Vascular aging as a driver of thrombosis in older adults: From mechanisms to gerotherapeutics
Georgios Zervas
1
,
Christina Konstantaki
1
,
Martin Sigl
2
,
Kimon Stamatelopoulos
1
,
Simon Tual-Chalot
3,*

,
Konstantinos Stellos
2,4,5,6,*
*Correspondence to:
Simon Tual-Chalot, Biosciences Institute, Vascular Biology and Medicine Theme, Faculty of Medical Sciences, Newcastle University, Newcastle Upon Tyne NE1 3BZ, UK.
E-mail: simon.tual-chalot@newcastle.ac.uk
Konstantinos Stellos, Department of Medicine VI, University Medical Centre Mannheim, Heidelberg University, Mannheim 68167, Germany; Department of Cardiovascular Research, European Center for Angioscience (ECAS), Medical Faculty Mannheim, Heidelberg University, Mannheim 68167, Germany; German Centre for Cardiovascular Research (DZHK), Partner Site Heidelberg/Mannheim, Mannheim 68167, Germany; Helmholtz-Institute for Translational AngioCardioScience (HI-TAC) of the Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC), Heidelberg University, Mannheim 68167, Germany. E-mail: konstantinos.stellos@medma.uni-heidelberg.de
Konstantinos Stellos, Department of Medicine VI, University Medical Centre Mannheim, Heidelberg University, Mannheim 68167, Germany; Department of Cardiovascular Research, European Center for Angioscience (ECAS), Medical Faculty Mannheim, Heidelberg University, Mannheim 68167, Germany; German Centre for Cardiovascular Research (DZHK), Partner Site Heidelberg/Mannheim, Mannheim 68167, Germany; Helmholtz-Institute for Translational AngioCardioScience (HI-TAC) of the Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC), Heidelberg University, Mannheim 68167, Germany. E-mail: konstantinos.stellos@medma.uni-heidelberg.de
Geromedicine. 2026;2:202513. 10.70401/Geromedicine.2025.0010
Received: September 23, 2025Accepted: December 03, 2025Published: December 15, 2025
Abstract
With increasing chronological age, the vascular system gradually loses its functional integrity, a process known as vascular aging. This decline is a major contributor to arterial and venous thromboembolic disorders, which represent one of the leading causes of mortality and morbidity among aged individuals. However, the precise biological mechanisms linking systemic aging to thrombotic susceptibility remain poorly understood, hindering the development of effective preventive and therapeutic strategies for age-related thrombotic diseases. Aging, as a fundamental determinant of vascular health, shifts the balance between prothrombotic and antithrombotic mechanisms through cumulative molecular and cellular alterations across vascular cells, red blood cells, platelets, and immune cells. These interconnected hallmarks collectively disrupt endothelial homeostasis, enhance platelet reactivity, and impair coagulation and fibrinolytic pathways. Emerging factors, including clonal hematopoiesis of indeterminate potential and environmental exposures, further exacerbate the thrombotic risk in older populations. Clinically, thrombosis management in the elderly requires careful calibration between protection against ischemia and bleeding risk, as age-associated changes are known to affect the safety and efficacy of antiplatelet and anticoagulant therapies. The development of geroscience-guided interventions, alongside optimized antithrombotic strategies, will be essential to reduce the thrombotic burden and improve outcomes in the aging population.
Keywords
Aging, thrombosis, arterial, venous
1. Introduction
Life expectancy continues to rise worldwide, but extended longevity is frequently accompanied by years of poor health and frailty[1]. Aging is a multifactorial biological process shaped by complex interactions among genetic, epigenetic, and biochemical mechanisms, which are collectively referred to as the hallmarks of aging[2]. It exerts profound effects on the vascular system, driving structural remodeling characterized by arterial stiffening, intimal and medial thickening, vascular calcification, and increased resistance[3]. Vascular aging is now widely recognized as an early and general predictor of unhealthy aging[3]. Given that the vascular system interlinks all organs, dysfunction within a single vascular bed can propagate to distant tissues, thereby accelerating systemic aging. Notably, recent longitudinal mapping proposes that blood vessels may function as upstream regulators of the aging trajectory[4].
Thrombosis represents one of the most clinically consequential outcomes of vascular aging-related dysfunction. Thromboembolic diseases, including myocardial infarction, ischemic stroke, and venous thromboembolism (VTE), remain leading causes of global mortality[5]. Although traditional cardiovascular risk factors such as hypertension, diabetes, and dyslipidemia contribute to thrombotic risk, they do not fully explain the steep rise in thromboembolic events with age[6]. This gap underlines the importance to explore aging itself as a biological driver of thrombosis. The incidence of both arterial and venous thrombotic events rises exponentially with age[7]. Arterial thrombosis typically results from atherothrombotic plaque rupture or erosion under high shear stress, while venous thrombosis develops in the context of stasis, hypercoagulability, and endothelial dysfunction[8]. Despite differences in hemodynamic context and thrombus composition, both processes are accelerated by age-related changes in endothelial cells, vascular smooth muscle cells, platelets, red blood cells, and immune cells, producing a vasoconstrictive, pro-inflammatory, and hypercoagulable environment[9]. These cellular alterations are compounded by systemic factors such as inflammaging, clonal hematopoiesis, and metabolic dysregulation, which further amplify thrombotic susceptibility. Emerging evidence also highlights environmental and molecular contributors to vascular aging and thrombosis. Micro- and nanoplastics, air pollution, and gut microbiota-derived metabolites such as trimethylamine-N-oxide (TMAO) have been linked to endothelial dysfunction and thrombo-inflammation[10-12]. Additionally, circulating biomarkers like amyloid-β1-40 (Aβ1-40) are elevated in older adults and may serve as indicators of vascular aging and thrombotic risk[13].
Despite its well-recognized clinical and societal impact, the biology of aging is not yet fully integrated into current thrombotic disease paradigms. Existing prevention and treatment strategies primarily focus on antiplatelet and anticoagulant therapies, with limited attention to aging-related molecular pathways. Emerging insights into aging-associated mechanisms and novel circulating biomarkers, however, offers opportunities for early detection of vascular dysfunction, risk stratification, and precision medicine. For instance, gerotherapeutics targeting the hallmarks of aging may complement conventional antithrombotic strategies by addressing the underlying drivers of the vascular dysfunction in older adults. This review summarizes current literature on the mechanisms by which aging promotes arterial and venous thrombosis, highlights emerging therapeutic strategies, particularly gerotherapeutics, and discusses their implications for clinical management in the elderly. By integrating insights from basic science, translational research, and clinical practice, we aim to clarify the mechanistic links between aging and thrombosis, and to explore more opportunities for mitigating the thrombotic risk in aging populations.
