Dynamics of muscle-bone crosstalk throughout lifespan
Chenxi Tang
1,2,3,#

,
Xiaona Yin
1,2,3,#
,
Hongbo Zhang
1,2,3,*
*Correspondence to:
Hongbo Zhang, The Key Laboratory for Stem Cells and Tissue Engineering, Ministry of Education, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou 510080, Guangdong, China.
E-mail: zhanghongbo@mail.sysu.edu.cn
Ageing Cancer Res Treat. 2026;3:202620. 10.70401/acrt.2026.0025
Received: April 27, 2026Accepted: June 11, 2026Published: June 11, 2026
Abstract
Operating as a physically and physiologically integrated unit, skeletal muscle and bone are fundamental to human mobility and metabolism. Their reciprocal crosstalk endures throughout life. In early embryonic development, with a primary focus on morphogenesis, skeletal muscle and bone actively drive the functional maturation of both tissues during development. When the interplay reaches relative homeostasis during adulthood, they reciprocally sustain the functional integrity and physiological homeostasis of one another. With aging, however, this intimate connection exacerbates reciprocal decline, initiating a pathological feed-forward loop that precipitates osteosarcopenia. As this crosstalk is orchestrated by a shifting matrix of mediators, from biomechanical loads to neuronal, immunological, and secretory signals, understanding their age-associated alterations may help provide a point of intervention for treatment. This review outlines dynamic changes in muscle-bone crosstalk throughout the lifespan, discusses longitudinal changes in aging, and provides stage-specific perspectives for intervention.
Keywords
Muscle-bone crosstalk, lifespan, biomechanical load, myokines, osteokines, extracellular vesicles, neural and immune regulation, lifelong intervention
1. Introduction
Skeletal muscle and bone constitute a morpho-functional unit, which is essential for human mobility, metabolism, and health[1-3]. Over the past two decades, emerging research has indicated muscle and bone as secretory organs that can exert local (autocrine and paracrine) and systemic (endocrine) control[4-7]. Accordingly, as the myokines (e.g., myostatin, irisin) secreted by skeletal muscle and the osteokines (e.g., osteocalcin (OCN), sclerostin) released by bone were increasingly examined, their role in muscle-bone crosstalk has also been gradually revealed[1,8-11]. For instance, myostatin can promote osteoclastogenesis through receptor activator of nuclear factor-kappa B (NF-κB) ligand (RANKL) related pathway[9], and exacerbate bone metabolic dysfunction in diabetes[10]. Conversely, bone-derived sclerostin competitively binds to low-density lipoprotein receptor-related protein 5/6 receptors, resulting in the reduction of the Wingless-related integration site (Wnt)/β-catenin signaling pathway and inhibition of myogenesis[11].
Studies across physiological and pathological states have not only elucidated the mediators’ network in muscle-bone crosstalk but also highlighted a high degree of co-variability across all life stages. Consequently, dysfunction in one tissue frequently causes impairment in the other[12]. During embryogenesis, the lack of mechanical loading in muscular dysgenesis severely disrupts bone formation and structural patterning[13,14]. This functional coupling persists during aging, underscored by the frequent clinical co-occurrence and the strongly correlated severities of sarcopenia and osteoporosis[15-17]. This bidirectional degeneration may be partially mediated by extracellular vesicles (EVs): atrophied muscles display a modified exosomal profile with elevated microRNAs (miRNAs) that suppress osteogenesis, whereas osteoclasts in osteoporotic bone release EVs containing miRNAs that may promote muscle proteolysis and catabolism[18]. The systemic unity of the two tissues is further demonstrated in therapeutic contexts, where treatments can elicit concomitant improvements in both bone and muscle, further supporting the paradigm of an integrated musculoskeletal unit[18-21].
Throughout the lifespan, age-associated alterations in muscle-bone crosstalk extend beyond changes in the intrinsic functions of individual mediators, encompassing dynamic shifts in their relative mechanistic weights. A representative example is mechanical loading, whose role shifts from directing embryonic patterning to supporting adult physiological adaptation, and later becomes compromised during aging[12,22,23]. While mechanical forces are crucial for shaping tissue morphology during embryogenesis[20], the adult phenotype depends on a strong interplay between mechanical loading and endocrine signaling to sustain homeostasis[24-26]. In aging, this interaction is disturbed, as chronic immuno-inflammatory insults (‘inflammaging’) emerge as a promoter for musculoskeletal decline[23,27]. Overall, the intricate network responsible for maintaining homeostasis in adulthood experiences bidirectional instability and change during aging[28]. Reduced physical activity impairs the secretion of protective myokines, such as irisin and cardiotrophin-like cytokine factor 1 (CLCF1), whereas age-related metabolic and inflammatory dysfunctions upregulate the release of catabolic cytokines[29-31]. Together, these age-specific phenotypes collectively underscore the spatiotemporal plasticity of muscle–bone crosstalk throughout the lifespan.
The temporal plasticity of the musculoskeletal axis dictates that pathological onset is highly stage-dependent, suggesting that treatment strategies need to be tailored to tackle the main regulatory issues that are unique to each stage of development or aging. Despite substantial progress in characterizing individual mediators, the field remains constrained by the absence of a comprehensive, lifespan-oriented framework. In an effort to bridge this gap, this review seeks to integrate stage-specific evidence to map the evolutionary dynamics of the muscle–bone unit. By defining key developmental and degenerative milestones, we offer a forward-looking perspective on temporally optimized interventions for musculoskeletal disease.
2. Embryonic Musculoskeletal Crosstalk
During embryogenesis, musculoskeletal development is not merely the concurrent expansion of two distinct tissues but a highly coordinated, bidirectional program driving mutual morphogenetic maturation. Driven by the fundamental nature of muscle contractility, this early embryonic crosstalk relies heavily on mechanotransductive signals[22,24]. Currently, due to the technical bottlenecks associated with in vivo spatiotemporal conditional knockout models, direct evidence regarding the reciprocal impact of early embryonic muscle and bone secretory functions remains elusive. Nevertheless, the potential for chemical communication between these developing tissues cannot be overlooked[25,32]. Concomitant with their own maturation, the immune and nervous systems modulate the musculoskeletal developmental microenvironment via paracrine signaling[33-36]. By providing the hematopoietic precursors for osteoclasts and the neural circuitry for muscle contraction, respectively, these extrinsic networks exert a secondary and supportive influence on muscle-bone crosstalk[37-39].
2.1 Mechanical loading
Following the onset of contractility, muscle-derived mechanical stress acts as a critical mediator of embryonic musculoskeletal crosstalk. Historical observations have shown that the absence of fetal movement (fetal akinesia) results in a simplified skeletal structure (including joint contractures, the degradation of bone ridges, and the formation of mechanically weaker, rounded bones), recognizing the necessity of mechanical loads[22,40]. Among the embryonic muscle-bone crosstalk, the mechanical loads imposed by muscles on bones are easier to understand and appreciate. The mechanical load exerted by bones on muscles originates from the elastic force accumulated by the deformation of bones under stress; however, it has not been precisely measured[25].
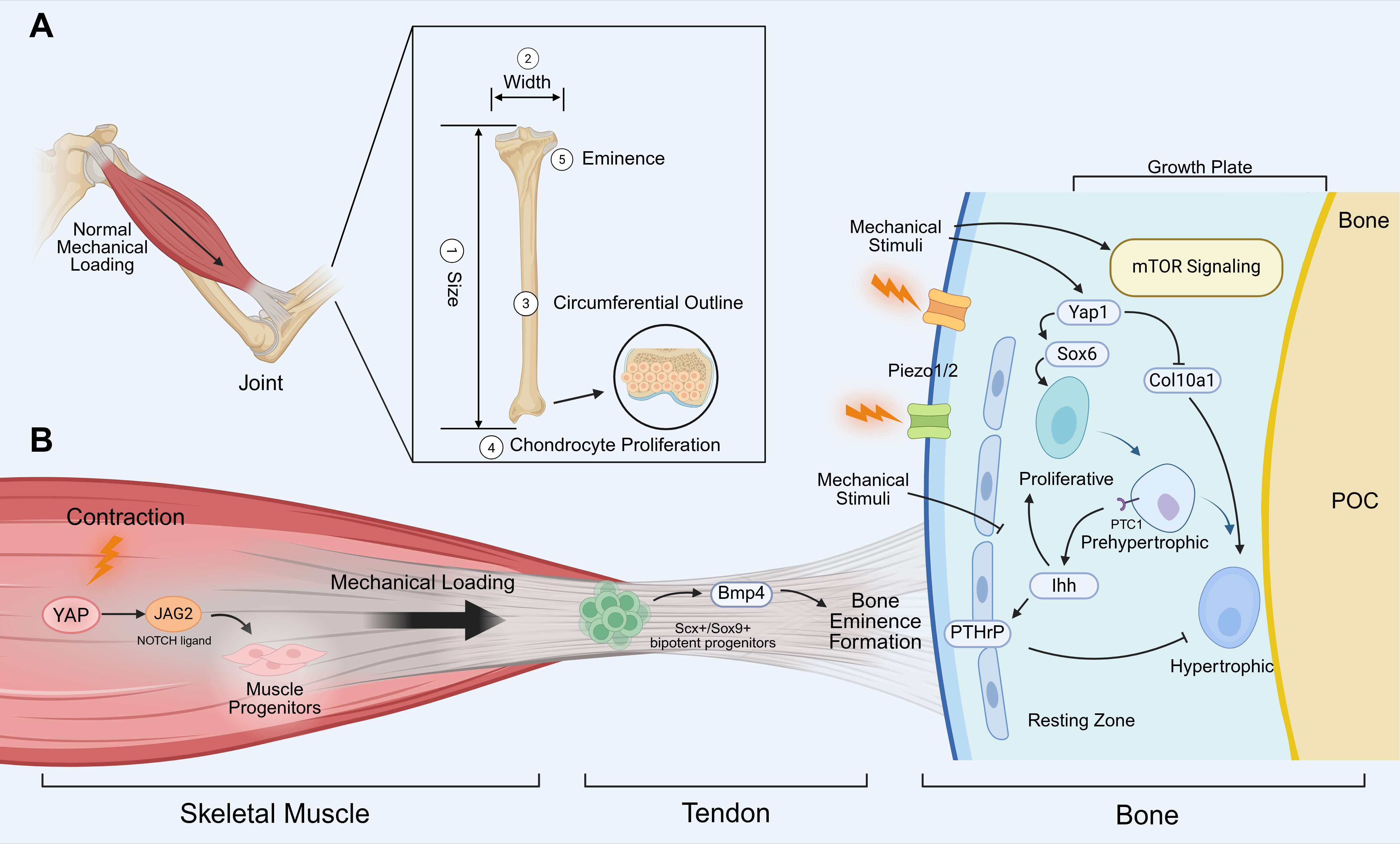
During embryonic development, the morphological impact of mechanical stress manifests across multiple spatial dimensions, governing bone size, width, circumferential outline, and so on (Figure 1A). In paralyzed chick and mouse embryos, various cartilaginous skeletal elements are shorter than normal. Studies in paralyzed chick embryos reported a reduction in the size of the proliferative zone, as well as in the number of proliferating chondrocytes[22]. To go a step further, this stimulatory effect of movement on chondrocyte proliferation may be site-specific, as the manipulation of movement led to alterations in the proportions between bones of the same limb[41]. This selective targeting is related to intrinsic mechanistic target of rapamycin (mTOR) pathway activity in individual growth plates[41]. Another molecular player that may regulate bone size in response to mechanical cues is the Indian hedgehog (Ihh)/parathyroid hormone-related protein (PTHrP) axis, which forms a negative-feedback loop that regulates chondrocyte proliferation and differentiation[42]. Specifically, in the growth plate, Ihh expressed by prehypertrophic and hypertrophic chondrocytes diffuses through the cartilage and stimulates cells in the resting zone to produce PTHrP. PTHrP then inhibits chondrocyte differentiation, thereby creating an Ihh/PTHrP feedback loop responsible for the maintenance of the growth plate[42,43]. Furthermore, recent studies emphasize that the timing of resting zone PTHrP expression is of crucial importance. The PTHrP expression must increase quickly at the onset of secondary ossification to prevent fusion of the primary and secondary ossification centers[44].
