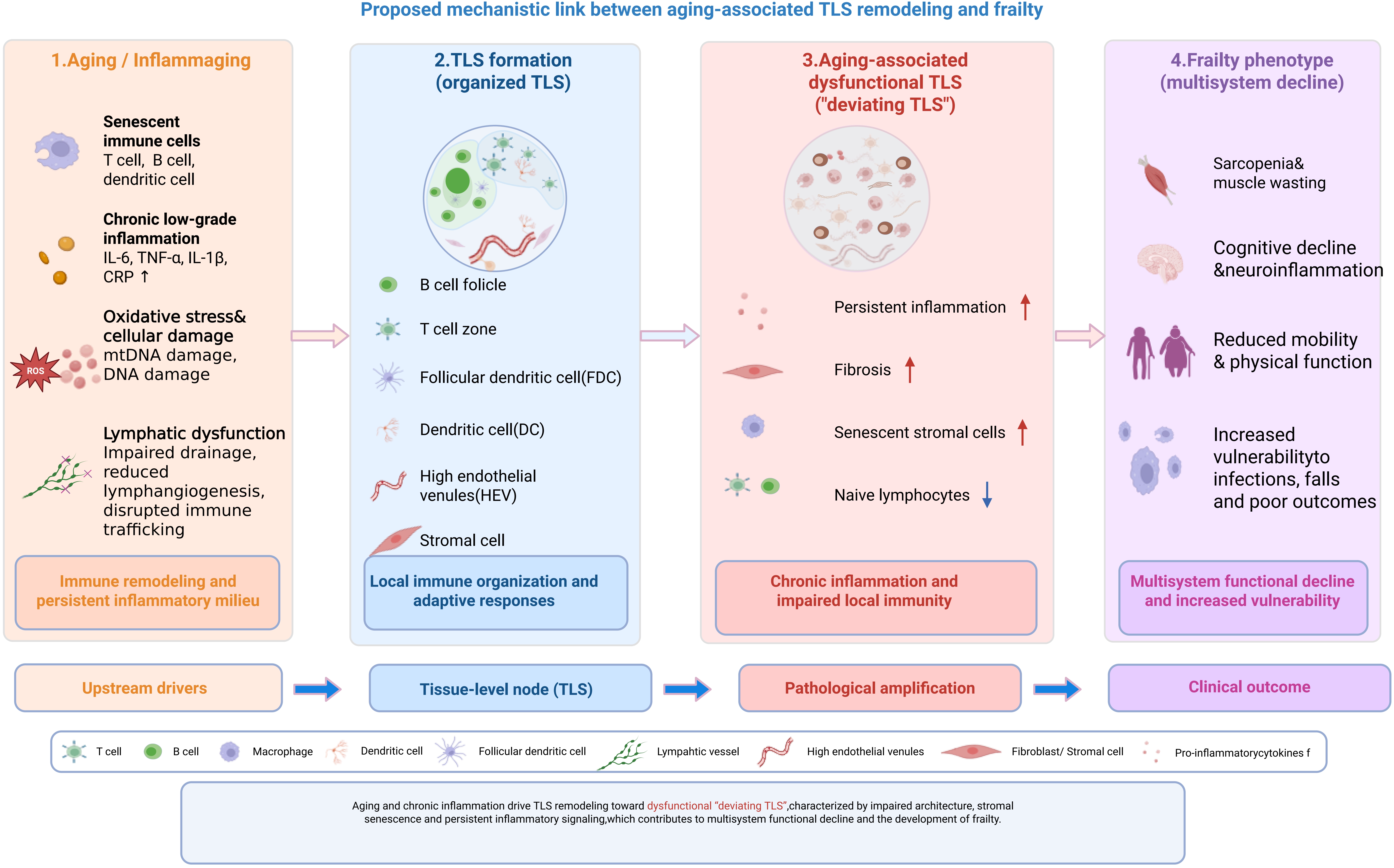
Figure 2. Comprehensive mechanistic framework of age-related TLS remodeling and its contribution to frailty pathogenesis. (A) Upstream drivers: Immunosenescence (including thymic involution, naive T cell depletion, and functional decline of innate/adaptive immune cells) and chronic low-grade inflammation (inflammaging) induce lymphatic and vascular dysfunction, collectively creating a pro-inflammatory milieu that initiates and shapes TLS remodeling; (B) TLS remodeling continuum: TLS undergo a progressive, irreversible transition during aging: from healthy/mature TLS (with clear T/B compartmentalization and functional germinal centers) to immature TLS, then to chronic inflammatory-skewed deviating TLS, and finally to fibrotic/inflamed dysfunctional TLS. Key changes in stromal components, vascular support, signaling pathways, and functional outcomes across each stage are summarized; (C) Organ-specific TLS alterations: TLS exhibit distinct tissue-specific phenotypes in aging, including meningeal TLS-like aggregates in the brain, disorganized iBALT in the lung, profibrotic TLS in vascular and hepatic tissues, and microbiota-driven mucosal TLS in the gastrointestinal tract, each contributing to organ-specific functional decline; (D) Downstream frailty phenotype: Chronic systemic inflammation combined with tissue-specific TLS dysfunction drives the development of multisystem frailty manifestations, including sarcopenia, osteoporosis, metabolic dysfunction, neurocognitive decline, impaired immunity, and increased risks of adverse clinical outcomes; (E) Potential intervention strategies: Targeted approaches to modulate TLS formation and function include molecular/cellular interventions targeting key signaling pathways, lifestyle modifications, and clinical/translational strategies leveraging TLS as biomarkers for patient stratification and precision intervention. Created in BioRender. Lin, Z. (2026) https://BioRender.com/prr472p. TLS: tertiary lymphoid structures.