2. Overview of the Mechanisms Governing Arterial and Venous Thrombosis
Atherothrombosis, defined as thrombus formation in association with an atherosclerotic plaque, underlies major adverse cardiovascular events (MACE), including myocardial infarction, stroke, and peripheral arterial ischemia[14] (Figure 1). Thrombus formation typically arises from either endothelial erosion or acute plaque rupture, both of which expose highly thrombogenic subendothelial components to circulating blood. Among these, von Willebrand factor (vWF) and collagen play pivotal roles in initial platelet adhesion[15]. At sites of vascular injury, platelets initially tether to vWF via the glycoprotein Ib-IX-V receptor complex, allowing engagement of the collagen receptor glycoprotein VI, a critical trigger for platelet activation. At the same time, tissue factor (TF), the key initiator of the extrinsic coagulation pathway, becomes exposed. In plaque rupture, TF primarily originates from lipid-rich necrotic core contents, whereas in plaque erosion, activated neutrophils promote TF release through the formation of neutrophil extracellular traps (NETs)[16]. NETs are extracellular web-like structures composed of decondensed chromatin, histones, proteolytic enzymes, reactive oxygen species (ROS), cytokines, and TF, released through a process known as neutrophil extracellular trap formation (NETosis)[17]. Once activated, TF forms a complex with coagulation factor VIIa, initiating the extrinsic coagulation cascade. This leads to the generation of thrombin, which not only converts fibrinogen to fibrin but also activates protease-activated receptors on platelets, amplifying platelet activation[18]. Thrombin also activates factors V and VIII, further propagating coagulation. Concurrently, secondary platelet-derived mediators, including adenosine diphosphate (ADP) binding to the P2Y purinoreceptor 12 (P2Y12), thromboxane A2 engaging the thromboxane receptor, and granule release products, recruit and activate additional platelets. These signals induce inside-out activation of the integrin αIIbβ3 (GPIIb/IIIa), facilitating fibrinogen and vWF binding, which is critical for stable platelet aggregation, especially under high arterial shear stress[19]. In healthy arteries, these procoagulant forces are counterbalanced by the natural anticoagulant properties of the endothelium. The thrombomodulin–endothelial protein C receptor (EPCR)–protein C axis exerts potent cytoprotective, barrier-stabilizing effects. Nevertheless, persistent exposure to coagulation proteases such as factor Xa and thrombin downregulates and induces shedding of thrombomodulin and EPCR, impairing protein C activation and promoting a procoagulant, pro-inflammatory endothelial phenotype[20]. This anticoagulant function declines progressively with aging, further reducing protein C pathway activation senescence and increasing susceptibility to thrombotic complications[21].
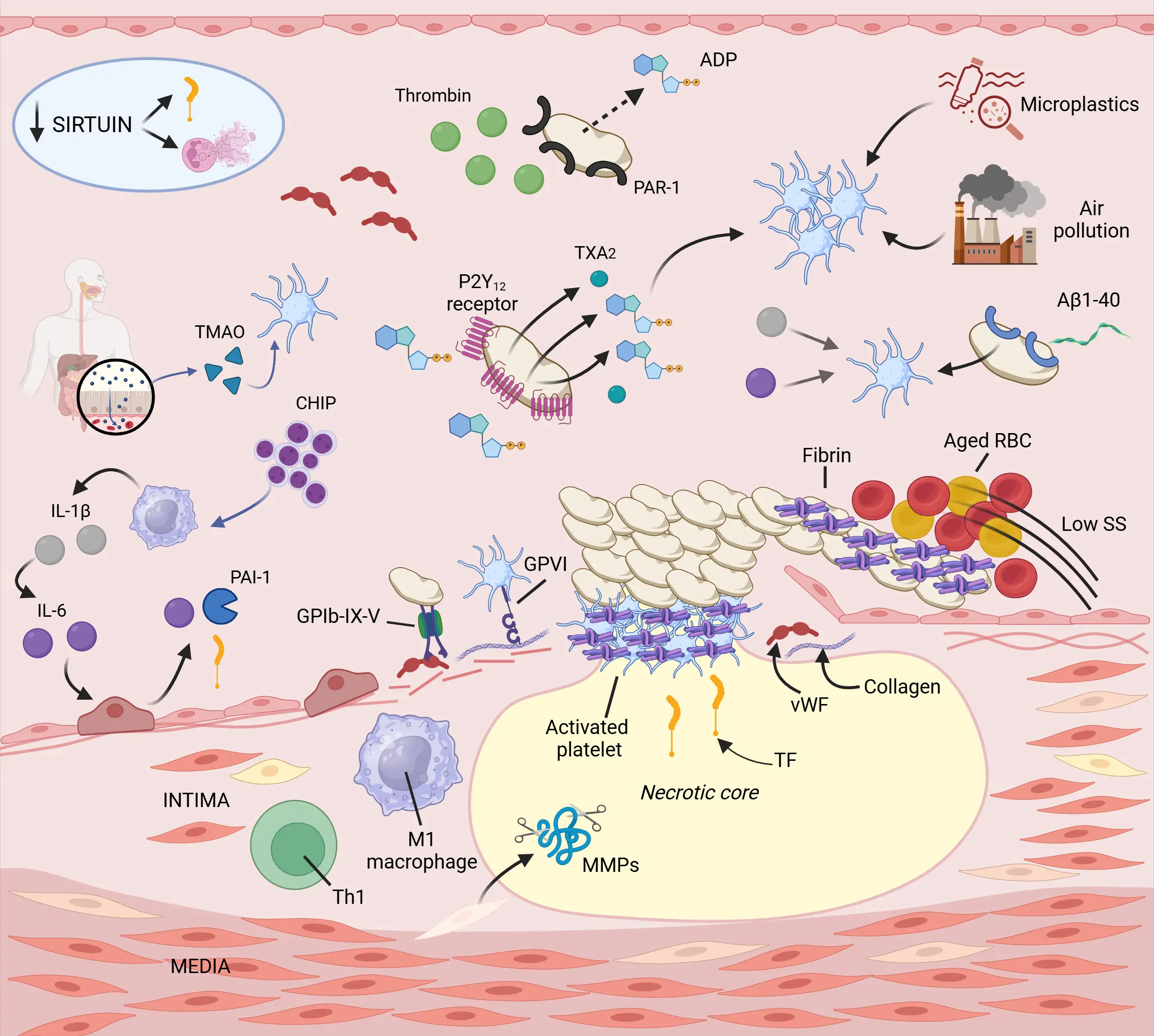
Figure 1. Arterial thrombosis in the elderly. With advancing age, arterial hemostasis is recalibrated from repair to persistence. The elderly are often characterized by sustained, low-grade inflammation, where innate immune cells, especially macrophages, release IL-1β, IL-6, and TNF-α. Although these molecules exert their prothrombotic effects by activating platelets, facilitating NETosis, and promoting endothelial senescence and dysfunction, chronic inflammation is maintained due to sustained stimuli. CHIP is characterized by mutations in HSCs that lead to hyperactive macrophages. During aging, there is a shift from gut symbiosis to dysbiosis, which acts as a source of inflammatory molecules. Simultaneously, increased gut permeability allows gut metabolites, such as TMAO, to enter the circulation and activate platelets. A remarkable characteristic of aged platelets is the “paradox” of ADP hypersensitivity combined with thrombin resistance. Reductions in sirtuin 1 levels lead to increased NETosis and tissue factor expression. Chemical pollutants, including microplastics and air pollution, accumulate over the years and thereby activates the coagulation cascade and platelets. Apart from these cellular changes, circulating prothrombotic factors, such as vWF and Aβ1-40, are present at elevated levels. Created in BioRender.com. IL: interleukin; TNF: tumor necrosis factor; CHIP: clonal hematopoiesis of intermediate potential; HSCs: hematopoietic stem cells; TMAO: trimethylamine-N-oxide; ADP: adenosine diphosphate; vWF: von Willebrand factor; β1-40: amyloid beta; TF: tissue factor; GPVI: glycoprotein VI; RBC: red blood cell; SS: shear stress; NETosis: neutrophil extracellular trap formation.
