Tau protein isoforms in neuropathological aging: Gerosuppressors, gerogenes or just travel companions Download PDF
Jesús Avila
1,2,*

,
Felix Hernández
1
,
José Viña
3,4
*Correspondence to:
Jesús Avila, Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid (CSIC-UAM), 28049 Madrid, Spain.
E-mail: javila@cbm.csic.es
Geromedicine. 2025;1:202503. 10.70401/Geromedicine.2025.0006
Received: July 15, 2025Accepted: October 15, 2025Published: October 17, 2025
Abstract
In recent years, the terms “gerosuppressors” and “gerogenes” have been introduced to describe factors that respectively delay or accelerate aging. These factors are present across various cell types. Specific proteins, such as tau predominantly expressed in neurons, may act as neuron-specific gerosuppressors or gerogenes. Tau exhibits a dual role influenced by its post-translational modifications, particularly phosphorylation. In this review, we discuss relevant examples of tau isoforms that demonstrate both roles, underscoring its dual influence on neuronal aging.
Keywords
Pathological aging, Alzheimer disease, tau protein, phosphorylation, folate receptor alpha
1. Introduction
Aging is commonly defined as the process of growing old, marked by a time-dependent decline in physiological and functional capabilities. More specifically, it refers to the progressive accumulation of cellular and tissue changes over time, resulting from the diminished ability to maintain homeostasis under stressful conditions. This decline ultimately increases the risk of disease and mortality.
Although this aging-related decline shares some similarities across the brain and peripheral organs, notable differences exist. Several hallmarks of aging have been identified that help characterize these shared features[1,2]. However, this review focuses on the distinctive aspects of brain aging, especially those driven by neuron-specific proteins such as Tau, which may be pivotal in neurodegenerative aging processes.
Gerontology, derived from the Greek “geron” meaning “old man”, focuses on the physiological and functional changes occurring in the human body during aging, either systemically or within specific organs. Some of these changes can be characterized by established hallmarks of aging. At the molecular level, recent research has identified two opposing genetic influences on aging: gerogenes, predominantly expressed in aged individuals and promote aging, and gerosuppressors, which may counteract or slow the aging process[3]. In this context, we examine brain-associated gerogenes and gerosuppressors, with particular emphasis on tau, a neuronal protein that may play a dual role in modulating brain aging.
However, aging does not affect all organs in the same way. Distinct differences between brain aging and peripheral aging have been observed, and specific biomarkers have been proposed to differentiate organ-specific aging patterns[4,5].
This study investigates two primary aspects: first, the differences in aging processes between the brain and peripheral tissues; and second, the potential dual role of the neuronal tau protein. Tau exists in multiple isoforms, some of which may function as gerogenes (phosphoisoforms), while others act as gerosuppressors (unphosphorylated isoforms).
2. Hallmarks of Brain Aging
Two well-cited review articles[1,2] have identified several hallmarks of aging that, with few exceptions, are common across numerous cell types. However, certain brain cells, particularly neurons, exhibit distinct characteristics that differentiate them from peripheral cells.
According to the Human Brain Proteome data from the Human Protein Atlas, the brain expresses a remarkably high proportion of proteins, approximately 76% of all human proteins. Some proteins, such as tau, are primarily localized in neurons[6]. Recently, a brain aging signature characterized by the specific expression of 64 genes was also identified[7].
Neurons exhibit distinct structural and functional features such as morphology, communication, proliferation, and energy consumption, which distinguish them from peripheral cells. Their asymmetric morphology is largely attributed to a high concentration of cytoskeletal proteins like tubulins[8], in contrast to the typically spherical shape of many peripheral cells. Neurons communicate via electrical signals, a specialized process that requires substantial energy. Although the brain accounts for only about 2% of total body mass, it consumes approximately 20% of the body’s energy[9]. This is supported by the high density of mitochondria in neurons[10], playing a crucial role in both neuronal function[11,12] and dysfunction[13,14]. Energy metabolism declines with brain aging[15], and a significant portion of neuronal energy is allocated to protein turnover[16]. As this process slows with age, post-translational modifications such as tau phosphorylation accumulate[17]. Glucose is the primary energy substrate for the brain, and regions exhibiting accelerated aging often display elevated glucose hypermetabolism[18].