{kind=link}
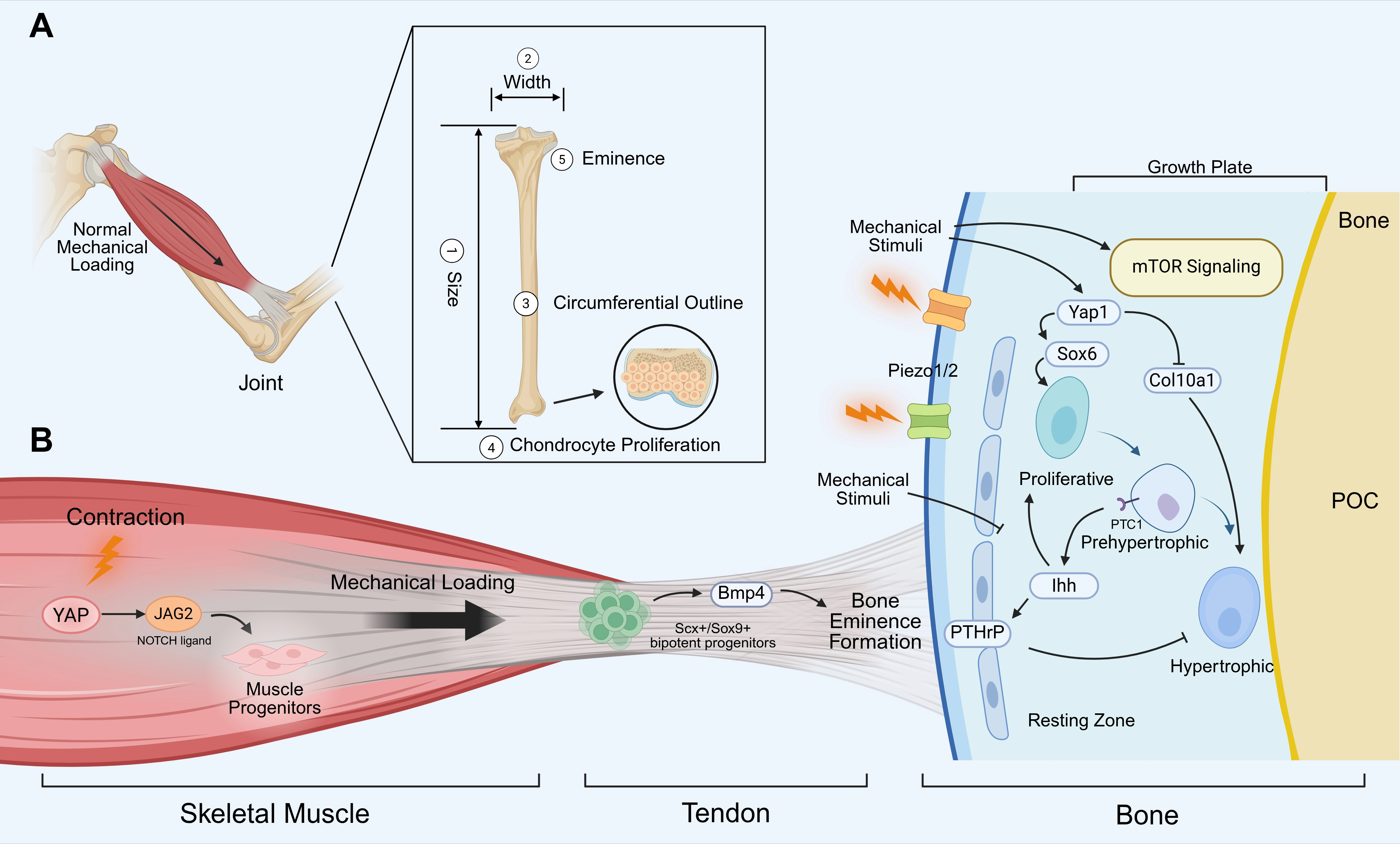
Figure 1. Mechanical loading in embryonic muscle-bone crosstalk. (A) The influence of mechanical stress on bone morphology. Mechanical stress from muscle contraction can regulate bone size, width, circumferential outline, and chondrocyte proliferation; (B) Mechanotransduction networks coordinating the developing musculoskeletal unit. Muscle contraction generates mechanical forces that maintain the fetal muscle progenitor pool via the YAP–JAG2 axis. These mechanical loads are transmitted through the tendon, where Scx+/Sox9+ bipotent progenitors at the enthesis, with Scx-stimulated Bmp4 being essential to drive bone eminence formation. In the adjacent skeletal compartment, mechanical stimuli are transduced by sensors such as Piezo1/2, leading to the activation of mTOR and Yap1 signaling. Yap1 can direct the regulation of Sox6 expression to promote chondrocyte proliferation, while inhibiting chondrocyte maturation by suppression of Col10a1 expression. The Ihh–PTHrP negative-feedback loop contributes to spatially orchestrating chondrocyte proliferation and hypertrophic differentiation within the growth plate. Ihh binds to its receptor PTC-1 to activate downstream signaling. Created in BioRender. Tang, C. (2026) https://biorender.com/cim0tgm. YAP: yes-associated protein; JAG2: jagged canonical notch ligand 2; Scx: scleraxis; Sox: SRY-Box transcription factor; Bmp4: bone morphogenetic protein 4; Piezo1/2: Piezo type mechanosensitive ion channel component 1/2; mTOR: mechanistic target of rapamycin; Col10a1: collagen type X alpha 1 chain; PTC1: patched-1; PTHrP: parathyroid hormone-related protein; Ihh: Indian hedgehog; POC: the primary ossification center.
The yes-associated protein (YAP) may represent a shared mechanosensitive pathway operating in both skeletal and muscle tissues (Figure 1B). In the skeletal compartment, Yap1 has been shown to promote chondrocyte proliferation and inhibit chondrocyte differentiation in vitro and in vivo[45]. Conversely, in the muscular compartment, YAP forced activity can prevent the loss of jagged canonical notch ligand 2 expression in muscle fibers and the decrease in the number of adjacent muscle progenitors in immobilized chick embryos[46]. Piezo type mechanosensitive ion channel component (Piezo) 1/2 is another important mechanosensitive pathway in embryonic bone development. Mice harboring a bone-specific deletion of Piezo1/2 exhibit inhibition of osteoblast differentiation and increased bone resorption, in which Yap1 activity was reduced[47]. Beyond embryonic function, Piezo1 expression in chondrocytes is required to generate the secondary spongiosa and thereby the trabecular bone compartment, a function that is relevant postnatally[48].
Additionally, muscle load was shown to regulate the bone width and circumferential outline. Mechanical load contributes to the process of mineral deposition; several studies show that the bones of paralyzed mice presented rounder bones than controls[49-51]. Finite element analysis further suggests that these rounder bones are mechanically inferior, supporting the concept that muscle-generated force contributes to the structural optimization of developing skeletal elements[51].
In addition to general skeletal scaling, mechanical loading specifically determines the localized morphology of bone. For instance, bone eminences are significantly smaller or completely lost in the absence of muscle activity[22]. The bone eminences serve as the insertion sites of tendons, constituting a critical structural interface for the transmission of mechanical forces between skeletal muscle and bone. While their maturation is synergistically regulated by muscle contraction and tendons, the initiation of formation is entirely independent of musculature[14]. Classical studies established that the transcription factor scleraxis (Scx) is requisite for tendinous anchoring, with connective tissue fibroblasts providing the essential spatial scaffold for muscle patterning[52,53]. Building on this, a recent study reveals that the establishment of attachment sites, including bone eminences, is orchestrated by resident Scx+/Sox9+ bipotent progenitors at the tissue interface (Figure 1B). The absence of Scx profoundly dysregulates the fate determination of these progenitor cells, leading to the developmental failure of the deltoid tuberosity and the formation of a rounded enthesis[54]. Collectively, these findings indicate that both muscle and tendons are essential for guiding the morphological maturation of specialized bone structures during embryonic development.
Besides its crucial role in the development of individual bones, mechanical strain governs the functional and structural integrity of the entire skeletal system by actively modifying periarticular matrices, tendons, and other connective tissues[22,55]. Taken together, these findings support a central role for mechanical stress in embryonic muscle-bone interactions and suggest that early biomechanical regulations help establish the structural basis for postnatal musculoskeletal function.
2.2 Secretory factors
Although the dominant influence of mechanical force makes it difficult to isolate the specific contribution of secreted factors during embryogenesis, biochemical signaling between developing musculoskeletal tissues likely also plays an important role. This is underscored by the fact that muscle-bone secretory signaling is established prior to functional contraction and is dynamically modulated by shifting cellular subtypes throughout development.
During early embryogenesis, myogenic progenitor cells exhibit secretory capabilities before the onset of muscle contractility. In the process of somite development, reciprocal signaling between the myotome and sclerotome orchestrates syndetome specification. Mechanistically, the fibroblast growth factor 8 (FGF8) signal emanating from the myotome’s center is received by the FGF receptor (FREK)-expressing cells at the anterior and posterior edges, and these cells, in turn, activate Scx in the sclerotome abutting the FREK-expressing myotome[56]. Beyond syndetome induction, FGF8 can stimulate sclerotomal proliferation and drive rib chondrogenesis; conversely, pharmacological blockade of FGF signaling causes deletions in developing ribs[57]. This suggests secreted factors primarily exert their morphogenetic effects during earlier developmental stages, which may represent an early form of musculoskeletal crosstalk.
The phenotypic comparison between the paralyzed models (retaining muscle tissue and paracrine secretions but lacking physical contraction) and the muscleless models (completely devoid of muscle tissue) provides a useful framework to uncouple the biochemical contributions of paracrine factors from the biomechanical effects of muscle contraction. Existing reviews have synthesized the divergent skeletal phenotypes between these two models. However, contrary to the expectation that the paralyzed model would exhibit a milder phenotype, it presents with severe developmental defects across nearly the entire skeleton. In contrast, the muscleless model demonstrates a selective preservation of certain skeletal elements, resulting in a less severe global phenotype[58]. Nowlan et al. discussed that this discrepancy may stem from differential exposure to external physical perturbations between chick (more paralyzed model) and mice (more muscleless model) embryos[58]. Besides, taking the correlation between muscle physiological contraction and secretory effects into consideration, the embryonic secretome in paralyzed models might be irregular. Subsequent evidence underscores that maternal exercise substantially rescued fetal akinesia-impaired joint and bone development and prevented disuse-induced resorption of the deltoid tuberosity[59]. These findings indicate that mechanical loading encompasses both internal and external sources, and its regulatory influence may outweigh that of secreted biochemical factors during the embryonic stage.
Several other myokines and osteokines hold potential regulatory roles in embryonic muscle-bone crosstalk. For instance, in the embryonic day 15 mouse fetus, Rankl mRNA has been detected in various endochondral skeletal sites, predominantly localized to the regions where bone formation and remodeling initially occur during skeletal development. Notably, strong levels of Rankl mRNA expression were present in the surrounding skeletal muscle[60]. Physiologically, RANKL acts in a paracrine manner to drive osteoclast lineage commitment and survival[60]. Emerging evidence indicates that RANKL signaling impairs muscle performance, and its inhibitor osteoprotegerin (OPG) can lessen this impact[61,62]. Therefore, such spatiotemporal alignment suggests the potential involvement of RANKL in embryonic muscle-bone crosstalk, although direct functional evidence for this role remains limited.
OCN acts as a profound pleiotropic hormone within skeletal muscle. By binding its particular receptor G protein-coupled receptor, class C, group 6, member A, OCN can promote myofiber proliferation and differentiation while enhancing insulin-independent glucose uptake[63-65]. Although embryonic skeletal expression of Ocn remains negligible until embryonic day E16.5, maternally derived OCN can traverse the placenta as early as E14.5[66]. While direct evidence connecting maternal OCN to embryonic muscle-bone crosstalk is currently lacking, its established function in brain development[66] implies a potential indirect pathway. Considering the essential role of motor innervation in promoting muscle structural development[67,68], maternal OCN might indirectly contribute to later musculoskeletal maturation.
Recent findings indicate that EVs, especially exosomes, may add a new dimension to musculoskeletal communication. During embryonic patterning, EVs presumably serve a supplementary function to the predominant signaling pathways (e.g., Wnt, Hh)[69,70]. However, while their roles in postnatal regeneration and aging-related pathologies, notably sarcopenia and osteoporosis[20,71,72], are well established, their precise in vivo contributions to early musculoskeletal assembly remain elusive.
2.3 Neural regulation and immune contribution of musculoskeletal system
Unlike mechanical stress and secreted factors, which primarily serve as intermediate mediators in embryonic muscle–bone crosstalk, the neural and immune systems modulate this interaction extrinsically. Concurrently, they exert an overarching regulatory effect on overall musculoskeletal development.
During embryonic development, nerves contribute to the maturation of both bone and muscle through distinct but interconnected mechanisms. The sensory nerve-bone interaction is governed by a distinct chemotactic loop, wherein nerve growth factor secreted by perichondrial cells attracts neurotrophic tyrosine kinase receptor type 1 (TrkA)-expressing sensory fibers to regions of active osteogenesis[73]. Disruption of TrkA signaling profoundly impairs innervation, coupled with delayed vascular invasion, a decrease in osteoprogenitors, and notably reduced long bone volume[73]. The paracrine release of other sensory neuropeptides, such as calcitonin gene-related peptide and substance P, can also promote osteogenesis through different pathways[74]. Collectively, these findings clarify a neuro-osteogenic axis, highlighting the sensory neural system as an important regulator of the initial skeletal framework.
The reciprocal interaction between motor neurons and skeletal muscle fundamentally dictates muscular maturity and structural integrity. While the early specification of myoblasts is largely autonomous of motor innervation, the subsequent differentiation depends on neurogenic trophic factors and nerve-induced electrical activity[75-78]. Their importance is underscored by data from aneural and paralyzed embryonic models. In paralyzed models, where innervation is physically present but functionally inactive, neuromuscular junctions (NMJs) can still develop, and myogenesis partially advances[79]. By contrast, the complete absence of neural innervation (aneural models) triggers far more than simple atrophy. It severely impairs muscle differentiation and precipitates progressive connective tissue replacement, indicating that both the trophic factors from the nerve and the nerve-evoked muscle activity are essential for the execution of the developmental program of the muscle[80]. The fibrosis process might be regulated by the persistent activation of signal transducer and activator of transcription 3 signaling in local fibro-adipogenic progenitors (FAPs), which redirects the microenvironment towards a fibrotic outcome[81]. Additionally, motor neurons can regulate bone development by stimulating muscle contraction, which is underscored by subsequent bone defects in the paralyzed embryo[58]. Therefore, the neural facilitation of embryonic bone and muscle development is primarily driven by paracrine signaling and electrical activity.