In the setting of frailty and immunosenescence, the key structural and cellular components that support TLS maturation and function, including DCs, FDCs, stromal networks, HEVs, and lymphatic circulation, undergo progressive degenerative changes[7,16,82,85,86].
These age-related impairments disrupt TLS architecture and immune orientation, shifting TLS from protective immune hubs toward inflammation-sustaining centers that amplify local tissue damage and accelerate frailty progression[8,9]. Exploring age-related TLS dysfunction in the context of frailty is therefore essential for elucidating the tissue-level immune mechanisms of frailty pathogenesis.
5.1 The impact of TLS on frailty
Age-related TLS dysfunction in the context of frailty arises from degenerative changes in its core cellular and structural components. These components undergo varying degrees of age-related damage, and their structural degeneration and dysregulated signaling networks directly impair local immune homeostasis, antigen responsiveness, and tissue repair mechanisms. Consequently, TLS alterations may represent more than passive manifestations of immunosenescence and could potentially contribute to frailty-associated immune dysregulation and tissue decline.
5.1.1 Senescence of DCs impairs TLS initiation capacity
DCs are the core initiators of TLS formation. By driving lymphotoxin signals (e.g., LTβ), recruiting lymphocytes, and activating stromal cells, DCs promote establishment of FDC networks, HEVs, and lymphoid tissue chemokine axes. In elderly individuals, DCs exhibit marked declines in migration capacity, antigen presentation efficiency, and responsiveness to inflammatory signals, resulting in delayed TLS formation and structural immaturity. This phenomenon has been confirmed in influenza infection models, where reduced activation capacity of DCs in aged mice led to significantly lower formation efficiency and structural maturation of iBALT/TLS[84,88-90]. Because TLS are critical for local anti-infection defense and initiation of adaptive immune responses, impaired DC function may represent an early event contributing to delayed immune responses and increased infection susceptibility in frail individuals.
5.1.2 Decline of FDCs results in inefficient B cell follicles and antibody responses
The FDC network serves as a critical scaffold for maintaining B cell follicle structure, GC reactions, and high-affinity antibody production. Aging can result in reduced FDC numbers, disruption of the network scaffold, and diminished antigen retention capacity, thereby impairing B cells’ ability to undergo effective selection and affinity maturation. In addition, the decreased secretion of CXCL13 by aged FDCs disrupts the organized aggregation of B cell zones, thereby impairing B cell responses within TLS. The diminished GC reactions and reduced antibody affinity observed in the elderly closely correlate with FDC functional decline[57,91,92]. Consequently, aging of the FDC network not only reduces antigen response efficiency within TLS but may also contribute to poor vaccine responses and reduced post-infection antibody protection, which are typical immune phenotypes observed in frail individuals.
5.1.3 Dysregulation of stromal cells and chemokine networks
5.1.3.1 (CCL19/CCL21/CXCL13) destroys TLS tissue structure
The structural integrity of TLS largely depends on stromal cells and the tissue chemokines they produce. With aging, both T zone reticular cells and fibroblastic reticular cells undergo degenerative transcriptional, metabolic, and cytoskeletal changes, resulting in reduced expression of CCL19, CCL21, and CXCL13, blurred T/B cell zone boundaries, and disrupted immune cell migration pathways. Studies have indicated that aged stromal cells may exhibit functional exhaustion, diminished proliferative capacity, and excessive activation of inflammatory signals, making TLS more susceptible to immaturity and functional instability[83]. This structural and signaling dysregulation contributes to impaired establishment of an effective local immune microenvironment in frail individuals, thereby delaying tissue repair.
5.1.4 Impaired function of HEVs limits immune cell entry into TLS
HEVs are critical vascular structures for lymphocyte entry into TLS, determining whether immune cells can efficiently migrate into tissues. In elderly individuals, HEV adhesion molecules (e.g., PNAd), glycosylation patterns, and intracellular signaling are all reduced, resulting in restricted lymphocyte trafficking. Studies have shown that the functional decline of HEV endothelial cells in aged lymph nodes involves mechanisms similar to the insufficient maturation of HEVs within TLS[70,93,94]. Such HEV defects lead to lower immune cell density within TLS, weakening the persistence and efficacy of local immune responses. In frail patients, this translates into impaired peripheral immune surveillance and delayed immune cell reconstitution following stress, increasing susceptibility to functional decline after infection or tissue injury.