Another distinctive feature of neurons is their limited capacity for proliferation. In adults, neurogenesis is restricted to specific regions, such as the dentate gyrus of the hippocampus[19]. Although adult neurogenesis decreases with age, it has been observed even in individuals up to 90 years old[20]. In contrast, glial cells, making up roughly 50% of brain cells, retain the ability to proliferate throughout life. Consequently, telomere length remains relatively stable in neurons, but progressively shortens in glial cells[21].
Telomere attrition is considered one of the hallmarks of aging[1,2]. In neurons, additional hallmarks include epigenetic alterations, loss of proteostasis, mitochondrial dysfunction, stem cell exhaustion, impaired intercellular communication, genomic instability, dysregulated nutrient sensing, and cellular senescence under stress or damage[1]. Subsequent studies have expanded this list to include macroautophagy dysfunction, chronic inflammation, and dysbiosis[2]. Most of these twelve hallmarks are evident in brain aging, though their manifestations may differ from those observed in peripheral tissues. For example, aging in brain microglia is marked by a phenotypic shift from anti-inflammatory to pro-inflammatory states[22].
Epigenetic alterations at histone modifications during aging have been associated with heterochromatin loss[23], and DNA methylation patterns in both heterochromatin and euchromatin are disrupted in aging brain cells[4,24]. Genomic instability is another key feature of aging; DNA double-strand breaks (DSBs) increase with age and may be implicated in the formation of inflammatory engrams[25]. The efficiency of DSB repair mechanisms declines over time, potentially resulting in the accumulation of senescent cells. Although these cells are non-proliferative, they can secrete pro-inflammatory factors that affect surrounding cells[26,27]. To address this, senolytic therapies are currently investigated to selectively eliminate senescent cells[28,29].
Nutritional changes, particularly those affecting folate metabolism, are also implicated in brain aging. Age-related intestinal malabsorption can lead to reduced folate levels, which have been associated with aging[30]. The consequences of folate reduction may vary among different brain cell types. In glial cells, folate is crucial for oligodendrocyte survival[31], while in astrocytes, a folate deficiency may contribute to telomere shortening[32]. The role of folate in neuronal aging will be discussed in the following section.
Given that aging is a major risk factor for numerous brain and peripheral diseases, reversing aged cells to a more youthful phenotype has become a central focus of current research.
3. Aging Reversion
Early studies centered on converting differentiated cells cultured in vitro back to an embryonic-like state. Notably, Yamanaka’s group demonstrated that a set of transcription factors, now referred to as the Yamanaka Factors (YFs), which are primarily expressed in embryonic cells, could induce this reprogramming process[33]. The expression of YFs has also been associated with mitochondrial remodeling[34], although additional research is required to clarify this relationship. This reprogramming has proven successful in peripheral cells, but it has not been observed in cultured, non-proliferating neurons[35].
Subsequent in vivo experiments in mice involving continuous expression of YFs, also demonstrated signs of cellular rejuvenation, but this was accompanied by tumor formation[36]. However, when YF expression was applied cyclically, cellular rejuvenation occurred in several peripheral organs without tumor development[37]. Remarkably, this cyclic YF expression in the brains of aged mice led to observable neuronal rejuvenation[38]. These findings suggest that YFs may reverse aging in both neuronal and peripheral tissues. Despite these promising results, translating such approaches from mice to humans remains a challenge.
To identify safer alternatives for inducing cell rejuvenation, researchers explored the use of small molecules. A cocktail of four metabolites, including the methyl donor methionine, was found to promote cellular rejuvenation in peripheral tissues and, to a lesser extent, in brain cells[38,39]. Interestingly, replacing methionine with folate resulted in a more pronounced rejuvenation effect in the brain cells of aged mice[40].
To elucidate the mechanism behind this effect, researchers focused on the previously reported interaction between folate and folate receptor alpha (FRα), a receptor expressed in neurons[40,41]. This interaction facilitates the nuclear translocation of FRα, where it acts as a transcription factor to promote YF expression[41]. Our studies confirmed that this folate-FRα-mediated mechanism operates in the brains of aged mice[38]. However, due to folate’s involvement in numerous metabolic pathways, its high-dose administration in elderly animals may result in undesirable side effects.
To mitigate this risk, researchers screened for peptides capable of binding FRα in a similar manner[42]. One such peptide was found to mimic the action of folate by binding to FRα without inducing toxic side effects[42]. Subsequently, a family of FRα-binding peptides with the ability to cross the blood-brain barrier (BBB) was identified[43]. These peptides have been shown to facilitate neuronal rejuvenation when administered systemically[43].