A critical unanswered question is whether immune cells directly take part in embryonic muscle–bone crosstalk. Current evidence suggests that immune cells are predominantly involved in embryonic morphogenesis[82,83], while the early interactions at the muscle-bone interface primarily rely on mechanical stress and local paracrine signaling. Future investigations utilizing spatial transcriptomics may elucidate whether a potential immune-mediated communication exists at the embryonic muscle-bone interface.
3. Muscle-Bone Crosstalk in Physiological Maturity
Postnatal musculoskeletal maturation marks a fundamental transition from generative morphogenesis to functional homeostasis. This shift is driven by the emergence of mechanical loading as a ubiquitous stimulus, coupled with the integration of maturing secretory networks, neural, and immune systems into muscle-bone crosstalk.
The musculoskeletal system’s relative homeostasis includes adaptation to a balanced metabolic network and physical activity. In adulthood, musculoskeletal pathology is predominantly driven by metabolic and biomechanical dysregulation. Chronic systemic disease or physical unloading is always the clinical cause. In conditions of Type 2 Diabetes Mellitus (T2DM), the muscle-bone unit can be impaired by systemic metabolic stressors. Myostatin plays a central role in this uncoupling. Its heightened expression in the skeletal muscle of individuals with T2DM can impede various pathways, such as phosphatidylinositol3-kinase (PI3K)/AKT/mTOR and Wnt/β-catenin, hindering osteoblast differentiation and bone mineralization[10]. Additionally, metabolic dysfunction-associated steatotic liver disease precipitates simultaneous muscle and bone wasting via shared pathogenic pathways, including insulin resistance, chronic inflammation, hormonal imbalance, and gut dysbiosis, which create a vicious cycle[84].
During this homeostatic phase, exercise acts as a major systemic regulator of musculoskeletal adaptation. The absence of physical activity, such as prolonged exposure to microgravity or long-term bed rest, can lead to osteoporosis and muscle atrophy[85,86]. Mechanosensitive pathways actively participate in it. In particular, Piezo1 activation can effectively attenuate muscle atrophy and bone resorption under pathological conditions[87]. In addition, it is now recognized that exercise can stimulate biochemical interactions between muscle and bone[88]. Indeed, irisin released by contracting muscles has been shown to promote osteoblast differentiation and proliferation in bone, increase bone density, improve bone quality, and enhance the mechanical support of cartilage by subchondral bone[89]. The exercise-induced muscle factor b-aminoisobutyric acid (BAIBA) is a bone-protective factor that prevents osteocyte cell death induced by reactive oxygen species[90]. Effects of exercise-induced myokines can also target the muscle itself, as the endogenous peptide apelin enhanced muscle function by triggering mitochondriogenesis, autophagy, and anti-inflammatory pathways in myofibers, as well as enhancing the regenerative capacity by targeting muscle stem cells[91]. Reciprocally, in response to physical activity, the skeleton secretes osteokines such as OCN, which is necessary to maintain muscle mass and can regulate muscle adaptation to exercise by favoring uptake and catabolism of glucose and fatty acids[92,93]. Together, these responses form a bidirectional mechanochemical loop, ensuring that tissue mass is dynamically adjusted to functional demands and thereby protecting against catabolic degradation.
In addition, exercise triggers a rapid release of EVs with the characteristic size of exosomes into the circulation[94]. Therefore, the reciprocal exosomal communication between muscle and bone can be stimulated[95], assisting in the maintenance of homeostasis in adulthood. EVs derived from skeletal muscle can transport lactate dehydrogenase A (LDHA), a key glycolytic enzyme, into bone marrow mesenchymal stem cells (BMSCs) to activate glycolysis, leading to increased osteogenic differentiation[96]. Although the amounts of LDHA-carrying EVs in response to exercise are not quantified, increased levels of LDHA after exercise are observed, and this is supported by a previous study that reported the presence of glycolytic enzymes in circulating EVs during incremental exercise[94]. Beyond transportation of functional miRNAs, EVs can package some myokines/osteokines[97,98], offering an unclassical secretory pathway into circulation.
In the adult stage, the maintenance of musculoskeletal integrity and mass is strictly related to neural and immune surveillance networks. Constitutive neural inputs via mature NMJs provide essential trophic support and electrical stimulation to muscle, preventing its denervation[99]. Resistance training can diminish functional loss in muscles despite only moderate increases in muscle mass, suggesting that the improvements are via neural adaptation[100]. In this process, the highly conserved Hippo pathway, with its main effectors YAP and transcriptional co-activator with PDZ-binding motif, is activated by exercise and interacts with various signaling pathways to increase skeletal muscle size[101-104]. Concurrently, the adult immune system plays an important dual role in skeletal muscle: hypertrophy in response to exercise and regeneration after injury[105,106]. Resident macrophages are an integral component of bone tissues and are important in bone homeostasis through regulating osteoblast function[107]. They positively influence fracture repair across multiple stages of the healing process[108].
In aggregate, adulthood represents a period of relative physiological stability compared to the embryonic and aging processes. The critical influence of mechanical loading persists from prenatal development into adulthood, where it operates in synergy with the mature secretory network to preserve musculoskeletal function and mass. The nervous and immune systems actively co-regulate this systemic network, while immune cells perform a secondary function by actively clearing damaged tissue and governing bone remodeling.
4. Impact of Aging on the Dynamics of Muscle–Bone Crosstalk
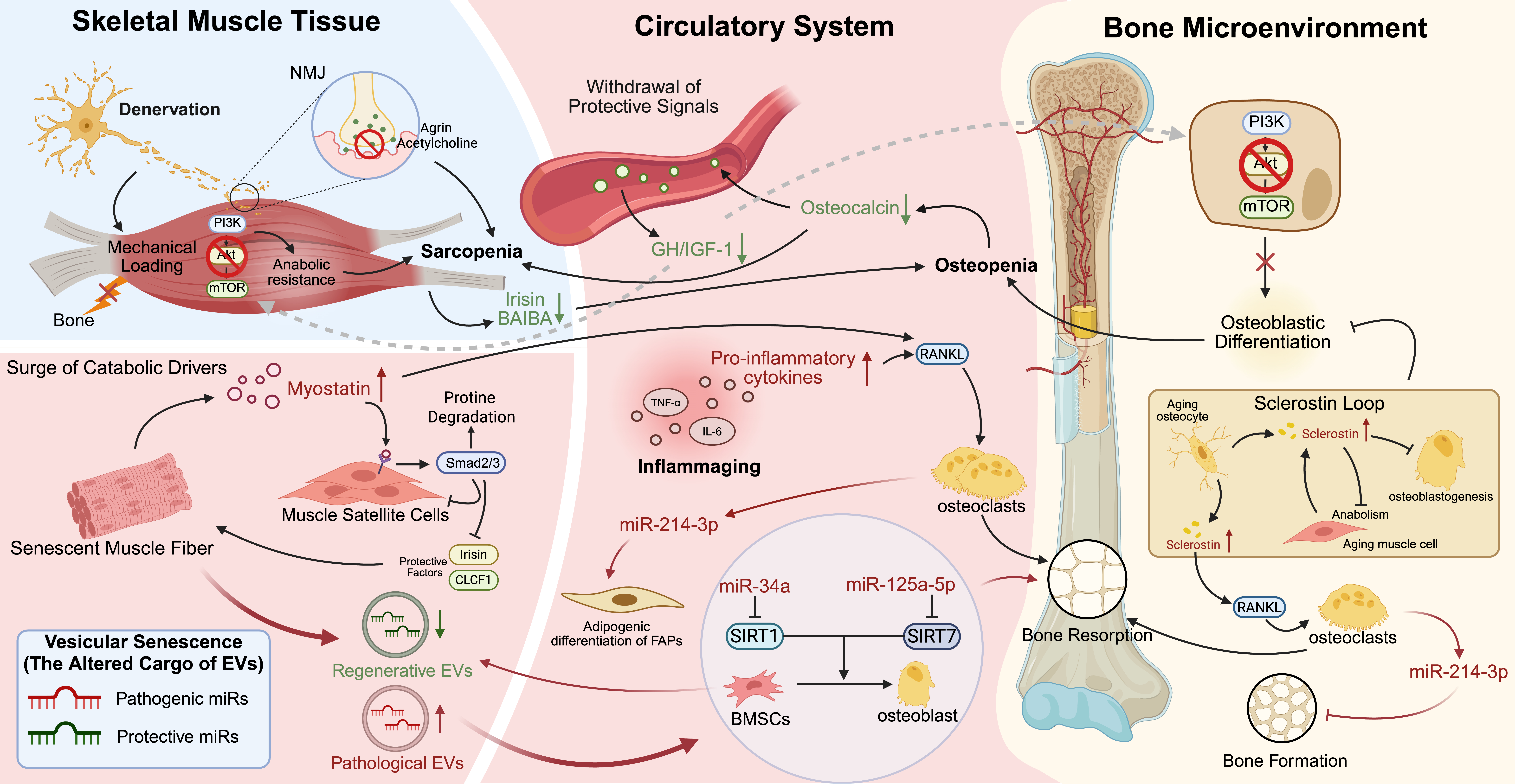
As the organism transitions from adult homeostasis to senescence, age-related dysregulation of the secretory network fundamentally disrupts muscle–bone crosstalk. This breakdown is further exacerbated by neurodegeneration and immunosenescence (Figure 2). Consequently, this systemic decline transforms their physiological synergy into a pathological, mutually detrimental feed-forward loop, ultimately accelerating the onset of osteosarcopenia[109].
{kind=link}
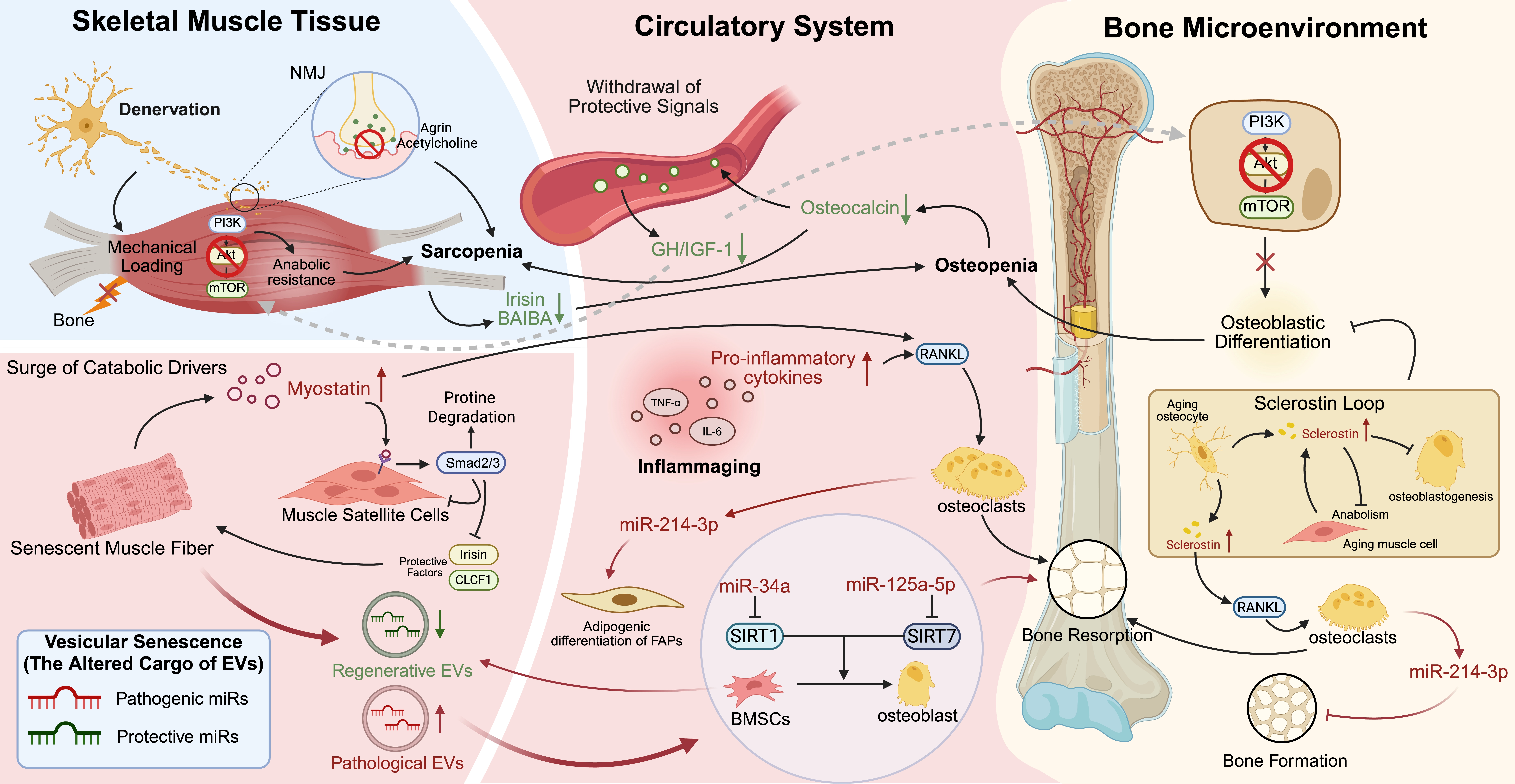
Figure 2. Aging induces a transition from homeostatic crosstalk to a mutually destructive feed-forward loop, driving osteosarcopenia. Denervation and anabolic resistance in skeletal muscle impair tissue maintenance, while senescent fibers release myostatin and pathological EVs. Systemically, protective myokines and osteokines (irisin and osteocalcin) are withdrawn, paralleled by a surge in pro-inflammatory cytokines (inflammaging) and pathological EVs. In the bone niche, these signals converge to suppress osteoblastogenesis (via Sirt1/7 blockade), hyperactivate osteoclasts, and fuel a vicious “sclerostin loop” that dually impairs bone and muscle anabolism. Red arrows highlight pathological microRNA signaling pathways. Grey arrows indicate that the impact of GH/IGF-1 axis dysregulation on the musculoskeletal system remains controversial. Created in BioRender. Tang, C. (2026) https://BioRender.com/444zkv6. NMJ: neuromuscular junction; GH: growth hormone; IGF-1: insulin-like growth factor 1; BAIBA: b-aminoisobutyric acid; PI3K: phosphatidylinositol3-kinase; mTOR: mechanistic target of rapamycin; CLCF1: cardiotrophin-like cytokine factor 1; TNF-α: tumor necrosis factor alpha; IL-6: interleukin 6; RANKL: receptor activator of NF-κB ligand; EVs: extracellular vesicles; FAPs: fibro-adipogenic progenitors; BMSCs: bone marrow mesenchymal stem cells.