Although venous thrombosis shares several molecular and cellular features with arterial thrombosis, it constitutes a distinct pathophysiological entity with unique characteristics in thrombus composition, location, and initiating mechanisms (Figure 2). Venous thrombi are typically fibrin-rich, contain abundant entrapped red blood cells (RBCs), and are commonly referred to as “red thrombi”[22]. Venous thrombosis primarily develops under conditions of low shear stress and venous stasis, where platelet adhesion via vWF is generally dispensable and overt endothelial disruption is usually absent. The pathogenesis of VTE is classically described by the Virchow’s Triad, comprising three interdependent elements: hypercoagulability, venous stasis, and endothelial dysfunction[23]. In a healthy venous system, the endothelium maintains an antithrombotic phenotype, partly through Krüppel-like factor 2-mediated transcriptional activation of thrombomodulin and other anticoagulant proteins[23]. Venous valve pockets, regions prone to disturbed and stagnant flow, are especially adapted to resist thrombus formation, with endothelial cells expressing high levels of thrombomodulin and low levels of vWF, in contrast to the prothrombotic environment observed in arterial high-shear zones. However, prolonged stasis or inflammation disrupts this balance, leading to endothelial activation characterized by upregulation of adhesion molecules such as intercellular adhesion molecule 1 and vascular cell adhesion molecule 1 (VCAM-1). This promotes leukocyte recruitment, particularly neutrophils, which may express TF upon activation[22]. In addition to endothelial-derived TF, circulating microvesicles from monocytes, platelets, and other blood cells further deliver TF to sites of thrombus initiation[24]. TF binds factor VIIa to form the extrinsic tenase complex, which activates factor X, and generates initial thrombin[25,26]. This small amount of thrombin is critical, as it activates platelets and factors like FVIII and FXI to form the intrinsic tenase complex, the major source of thrombin during the amplification phase of coagulation[26]. Interestingly, NETs are also found in venous thrombi[27], suggesting that some mechanisms are shared across vascular beds.
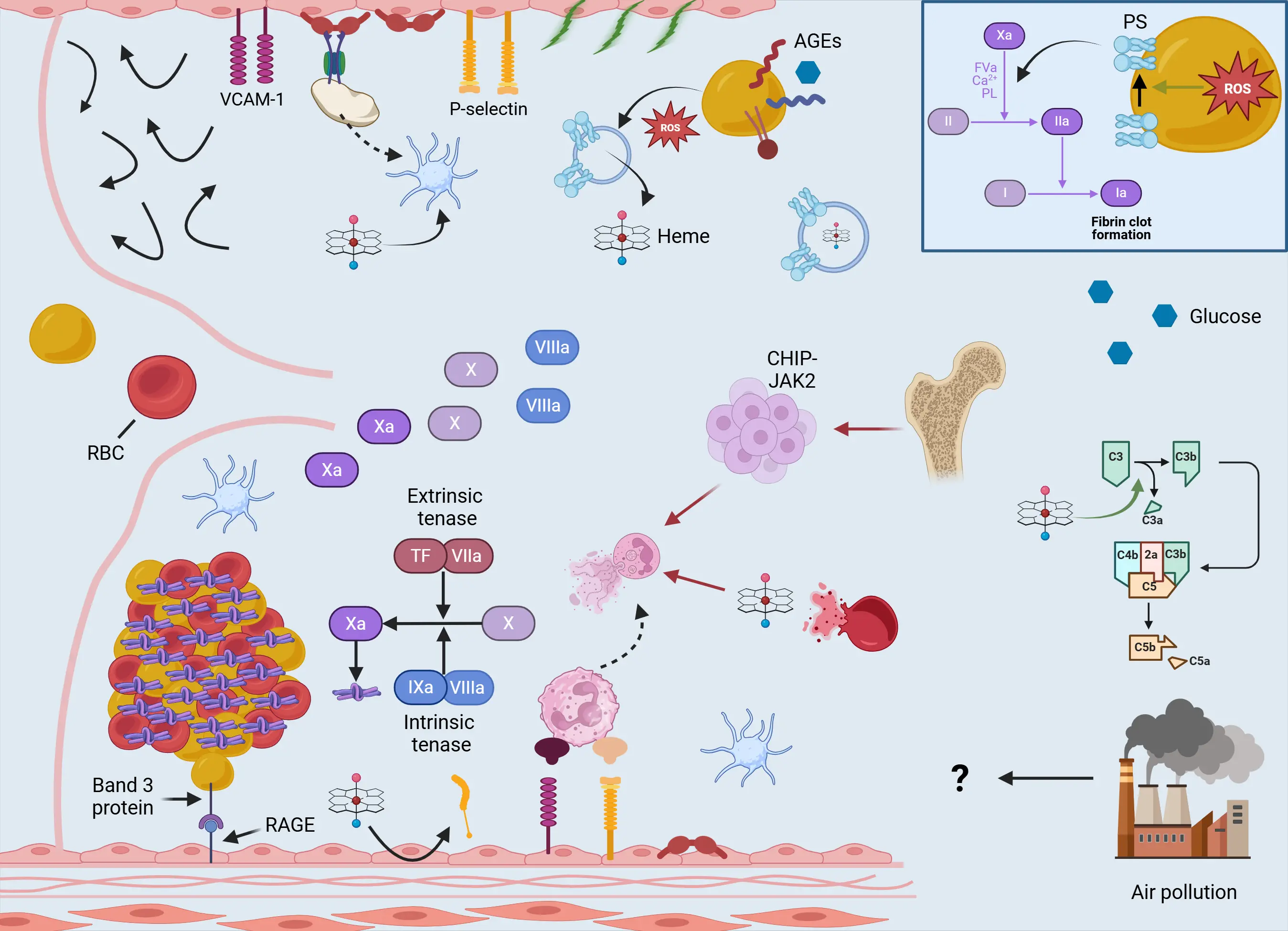
Figure 2. Venous thrombosis in the elderly. Aging modifies the venous thrombotic field, as endothelial activation and dysfunction predominate, leading to increased expression of adhesion molecules such as VCAM-1 and P-selectin. Aged venous valve pockets and age-related immobility create regions of low shear stress and stasis, predisposing to thrombus formation. Aging is associated with increased circulating levels of procoagulant factors (e.g., factors VIII, IX, and X; von Willebrand factor; and PAI-1), thus promoting a shift toward thrombosis through alterations in both coagulation and fibrinolytic pathways. RBCs emerge as key regulators of venous thrombosis in aged individuals, as abundant circulating ROS enhance a distinct phenotype in which RBCs expose PS, a critical component of the prothrombinase complex, and secrete thrombogenic MVs containing heme. In turn, heme, derived from MVs or lysed erythrocytes, exerts multiple effects, including increased NETosis, platelet activation, complement activation, and tissue factor expression from endothelial cells. Hyperglycemic conditions, such as those in diabetic patients, further augment erythrocyte dysfunction. In addition, mutations leading to CHIP, particularly the JAK2 mutation, amplify the formation of NETs, contributing to thrombus growth. As mentioned in the main text, older individuals have accumulated exposure to chemical pollution. Although the role of air pollution is well established in arterial thrombosis, its contribution to venous thrombosis remains less clear. Created in BioRender.com. VCAM-1: vascular cell adhesion molecule 1; RBCs: red blood cells; ROS: reactive oxygen species; PS: phosphatidylserine; MVs: microvesicles; CHIP: clonal hematopoiesis of intermediate potential; NETs: neutrophil extracellular traps; AGEs: advanced glycation end-products; RAGE: receptor for advanced glycation end products; TF: tissue factor; NETosis: neutrophil extracellular trap formation.