The data discussed above support the following hypothesis for a neuronal rejuvenation pathway: FRα-binding peptides interact with membrane-bound FRα on neurons, initiating its translocation to the nucleus. Once inside the nucleus, FRα may function as a transcription factor, promoting the expression of YFs and thereby facilitating the restoration of a youthful neuronal phenotype. Further research is required to experimentally validate this proposed mechanism. The next critical step is to evaluate the clinical potential of these FRα-binding peptides in human trials.
Concerning neuronal aging, we will discuss the role of neuronal proteins such as tau.
4. Neuropathological Aging: Role of Tau Protein
Tau is a neuronal microtubule-associated protein expressed in the brain as multiple isoforms, generated through alternative splicing[44], intron retention[45], and post-translational modifications such as phosphorylation[6]. Its significance is underscored by its central role in Alzheimer’s disease (AD), the most well-characterized tauopathy. One of the major pathological features of this disease is the accumulation of tau aggregates[5]. Given that aging is the primary risk factor for AD, the pathological effects of specific tau isoforms, especially phosphorylated forms, may stem from their ability to accelerate neuronal aging.
The presence of amyloid may promote oxidative stress and activate specific tau kinases[46] (Figure 1), leading to tau modifications that accelerate pathological aging. Notably, synaptic loss, which is closely associated with cognitive decline in pathological aging and AD follows a pattern shaped by the brain’s connectome and is influenced by the degree of tau phosphorylation[47].
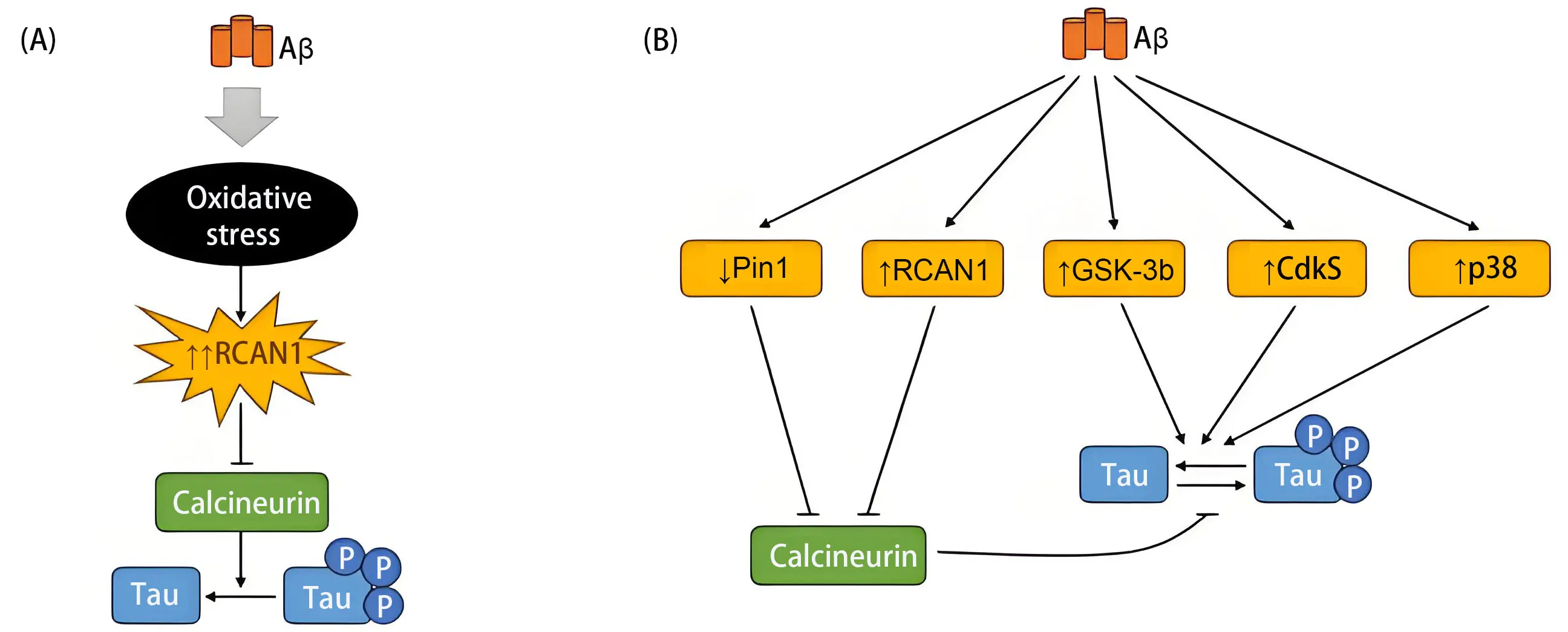
Figure 1. Amyloid pathology promotes tau phosphorylation. (A) Aggregated amyloid peptides may inhibit tau phosphatases, leading to the sustained presence of phosphorylated tau; (B) Amyloid aggregates can also promote the activation of specific tau kinases, enhancing tau phosphorylation. Aβ: amyloid beta; RCAN1: regulator of calcineurin 1; GSK-3b: glycogen synthase kinase 3β; Cdk5: cyclin-dependent kinase 5; Pin1: peptidyl-prolyl cis-trans isomerase NIMA-interacting 1; p38: p38 mitogen-activated protein kinase.