4.1 The withdrawal of protective anabolic signals
A defining hallmark of aging is the systemic withdrawal of protective, tissue-derived signals that sustain musculoskeletal integrity. As an important mediator in muscle-bone crosstalk, bone-derived OCN signaling in myofibers is indispensable for mass preservation[92]. It is also an essential pathway for muscle adaptation to exercise by favoring the uptake and catabolism of glucose and fatty acids[93]. Data from mice, monkeys, and humans show a similar age-related decline of serum OCN, in synergy with declined exercise capacity[93]. Similarly, clinical data show that the irisin serum levels are negatively correlated with age[110]. In patients with osteopenia/osteoporosis, the irisin serum levels were significantly lower, and treatment with irisin decreased the expression of p21 in murine osteoblasts, suggesting a potential senolytic action of this myokine[110]. Furthermore, the exercise-induced protective factors, like BAIBA and apelin mentioned above, are dysregulated in aging. Within the mice model, the protective effect of BAIBA was found to be lost with age, not due to loss of the muscle capacity to produce BAIBA but likely due to inhibited mas-related G protein-coupled receptor type D expression with aging, a signaling preventing mitochondrial breakdown in osteocytes[90]. The release of apelin is reduced in an age-dependent manner in humans and rodents and is positively associated with the beneficial effects of exercise in older persons[91]. Another exercise-induced protective factor, CLCF1, whose upregulation in plasma levels after exercise decreases with age in both humans and rodents, might be attributable to age-related alterations in cellular signaling, receptor sensitivity, or transcriptional regulation[30].
Aging is associated with a decrease in growth hormone (GH) and insulin-like growth factor 1 (IGF1) secretion. This age-associated attenuation of the GH/IGF-1 axis was recognized as a potential contributor to osteosarcopenia[111]. IGF-1/PI3K/Akt/mTOR signaling functions as a classical anabolic pathway. However, in catabolic situations, like aging, the absence of IGF-1 results in activation of forkhead box protein O3, which regulates the expression of its downstream targets atrogin-1 and muscle-specific RING finger protein 1 (Murf-1) that cause proteasomal degradation of proteins[112]. Specifically, within muscle tissue, IGF-1 plays a central role in muscle growth, differentiation, and regeneration[113], and aging appears to result in a mTOR complex 1 signaling decline in some specific muscles[114]. Simultaneously, bone matrix IGF-1 stimulates the same PI3K/Akt/mTOR axis to promote osteoblastic differentiation of recruited mesenchymal stem cells, thus maintaining proper bone microarchitecture and mass[115]. Suppression of Akt activity in skeletal muscle of mammals could accelerate osteosarcopenia and consequently reduce lifespan[116]. Consequently, the dysregulation of this upstream axis may contribute to a synchronous failure in muscle and bone.
4.2 The surge of catabolic drivers
During aging, the detrimental upregulation of growth inhibitors and catabolic drivers disrupts muscle–bone crosstalk. Originally identified as a potent negative regulator of muscle mass, myostatin signaling is aberrantly upregulated in aging and sarcopenia[28]. Myostatin binds to Activin receptor type II on muscle cell membranes and activates the Smad2/3 signaling pathway, which can suppress myoblast proliferation and differentiation[117,118]. Besides, activated Smad2/3 upregulates the expression of atrogin-1 and Murf-1, thereby accelerating the degradation of muscle fiber proteins[119]. Furthermore, myostatin directly inhibits transcription of protective myokines like irisin and CLCF1 in muscle cells, thereby deepening the catabolic state[28]. Beyond causing muscle wasting, dysregulation of myostatin directly targets bone. It promotes osteoclastogenesis by regulating the RANKL-induced NF-κB pathway and inhibits osteoblast differentiation[9,10]. This transforms skeletal muscle from a supportive scaffold into an osteotoxic source.
Parallel to this, the osteocyte-secreted Wnt inhibitor, sclerostin, is often upregulated in aging bone, particularly under conditions of mechanical unloading[28,120,121]. Under physiological conditions, sclerostin regulates bone remodeling by inhibiting Wnt-related pathways. However, aging-induced sclerostin overexpression disrupts this balance. By blocking activation of osteogenic transcription factors like Runx family transcription factor 2 (Runx2) downstream of Wnt signaling, sclerostin inhibits osteoblast differentiation and bone matrix deposition to cause bone loss[121,122]. The role of osteocyte-derived sclerostin in muscle homeostasis during aging is controversial[1]. Notably, skeletal muscle secretes sclerostin, which might work synergistically with bone-derived sclerostin to strengthen the negative regulatory mechanism of osteogenesis[123]. Emerging data demonstrated aging-associated up-regulation of sclerostin expression in mouse skeletal muscles, which is negatively correlated with skeletal muscle weight, suggesting that skeletal muscle-derived sclerostin may play a role in aging-associated skeletal muscle atrophy[124].
4.3 Immune and neural dysregulation
Aging is characterized by chronic, low-grade, sterile inflammation, driven predominantly by the senescence-associated secretory phenotype (SASP)[31]. Highlighting this, recent single-cell analysis has identified a subset of lipid-associated macrophages (triggering receptor expressed on myeloid cells 2, TREM2+ Macs) in both aged muscle and bone, indicating immune landscape remodeling underlies synchronized muscle and bone aging[125]. Age-related pro-inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6), potently upregulate RANKL production while inhibiting its decoy receptor, OPG. This imbalance stimulates osteoclast activity and bone resorption[28,126]. The roles of RANK/RANKL signaling in muscle are primarily detrimental. For instance, transgenic RANKL overexpression can decrease muscle mass, force, and glucose uptake, while anti-RANKL treatments always improve muscle function[127,128]. Notably, under physiological conditions, RANKL regulates both bone and muscle by inducing mitochondrial biogenesis and oxidative metabolism in skeletal muscle fibers[129].
IL-6 is a candidate for promoting cell senescence and an important part of SASP[31]. While transient IL-6 is anabolic during exercise[130], chronic basal elevation of IL-6 in aging acts as a catabolic factor, promoting both muscle atrophy and bone resorption[81,131,132]. Recent frontiers have expanded this immune-muscle-bone axis beyond the classic IL-6 paradigm. In humans, lower levels of interleukin-33 (IL-33) were linked to an increased risk of co-occurrence of sarcopenia and osteoporosis, indicating its potential function in musculoskeletal aging[133]. However, the effects of IL-33 are context-dependent. On the one hand, IL-33 exerts a protective effect on bone formation. IL-33 can inhibit osteoclastic differentiation through the IL-33/miR-34a-5p/Notch1 pathway and suppress bone resorption induced by TNF-α[134,135]. On the other hand, IL-33 may have adverse effects on bone metabolism. As a pro-inflammatory cytokine, IL-33 participates in inflammatory bone loss, causing the production of bone resorption factors[136].
During aging, the musculoskeletal unit suffers from a progressive failure of the peripheral nervous system[100]. NMJs serve as the nexus between the nervous and muscular systems. Therefore, NMJ fragmentation and transmission failure in aging can subsequently lead to loss of muscle mass and weakness[137]. This synaptic detachment can exacerbate sarcopenia by the loss of mitochondrial catalase and increased oxidative stress[138]. As an important component of NMJ, active acetylcholine receptors have been shown to prevent muscle atrophy[139], but their numbers and function decline with age. Specifically, motor innervation of muscle will subsequently leads to progressive skeletal atrophy[140]. A recent study demonstrates that sensory nerves constitute a critical component of the skeletal stem cell (SSC) niche, regulate SSC self-renewal and osteogenic differentiation[141]. In aggregate, age-related neural denervation can halt both the biochemical and the biomechanical signals, thereby contributing to osteosarcopenia.
4.4 The altered cargo of EVs
Recent studies have identified EVs, especially exosomes, as essential carriers of the senescence-associated signals. In contrast to soluble cytokines, EVs contain intricate genetic and protein materials, enabling them to transmit aging signals to distant tissues[142]. In the aging musculoskeletal unit, the EV secretory profile undergoes a pathological shift from “regenerative mediators” to “senescence messengers”. For instance, miR-34a was identified as a potent accelerator of age-related dysfunction. Aged myotubes secrete EVs that are significantly enriched in miR-34a compared to those secreted by younger myotubes[143]. Upon uptake by BMSCs, these muscle-derived EVs suppress the expression of the pro-osteogenic factor SIRT1, thereby inducing cellular senescence and inhibiting osteoblast differentiation[143]. This suggests that EVs from aging muscle actively influence the bone microenvironment, propagating a senescent signal rather than merely failing to provide support.
This pathological degeneration of function is compounded by the loss of protective or regenerative cargoes. MiR-181a, an exercise-induced miRNA, has been predicted to regulate transcription factors and co-activators involved in the adaptive response of muscle to exercise[144]. Under physiological conditions, miR-181a also contributes to osteogenesis[145]. Downregulation of miR-181a with age was associated with an accumulation of autophagy-related proteins and abnormal mitochondria in muscle[146]. The regenerative capacity of bone-derived exosomes also declines. In the healthy state, miR-126-5p derived from BMSC exosomes promotes skeletal muscle regeneration by regulating F-box protein 32 (FBXO32)/MyoD signaling[72]. In post-menopausal osteoporotic women, miR-126-5p is specifically associated with muscle mass wasting[147].
Conversely, the pathological EVs emerge with age, contributing to musculoskeletal degeneration. Specific miRNAs derived from muscle (myomiRs), miR-1 and miR-133a, were found to increase with age[148]. The age-related alteration of miR-133a is negatively correlated with serum response factor expression, which is vital for muscle regeneration after injury[148]. In skeletal tissue, miR-133a can target Runx2 to inhibit osteogenesis[144]. Similarly, atrophic skeletal muscle secretes abundant miR-125a-5p. It directly targets SIRT7 in preosteoblasts, abrogating SIRT7-mediated histone deacetylation at the Sp7 promoter to suppress Sp7 transcription and subsequent osteogenic differentiation, leading to sarcopenia-related osteoporosis[149]. In aging, increased osteoclastic miR-214-3p associates with both elevated serum exosomal miR-214-3p and reduced bone formation in elderly women with fractures[150]. It can be taken up by skeletal muscle FAPs, promoting adipogenic differentiation, thereby increasing intermuscular adipose tissue infiltration and decreasing muscle strength[151].
Beyond these local interactions, the composition of circulating EVs serves as a systemic indicator of biological age. A recent study revealed that the protein cargo of EVs correlates strongly with the epigenetic aging clock, suggesting that EV-mediated signaling is intrinsically tied to the organism’s systemic metabolic and epigenetic state[152]. This “vesicular clock” likely coordinates the synchronized decline of muscle and bone by circulating pro-inflammatory and anti-regenerative factors throughout the musculoskeletal system, reinforcing the concept that osteosarcopenia is driven by a systemic failure of vesicular communication[1,20,142,153].
5. Changes of Muscle-bone Crosstalk Across the Lifespan
Throughout the lifespan, the paradigm of muscle-bone crosstalk transitions from synergistic co-development during embryogenesis to relative homeostatic maintenance in adulthood, and ultimately to a pathological vicious cycle during aging (Figure 3). Fundamentally, these profound physiological shifts are driven by dynamic alterations in the intrinsic functions, synergistic interactions, and relative contributions of key mediators: mechanical loading, the secretory network, and neuro-immune regulation. During embryonic development, mechanical stress predominates in driving the positive muscle-bone interplay. While the precise role of the secretory network remains elusive but holds immense potential, neural and immune factors orchestrate regulation at the systemic level. In adulthood, persistent mechanical loading remains central, while secretory, neural, and immune pathways act together to maintain tissue adaptation and homeostasis. Conversely, during aging, the secretory network becomes highly dysregulated. The inherently high interactivity between muscle and bone underpins a vicious cycle of senescence, which is further exacerbated by neurodegeneration and immunosenescence, collectively accelerating the deterioration of the musculoskeletal system.