3. Aging as a Driver of Pro-Thrombotic Cellular Phenotypes
Hallmarks of aging[2], particularly those associated with cardiovascular aging[28], have been extensively described in vascular cells and are known to directly or indirectly modulate thrombus formation in older individuals (Table 1). Beyond these established hallmarks, several additional mechanisms have been identified and may substantially contribute to the heightened thrombotic risk in the elderly (Figure 3).
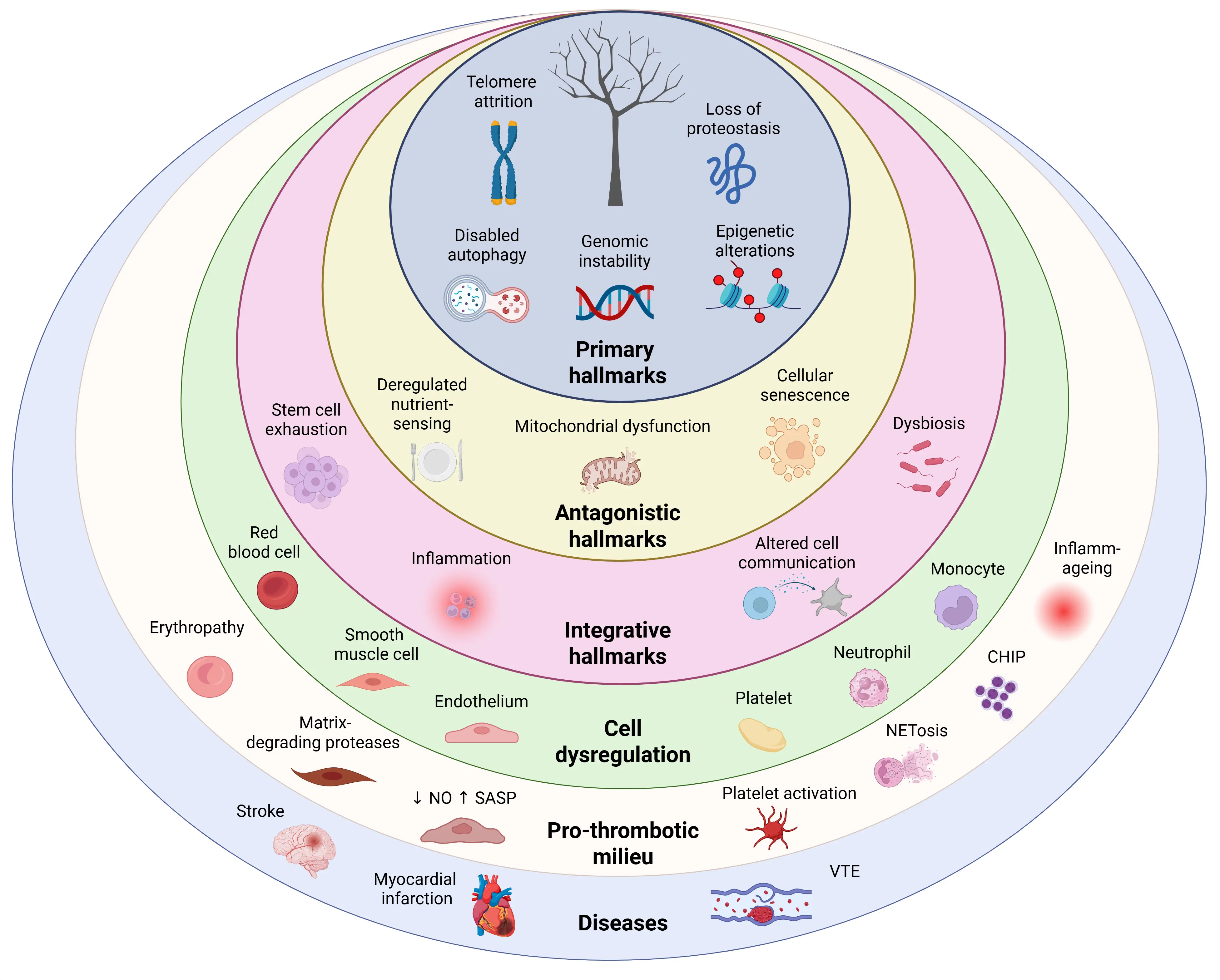
Figure 3. Hallmarks of aging: key regulators of thrombotic disease. In the innermost ring, primary hallmarks of aging (telomere attrition, genomic instability, loss of proteostasis, epigenetic alterations and defective autophagy), which represent the initial sources of damage, drive the development of antagonistic hallmarks (cellular senescence, mitochondrial dysfunction and deregulated nutrient sensing), reflecting adaptive responses to this damage. These changes give rise to integrative hallmarks (stem cell exhaustion, chronic systemic inflammation, altered intercellular communication and gut dysbiosis), which in turn cause dysregulation of vascular and blood cells (endothelium, smooth muscle cells, red blood cells, platelets, neutrophils and monocytes), accumulation of CHIP, matrix-degrading proteases, NETosis and inflammaging. Together, these alterations promote a pro-thrombotic milieu, ultimately contributing to arterial and venous thrombotic diseases, including myocardial infarction, stroke and VTE. Created in BioRender.com. CHIP: clonal haematopoiesis of indeterminate potential; VTE: venous thromboembolism; SASP: senescence-associated secretory phenotype; NETosis: neutrophil extracellular trap formation.