Tau pathology is closely linked to aging-related declines in cellular energy metabolism[4], which impair protein turnover and promote intracellular Ttau accumulation. This buildup may (Figure 2):

Figure 2. Tau protein in brain aging: from physiological decline to molecular duality. (A) Key processes involved in physiological brain aging include structural and functional neuronal alterations, a decline in cellular energy metabolism, loss of heterochromatin integrity, increased permeability of BBB, and age-related changes in nutrient availability; (B) The tau protein plays dual roles in brain aging, acting either as a gerosuppressor or a gerogene. Unphosphorylated tau isoforms function as gerosuppressors, while phosphorylated forms tend to promote gerogenic activity. Impaired tau turnover, often resulting from persistent phosphorylation, contributes to aging-related pathologies. Within the nucleus, unphosphorylated tau may help preserve genomic integrity, reinforcing its gerosuppressive role. In contrast, aggregated P-tau fibrils can compromise BBB integrity, accelerating brain aging and positioning tau as a GG; (C) The transition of tau from acting as a GS to becoming a GG triggers changes that represent important hallmarks of tau pathology in neurodegenerative diseases. BBB: blood-brain barrier; P-tau: phosphorylated tau; GG: gerogen; GS: gerosuppressor.
a) Enhance tau phosphorylation, impairing its cytoskeletal functions related to neuronal shape and synaptic transmission;
b) Facilitate tau aggregation, exacerbated by impaired clearance;
c) Promote tau translocation to the nucleus;
d) Lead to secretion of extracellular tau.
Additionally, tau-related neuroaging may involve altered adult neurogenesis[48] in the dentate gyrus and increased BBB permeability, both associated with aging[49]. We investigated tau’s role in neuronal aging by examining its impact on cytoplasmic and nuclear processes, BBB integrity, and adult neurogenesis.
In the cytoplasm, phosphorylated tau disrupts dendritic spine structure and synaptic communication. Tau phosphorylation, prominent in AD[50], occurs in both the soma[51] and dendrites, affecting neuronal morphology and function[51-53]. Aging also induces the relocation of N-methyl-D-aspartate (NMDA) receptors from synaptic to extrasynaptic sites[54]. Tau may participate in this shift, further acting as a gerogen[55].
In AD, phosphorylated and aggregated tau contributes to the formation of a “pathological connectome,” which aligns with the progression of Braak stages[56]. These stages are characterized by sequential tau phosphorylation events, from pre-fibrillar forms (pTau262, pTau356) to early/mid-stage markers (pTau196, pTau202), and finally to late-stage paired helical filaments (pTau396, pTau401).
In the nucleus, tau localizes to the nucleolus and pericentromeric heterochromatic regions, where it binds nucleic acids[57,58], potentially protecting DNA from damage and acting as a gerosuppressor. However, age-associated tau phosphorylation reduces its DNA-binding capacity, thereby weakening its protective role. Under physiological conditions, tau may prevent DSBs, particularly those arising during inflammatory neural activity (“inflammatory engrams”)[25].
Conversely, tau’s presence in heterochromatin may prevent its decondensation[59], contributing to heterochromatin loss, a recognized aging mechanism[23]. Tau influences the transport of the Su(var)3-9 to histone H3 via HP1-α, affecting H3K9me3 methylation levels[59]. When tau is absent[59] or phospho-tau is present[60], heterochromatin loss may be promoted, highlighting tau’s role as a gerosuppressor under normal conditions. This chromatin reorganization can activate repetitive DNA elements, such as transposons, causing cytotoxicity[61,62], and further implicating tau as a gerogen.