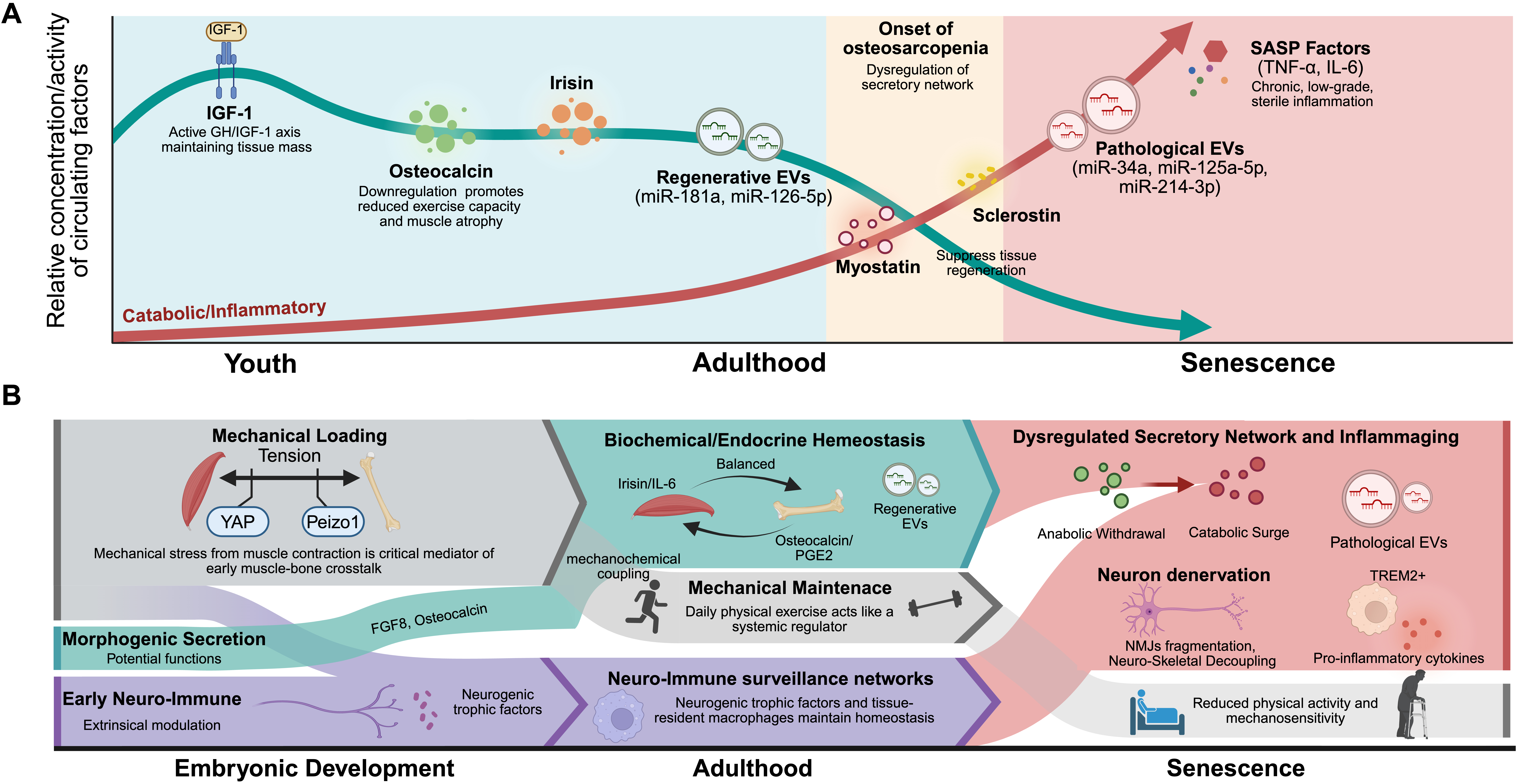{kind=link}
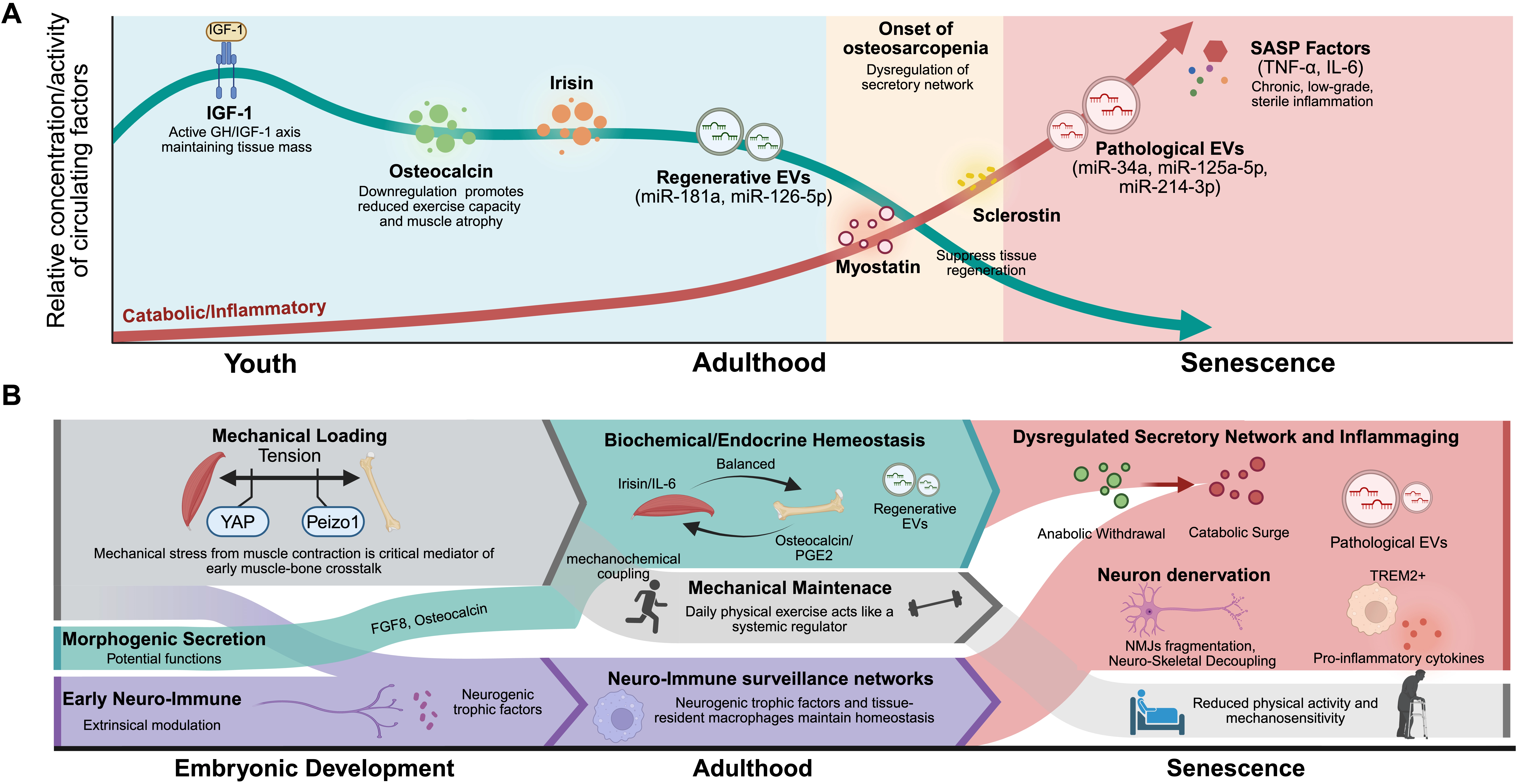
Figure 3. Lifespan trajectories of muscle–bone crosstalk and the pathogenesis of osteosarcopenia. (A) Temporal shifts in the systemic secretome. The age-related pathogenesis of osteosarcopenia is driven by the gradual withdrawal of anabolic/regenerative networks and a corresponding upsurge in catabolic/inflammatory signaling; (B) Stage-specific mechanistic transitions within the musculoskeletal unit. Early muscle–bone crosstalk relies on mechanical tension and morphogenetic secretion for reciprocal development. During adulthood, continuous mechanical maintenance and biochemical homeostasis (e.g., balanced myokines and osteokines) preserve tissue integrity. Ultimately, aging disrupts this homeostatic plateau, triggering a vicious cycle characterized by neuroskeletal decoupling, secretory dysregulation, and inflammaging. Created in BioRender. Tang, C. (2026) https://BioRender.com/lmer6pc. GH: growth hormone; IGF-1: insulin-like growth factor 1; EVs: extracellular vesicles; SASP: senescence-associated secretory phenotype; TNF-α: tumor necrosis factor alpha; IL-6: interleukin 6; YAP: yes-associated protein; FGF8: fibroblast growth factor 8; PGE2: prostaglandin E2; TREM2: triggering receptor expressed on myeloid cells 2; NMJ: neuromuscular junction.
6. Muscle-Bone Crosstalk in Cancer
As the understanding of muscle-bone crosstalk deepens, increasing attention has shifted toward its role in cancer, a condition with an incidence that rises significantly with age. The intimate coupling between muscle and bone persists pathologically during cancer progression, where bone metastasis is frequently accompanied by profound skeletal muscle defects[154]. Recent studies have identified various tumor-derived secreted molecules and EVs that not only facilitate metastasis but simultaneously drive bone destruction and muscle atrophy. Furthermore, systemic metabolic disruptions, including hepatic and mitochondrial dysfunctions, severely impair muscle regeneration and exacerbate muscle loss[154]. Mechanistically, transforming growth factor beta (TGFβ) acts as a central mediator in this pathological crosstalk. Osteolytic cancer in the bone leads to release of TGFβ from the bone, which can induce skeletal muscle weakness via the TGFβ-NADPH oxidase 4-ryanodine receptor (TGFβ-Nox4-RyR1) axis[155].
Beyond the direct effects of cancer cachexia, anti-cancer treatments severely compromise musculoskeletal integrity, profoundly diminishing patients’ quality of life[156]. Chemotherapy-induced bone loss can cause muscle weakness by disrupting both endocrine signaling and mechanical loading[156]. For instance, estrogen deprivation therapy in breast cancer accelerates bone turnover, which cascades into muscle wasting and dysfunction[157]. Consequently, developing strategies to preserve muscle-bone homeostasis during chemotherapy needs further investigation.
7. Intervention Strategies Targeting Osteosarcopenia
Current interventions for osteosarcopenia primarily encompass pharmacological treatments and physical exercise, while several novel therapeutic modalities are also emerging. To effectively mitigate disease progression[158], it is essential to implement stage-specific and personalized strategies that address the age-dependent alterations in muscle-bone crosstalk.
7.1 Pharmacological interventions
Current management of osteosarcopenia largely relies on pharmacological therapies targeting its skeletal component, together with broader strategies aimed at preserving muscle mass and function[159]. Moreover, pharmacological approaches are shifting towards dual-action agents that simultaneously target both tissues. Given that elevated myostatin acts as a shared catabolic mediator in both muscle and bone during aging and diabetes, targeting this pathway potentially offers dual musculoskeletal benefits[9,10,117]. Recent studies utilizing myostatin antibodies have demonstrated improvements in muscle and bone mass, as well as strength, in a mouse model of insulin‐deficient diabetes[160]. To date, the substantial number of myostatin inhibitors entering the preclinical pipeline underscores its pivotal role in the management of osteosarcopenia[161]. While primarily osteo-anabolic, new sclerostin inhibitors also exhibit potential myo-protective effects[121,162]. Hormonal therapies hold promising potential in promoting both muscle and bone health. However, the adverse effects associated with hormone replacement therapies remain a concern, limiting their widespread use[163].
7.2 Exercise interventions
In the early stages of age-related musculoskeletal decline, exercise serves as a “physiological polypill”, restoring mechanical input and partially correcting mediator dysregulation (Figure 4). Resistance training may reactivate the mechanosensitive pathways, including Piezo1-related signals in osteocytes and satellite cells, which are diminished with aging[85,164]. This mechanical loading can suppress the expression of sclerostin and RANKL in osteoblastic lineage cells and may help shift the bone microenvironment from resorption to formation[165].