Table 1. Hallmarks of vascular aging across cell types and their thrombotic consequences.
| Hallmark of CVD aging | Intervention | Specificity, Experimental model | Cell type | Functional changes |
| Genomic instability[29] | Trf2-/- | Global, mice | Endothelial cells | Accelerated endothelial dysfunction, ↓ NO bioavailability |
| Genomic instability[30] | Ercc1(d/-), Xpd(TTD) | Global, mice | Endothelial cells | Accelerated endothelial dysfunction, ↓ endothelial eNOS levels |
| Genomic instability[31] | CHIP | Single-molecule sequencing in human blood | Hematopoietic stem cells | ↑ ischemic stroke and VTE |
| Disabled macroautophagy[32] | CD44 overexpression | Endothelial knock-in mice | Endothelial cells | Endothelial dysfunction, ↓ NO |
| Disabled macroautophagy[33] | Atg5 inhibition | Global, mice | Neutrophils | ↓ NETosis, ↑ apoptosis |
| Epigenetic alterations[34] | Decreased expression of MEG3 | HUVECs, mice | Endothelial cells | Endothelial senescence |
| Epigenetic alterations[35] | H19-/- | HUVECs, mice | Endothelial cells | Endothelial senescence, pro-inflammatory phenotype |
| Epigenetic alterations[36] | CSE-/- | Endothelial cells, mice/mesenteric endothelial cells, human | Endothelial cells | Telomere attrition, endothelial senescence |
| Mitochondrial dysfunction[37] | Prkaa-/- | Global or endothelial cell specific, mice | Endothelial cells | Endothelial senescence, oxidative stress, ↓ NO bioavailability |
| Mitochondrial dysfunction[38] | mTORC1 overexpression | Megakaryocytes and platelets specific, mice | Platelets | ↑ MK size, ↑ MPV, ↑ platelet activation, ↑ venous thrombosis |
| Mitochondrial dysfunction[39] | TLR9 activation | Plasma, human/mice | Neutrophils | ↑ NET formation, NETs more oxidized and resistant to DNase degradation |
| Cell senescence[40] | Replicative senescence | Endothelial cell specific, porcine | Endothelial cells | ↑ TF expression and activity, platelet aggregation |
| Cell senescence[41] | PAI-1 | Endothelial cell line, human | Endothelial cells | ↓ Fibrinolysis |
| Cell senescence[42] | Replicative senescence/physiological aging | HUVECs/mice | Endothelial cells | ↑ VWF expression, senescence-driven prothrombotic phenotype |
| Cell senescence[43] | CMV seropositivity | CD4+/CD8+, human | T-cells | ↑ cardiovascular mortality (MI, stroke), ↑ Senescent CD4+ and CD8+ T cells |
| Cell senescence[44] | CD8(+)CD56(+) accumulation | CAD, human | T-cells | ↑ IFN-γ → chronic vascular inflammation, plaque destabilization, pro-thrombotic milieu |
| Cell senescence[45] | Replicative senescence, DNA damage-induced senescence | Carotid plaque VSMC, human | Vascular smooth muscle cells | ↑ secretion of matrix-degrading proteases, plaque vulnerability |
| Deregulated nutrient-sensing[46] | Hypomagnesimia (during normal aging) | Plasma/erythrocytes/platelets, human | Platelets | ↓ Intraplatelet magnesium (especially in elderly diabetics) → possible ↑ platelet hyperreactivity |
| Altered cell communication[47] | Serotonin stimulation | Platelets, human | Platelets | Platelet aggregation, amplification of ADP/adrenaline responses |
| Inflammation[48] | IL-1β increase | Humans | NA | ↑ NETosis |
| Inflammation[49] | IL-6 increase | Humans | NA | HR = 1.37 for AF |
CAD: coronary artery disease; CHIP: clonal haematopoiesis of potential; CVD: cardiovascular; eNOS: endothelial nitric oxide synthase; HUVECs: human umbilical vein endothelial cells; IVC: inferior vena cava; MPV: mean platelet volume; MI: myocardial infarction; MK: megakaryocyte; NET: neutrophil extracellular trap; NO: nitric oxide; PAI-1: plasminogen activator inhibitor-1; TF: tissue factor; TLR9: toll-like receptor 9; VSMC: vascular smooth muscle cell; VTE: venous thromboembolism; ADP: adenosine diphosphate; NETosis: neutrophil extracellular trap formation; AF: atrial fibrillation; NA: not available; vWF: von Willebrand factor; ↑: increased; ↓: decreased; →: caused.
3.1 Endothelial cells
Under physiological conditions, the endothelium exerts a central antiplatelet and anticoagulant function through the production of key inhibitory mediators such as nitric oxide (NO), prostacyclin, and the ectonucleotidase CD39, which collectively suppress platelet activation and aggregation[50]. It also synthesizes antithrombotic factors, including antithrombin, protein C and S, and thrombomodulin, which modulate coagulation and confer protection against venous thrombosis[51]. On the endothelial surface, thrombomodulin binds thrombin, redirecting its activity from clot formation to the activation of protein C. EPCR binds protein C and enhances its activation by the thrombin–thrombomodulin complex, thereby boosting local generation of activated protein C (APC). APC then inactivates the procoagulant cofactors Va and VIIIa while delivering potent anti-inflammatory and cytoprotective signals[52,53]. Decreased expression or availability of these components, caused by inflammation, oxidative stress, or protease-mediated shedding, impairs APC production, shifting the local environment toward a prothrombotic and proinflammatory state. Aging is a well-established driver of endothelial dysfunction, featuring reduced NO bioavailability, impaired vasodilation, elevated oxidative stress, and DNA damage[54,55]. NO inhibits platelet adhesion[56], suppresses TF expression[57,58], reduces fibrinogen binding to platelet glycoprotein IIb/IIIa[59], and scavenges reactive oxygen species[60]. Autophagy, a critical housekeeping mechanism for cellular homeostasis, declines with aging[61]. This decline results in reduced NO levels, thickened arterial wall, and upregulated senescence markers including p16, p21, and SA-β-gal[32]. Weibel-Palade bodies, which store vWF, colocalize with autophagosomes, and inhibition of autophagic genes Atg5 or Atg7 impairs vWF secretion. These findings indicate that autophagy regulates endothelial VWF secretion, and transient pharmacological inhibition of autophagic flux may represent a potential therapeutic strategy to prevent thrombotic events[62]. Nonetheless, senescent endothelial cells and aged mice exhibited increased vWF mRNA and cellular protein levels in the brain, lungs, and liver, accompanied by significantly higher plasma concentrations[42]. This indicates that these mechanisms may prevail over impaired autophagy and may contribute to the increased plasma vWF observed in aging[63,64]. Similarly, porcine endothelial senescent cells showed upregulation of two major pro-oxidant enzymes, NADPH oxidase and cyclooxygenase 2 (COX-2), leading to oxidative stress, decreased NO bioavailability and increased TF expression and activity. This pro-thrombotic response can be attenuated by pharmacological inhibition with VAS-2870 (NADPH oxidase inhibitor) or indomethacin[40].