Nuclear oligomeric tau, particularly in the nucleolus, can disrupt RNA metabolic processes, such as ribosome biogenesis and splicing[63], thus further accelerating cellular aging and reinforcing tau’s role as a gerogen.
Moreover, aging increases BBB permeability. Extracellular tau, secreted due to intracellular accumulation, may exacerbate BBB dysfunction by altering endothelial cell metabolism[64], representing another pathological facet of tau’s role in aging.
During adult hippocampal neurogenesis in the dentate gyrus, newborn DCX-positive cells migrate from the subgranular zone toward the upper layers and project axons into the CA3 region. Comparative studies between wild-type and tau knockout mice have shown that tau, acting as a gerosuppressor, facilitates this migratory process[48], likely by promoting microtubule dynamics via its 3R isoform specifically expressed in newborn neurons[65].
On the other hand, although tau-related neuroaging has been discussed, the aging-associated changes in tau itself have rarely been mentioned. In this context, the inclusion of certain exons in tau protein increases with age. In fact, this has been reported as a common feature in several proteins during human aging[66]. For Ttau, there is an age-related increase in the inclusion of exons 2 and 10[67]. Moreover, as previously indicated, the age-dependent increase in nuclear tau[68], along with reduced tau turnover, facilitates its aggregation (Figure 2).
5. Conclusion
In summary, tau is essential for normal neuronal function, as evidenced by behavioral and cognitive differences observed in wild-type vs. tau knockout models[69]. In young animals, tau acts as a gerosuppressor. However, during aging, energy deficits impair tau turnover, resulting in pathological phosphorylation by multiple kinases[70] and the emergence of gerogenic phospho-tau isoforms. Although kinase inhibition strategies have been explored[71], enhancing neuronal energy metabolism to restore tau turnover seems to be a more effective yet complex therapeutic approach. This consideration raises the question of whether neuronal energy hypometabolism should be regarded as a distinct hallmark of aging or incorporated into existing categories such as proteostasis.
In any case, these findings highlight key differences between neuronal and peripheral aging, with neuron-specific proteins like tau playing a critical role. During physiological aging, tau transitions from acting as a gerosuppressor to acting as a gerogen, resembling a “travel companion” throughout brain aging.
Acknowledgements
We thank Dr. Carlos López-Otín for his insightful comments and valuable suggestions that contributed to the development of this manuscript. We also acknowledge Nuria de la Torre for her generous assistance with the editing and preparation of the text.
Authors contribution
Avila J, Hernández F, Viña J: Conceptualization, writing-original draft, writing-review & editing.
Conflicts of interest
Jesús Avila is an Editorial Board Member of Geromedicine. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
None.
Copyright
© The Author(s) 2025.
References
-
1. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The Hallmarks of Aging. Cell. 2013;153(6):1194-1217.[DOI]
-
2. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell. 2023;186(2):243-278.[DOI]
-
3. Kroemer G, Maier AB, Cuervo AM, Gladyshev VN, Ferrucci L, Gorbunova V, et al. From geroscience to precision geromedicine: Understanding and managing aging. Cell. 2025;188(8):2043-2062.[DOI]
-
4. Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19(6):371-384.[DOI]
-
5. Oh HSH, Rutledge J, Nachun D, Pálovics R, Abiose O, Moran-Losada P, et al. Organ aging signatures in the plasma proteome track health and disease. Nature. 2023;624(7990):164-172.[DOI]
-
6. Avila J, Lucas JJ, Perez MA, Hernandez F. Role of tau protein in both physiological and pathological conditions. Physiol Rev. 2004;84(2):361-384.[DOI]