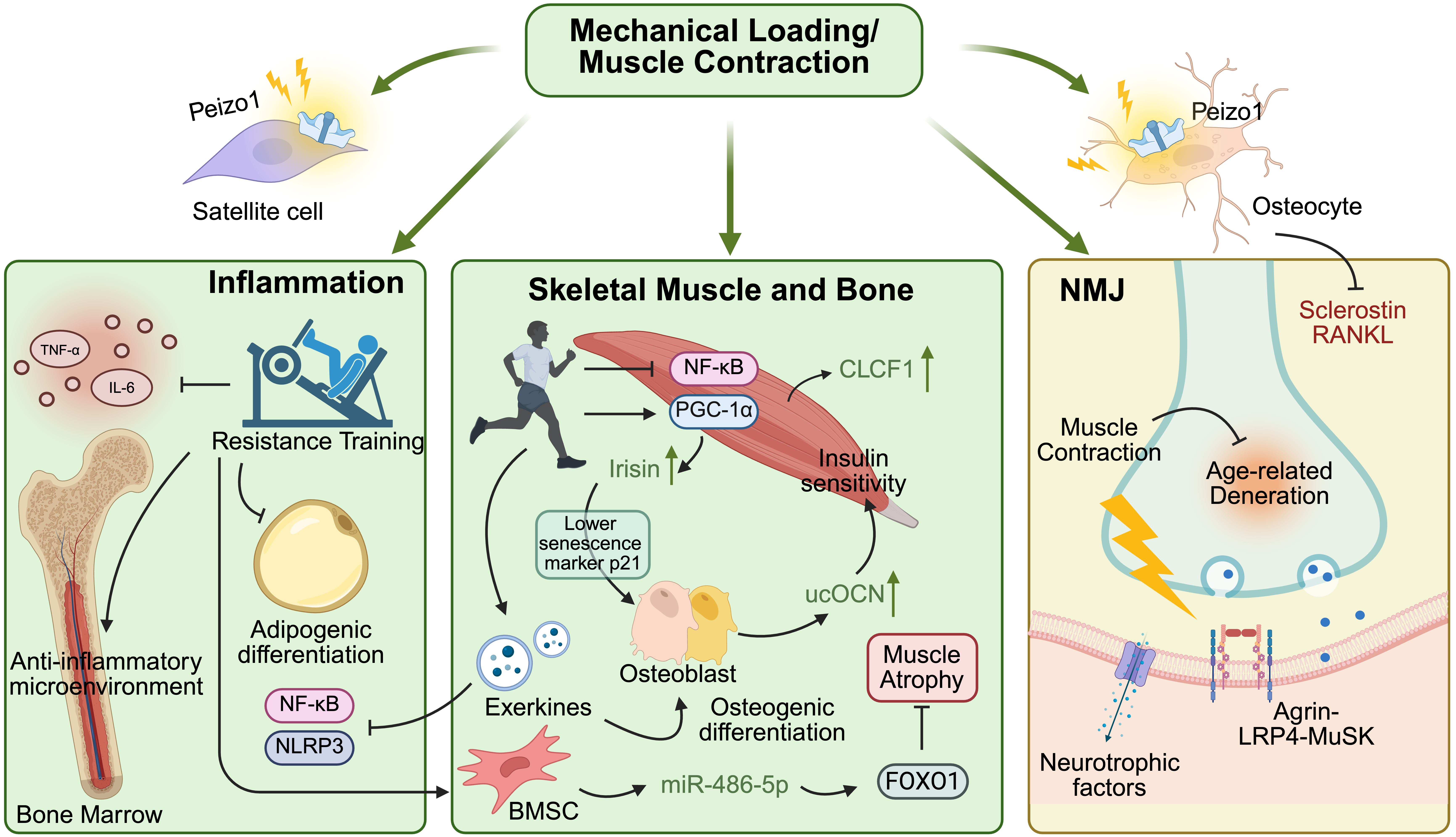{kind=link}
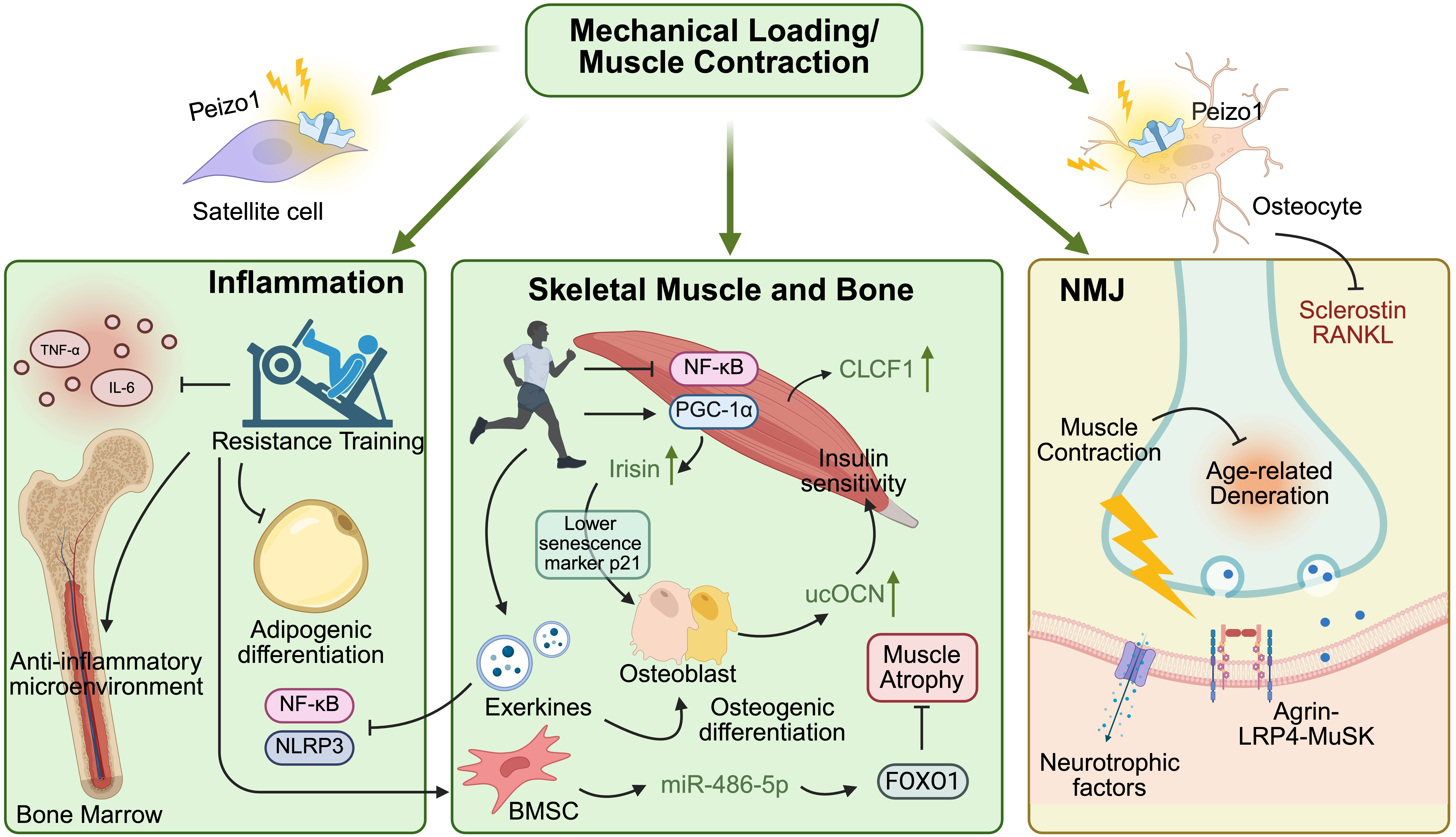
Figure 4. Pleiotropic effects of exercise on the aging musculoskeletal system. Muscle contraction and mechanical loading activate cellular mechanosensors to counteract age-related tissue decline via multiple systemic networks. In the bone marrow compartment, physical exertion prevents adipogenesis and limits inflammaging by suppressing the NF-κB and NLRP3 pathways, reducing local TNF-α and IL-6 levels. Within the muscle–bone unit, exercise shifts the biochemical balance toward anabolism: exerkines (irisin, CLCF1) and BMSC-derived miR-486-5p promote osteogenesis and inhibit FOXO1-mediated muscle atrophy, while catabolic signals (myostatin, sclerostin) are concurrently suppressed. At the nervous interface, continuous mechanical input sustains NMJ structural integrity and delays synaptic degeneration. Created in BioRender. Tang, C. (2026) https://BioRender.com/jwl3j3j. TNF-α: tumor necrosis factor alpha; IL-6: interleukin 6; Piezo1: Piezo type mechanosensitive ion channel component 1; NF-κB: nuclear factor-kappa B; RANKL: receptor activator of NF-κB ligand; NLRP3: NOD-, LRR-, and pyrin domain-containing protein 3; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; CLCF1: cardiotrophin-like cytokine factor 1; ucOCN: undercarboxylated osteocalcin; FOXO1: forkhead box protein O1; NMJ: neuromuscular junction; LRP4: low-density lipoprotein receptor-related protein 4; MuSK: muscle-specific kinase; BMSC: bone marrow mesenchymal stem cell.
Exercise remodels the muscle-bone crosstalk in aging by activating protective pathways and inhibiting pro-degenerative signals: activating irisin and CLCF1 secretion[30,89]; inducing muscle-derived IL-6 acting in osteoblasts that express OCN[1,166]; inhibiting NF-κB pathway activity, which can contribute to osteoclastogenesis and osteoclast activation[1,167]; reducing expression of sclerostin in osteocytes[168]. These factors enter a positive feedback loop: Irisin lowers the osteoblast senescence marker p21, while bone-derived ucOCN can enhance muscle insulin sensitivity and myofiber adaptation[1,109].
Exercise may also influence muscle-bone crosstalk primarily by reprogramming the cargo of skeletal muscle-derived extracellular vesicles (SKM-EVs) during aging[169,170]. Mechanistically, contraction-induced mechanical stress alters intracellular sorting pathways, selectively enriching SKM-EVs with pro-regenerative “exerkines”[169-171]. For example, following high-intensity resistance exercise in humans, skeletal muscle pri-miR-1a increased ~2.5-fold while EV-associated miR-1 in circulation increased during recovery, consistent with enhanced export of a muscle-derived exerkine signal[172]. Upon release, these exercise-induced SKM-EVs may exert dual anti-aging effects: within the vascular microenvironment, these vesicles target endothelial cells to promote proliferation and tubulogenesis, thereby protecting against tube formation impairments and induction of cellular senescence following acute oxidative stress[173]; concurrently, within the skeletal system, SKM-EVs carrying specific miRNAs reach the bone matrix via the systemic circulation and are internalized by osteoblasts, directly reshaping their downstream transcriptional responses to stimulate osteogenic differentiation[170]. Reciprocally, exercise can increase the release of bone exosomal miRNAs that help to preserve muscle mass. A specific example is the mechanical loading of bone tissue, increasing the release of exosomal miR-486-5p from BMSCs, which targets the muscle forkhead box protein O1 pathway to prevent muscle atrophy[174]. Consequently, exercise partially disrupts the vicious cycle of osteosarcopenia through the muscle-vascular-bone vesicular communication network.
In addition, exercise serves as a potent intervention to mitigate immunosenescence[175,176]. Regular exercise in older adults can enhance physical performance, decrease systemic inflammatory mediators associated with the SASP (such as IL-6 and TNF-α), and reduce markers of T-cell senescence and immune dysfunction[177]. Through systemic cellular crosstalk, exercise-induced myokines and circulating EVs may contribute to shaping anti-aging immune balance through direct immunomodulation (polarizing macrophages and T cells), metabolic regulation (optimizing glucose/lipid metabolism to reduce metabolic stress on immune cells), and systemic anti-inflammatory signaling (suppressing NF-κB pathways)[176,178]. Importantly, exercise benefits extend beyond the peripheral circulation to optimize the microenvironment of the bone marrow. A study found that a 5-week aerobic exercise increased BMSC count and osteogenic potential while suppressing adipogenic tendencies in mice[179]. Moreover, resistance exercise can improve bone strength in older female rats by increasing the osteogenic potential of BMSCs and decreasing adipogenic differentiation and pro-inflammatory cytokine levels[180]. As the bone marrow coordinates both hematopoiesis and bone remodeling, this exercise-driven reduction in systemic SASP directly protects the musculoskeletal system from inflammatory damage, thereby delaying its synchronized age-related decline.
Concurrently, the progressive degeneration of the NMJ and the subsequent denervation of muscle fibers exacerbate the situation[137,140]. Modified and regular exercise interventions in older adults may partially counteract this degenerative progression by promoting compensatory reinnervation, which rescues denervated muscle fibers and preserves the structural integrity of motor units[181]. Exercise-induced repeated muscle contractions significantly sustain the key signaling pathways’ activities at the postsynaptic membrane, such as the agrin-LRP4-muscle-specific kinase axis[182], and locally upregulate the secretion of neurotrophic factors, thereby stabilizing the NMJ both structurally and biochemically[182]. Consequently, by successfully maintaining the integrity of nerve-muscle communication, exercise sustains muscle functions, ultimately disrupting the cascade of age-induced musculoskeletal decline[183].
7.3 Potential next-generation strategies
For frail older adults suffering from severe anabolic resistance, traditional exercise interventions are often unfeasible. Here, vesicular engineering acts as a potential next-generation therapy, moving towards engineering the extracellular environment to mimic the “youthful” secretome. Restoring miR-181a levels in old mice prevented the accumulation of aging markers and improved mitochondrial quality and muscle function[146]. Exosomes derived from healthy BMSCs can deliver miR-126-5p to aged muscle. This cargo targets and downregulates the atrophy gene FBXO32, stabilizing MyoD and promoting muscle regeneration[72]. Compared to BMSCs, exosomes derived from human fetal cartilage-derived progenitor cells (F-Exo) exhibited superior muscle regeneration capabilities. In vitro, F-Exo upregulated myogenic markers and enhanced myotube formation in myoblasts; in vivo, it significantly increased muscle mass and fiber area in sarcopenic rats. Mechanistically, miR-145 participates in it to activate the Wnt signaling pathway; however, the precise underlying mechanisms further need to be investigated[184]. Consequently, the exogenous supplementation of pro-regenerative EVs provides a promising therapeutic strategy (Table 1).