3.2 Vascular smooth muscle cells (VSMCs)
Aging affects VSMCs’ contractility, plasticity, and stiffness[65]. VSMCs play a pivotal role in maintaining plaque stability, whereas their loss or dysfunction contributes to atherogenesis and plaque progression[66]. As the principal source of collagen production in the fibrous cap, VSMCs provide structural integrity that prevents plaque rupture. Apoptosis of VSMCs, induced by pro-inflammatory cytokines, oxidized low-density lipoprotein, or mechanical stress, exposes phosphatidylserine on the cell surface, thereby promoting local thrombin generation and enhancing plaque thrombogenicity[67]. Apoptotic VSMC remnants can also initiate microcalcification, increasing mechanical stress on the fibrous cap and heightening the risk of rupture. Inadequate clearance of apoptotic cells, particularly under hyperlipidemic conditions, leads to secondary necrosis and the release of pro-inflammatory cytokines, further amplifying plaque inflammation[68]. Senescent VSMCs, triggered by DNA damage, oxidative stress, or telomere attrition, contribute to fibrous cap thinning by reducing collagen synthesis and increasing the secretion of extracellular matrix-degrading proteases such as matrix metalloproteinases. Simultaneously, they exacerbate inflammation via an interleukin (IL)-1α that drives senescence-associated secretory phenotype[45].
Beyond their structural functions, VSMCs actively participate in thrombosis formation. Specifically, in arterial thrombosis, these cells can express TF and release chemokines that recruit platelets and leukocytes, amplifying thrombus formation after plaque rupture[69,70]. Their extracellular matrix not only stabilizes the plaque but also provides a scaffold that promotes platelet adhesion and aggregation in the high-shear arterial environment[71]. In contrast, in venous thrombosis, VSMCs appear to play a more supportive role by modulating endothelial function, contributing to extracellular matrix remodeling, and expressing pro-coagulant molecules under inflammatory conditions, thereby indirectly promoting fibrin-rich thrombus formation in the low-shear venous environment[72,73].
3.3 Platelets
Aging profoundly reshapes platelet biology, disrupting hemostatic balance and increasing susceptibility to thrombotic events. Epidemiological studies reveal a decline in platelet counts with age, particularly in men[74,75], possibly reflecting exhaustion of hematopoietic stem cell (HSC) reserves[76] Despite this decline, platelets from older individuals display enhanced functional activity, reflected by increased sensitivity to activation agonists such as ADP, which is attributed to raised surface density of ADP-specific P2Y12 receptor on aged platelets[77]. In elderly volunteers, platelets demonstrated elevated P-selectin (a marker of α-granule release), CD63 (a marker of dense granule release), and PAC-1 binding (an indicator of conformationally active GPIIb/IIIa at the fibrinogen-binding site), all consistent with enhanced basal activation[78]. These conformational changes, associated with elevated levels of inflammatory cytokines (IL-1β, IL-6), result in more intensive platelet aggregation. Furthermore, aged mice seem to adopt an alternative differentiation pathway wherein HSCs generate Tom+ megakaryocyte progenitors that produce hyper-reactive platelets, thus exacerbating thrombocytosis and thrombosis[79]. Similarly, the pro-inflammatory milieu of aging drives megakaryocyte reprogramming, mitochondrial dysfunction, and platelet hyperreactivity, effects that can be reversed by TNF-α blockade[80].
Paradoxically, in contrast to ADP hyperreactivity, platelets from older adults were found to be less responsive, or “resistant” to thrombin. This resistance has been attributed to reduced expression of glycoprotein Ibα, impaired secondary ADP release due to diminished ADP storage (indicating that hyperreactivity is not driven by post-activation dense granule release), and blunted calcium flux caused by desensitization of thrombin receptors (PAR-1 and PAR-4). Notably, PAR-1 levels remain unchanged, while PAR-4 expression is increased, a phenomenon also observed in thrombo-inflammatory conditions[81].
ROS are among the major drivers of platelet activation[50]. Endogenously generated ROS within platelets act as key second messengers, modulating pathways regulating activation, aggregation, and thrombus formation. In aged mice lacking platelet-specific superoxide dismutase, platelet-dependent thrombin generation was markedly increased, rendering these mice more susceptible to carotid artery and pulmonary thrombosis compared with age-matched controls[82]. Similarly, in aged C57BL/6J mice, platelet-derived ROS promoted hyperactivation and enhanced both arterial and venous thrombosis. ROS also activate mechanistic target of rapamycin complex 1 (mTORC1) in megakaryocytes and platelets of aged mice, leading to increased mean platelet volume and platelet hyperactivation, thereby promoting venous thrombosis[38]. This mechanism also seems to be preserved in humans[83]. mTORC1 integrates nutrient and growth factor signals to regulate cell growth, protein synthesis, and metabolism. Its pharmacological inhibition reduces thrombus formation, highlighting its role as a driver of age-related thrombotic susceptibility[38]. Moreover, increased ROS production increases lipid peroxidation and drives free radical-mediated transformation of arachidonic acid into bioactive isoprostanes[84,85], which in turn trigger platelet adhesion and activation[50,86]. Interestingly, metabolic disorders such as type 2 diabetes or visceral obesity further augment ROS generation, resulting in platelet activation[87,88]. In addition, non-insulin dependent diabetes combined with aging lowers intraplatelet magnesium in elderly patients, promoting calcium overload and potentially fostering platelet hyperaggregability[46].
3.4 Red blood cells
Until recently, RBCs were considered passive participants in thrombosis; however, emerging evidence reveals their active involvement in both VTE and atherothrombosis. These cells are uniquely adapted for oxygen transport, lacking organelles to maximize hemoglobin content and membrane flexibility. These features render them atypical in aging biology, as many primary hallmarks of aging are not applicable. Nevertheless, aging alters RBC function in ways that promote thrombosis. Aged RBCs exhibit increased cytosolic viscosity, which may disrupt microcirculatory flow[89]. Aging also elevates ROS levels systemically[90], and RBCs, lacking mitochondria, rely solely on enzymatic balance to maintain redox homeostasis[91]. Excess ROS triggers phosphatidylserine externalization[92], a procoagulant signal that enhances thrombin generation and fibrin formation[93]. Moreover, histones released from NETs bind RBCs, inducing phosphatidylserine exposure and amplifying coagulation[94]. RBCs also release microvesicles in response to ROS[95], many of which expose phosphatidylserine and carry heme[96,97], a potent prothrombotic molecule that activates endothelial cells to express TF[98], activates platelets[99], induces NETs formation[100], and triggers complement activation[101]. These features define a condition termed “erythropathy”[102] which is linked to aging[103] and metabolic diseases like type 2 diabetes and obesity[104]. In type 2 diabetes, RBCs show elevated arginase 1[105], which competes with NO synthesis under oxidative and hyperglycemic conditions[106], further impairing endothelial function[107,108]. Hyperglycation of RBC proteins, such as Band 3, increases RBCs aggregability and adhesion to endothelial cells via receptor for advanced glycation end products (AGEs)[109,110]. These data could potentially explain the increased VTE risk in type 2 diabetes[111]. Epidemiological studies also associate altered hematocrit with thrombotic risk. For instance, the Framingham Study identified a relationship between hematocrit levels and stroke/transient ischemic attack in older women[112], while the Tromso Study displayed an elevated risk of first VTE in individuals with high vs. low hematocrit levels, markedly in males but not in females[113]. These findings indicate that RBCs are not simple bystanders, but active contributors to thrombus formation and propagation.