-
7. Jin K, Yao Z, van Velthoven CTJ, Kaplan ES, Glattfelder K, Barlow ST, et al. Brain-wide cell-type-specific transcriptomic signatures of healthy ageing in mice. Nature. 2025;638(8049):182-196.[DOI]
-
8. Matus A. Stiff microtubules and neuronal morphology. Trends Neurosci. 1994;17(1):19-22.[DOI]
-
9. Balasubramanian V. Brain power. Proc Natl Acad. U.S.A. 2021;118(32):e2107022118.[DOI]
-
10. Misgeld T, Schwarz TL. Mitostasis in neurons: Maintaining mitochondria in an extended cellular architecture. Neuron. 2017;96(3):651-666.[DOI]
-
11. Rangaraju V, Lewis TL, Hirabayashi Y, Bergami M, Motori E, Cartoni R, et al. Pleiotropic mitochondria: The influence of mitochondria on neuronal development and disease. J Neurosci. 2019;39(42):8200-8208.[DOI]
-
12. Devine MJ, Kittler JT. Mitochondria at the neuronal presynapse in health and disease. Nat Rev Neurosci. 2018;19(2):63-80.[DOI]
-
13. Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G. Mitochondrial dysfunction is a trigger of Alzheimer's disease pathophysiology. Biochim Biophys Acta Mol Basis Dis. 2010;1802(1):2-10.[DOI]
-
14. Wang W, Zhao F, Ma X, Perry G, Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer's disease: Recent advances. Mol Neurodegeneration. 2020;15(1):30.[DOI]
-
15. Błaszczyk JW. Energy metabolism decline in the aging brain—pathogenesis of neurodegenerative disorders. Metabolites. 2020;10(11):450.[DOI]
-
16. Bergmann C, Mousaei K, Rizzoli SO, Tchumatchenko T. How energy determines spatial localisation and copy number of molecules in neurons. Nat Commun. 2025;16(1):1424.[DOI]
-
17. Hernández F, Avila J. Tauopathies. Cell Mol Life Sci. 2007;64(17):2219-2233.[DOI]
-
18. Antal BB, van Nieuwenhuizen H, Chesebro AG, Strey HH, Jones DT, Clarke K, et al. Brain aging shows nonlinear transitions, suggesting a midlife “critical window” for metabolic intervention. Proc Natl Acad Sci U.S.A. 2025;122(10):e2416433122.[DOI]
-
19. Gage FH. Adult neurogenesis in the human dentate gyrus. Hippocampus. 2024;35(1):e23655.[DOI]
-
20. Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, Rábano A, Cafini F, Pallas-Bazarra N, et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med. 2019;25(4):554-560.[DOI]
-
21. Tomita KI, Aida J, Izumiyama‐Shimomura N, Nakamura KI, Ishikawa N, Matsuda Y, et al. Changes in telomere length with aging in human neurons and glial cells revealed by quantitative fluorescence in situ hybridization analysis. Geriatrics Gerontology Int. 2018;18(10):1507-1512.[DOI]
-
22. Antignano I, Liu Y, Offermann N, Capasso M. Aging microglia. Cell Mol Life Sci. 2023;80(5):126.[DOI]
-
23. Villeponteau B. The heterochromatin loss model of aging. Exp Gerontol. 1997;32(4-5):383-394.[DOI]
-
24. Lee JH, Kim EW, Croteau DL, Bohr VA. Heterochromatin: An epigenetic point of view in aging. Exp Mol Med. 2020;52(9):1466-1474.[DOI]
-
25. Jovasevic V, Wood EM, Cicvaric A, Zhang H, Petrovic Z, Carboncino A, et al. Formation of memory assemblies through the DNA-sensing TLR9 pathway. Nature. 2024;628(8006):145-153.[DOI]
-
26. González‐Gualda E, Baker AG, Fruk L, Muñoz‐Espín D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2020;288(1):56-80.[DOI]
-
27. Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155(5):1104-1118.[DOI]
-
28. Lagoumtzi SM, Chondrogianni N. Senolytics and senomorphics: Natural and synthetic therapeutics in the treatment of aging and chronic diseases. Free Radic Biol Med. 2021;171:169-190.[DOI]
-
29. Zhang L, Pitcher LE, Prahalad V, Niedernhofer LJ, Robbins PD. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 2022;290(5):1362-1383.[DOI]
-
30. Araújo JR, Martel F, Borges N, Araújo JM, Keating E. Folates and aging: Role in mild cognitive impairment, dementia and depression. Ageing Res Rev. 2015;22:9-19.[DOI]
-
31. Weng Q, Wang J, Wang J, Tan B, Wang J, Wang H, et al. Folate metabolism regulates oligodendrocyte survival and differentiation by modulating AMPKα activity. Sci Rep. 2017;7(1):1705.[DOI]