Table 1. Different interventions targeting osteosarcopenia.
| Type of interventions | Specific mediators | Main functions in osteosarcopenia | References |
| Pharmacological Interventions | Myostatin inhibitors | Enhance skeletal muscle mass, skeletal functions, and bone properties. | [160,161] |
| Sclerostin inhibitors | The novel aptamers could significantly increase bone mineral density and improve bone microarchitecture, bone biomechanics, muscle function and histological properties of muscle and bone, without adverse cardiovascular effects. | [162] | |
| Hormone replacement | The clinical benefits have not always been consistently observed and the adverse effects restrict their widespread use. | [163] | |
| Exercise Interventions | Resistance exercise | Increase in circulating plasma CLCF1 levels; increase serum irisin concentrations; activate Piezo1 activity; raise ucOCN; increase miR-1 in circulation during recovery, consistent with enhanced export of a muscle-derived exerkine signal; improve bone strength in older female rats. | [30,89,164,166,172,180] |
| Interval exercise | Increase in circulating plasma CLCF1 levels; raise ucOCN; decrease both bone resorption and inhibition of bone formation (Sost mRNA). | [30,166,168] | |
| Regular exercise | Decrease mediators associated with the SASP, and reduce markers of T-cell senescence and immune dysfunction; counteract NMJ degenerative progression. | [177,181] | |
| Aerobic exercise | Increased BMSC count and osteogenic potential. | [179] | |
| Moderate-intensity exercise | Improve inflammation by inhibiting the PI3K/Akt/NF-κB and further NLRP3/caspase-1/GSDMD signaling pathways. | [167] | |
| Potential Next-generation Strategies | Pro-regenerative EVs | Showcases significant potential, though further rigorous validation is warranted. | [72,146,184] |
ucOCN: undercarboxylated osteocalcin; Piezo1: Piezo type mechanosensitive ion channel component 1; CLCF1: cardiotrophin-like cytokine factor 1; SASP: senescence-associated secretory phenotype; NMJ: neuromuscular junction; PI3K: phosphatidylinositol3-kinase; NF-κB: nuclear factor-kappa B; NLRP3: NOD-, LRR-, and pyrin domain-containing protein 3; GSDMD: gasdermin D; EVs: extracellular vesicles.
8. Conclusion
Muscle–bone crosstalk is not a static physiological state, but a dynamic, lifelong continuum. Mechanical, secretory, neural, and immune modes of communication shift in their relative prominence from embryogenesis to adulthood and aging. During embryogenesis, mechanical coupling is especially important for morphogenesis. In adulthood, these regulatory systems remain relatively balanced to support homeostasis. In aging, the cytokine- and EV-mediated secretory networks shift from trophic to deleterious, driving the concurrent tissue degeneration characteristic of osteosarcopenia.
Despite these insights, understanding these complex dynamics highlights substantial knowledge gaps. The upstream mechanisms driving the spatiotemporal shifts of these crosstalk mediators, and their precise interplay with the hallmarks of aging, remain poorly defined. Furthermore, methodological constraints continue to obscure the complete embryonic secretory interactome. There is a critical need to delineate these downstream signaling cascades and systematically map the muscle-bone interactome across life stages. Similarly, profiling the comprehensive landscape of EVs and deciphering their specific pathogenic roles in aging requires the development of novel in vivo tracking models. Clinically, while numerous translational strategies for osteosarcopenia are progressing through clinical trials, the field still lacks standardized and personalized therapeutic paradigms.
Ultimately, conceptualizing the musculoskeletal system as an integrated functional unit is the cornerstone of future therapeutic development. By shifting the clinical focus toward targeting shared signaling networks rather than isolated tissue interventions, the field can develop more effective strategies to mitigate age-related physical frailty and promote healthy longevity.
Acknowledgements
The authors declare that Gemini 3.1 PRO was used solely for language polishing during the manuscript preparation process. The authors take full responsibility for the integrity, originality, and accuracy of the work.
Authors contribution
Tang C, Yin X: Investigation, data curation, formal analysis, writing-original draft, writing review & editing.
Zhang H: Writing review & editing.
Conflicts of interest
Hongbo Zhang is an Editorial Board Member of Ageing and Cancer Research & Treatment. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the Ministry of Science and Technology of China (Grant Nos. 2022YFA1104900 and 2024ZD0530500), the National Natural Science Foundation of China (Grant Nos. W2511023 and 31871370), the Open Fund of State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University, China (Grant No. KF-202503), the Guangzhou Science and Technology Planning Project (Grant No. 2024A04J4614) and the Fundamental Research Funds for the Central Universities (Grant No. 24ykzy002).
Copyright
© The Author(s) 2026.
References
-
2. Frontera WR, Ochala J. Skeletal muscle: A brief review of structure and function. Calcif Tissue Int. 2015;96(3):183-195.[DOI]
-
3. Su N, Yang J, Xie Y, Du X, Chen H, Hong Z, et al. Bone function, dysfunction and its role in diseases including critical illness. Int J Biol Sci. 2019;15(4):776-787.[DOI]
-
5. Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130(3):456-469.[DOI]
-
6. Aoi W, Tanimura Y. Roles of skeletal muscle-derived exosomes in organ metabolic and immunological communication. Front Endocrinol. 2021;12:697204.[DOI]
-
8. Brotto M, Bonewald L. Bone and muscle: Interactions beyond mechanical. Bone. 2015;80:109-114.[DOI]
-
9. Zhi X, Chen Q, Song S, Gu Z, Wei W, Chen H, et al. Myostatin promotes osteoclastogenesis by regulating Ccdc50 gene expression and RANKL-induced NF-κB and MAPK pathways. Front Pharmacol. 2020;11:565163.[DOI]
-
11. Aryana IGPS, Rini SS, Soejono CH. Importance of sclerostin as bone-muscle mediator crosstalk. Ann Geriatr Med Res. 2022;26(2):72-82.[DOI]
-
15. Drey M, Sieber CC, Bertsch T, Bauer JM, Schmidmaier R. Osteosarcopenia is more than sarcopenia and osteopenia alone. Aging Clin Exp Res. 2016;28(5):895-899.[DOI]
-
16. Pang BWJ, Wee SL, Chen KK, Lau LK, Jabbar KA, Seah WT, et al. Coexistence of osteoporosis, sarcopenia and obesity in community-dwelling adults–The Yishun Study. Osteoporos Sarcopenia. 2021;7(1):17-23.[DOI]
-
17. Xue M, You XH, Wang XY, Xu YJ. Pectoral muscle cross-sectional area correlates with bone mineral density in postmenopausal women. Front Med. 2026;13:1781151.[DOI]
-
18. Zhang B, Chen Y, Chen Q, Zhang H. Exosomal miRNAs in muscle-bone crosstalk: Mechanistic links, exercise modulation and implications for sarcopenia, osteoporosis and osteosarcopenia. Metabolism. 2025;170:156333.[DOI]
-
21. Farsani MA, Banitalebi E, Faramarzi M, Bakhtiari N, Rahimi M, Duque G. Bone–muscle crosstalk following exercise plus Ursolic acid by myomiR-133a/Cx43-Runx2 axis in aged type 2 diabetes rat models. Chem Biol Interact. 2023;370:110315.[DOI]
-
22. Felsenthal N, Zelzer E. Mechanical regulation of musculoskeletal system development. Development. 2017;144(23):4271-4283.[DOI]
-
23. Leser JM, Harriot A, Buck HV, Ward CW, Stains JP. Aging, osteo-sarcopenia, and musculoskeletal mechano-transduction. Front Rehabil Sci. 2021;2:782848.[DOI]
-
24. Nowlan NC, Murphy P, Prendergast PJ. Mechanobiology of embryonic limb development. Ann N Y Acad Sci. 2007;1101(1):389-411.[DOI]
-
25. Sui H, Dou J, Shi B, Cheng X. The reciprocity of skeletal muscle and bone: An evolving view from mechanical coupling, secretory crosstalk to stem cell exchange. Front Physiol. 2024;15:1349253.[DOI]
-
26. Zhao Z, Yan K, Guan Q, Guo Q, Zhao C. Mechanism and physical activities in bone-skeletal muscle crosstalk. Front Endocrinol. 2024;14:1287972.[DOI]
-
27. Gomarasca M, Banfi G, Lombardi G. Myokines: The endocrine coupling of skeletal muscle and bone. Adv Clin Chem. 2020;94:155-218.[DOI]
-
28. Gao S, Li N, Qi L, Li X, Zhang J, Zhang Z, et al. Myokines and osteokines in aging-related degenerative diseases: Regulatory networks in the muscle-bone-brain axis. Cytokine Growth Factor Rev. 2025;86:17-28.[DOI]
-
34. Zhou A, Wu B, Yu H, Tang Y, Liu J, Jia Y, et al. Current understanding of osteoimmunology in certain osteoimmune diseases. Front Cell Dev Biol. 2021;9:698068.[DOI]
-
36. Liu S, Liu S, Li S, Liang B, Han X, Liang Y, et al. Nerves within bone and their application in tissue engineering of bone regeneration. Front Neurol. 2023;13:1085560.[DOI]
-
37. Malvandi AM, Gerosa L, Banfi G, Lombardi G. The bone-muscle unit: From mechanical coupling to soluble factors-mediated signaling. Mol Aspects Med. 2025;103:101367.[DOI]
-
40. Hall JG. In utero movement and use of limbs are necessary for normal growth: A study of individuals with arthrogryposis. Prog Clin Biol Res. 1985;200:155-162.[PubMed]
-
46. Esteves de Lima J, Bonnin MA, Birchmeier C, Duprez D. Muscle contraction is required to maintain the pool of muscle progenitors via YAP and NOTCH during fetal myogenesis. eLife. 2016;5:e15593.[DOI]
-
47. Zhou T, Gao B, Fan Y, Liu Y, Feng S, Cong Q, et al. Piezo1/2 mediate mechanotransduction essential for bone formation through concerted activation of NFAT-YAP1-ß-catenin. eLife. 2020;9:e52779.[DOI]
-
53. Kardon G. Muscle and tendon morphogenesis in the avian hind limb. Development. 1998;125(20):4019-4032.[DOI]
-
55. Kahn J, Shwartz Y, Blitz E, Krief S, Sharir A, Breitel DA, et al. Muscle contraction is necessary to maintain joint progenitor cell fate. Dev Cell. 2009;16(5):734-743.[DOI]
-
59. Panebianco CJ, Huang Y, Khatib N, Gottlieb DC, Essaidi M, Ahmed S, et al. Maternal exercise rescues fetal akinesia-impaired joint and bone development. FASEB J. 2025;39(24):e71341.[DOI]
-
60. Kartsogiannis V, Zhou H, Horwood NJ, Thomas RJ, Hards DK, Quinn JMW, et al. Localization of RANKL (receptor activator of NFκB ligand) mRNA and protein in skeletal and extraskeletal tissues. Bone. 1999;25(5):525-534.[DOI]
-
64. Lin X, Parker L, McLennan E, Zhang X, Hayes A, McConell G, et al. Recombinant uncarboxylated osteocalcin per Se enhances mouse skeletal muscle glucose uptake in both extensor digitorum longus and soleus muscles. Front Endocrinol. 2017;8:330.[DOI]
-
65. Liu S, Gao F, Wen L, Ouyang M, Wang Y, Wang Q, et al. Osteocalcin induces proliferation via positive activation of the PI3K/Akt, P38 MAPK pathways and promotes differentiation through activation of the GPRC6A-ERK1/2 pathway in C2C12 myoblast cells. Cell Physiol Biochem. 2017;43(3):1100-1112.