5.1.5 Overall decline of TLS microenvironment reinforces chronic low-grade inflammation and exacerbates frailty phenotype
In the context of frailty, the functional balance of TLS may shift from protective immune regulation toward a more pro-inflammatory and tissue-damaging immune microenvironment. Rather than supporting effective adaptive immunity and tissue repair, age-related TLS dysfunction in the context of frailty reinforces the core pathogenic mechanism of frailty, immunosenescence coupled with chronic low-grade inflammation (inflammaging). Evidence indicates that dysfunctional TLS are prone to sustaining persistent inflammation rather than mounting effective adaptive immune responses, leaving local tissues in a prolonged state of inflammation–repair imbalance[17,32,95]. This chronic inflammatory microenvironment can drive muscle wasting, metabolic dysregulation, organ functional decline, and reduced physical performance, forming a key biological basis for the onset of frailty. This establishes a self-perpetuating feed-forward loop: upstream immunosenescence and inflammaging drive TLS dysfunction, which in turn amplifies chronic inflammation and multi-organ damage, ultimately accelerating frailty progression. Thus, age-related TLS alterations in the context of frailty may represent an important tissue-level interface within the pathological processes associated with frailty.
5.2 Clinical application of TLS as a target for frailty intervention
Targeting age-related TLS alterations in the context of frailty represents a promising, although still largely preclinical, conceptual strategy that may help modulate immune homeostasis, mitigate chronic inflammatory remodeling, and potentially support physiological resilience. The dynamic changes of TLS not only reflect immune remodeling during frailty progression but also highlight candidate targets for clinical intervention.
Notably, most strategies discussed herein are indirect modulators that shape TLS formation and function by improving lymphatic drainage, resolving chronic inflammation, or restoring immune microenvironmental homeostasis, rather than direct and selective TLS-targeted interventions. Strictly specific interventions that directly act on TLS structural assembly or maturation, such as modulation of LTβR signaling and the CXCL13/CCL19/CCL21 chemokine axes, remain exclusively at the preclinical stage. This distinction ensures cautious interpretation of translational potential in frail populations. Given the limited direct evidence from dedicated frailty models, all interventions are discussed with rigorous caution, emphasizing safety, organ specificity, and potential risks in older adults. By targeting TLS formation, enhancing lymphatic function, and optimizing the immune microenvironment, it may be possible to alleviate immunosenescence and improve systemic physiological resilience.
5.2.1 Molecular and signaling pathway regulation
TLS formation depends on the LTβR, IL-7, IL-22, and CXCL13/CCL19/CCL21 signaling axes. Barone et al. demonstrated that IL-22 can stimulate fibroblasts to secrete lymphoid chemokines CXCL13 and CCL19, thereby promoting TLS assembly and immune activation[70]. In elderly or frailty models, IL-22 levels are decreased, potentially impairing TLS maturation. Restoration of the IL-22–STAT3 signaling pathway may improve the TLS microenvironment and enhance local antigen presentation and immune cell recruitment efficiency. Moreover, LTβR agonists and specific cytokine combinations (e.g., IL-7 + CXCL13) have been shown in animal studies to facilitate TLS reconstruction, indicating their therapeutic potential. However, systemic administration may carry pro-inflammatory or pro fibrotic risks in elderly individuals, requiring tissue targeted delivery and dose optimization.
5.2.2 Improving lymphatic circulation and immune microenvironment
Frail individuals often exhibit impaired lymphatic flow and persistent tissue inflammation, which are closely associated with dysregulated immune circulation. Studies have shown that activation of the VEGF-C/VEGFR-3 signaling pathway can promote lymphangiogenesis, improve lymphatic drainage, and facilitate the resolution of chronic inflammation, thereby contributing to the maintenance of lymphatic circulation and local immune homeostasis[96]. In addition, sphingosine 1 phosphate (S1P) and its receptors play a crucial role in maintaining lymphatic endothelial barrier integrity and regulating immune cell migration[97]. These mechanisms may collectively delay the progression of frailty through a “lymphatic function improvement→TLS structural remodeling→restoration of immune homeostasis” pathway. Notably, excessive VEGF-C activation may induce abnormal lymphatic proliferation and disrupt tissue fluid homeostasis in elderly individuals. It should be emphasized that most existing evidence for these strategies is derived from preclinical models of inflammatory bowel disease and skin inflammation, and the efficacy and safety in frail populations remain to be verified in dedicated clinical studies. Care should be taken to strictly control the intensity of intervention to avoid disrupting physiological tissue fluid balance.
5.2.3 Organ-specific TLS-targeted intervention strategies
Given the tissue-specific characteristics of TLS in aging, organ-targeted interventions may offer more precise and effective approaches to mitigating frailty, integrating both pharmacological and non-pharmacological strategies, with strict distinction between promoting the maturation of protective TLS and inhibiting the formation of inflammatory deviating TLS:
Lung: Pharmacologically, inhibit the NF-κB/IL-17 pathway to suppress the formation of inflammatory deviating TLS/iBALT; non-pharmacologically, enhance respiratory muscle training and pulmonary hygiene to improve lymphatic drainage, alleviating chronic pulmonary inflammation and reducing postoperative infection risk[98]. Notably, excessive inhibition of the NF-κB/IL-17 pathway may reduce the formation of protective iBALT, decreasing the anti-infection ability of the elderly lung.