3.5 Clonal hematopoiesis of intermediate potential (CHIP)
Aging also impacts HSCs, forcing myelopoiesis at the expense of lymphopoiesis[114]. This myeloid bias, coupled with increased replicative activity of HSCs, renders HSCs susceptible to somatic mutations. This phenomenon may lead to CHIP, a precursor to hematologic malignancies increasingly prevalent in individuals over 70 years of age[115]. CHIP is associated with higher risk of cardiovascular mortality, including coronary artery disease (CAD) and ischemic stroke[116,117]. Mutations in genes like DNMT3A, TET2, ASXL1 and JAK2 drive this risk. DNMT3A, TET2, and ASXL1 mutations double the risk of coronary heart disease, while the JAK2V617F mutation was related to a 12-fold increased risk of CAD. These mutations promote pro-inflammatory and/or pro-thrombotic leukocyte phenotype, depending on their ancestral variant[118,119]. Additionally, these mutations are strongly associated with polycythemia vera, a myeloproliferative neoplasm characterized by increased red blood cell mass and elevated thrombotic risk[120]. TET2 mutation accelerates atherosclerosis by enhancing IL-1β and IL-6 secretion through NLRP3 inflammasome activation in macrophages[121]. TET2 is also a risk factor for VTE[122], in connection with ischemic stroke[123] and pulmonary embolism[124]. The prevalence of JAK2V617F rises progressively with age[125], and JAK2 is frequently mutated in myeloproliferative neoplasms[126], provoking both arterial and venous thrombosis[127]. In older women, JAK2 was the only mutation clearly associated with VTE and ischemic stroke, increasing their risk by 2-fold[31]. In contrast to TET2, JAK2 mutations are rare in the general population[115,128], which poses challenges for comparative analyses due to small sample sizes. However, in younger adults, the mutations confer a stronger thromboembolic risk than TET2 mutations[124,129]. This difference may be explained by the fact that the gain-of-function JAK2V617F variant affects all myeloid progenitor cells[130] (i.e., macrophages, erythrocytes, neutrophils, and platelets) compared to TET2 which mainly controls macrophages[131]. Additionally, JAK2 promotes erythrocytosis[120], increasing blood viscosity and exacerbating a pro-inflammatory macrophage phenotype through AIM2 inflammasome activation[132]. These mutations also activate β1 and β2 integrins, key regulators of leukocytes adhesion to endothelial cells, thereby amplifying venous thrombosis[133]. Platelets carrying JAK2 mutations can be activated by mediating thromboxane cross talk with normal platelets and neutrophils[134]. Finally, the mutations facilitate plaque erosion and thrombosis in both mice[135] and younger adults[136], making it the most consequential CHIP mutation for thrombotic risk.
3.6 Inflammaging
Aging is closely linked to inflammaging, a persistent, sterile, low-grade inflammation occurring in the absence of acute infection or overt stimuli[137]. This chronic pro-inflammatory state is characterized by activation of innate immune cells, including monocytes, macrophages, and neutrophils, and is accompanied by elevated levels of IL-1 β, IL-6, TNF-α, and C-reactive protein (CRP)[138,139]. These cytokines actively promote thrombosis, either individually or synergistically, in preclinical models and in humans[140-143]. IL-1β, for instance, induces IL-6 production, stimulating hepatic synthesis of fibrinogen and plasminogen activator inhibitor-1 (PAI-1)[144]. In older patients with ST-elevation myocardial infarction (STEMI), IL-1β primes neutrophils for NETosis, releasing NETs which carry TF, thus driving thrombin generation, platelet activation, and fibrin formation[48]. Aging also alters NETosis. To be specific, mitochondrial stress enhances oxidized NETs formation[39], while autophagy defects (e.g., Atg5 deficiency) impair NET release[33].
Intestinal microbiota represents another critical driver of age-related inflammation. Aging promotes a decline in protective microorganisms[145], such as Coprococcus, and fosters dysbiosis[11]. Dysbiosis increases gut permeability, permitting bacteria and their derivatives such as pathogen-associated, damage-associated, and microbial-associated molecular patterns to enter the bloodstream[146]. Centenarians exhibit a characteristic shift in gut microbiota, with increased Proteobacteria and reduced butyrate-producing bacteria, correlating with elevated systemic IL-6 levels[147]. Beyond contributing to inflammaging, the gut microbiome has been implicated in maintaining a prothrombotic milieu[148]. Metabolites can hyperactivate platelets[149], stimulate hepatic production of vWF[150], and trigger NETosis in neutrophils[151]. For instance, the gut microbial metabolite TMAO and its precursors are associated with increased risks of MACE and all-cause mortality in patients with various cardiovascular diseases[152], likely via NLRP-3 inflammasome stimulation[153], and platelet activation[10]. Although TMAO associations are independent of age[152], older adults tend to exhibit elevated plasma TMAO levels due to dietary and microbial shifts[154].
Finally, aging enhances trained immunity, a non-antigen-specific heightened immune response upon re-stimulation, mediated by epigenetic and metabolic reprogramming[155]. Activation of NLRP-3 inflammasome, a central part of the innate immune response, increases with age[156] and can also be triggered by Western diet[157]. Trained immunity may be further amplified by microbial agents and endogenous factors such as hyperglycemia in type 2 diabetes[158]. Older patients show an increased risk of recurrent ischemic events[159], and recent evidence indicates that monocyte trained immunity accelerates atherosclerosis after STEMI in patients with advanced coronary lesions[160]. Considering that trained immunity promotes hypercoagulability through TF production and PAI-1 from macrophages[161], it might be a promising target for preventing thrombosis in the elderly (Figure 4).