-
67. Santoso JW, Do SK, Verma R, Do AV, Hendricks E, Ichida JK, et al. Human iPSC-derived motor neuron innervation enhances the differentiation of muscle bundles engineered with benchtop fabrication techniques. ACS Biomater Sci Eng. 2025;11(3):1731-1740.[DOI]
-
71. Xing Z, Guo L, Li S, Huang W, Su J, Chen X, et al. Skeletal muscle-derived exosomes prevent osteoporosis by promoting osteogenesis. Life Sci. 2024;357:123079.[DOI]
-
74. Tao R, Mi B, Hu Y, Lin S, Xiong Y, Lu X, et al. Hallmarks of peripheral nerve function in bone regeneration. Bone Res. 2023;11:6.[DOI]
-
78. Harris AJ, Harris A. Embryonic growth and innervation of rat skeletal muscles I. Neural regulation of muscle fibre numbers. Philos Trans R Soc Lond B Biol Sci. 1981;293(1065):257-277.[DOI]
-
83. Wood W, Martin P. Macrophage functions in tissue patterning and disease: New insights from the fly. Dev Cell. 2017;40(3):221-233.[DOI]
-
84. Fan A, Zhang J, Ye Q. Osteosarcopenia in metabolic dysfunction–associated steatotic liver disease: From mechanisms to management. Front Endocrinol. 2025;16:1706068.[DOI]
-
86. Lei L, Wen Z, Cao M, Zhang H, Ling SK, Fu BS, et al. The emerging role of Piezo1 in the musculoskeletal system and disease. Theranostics. 2024;14(10):3963-3983.[DOI]
-
87. Wang L, You X, Zhang L, Zhang C, Zou W. Mechanical regulation of bone remodeling. Bone Res. 2022;10:16.[DOI]
-
88. Kirk B, Feehan J, Lombardi G, Duque G. Muscle, bone, and fat crosstalk: The biological role of myokines, osteokines, and adipokines. Curr Osteoporos Rep. 2020;18(4):388-400.[DOI]
-
89. Ning K, Wang Z, Zhang XA. Exercise-induced modulation of myokine irisin in bone and cartilage tissue: Positive effects on osteoarthritis: A narrative review. Front Aging Neurosci. 2022;14:934406.[DOI]
-
92. Mera P, Laue K, Wei J, Berger JM, Karsenty G. Osteocalcin is necessary and sufficient to maintain muscle mass in older mice. Mol Metab. 2016;5(10):1042-1047.[DOI]
-
94. Frühbeis C, Helmig S, Tug S, Simon P, Krämer-Albers EM. Physical exercise induces rapid release of small extracellular vesicles into the circulation. J Extracell Vesicle. 2015;4:28239.[DOI]
-
100. Iyer SR, Shah SB, Lovering RM. The neuromuscular junction: Roles in aging and neuromuscular disease. Int J Mol Sci. 2021;22(15):8058.[DOI]
-
101. Watt KI, Turner BJ, Hagg A, Zhang X, Davey JR, Qian H, et al. The Hippo pathway effector YAP is a critical regulator of skeletal muscle fibre size. Nat Commun. 2015;6:6048.[DOI]
-
102. Goodman CA, Dietz JM, Jacobs BL, McNally RM, You JS, Hornberger TA. Yes-Associated Protein is up-regulated by mechanical overload and is sufficient to induce skeletal muscle hypertrophy. FEBS Lett. 2015;589(13):1491-1497.[DOI]
-
103. Yang Z, Nakagawa K, Sarkar A, Maruyama J, Iwasa H, Bao Y, et al. Screening with a novel cell-based assay for TAZ activators identifies a compound that enhances myogenesis in C2C12 cells and facilitates muscle repair in a muscle injury model. Mol Cell Biol. 2014;34(9):1607-1621.
-
106. Tidball JG. Regulation of muscle growth and regeneration by the immune system. Nat Rev Immunol. 2017;17(3):165-178.[DOI]
-
116. Sasako T, Umehara T, Soeda K, Kaneko K, Suzuki M, Kobayashi N, et al. Deletion of skeletal muscle Akt1/2 causes osteosarcopenia and reduces lifespan in mice. Nat Commun. 2022;13:5655.[DOI]
-
117. Cano-Martínez A, Méndez-Castro JA, García-Vázquez VE, Carreón-Torres E, Díaz-Díaz E, Sánchez-Aguilar M, et al. A combination of resveratrol and quercetin prevents sarcopenic obesity: Its role as a signaling inhibitor of Myostatin/ActRIIA and ActRIIB/Smad and as an enhancer of insulin actions. Int J Mol Sci. 2025;26(10):4952.[DOI]
-
120. Koide M, Kobayashi Y. Regulatory mechanisms of sclerostin expression during bone remodeling. J Bone Miner Metab. 2019;37(1):9-17.[DOI]
-
121. Iwamoto R, Koide M, Udagawa N, Kobayashi Y. Positive and negative regulators of sclerostin expression. Int J Mol Sci. 2022;23(9):4895.[DOI]
-
122. Oh WT, Yang YS, Xie J, Ma H, Kim JM, Park KH, et al. WNT-modulating gene silencers as a gene therapy for osteoporosis, bone fracture, and critical-sized bone defects. Mol Ther. 2023;31(2):435-453.[DOI]
-
123. Kobayashi T, Yokoyama S, Asakura A, Goto K. A possible physiological role of sclerostin in aging-associated skeletal muscle atrophy. Physiology. 2024;39(S1):2294.[DOI]
-
124. Magarò MS, Bertacchini J, Florio F, Zavatti M, Potì F, Cavani F, et al. Identification of sclerostin as a putative new myokine involved in the muscle-to-bone crosstalk. Biomedicines. 2021;9(1):71.[DOI]
-
127. Marcadet L, Bouredji Z, Argaw A, Frenette J. The roles of RANK/RANKL/OPG in cardiac, skeletal, and smooth muscles in health and disease. Front Cell Dev Biol. 2022;10:903657.[DOI]
-
131. Wang J, Leung KS, Chow SK, Cheung WH. Inflammation and age-associated skeletal muscle deterioration (sarcopaenia). J Orthop Transl. 2017;10:94-101.[DOI]
-
132. Li Y, Lu L, Xie Y, Chen X, Tian L, Liang Y, et al. Interleukin-6 knockout inhibits senescence of bone mesenchymal stem cells in high-fat diet-induced bone loss. Front Endocrinol. 2021;11:622950.[DOI]
-
133. Liu M, Han Y, Wang J, Zhu Y, Zhang Y, Chu Q, et al. Skeletal muscle-derived IL-33 mediates muscle-to-bone crosstalk and regulates bone metabolism via CD8+ T cell-secreted CCL5. eBioMedicine. 2025;122:106024.[DOI]
-
134. Kang H, Yang K, Xiao L, Guo L, Guo C, Yan Y, et al. Osteoblast hypoxia-inducible factor-1α pathway activation restrains osteoclastogenesis via the interleukin-33-MicroRNA-34a-Notch1 pathway. Front Immunol. 2017;8:1312.[DOI]
-
135. Ohori F, Kitaura H, Ogawa S, Shen WR, Qi J, Noguchi T, et al. IL-33 inhibits TNF-α-induced osteoclastogenesis and bone resorption. Int J Mol Sci. 2020;21(3):1130.[DOI]
-
136. Cordero da Luz FA, Lima Oliveira AP, Borges D, Cristina Brígido P, Barbosa Silva MJ. The physiopathological role of IL-33: New highlights in bone biology and a proposed role in periodontal disease. Mediat Inflamm. 2014;2014:342410.[DOI]
-
137. Arnold WD, Clark BC. Neuromuscular junction transmission failure in aging and sarcopenia: The nexus of the neurological and muscular systems. Ageing Res Rev. 2023;89:101966.[DOI]
-
142. Alfonzo MC, Al Saedi A, Fulzele S, Hamrick MW. Extracellular vesicles as communicators of senescence in musculoskeletal aging. JBMR Plus. 2022;6(11):e10686.[DOI]
-
144. Borja-Gonzalez M, Casas-Martinez JC, McDonagh B, Goljanek-Whysall K. Aging Science Talks: The role of miR-181a in age-related loss of muscle mass and function. Transl Med Aging. 2020;4:81-85.[DOI]
-
148. Mytidou C, Koutsoulidou A, Zachariou M, Prokopi M, Kapnisis K, Spyrou GM, et al. Age-Related Exosomal and Endogenous Expression Patterns of miR-1, miR-133a, miR-133b, and miR-206 in skeletal muscles. Front Physiol. 2021;12:708278.[DOI]
-
149. Shao X, Zhang P, Fan Z, Lin J, Chen X, Liu N, et al. Atrophic skeletal muscle-derived extracellular vesicles transfer miR-125a-5p to inhibit bone formation in osteoporosis during aging. Adv Sci. 2026;13(25):e15362.[DOI]
-
150. Li D, Liu J, Guo B, Liang C, Dang L, Lu C, et al. Osteoclast-derived exosomal miR-214-3p inhibits osteoblastic bone formation. Nat Commun. 2016;7:10872.[DOI]
-
153. Gatti M, Beretti F, Malenchini M, Bertucci E, Ceneri E, Follo MY, et al. Mesenchymal stromal cell-derived extracellular vesicles as a therapeutic treatment for osteosarcopenia: Crosstalk among neurons, muscle, and bone. Int J Mol Sci. 2025;26(16):7875.[DOI]
-
155. Regan JN, Trivedi T, Guise TA, Waning DL. The role of TGFβ in bone-muscle crosstalk. Curr Osteoporos Rep. 2017;15(1):18-23.[DOI]
-
157. Ballinger TJ, Thompson WR, Guise TA. The bone-muscle connection in breast cancer: Implications and therapeutic strategies to preserve musculoskeletal health. Breast Cancer Res. 2022;24(1):84.[DOI]
-
158. Azzolino D, Spolidoro GCI, Saporiti E, Luchetti C, Agostoni C, Cesari M. Musculoskeletal changes across the lifespan: Nutrition and the life-course approach to prevention. Front Med. 2021;8:697954.[DOI]
-
163. Yi YT, Zhao HF, Wang WZ, Li X. Osteosarcopenia: Epidemiology, molecular mechanisms, and management. Front Endocrinol. 2025;16:1577758.[DOI]
-
165. Du Y, Xu B, Li Q, Peng C, Yang K. The role of mechanically sensitive ion channel Piezo1 in bone remodeling. Front Bioeng Biotechnol. 2024;12:1342149.[DOI]
-
167. Liu J, Jia S, Yang Y, Piao L, Wang Z, Jin Z, et al. Exercise induced meteorin-like protects chondrocytes against inflammation and pyroptosis in osteoarthritis by inhibiting PI3K/Akt/NF-κB and NLRP3/caspase-1/GSDMD signaling. Biomed Pharmacother. 2023;158:114118.[DOI]
-
168. Boudenot A, Pallu S, Uzbekov R, Dolleans E, Toumi H, Lespessailles E. Free-fall landing and interval running have different effects on trabecular bone mass and microarchitecture, serum osteocalcin, biomechanical properties, SOST expression and on osteocyte-related characteristics. Appl Physiol Nutr Metab. 2021;46(12):1525-1534.
-
169. Li Y, Wu Q, Wang J, Ding J, He J. Exerkine-loaded exosomes in muscle aging: A nexus of exercise, regeneration, and crosstalk. Front Cell Dev Biol. 2026;14:1706977.[DOI]
-
170. Jia J, Wang L, Zhou Y, Zhang P, Chen X. Muscle-derived extracellular vesicles mediate crosstalk between skeletal muscle and other organs. Front Physiol. 2025;15:1501957.[DOI]
-
174. Li Z, Liu C, Li S, Li T, Li Y, Wang N, et al. BMSC-derived exosomes inhibit dexamethasone-induced muscle atrophy via the miR-486-5p/FOXO1 axis. Front Endocrinol. 2021;12:681267.[DOI]
-
175. Müller L, Di Benedetto S. Immunosenescence and inflammaging: Mechanisms and modulation through diet and lifestyle. Front Immunol. 2025;16:1708280.[DOI]
-
176. Xiao H, Ren J. Exercise ameliorates immunosenescence: From mechanisms to interventions. Biology. 2026;15(1):58.[DOI]
-
177. Englund DA, Sakamoto AE, Fritsche CM, Heeren AA, Zhang X, Kotajarvi BR, et al. Exercise reduces circulating biomarkers of cellular senescence in humans. Aging Cell. 2021;20(7):e13415.[DOI]
-
178. Garza AP, Morton L, Motsch AL, Puta C, Stiebler M, Lading Y, et al. Acute exercise alters immune responses in older adults, with extracellular vesicle changes observed in a high-intensity intervention. Front Immunol. 2025;16:1661161.[DOI]
-
179. Marędziak M, Śmieszek A, Chrząstek K, Basinska K, Marycz K. Physical activity increases the total number of bone-marrow-derived mesenchymal stem cells, enhances their osteogenic potential, and inhibits their adipogenic properties. Stem Cells Int. 2015;2015:379093.[DOI]
-
180. Singulani MP, Stringhetta-Garcia CT, Santos LF, Morais SRL, Louzada MJQ, Oliveira SHP, et al. Effects of strength training on osteogenic differentiation and bone strength in aging female Wistar rats. Sci Rep. 2017;7:42878.[DOI]
-
182. Wang Q, Cui C, Zhang N, Lin W, Chai S, Chow SK, et al. Effects of physical exercise on neuromuscular junction degeneration during ageing: A systematic review. J Orthop Transl. 2024;46:91-102.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Tang C, Yin X, Zhang H. Dynamics of muscle-bone crosstalk throughout lifespan. Ageing Cancer Res Treat. 2026;3:202620. https://doi.org/10.70401/acrt.2026.0025
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Embryonic Musculoskeletal Crosstalk
- 3. Muscle-Bone Crosstalk in Physiological Maturity
- 4. Impact of Aging on the Dynamics of Muscle–Bone Crosstalk
- 5. Changes of Muscle-bone Crosstalk Across the Lifespan
- 6. Muscle-Bone Crosstalk in Cancer
- 7. Intervention Strategies Targeting Osteosarcopenia
- 8. Conclusion
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Tang C, Yin X, Zhang H. Dynamics of muscle-bone crosstalk throughout lifespan. Ageing Cancer Res Treat. 2026;3:202620. https://doi.org/10.70401/acrt.2026.0025
copy
Share Link
copy