-
32. Li Z, Zhou D, Zhang D, Zhao J, Li W, Sun Y, et al. Folic acid inhibits aging-induced telomere attrition and apoptosis in astrocytes in vivo and in vitro. Cereb Cortex. 2021;32(2):286-297.[DOI]
-
33. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663-676.[DOI]
-
34. Xu X, Duan S, Yi F, Ocampo A, Liu GH, Izpisua Belmonte JC. Mitochondrial regulation in pluripotent stem cells. Cell Metab. 2013;18(3):325-332.[DOI]
-
35. Kim J, Lengner CJ, Kirak O, Hanna J, Cassady JP, Lodato MA, et al. Reprogramming of postnatal neurons into induced pluripotent stem cells by defined factors. Stem Cells. 2011;29(6):992-1000.[DOI]
-
36. Abad M, Mosteiro L, Pantoja C, Cañamero M, Rayon T, Ors I, et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature. 2013;502(7471):340-345.[DOI]
-
37. Ocampo A, Reddy P, Martinez-Redondo P, Platero-Luengo A, Hatanaka F, Hishida T, et al. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell. 2016;167(7):1719-1733.e12.[DOI]
-
38. Antón-Fernández A, Cuadros R, Peinado-Cahuchola R, Hernández F, Avila J. Role of folate receptor α in the partial rejuvenation of dentate gyrus cells: Improvement of cognitive function in 21-month-old aged mice. Sci Rep. 2024;14(1):6915.[DOI]
-
39. Hernandez-Benitez R, Wang C, Shi L, Ouchi Y, Zhong C, Hishida T, et al. Intervention with metabolites emulating endogenous cell transitions accelerates muscle regeneration in young and aged mice. Cell Rep Med. 2024;5(3):101449.[DOI]
-
40. Boshnjaku V, Shim KW, Tsurubuchi T, Ichi S, Szany EV, Xi G, et al. Nuclear localization of folate receptor alpha: A new role as a transcription factor. Sci Rep. 2012;2(1):980.[DOI]
-
41. Mohanty V, Shah A, Allender E, Siddiqui MR, Monick S, Ichi S, et al. Folate receptor alpha upregulates Oct4, Sox2 and Klf4 and downregulates miR-138 and miR-let-7 in cranial neural crest cells. Stem Cells. 2016;34(11):2721-2732.[DOI]
-
42. Hulin-Curtis SL, Davies JA, Nestić D, Bates EA, Baker AT, Cunliffe TG, et al. Identification of folate receptor α (FRα) binding oligopeptides and their evaluation for targeted virotherapy applications. Cancer Gene Ther. 2020;27(10-11):785-798.[DOI]
-
43. Anton-Fernandez A, Domene-Serrano I, Cuadros R, Peinado-Cahuchola R, Sanchez-Pece M, Hernandez F, et al. Peptide family promotes brain cell rejuvenation and improved cognition through peripheral delivery. ACS Omega. 2025;10(13):13236-13250.[DOI]
-
44. Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta Mol Basis Dis. 2005;1739(2-3):91-103.[DOI]
-
45. García-Escudero V, Ruiz-Gabarre D, Gargini R, Pérez M, García E, Cuadros R, et al. A new non-aggregative splicing isoform of human Tau is decreased in Alzheimer’s disease. Acta Neuropathol. 2021;142(1):159-177.[DOI]
-
46. Viña J, Borrás C, Mas-Bargues C. Free radicals in Alzheimer’s disease: From pathophysiology to clinical trial results. Free Radic Biol Med. 2024;225:296-301.[DOI]
-
47. Luan Y, Wang W, Huang Q, Wang Y, Nussbaumer J, Wang J, et al. Synaptic loss pattern is constrained by brain connectome and modulated by phosphorylated tau in Alzheimer's disease. Nat Commun. 2025;16(1):6356.[DOI]
-
48. Fuster-Matanzo A, Llorens-Martín M, Jurado-Arjona J, Avila J, Hernández F. Tau protein and adult hippocampal neurogenesis. Front Neurosci. 2012;6:[DOI]
-
49. Knox EG, Aburto MR, Clarke G, Cryan JF, O'Driscoll CM. The blood-brain barrier in aging and neurodegeneration. Mol Psychiatry. 2022;27(6):2659-2673.[DOI]
-
50. Avila J. Tau phosphorylation and aggregation in Alzheimer’s disease pathology. FEBS Lett. 2006;580(12):2922-2927.[DOI]
-
51. Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133(5):665-704.[DOI]
-
52. Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: The story so far. Nat Rev Neurol. 2016;12(1):15-27.[DOI]
-
53. Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2(7):a006247.[DOI]
-