Brain: Pharmacologically, modulate the IL-6/STAT3 pathway to suppress meningeal deviating TLS-mediated neuroinflammation; complement with cognitive training and sleep optimization to reduce neuroinflammatory burden, potentially lowering the incidence of postoperative cognitive dysfunction in frail surgical patients[99]. It should be noted that long-term inhibition of the IL-6/STAT3 pathway may impair the normal immune surveillance function of protective meningeal TLS, increasing the risk of intracranial infection in elderly individuals.
Cardiovascular system: Pharmacologically, target the LTβR/CCL21 pathway to inhibit the accumulation of pro-fibrotic deviating TLS in vascular lesions and improve endothelial function; combine with moderate aerobic exercise (e.g., brisk walking, cycling) to enhance cardiovascular reserve in aged individuals[32]. Excessive LTβR signaling inhibition may disrupt the formation of protective TLS in normal vascular tissues, impairing local immune surveillance.
Gastrointestinal tract: Pharmacologically and nutritionally, regulate gut microbiota via probiotics/prebiotics to reduce chronic antigenic stimulation and mitigate deviating TLS-mediated gut barrier dysfunction; support with a high-fiber, anti-inflammatory diet to suppress systemic inflammation[100,101]. Non-targeted microbiota modulation may induce dysbiosis and aberrant TLS formation, exacerbating intestinal inflammation in elderly individuals.
Liver/Kidney: Pharmacologically, inhibit pro-fibrotic pathways (e.g., TGF-β/Smad, CCL21/CCR7) to prevent pro-fibrotic deviating TLS-associated tissue fibrosis; adopt lifestyle modifications such as sodium restriction (kidney) and alcohol abstinence (liver) to preserve organ function[101]. Over-inhibition of these pro-fibrotic pathways may impair physiological tissue repair and the formation of protective TLS in the context of acute organ injury.
These organ-specific strategies integrate modulating the structural and functional remodeling of TLS via molecular targeting with tissue-adapted interventions, addressing both local TLS dysfunction and organ-specific functional decline, thereby complementing systemic general intervention approaches.
5.2.4 Combination of TLS function restoration and immunotherapy
The maturity of TLS is closely associated with immunotherapy efficacy. Vanhersecke et al. found that the presence of mature TLS can independently predict the therapeutic response to immune checkpoint inhibitors, suggesting that TLS functional status serves as a histological indicator of immune system health[102]. In frail populations, promoting TLS maturation or functional restoration may enhance vaccine responsiveness and defense against infections. In the future, TLS-targeting drugs or biologics may be combined with immune enhancers to establish a comprehensive “immune reconstruction + inflammation control” treatment strategy, with close monitoring of immune-related adverse events in elderly patients.
5.2.5 Lifestyle and non-pharmacological interventions
Non-pharmacological approaches are also critical for modulating TLS function. Regular aerobic exercise can enhance lymphatic return and promote immune cell migration, while resistance training improves muscle metabolism and local microcirculation, thereby supporting TLS homeostasis. Nutritional interventions, such as vitamin D supplementation, ω-3 fatty acids, and antioxidant polyphenols, can reduce systemic inflammation and improve immune cell metabolism[103]. Several studies indicate that exercise combined with an anti-inflammatory diet can significantly improve vaccine responses in the elderly, indirectly reflecting enhanced TLS function[87,104,105]. These interventions are generally safe and suitable for long-term application in frail populations.
5.2.6 Clinical prospects and biomarker value of TLS
As a structural indicator of immune status in frailty, TLS hold promising clinical applications. Future studies may utilize TLS maturity, cellular composition, and chemokine profiles as biomarkers to evaluate frailty-associated immune function. The integration of radiomics and spatial transcriptomics technologies could enable dynamic monitoring and graded assessment of TLS. Furthermore, AI-based histopathological analysis can automatically detect changes in TLS distribution and density, providing technical support for precise phenotyping. Monitoring immune health through TLS-related metrics may facilitate the development of an integrated system for “early frailty detection, intervention evaluation, prognosis prediction”.
Collectively, all TLS-related intervention strategies for frailty discussed in this section are still in preclinical exploration, and the specificity, efficacy and safety in frail elderly populations need to be verified in dedicated clinical trials. The functional duality of TLS in both immune protection and pathological progression should be fully considered in the development of targeted interventions, to maximize therapeutic benefits while minimizing potential risks in vulnerable elderly populations.
6. Summary and Future Research Directions
Taken together, the evidence synthesized in this review supports a unifying mechanistic framework: immunosenescence drives lymphatic dysfunction, which promotes TLS dysregulation and ultimately accelerates frailty progression. Frailty is a prominent syndrome in geriatric medicine that reflects multisystem functional decline, with its development closely associated with immunosenescence, chronic low-grade inflammation, and lymphatic system dysfunction. TLS, as candidate hubs for local immune responses and tissue microenvironment remodeling, have been shown to exert candidate immunoregulatory roles in chronic inflammation, infection, and tumor contexts. Current evidence suggests that the formation, maturation, and functional abnormalities of TLS may constitute important structural manifestations of immune system aging and frailty-related immune phenotypes[16-18].