54. Potier B, Billard JM, Riviere S, Sinet PM, Denis I, Champeil-Potokar G, et al. Reduction in glutamate uptake is associated with extrasynaptic NMDA and metabotropic glutamate receptor activation at the hippocampal CA1 synapse of aged rats. Aging Cell. 2010;9(5):722-735.[DOI]
-
55. Pallas-Bazarra N, Draffin J, Cuadros R, Antonio Esteban J, Avila J. Tau is required for the function of extrasynaptic NMDA receptors. Sci Rep. 2019;9(1):9116.[DOI]
-
56. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239-259.[DOI]
-
57. Camero S, Benitez MJ, Cuadros R, Hernandez F, Avila J, Jimenez JS. Thermodynamics of the interaction between Alzheimer’s disease related tau protein and DNA. PLoS One. 2014;9(8):e104690.[DOI]
-
58. Sultan A, Nesslany F, Violet M, Begard S, Loyens A, Talahari S, et al. Nuclear tau, a key player in neuronal DNA protection. J Biol Chem. 2011;286(6):4566-4575.[DOI]
-
59. Mansuroglu Z, Benhelli-Mokrani H, Marcato V, Sultan A, Violet M, Chauderlier A, et al. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci Rep. 2016;6:33047.[DOI]
-
60. Ramirez P, Zuniga G, Sun W, Beckmann A, Ochoa E, DeVos SL, et al. Pathogenic tau accelerates aging-associated activation of transposable elements in the mouse central nervous system. Prog Neurobiol. 2022;208(2022):102181.[DOI]
-
61. Frost B, Dubnau J. The role of retrotransposons and endogenous retroviruses in age-dependent neurodegenerative disorders. Annu Rev Neurosci. 2024;47(1):123-143.[DOI]
-
62. Ochoa E, Ramirez P, Gonzalez E, De Mange J, Ray WJ, Bieniek KF, et al. Pathogenic tau-induced transposable element-derived dsRNA drives neuroinflammation. Sci Adv. 2023;9(1):eabq5423.[DOI]
-
63. Puangmalai N, Aday AE, Samples M, Bhatt N, Cascio FL, Marcatti M, et al. Pathogenic oligomeric Tau alters neuronal RNA processes through the formation of nuclear heteromeric amyloids with RNA-binding protein Musashi1. Prog Neurobiol. 2025;247:102742.[DOI]
-
64. Guzman-Hernandez R, Fossati S. Fibrillar tau alters cerebral endothelial cell metabolism, vascular inflammatory activation, and barrier function in vitro and in vivo. Alzheimers Dement. 2025;21(3):e70077.[DOI]
-
65. Llorens-Martin M, Teixeira CM, Fuster-Matanzo A, Jurado-Arjona J, Borrell V, Soriano E, et al. Tau isoform with three microtubule binding domains is a marker of new axons generated from the subgranular zone in the hippocampal dentate gyrus: Implications for Alzheimer’s disease. J Alzheimers Dis. 2012;29(4):921-930.[DOI]
-
66. Tollervey JR, Wang Z, Hortobagyi T, Witten JT, Zarnack K, Kayikci M, et al. Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res. 2011;21(10):1572-1582.[DOI]
-
67. Avila J, de Barreda EG, Pallas-Bazarra N, Hernandez F. Tau and neuron aging. Aging Dis. 2013;4(1):23-28.[PMC]
-
68. Anton-Fernandez A, Valles-Saiz L, Avila J, Hernandez F. Neuronal nuclear tau and neurodegeneration. Neuroscience. 2023;518:178-184.[DOI]
-
69. Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, et al. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369(6480):488-491.[DOI]
-
70. Hanger DP, Anderton BH, Noble W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15(3):112-119.[DOI]
-
71. Bhat R, Xue Y, Berg S, Hellberg S, Ormo M, Nilsson Y, et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem. 2003;278(46):45937-45945.[DOI]
Copyright
© The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Avila J, Hernández F, Viña J. Tau protein isoforms in neuropathological aging: Gerosuppressors, gerogenes or just travel companions. Geromedicine. 2025;1:202503. https://doi.org/10.70401/Geromedicine.2025.0006
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Hallmarks of Brain Aging
- 3. Aging Reversion
- 4. Neuropathological Aging: Role of Tau Protein
- 5. Conclusion
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- Copyright
Science Exploration Style
Avila J, Hernández F, Viña J. Tau protein isoforms in neuropathological aging: Gerosuppressors, gerogenes or just travel companions. Geromedicine. 2025;1:202503. https://doi.org/10.70401/Geromedicine.2025.0006
copy