In frailty, the key components of TLS, including DCs, FDCs, stromal cells, and HEVs, undergo functional decline, accompanied by dysregulation of chemokine networks and reduced lymphocyte homing efficiency. These alterations result in unstable TLS architecture, decreased antigen presentation efficiency, and altered immune regulation direction[106,107]. Consequently, TLS may shift from “protective immune microenvironments” to “chronic inflammation-sustaining hubs”, exacerbating local immune imbalance, impairing tissue repair, and promoting the development of systemic frailty phenotypes[108,109].
Although direct studies on TLS in frail populations remain limited, evidence from animal models and histological analyses indicates that lymphatic drainage impairment, stromal cell functional decline, and downregulation of key signaling pathways, such as IL-22 and LTβR, may contribute to frailty pathogenesis through effects on the TLS microenvironment[70,110]. Future research should focus on the dynamic changes and reversibility of TLS across different frailty stages, employing multi-omics approaches, single-cell, and spatial transcriptomic analyses to systematically elucidate the role of TLS abnormalities in immunosenescence. The causal role, directional effect, and tissue specificity of TLS in frailty remain to be verified in dedicated models and human cohorts.
At the intervention level, strategies that improve lymphatic function, modulate TLS microenvironments, and control chronic inflammation hold putative therapeutic value. Targeting signaling pathways including VEGF-C/VEGFR-3, S1P, IL-22, IL-7, and LTβR has demonstrated the potential to enhance lymphatic drainage and restore local immune homeostasis in preclinical studies[111,112]. In parallel, lifestyle interventions, such as structured exercise, optimized nutrition, and anti-inflammatory regimens, can provide non-pharmacological support for TLS functional recovery by mitigating chronic low-grade inflammation and improving immune microenvironments[113,114]. Interventions should prioritize safety, organ specificity, and avoidance of pro-inflammatory or pro-fibrotic side effects in elderly individuals.
Overall, TLS are poised to serve as a candidate histological node linking immunosenescence, lymphatic dysfunction, and frailty. Histologically, abnormal TLS formation and functional imbalance may reflect the peripheral tissue manifestations of immunosenescence, amplifying local inflammatory signals while limiting resolution of immune responses. These processes contribute to multi-organ functional decline and provide a novel tissue-level perspective for understanding frailty-associated immune phenotypes[16]. Incorporating TLS compositional features, maturity, and chemokine profiles into frailty immune assessments may offer a new theoretical framework for early detection, risk stratification, and individualized interventions. Future research should integrate basic immunology with clinical geriatrics to establish an integrated model of “frailty, immunosenescence, lymphatic dysfunction, TLS abnormalities”. Multidisciplinary, multi-level studies will elucidate the emblematic role of TLS in immune aging and explore their potential as candidate biomarkers in frailty evaluation. Furthermore, TLS may represent a novel target for correcting immune imbalance, supporting both early detection of immune decline and guidance of precise phenotyping and individualized interventions, thereby providing foundational insights for frailty prevention and management.
Acknowledgements
The authors would like to thank all the researchers whose work is included in this review. We acknowledge the use of AI-assisted tools, including ChatGPT (OpenAI, USA) for language editing and polishing of the manuscript, and BioRender.com for the creation and optimization of the schematic figures. All authors have full control over the content of the manuscript, have thoroughly reviewed and revised all parts of the manuscript, and take full responsibility for all content of the manuscript.
Authors contribution
Zheng L, Wen S: Conceptualization, supervision.
Lin Z: Investigation, formal analysis.
Chen J, Liu S: Writing-review & editing.
Conflicts of interest
Shaodi Wen is a Youth Editorial Board of Ageing and Cancer Research & Treatment. The other author declares no conflicts of interest.
Ethical approval
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
None.
Copyright
© The Author(s) 2026.
References
-
1. Fried LP, Tangen CM, Walston J, Newman AB, Hirsch C, Gottdiener J, et al. Frailty in older adults: Evidence for a phenotype. J Gerontol A Biol Sci Med Sci. 2001;56(3):M146-M157.[DOI]
-
2. Collard RM, Boter H, Schoevers RA, Oude Voshaar RC. Prevalence of frailty in community-dwelling older persons: A systematic review. J Am Geriatr Soc. 2012;60(8):1487-1492.[DOI]
-
4. Clegg A, Young J, Iliffe S, Rikkert MO, Rockwood K. Frailty in elderly people. Lancet. 2013;381(9868):752-762.[DOI]
-
5. Kojima G, Iliffe S, Walters K. Frailty index as a predictor of mortality: A systematic review and meta-analysis. Age Ageing. 2018;47(2):193-200.[DOI]
-
6. Hoogendijk EO, Afilalo J, Ensrud KE, Kowal P, Onder G, Fried LP. Frailty: Implications for clinical practice and public health. Lancet. 2019;394(10206):1365-1375.[DOI]
-
7. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69:S4-S9.[DOI]
-
8. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and inflamm-aging as two sides of the same coin: Friends or foes? Front Immunol. 2018;8:1960.[DOI]
-
10. Soysal P, Stubbs B, Lucato P, Luchini C, Solmi M, Peluso R, et al. Inflammation and frailty in the elderly: A systematic review and meta-analysis. Ageing Res Rev. 2016;31:1-8.[DOI]
-
11. Palmer DB. The effect of age on thymic function. Front Immunol. 2013;4:316.[DOI]
-
12. Liang Z, Dong X, Zhang Z, Zhang Q, Zhao Y. Age-related thymic involution: Mechanisms and functional impact. Aging Cell. 2022;21(8):e13671.[DOI]
-
13. Turner VM, Mabbott NA. Influence of ageing on the microarchitecture of the spleen and lymph nodes. Biogerontology. 2017;18(5):723-738.[DOI]
-
16. Sato Y. Immune aging and its implication for age-related disease progression. Physiology. 2025;40(4):363-373.[DOI]
-
17. Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19(6):307-325.[DOI]
-
18. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol. 2014;14(7):447-462.[DOI]
-
21. Shang T, Liang J, Kapron CM, Liu J. Pathophysiology of aged lymphatic vessels. Aging. 2019;11(16):6602-6613.[DOI]
-
22. Kataru RP, Park HJ, Shin J, Baik JE, Sarker A, Brown S, et al. Structural and functional changes in aged skin lymphatic vessels. Front Aging. 2022;3:864860.[DOI]
-
23. Fulop T, McElhaney J, Pawelec G, Cohen AA, Morais JA, Dupuis G, et al. Frailty, inflammation and immunosenescence. Interdiscip Top Gerontol Geriatr. 2015;41:26-40.[DOI]
-
24. Teillaud JL, Houel A, Panouillot M, Riffard C, Dieu-Nosjean MC. Tertiary lymphoid structures in anticancer immunity. Nat Rev Cancer. 2024;24(9):629-646.[DOI]
-
25. Zhang Q, Wu S. Tertiary lymphoid structures are critical for cancer prognosis and therapeutic response. Front Immunol. 2023;13:1063711.[DOI]
-
26. Jia H, Huang W, Liu C, Tang S, Zhang J, Chen C, et al. Immunosenescence is a therapeutic target for frailty in older adults: A narrative review. Ann Transl Med. 2022;10(20):1142.[DOI]
-
27. Soysal P, Stubbs B, Lucato P, Luchini C, Solmi M, Peluso R, et al. Corrigendum to “inflammation and frailty in the elderly: A systematic review and meta-analysis” [Ageing Res Rev. 31 (2016) 1-8]. Ageing Res Rev. 2017;35:364-365.[DOI]
-
28. Li X, Zhang X, Cao Z, Guan J, Qiu F, Zhang Q, et al. Tertiary lymphoid structures: Allies of cancer immunotherapy. Immunology. 2025.[DOI]
-
29. Taniguchi K, Yoshikawa T, Yanagita M. The roles of tertiary lymphoid structures in orchestrating immune responses in peripheral organs. Inflamm Regen. 2025;45:28.[DOI]
-
30. Ji RC. The emerging importance of lymphangiogenesis in aging and aging-associated diseases. Mech Ageing Dev. 2024;221:111975.[DOI]
-
31. Van Hoi ET, De Glas NA, Portielje JEA, Van Heemst D, Van Den Bos F, Jochems SP, et al. Biomarkers of the ageing immune system and their association with frailty–A systematic review. Exp Gerontol. 2023;176:112163.[DOI]
-
32. Zhao L, Jin S, Wang S, Zhang Z, Wang X, Chen Z, et al. Tertiary lymphoid structures in diseases: Immune mechanisms and therapeutic advances. Sig Transduct Target Ther. 2024;9:225.[DOI]
-
33. Sato Y, Silina K, van den Broek M, Hirahara K, Yanagita M. The roles of tertiary lymphoid structures in chronic diseases. Nat Rev Nephrol. 2023;19(8):525-537.[DOI]
-
35. Feng D, Xiao Y, Wang J, Wu R, Tuo Z, Yoo KH, et al. Unraveling links between aging, circadian rhythm and cancer: Insights from evidence-based analysis. Chin J Cancer Res. 2024;36(2):341-350.[DOI]
-
36. Zhang H, Xu X, Xie Z, Zhao Y, Chen Y, Ye Y, et al. Endothelial derived microvesicles modulate cerebrovascular and brain aging via miR-17-5p and the biomarker potential in vascular aging. Interdiscip Med. 2025;3(6):e70072.[DOI]
-
37. Dent E, Kowal P, Hoogendijk EO. Frailty measurement in research and clinical practice: A review. Eur J Intern Med. 2016;31:3-10.[DOI]
-
38. Rockwood K, Mitnitski A. Frailty in relation to the accumulation of deficits. J Gerontol A Biol Sci Med Sci. 2007;62(7):722-727.[DOI]
-
39. Cruz-Jentoft AJ, Bahat G, Bauer J, Boirie Y, Bruyère O, Cederholm T, et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing. 2019;48(1):16-31.[DOI]
-
41. Maggio M, Lauretani F, Ceda GP, Bandinelli S, Basaria S, Ble A, et al. Association between hormones and metabolic syndrome in older Italian men. J Am Geriatr Soc. 2006;54(12):1832-1838.[DOI]
-
42. Cappola AR, Xue QL, Fried LP. Multiple hormonal deficiencies in anabolic hormones are found in frail older women: The women’s health and aging studies. J Gerontol A Biol Sci Med Sci. 2009;64A(2):243-248.[DOI]
-
44. Li D, Yu Q, Wu R, Tuo Z, Wang J, Ye L, et al. Interactions between oxidative stress and senescence in cancer: Mechanisms, therapeutic implications, and future perspectives. Redox Biol. 2024;73:103208.[DOI]
-
45. Ji LL. Exercise at old age: Does it increase or alleviate oxidative stress? Ann N Y Acad Sci. 2001;928(1):236-247.[DOI]
-
46. Buchman AS, Boyle PA, Wilson RS, Tang Y, Bennett DA. Frailty is associated with incident Alzheimer’s disease and cognitive decline in the elderly. Psychosom Med. 2007;69(5):483-489.[DOI]
-
47. Vaughan L, Corbin AL, Goveas JS. Depression and frailty in later life: A systematic review. Clin Interv Aging. 2015;10:1947-1958.[DOI]
-
48. Goronzy JJ, Weyand CM. Successful and maladaptive T cell aging. Immunity. 2017;46(3):364-378.[DOI]
-
49. Sauce D, Larsen M, Fastenackels S, Duperrier A, Keller M, Grubeck-Loebenstein B, et al. Evidence of premature immune aging in patients thymectomized during early childhood. J Clin Invest. 2009;119(10):3070-3078.[DOI]
-
50. Bektas A, Schurman SH, Sen R, Ferrucci L. Human T cell immunosenescence and inflammation in aging. J Leukoc Biol. 2017;102(4):977-988.[DOI]
-
51. Frasca D, Blomberg BB. Aging affects human B cell responses. J Clin Immunol. 2011;31(3):430-435.[DOI]
-
53. Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol. 2013;13(12):875-887.[DOI]
-
55. Randall TD, Carragher DM, Rangel-Moreno J. Development of secondary lymphoid organs. Annu Rev Immunol. 2008;26:627-650.[DOI]
-
57. Turner VM, Mabbott NA. Structural and functional changes to lymph nodes in ageing mice. Immunology. 2017;151(2):239-247.[DOI]
-
58. Van Gool F, Nguyen MLT, Mumbach MR, Satpathy AT, Rosenthal WL, Giacometti S, et al. A mutation in the transcription factor Foxp3 drives T helper 2 effector function in regulatory T cells. Immunity. 2019;50(2):362-377.[DOI]
-
60. Dieu-Nosjean MC, Goc J, Giraldo NA, Sautès-Fridman C, Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 2014;35(11):571-580.[DOI]
-
61. Carragher DM, Rangel-Moreno J, Randall TD. Ectopic lymphoid tissues and local immunity. Semin Immunol. 2008;20(1):26-42.[DOI]
-
62. Chen Y, Wu Y, Yan G, Zhang G. Tertiary lymphoid structures in cancer: Maturation and induction. Front Immunol. 2024;15:1369626.[DOI]
-
65. Tan C, Huang J, Gao N, Wu B, Juliet M, Xiao J, et al. Dynamic remodeling of tertiary lymphoid structures in response to cancer therapy: A recent review. Cancer Immunol Immunother. 2025;74(10):313.[DOI]
-
67. Chen K, Bao Z, Tang P, Gong W, Yoshimura T, Wang JM. Chemokines in homeostasis and diseases. Cell Mol Immunol. 2018;15(4):324-334.[DOI]
-
70. Barone F, Nayar S, Campos J, Cloake T, Withers DR, Toellner KM, et al. IL-22 regulates lymphoid chemokine production and assembly of tertiary lymphoid organs. Proc Natl Acad Sci U S A. 2015;112(35):11024-11029.[DOI]
-
73. Ruddle NH. Lymphatic vessels and tertiary lymphoid organs. J Clin Invest. 2014;124(3):953-959.[DOI]
-
75. Thomas R, Wang W, Su DM. Contributions of age-related thymic involution to immunosenescence and inflammaging. Immun Ageing. 2020;17:2.[DOI]
-
76. Lancaster JN. Aging of lymphoid stromal architecture impacts immune responses. Semin Immunol. 2023;70:101817.[DOI]
-
77. Ciccimarra R, Zoboli M, Ragionieri L, Cacchioli A, Gazza F, Saleri R, et al. Histological characterization and spatial profiling of age-induced tertiary lymphoid structures in mouse lung parenchyma. Ann Anat. 2025;260:152660.[DOI]
-
80. Vaccaro A, de Alves Pereira B, van de Walle T, Dimberg A. Tertiary lymphoid structures in central nervous system disorders. Methods Mol Biol. 2025;2864:21-42.[DOI]
-
81. Hwang JY, Silva-Sanchez A, Carragher DM, de la Luz Garcia-Hernandez M, Rangel-Moreno J, Randall TD. Inducible bronchus–associated lymphoid tissue (iBALT) attenuates pulmonary pathology in a mouse model of allergic airway disease. Front Immunol. 2020;11:570661.[DOI]
-
84. Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol. 2004;5(2):133-139.[DOI]
-
86. Wallace LMK, Theou O, Godin J, Andrew MK, Bennett DA, Rockwood K. Investigation of frailty as a moderator of the relationship between neuropathology and dementia in Alzheimer’s disease: A cross-sectional analysis of data from the Rush Memory and Aging Project. Lancet Neurol. 2019;18(2):177-184.[DOI]
-
87. Dinas PC, Koutedakis Y, Ioannou LG, Metsios G, Kitas GD. Effects of exercise and physical activity levels on vaccination efficacy: A systematic review and meta-analysis. Vaccines. 2022;10(5):769.[DOI]
-
88. Goonetilleke N, Liu MKP, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009;206(6):1253-1272.[DOI]
-
89. Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol. 2011;12(7):639-646.[DOI]
-
90. Shaw AC, Joshi S, Greenwood H, Panda A, Lord JM. Aging of the innate immune system. Curr Opin Immunol. 2010;22(4):507-513.[DOI]
-
92. Frasca D, Diaz A, Romero M, Garcia D, Blomberg BB. B cell immunosenescence. Annu Rev Cell Dev Biol. 2020;36:551-574.[DOI]
-
93. Thompson HL, Smithey MJ, Surh CD, Nikolich-Žugich J. Functional and homeostatic impact of age-related changes in lymph node stroma. Front Immunol. 2017;8:706.[DOI]
-
95. Pipi E, Nayar S, Gardner DH, Colafrancesco S, Smith C, Barone F. Tertiary lymphoid structures: Autoimmunity goes local. Front Immunol. 2018;9:1952.[DOI]
-
97. Robertson TF, Chengappa P, Gomez Atria D, Wu CF, Avery L, Roy NH, et al. Lymphocyte egress signal sphingosine-1-phosphate promotes ERM-guided, bleb-based migration. J Cell Biol. 2021;220(6):e202007182.[DOI]
-
99. Mou Y, Du Y, Zhou L, Yue J, Hu X, Liu Y, et al. Gut microbiota interact with the brain through systemic chronic inflammation: Implications on neuroinflammation, neurodegeneration, and aging. Front Immunol. 2022;13:796288.[DOI]
-
100. Cristofori F, Dargenio VN, Dargenio C, Miniello VL, Barone M, Francavilla R. Anti-inflammatory and immunomodulatory effects of probiotics in gut inflammation: A door to the body. Front Immunol. 2021;12:578386.[DOI]
-
101. Kim JY. Gut microbiota, probiotics, and aging: Molecular mechanisms and implications for healthy aging. J Microbiol Biotechnol. 2026;36:e2511046.[DOI]
-
102. Vanhersecke L, Brunet M, Guégan JP, Rey C, Bougouin A, Cousin S, et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat Cancer. 2021;2:794-802.[DOI]
-
103. Ticinesi A, Meschi T, Lauretani F, Felis G, Franchi F, Pedrolli C, et al. Nutrition and inflammation in older individuals: Focus on vitamin D, n-3 polyunsaturated fatty acids and whey proteins. Nutrients. 2016;8(4):186.[DOI]
-
107. van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10(9):664-674.[DOI]
-
109. Nagy Á, Lánczky A, Menyhárt O, Győrffy B. Author Correction: Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. 2018;8:11515.[DOI]
-
110. Hammitzsch A, Tallant C, Fedorov O, O’Mahony A, Brennan PE, Hay DA, et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc Natl Acad Sci U S A. 2015;112(34):10768-10773.[DOI]
-
111. D’Alessio S, Correale C, Tacconi C, Gandelli A, Pietrogrande G, Vetrano S, et al. VEGF-C-dependent stimulation of lymphatic function ameliorates experimental inflammatory bowel disease. J Clin Invest. 2014;124(9):3863-3878.[DOI]
-
112. Schwager S, Detmar M. Inflammation and lymphatic function. Front Immunol. 2019;10:308.[DOI]
-
113. Li Y, Meng Q, Luo B, Li M, Fang J, Allred SR, et al. Exercises in activating lymphatic system on fluid overload symptoms, abnormal weight gains, and physical functions among patients with heart failure: A randomized controlled trial. Front Cardiovasc Med. 2023;10:1094805.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Lin Z, Chen J, Liu S, Wen S, Zheng L. Frailty and tertiary lymphoid structures in immunosenescence: Emerging associations, mechanistic clues and future perspectives. Ageing Cancer Res Treat. 2026;3:202606. https://doi.org/10.70401/acrt.2026.0024
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Definition and Clinical Features of Frailty
- 3. Relationship Between Immunosenescence and Frailty
- 4. Tertiary Lymphoid Structures and Their Functions
- 5. The Role and Clinical Significance of TLS in Frailty
- 6. Summary and Future Research Directions
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Lin Z, Chen J, Liu S, Wen S, Zheng L. Frailty and tertiary lymphoid structures in immunosenescence: Emerging associations, mechanistic clues and future perspectives. Ageing Cancer Res Treat. 2026;3:202606. https://doi.org/10.70401/acrt.2026.0024
copy
Share Link
copy