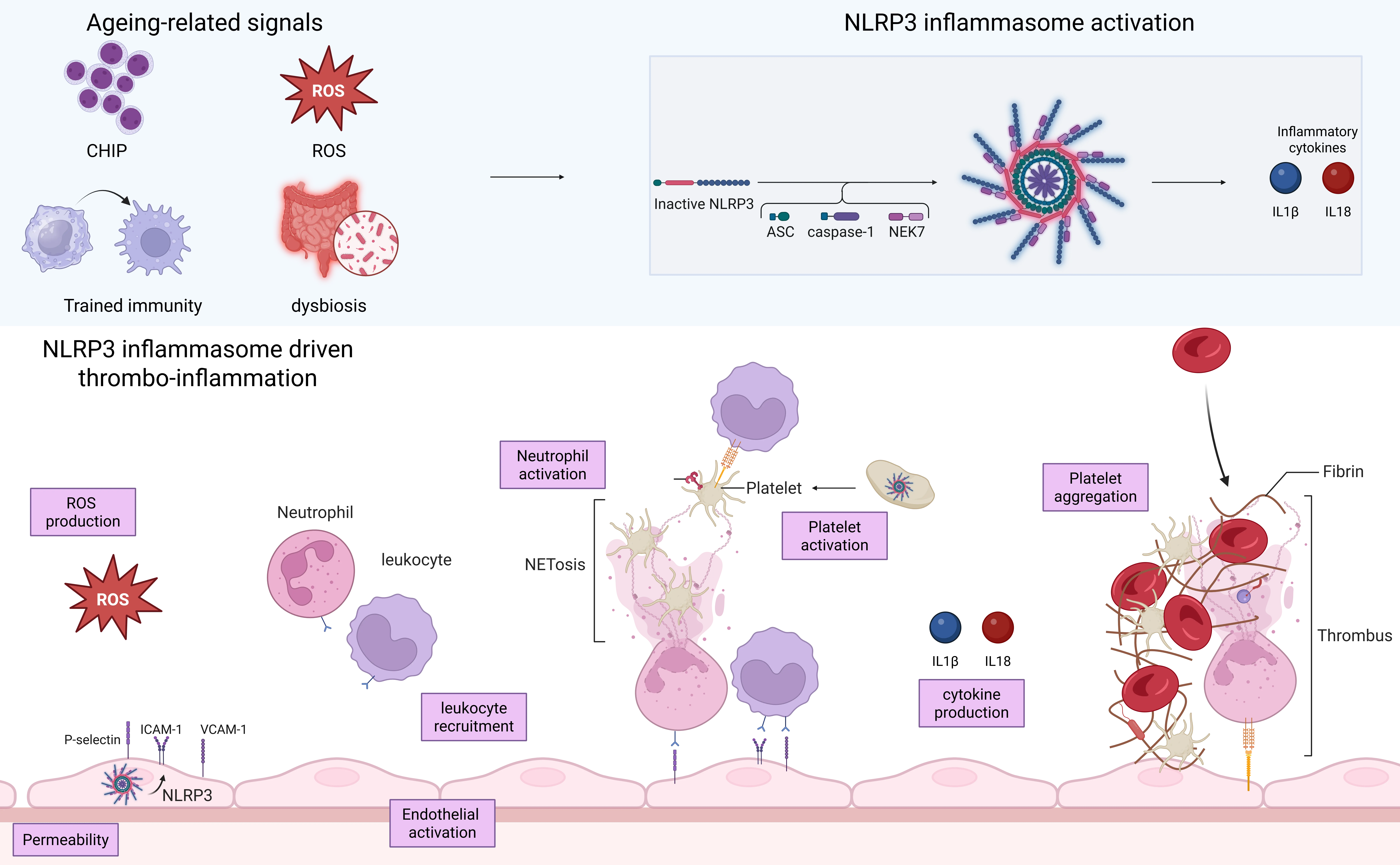
Figure 4. NLRP3 inflammasome activation as a driver of age-related platelet and endothelial dysfunction. Ageing-associated stimuli, including CHIP, increased ROS, trained immunity and gut dysbiosis, converge to activate the NLRP3 inflammasome. Assembly of the NLRP3 complex with ASC, caspase-1 and NEK7 leads to the maturation and release of the pro-inflammatory cytokines IL-1β and IL-18 (top panel). In the vasculature (bottom panel), NLRP3 activation in endothelial cells increase vascular permeability, increase ROS production, and promote endothelial activation with up-regulation of adhesion molecules, favouring leukocyte recruitment and neutrophil activation. Activated neutrophils undergo NETosis and produce cytokines that further amplify inflammation. At the same time, NLRP3 is activated intrinsically within platelets, enhancing platelet activation and aggregation. Together with fibrin deposition, these processes culminate in thrombus formation. Created in BioRender.com. ASC: apoptosis-associated speck-like protein; CHIP: clonal haematopoiesis of indeterminate potential; IL: interleukin; NETosis: neutrophil extracellular trap formation; NLRP3: NOD-, LRR- and pyrin domain-containing protein 3; ROS: reactive oxygen species.
4. Age-Driven Shifts in Hemostatic Balance
4.1 Coagulation factors
Aging favors prothrombotic imbalance through mechanisms impacting coagulation and fibrinolysis (Table 2). Increased fibrinogen and vWF, elevated prothrombin fragments and factors V, VII, VIII, IX, and impaired fibrinolysis, have been observed in both aged men and women, suggesting a persistent impact of aging on prothrombotic states and the heightened cardiovascular risk[162-164]. Aging also directly alters thrombin generation. Population-based studies show that with advancing age, men experience an increase in peak thrombin and endogenous thrombin potential, while these parameters tend to decrease or remain stable in women[165]. Women generally display shorter lag times and higher velocity indices than men, suggesting a more procoagulant profile, particularly at older ages[166]. In addition, the sensitivity of the APC anticoagulant pathway to thrombomodulin diminishes with age in both sexes, leading to reduced downregulation of thrombin generation. This age-related desensitization of the APC pathway is more pronounced in women, contributing to the observed sex-specific differences in thrombotic risk. These findings obtained from calibrated thrombin generation assays provide a comprehensive functional assessment of coagulation, integrating the net effect of multiple pro- and anticoagulant proteases, effects not captured by conventional prothrombin time or activated partial thromboplastin time tests. This further underscores the complexity of coagulation changes with aging and the necessity for sex- and age-specific reference values for TG parameters in clinical risk assessment.
Table 2. Alterations in prothrombotic factors in the elderly population.
| Biomarker | Biological source | Thrombosis impact | Direction of change with aging |
| Fibrinogen | Liver[176] | Conversion to fibrin (by thrombin), bridging of activated platelets[176] | ↑ [176] |
| II (thrombin) | Liver (produced as prothrombin, the inactive precursor of thrombin)[177] | Conversion of fibrinogen to fibrin[177] | ↑ [165,166] |
| V | Liver & megakaryocytes[178] | Conversion of prothrombin to thrombin (cofactor in the prothrombinase complex)[178] | ↑ [163] |
| VIII | Mainly by liver endothelial sinusoidal cells & lymphatic endothelial cells[179,180] | Conversion of X to Xa (as a complex with IXa)[179,180] | ↑ [64,163,171] |
| X | Liver[181] | Conversion of prothrombin to thrombin (cofactor in the prothrombinase complex)[181] | = [163] |
| XI | Liver[182] | activates factor IX, → formation of the intrinsic tenase complex (factors IXa and VIIIa)[183] | ↑ [184] |
| vWF | Endothelial cells & megakaryocytes[185] | Platelet adhesion to connective tissue, carrier of VIII[185] | ↑ [63,64,164] |
| Protein C | Liver (produced as proprotein C, the inactive precursor of protein C)[186] | Inactivation of Va & VIIIa as a cofactor with protein S[186] | = [165,167,168] ↑ [169,170] ↑ only in women[171,172] |
| Protein S | Liver[187] | Inactivation of Va & VIIIa as a cofactor with protein C[187] | = [167,168] ↑ [170,173] ↑ only in women[171,172] = in women ↓ in men[174] |
| PAI-1 | Vascular endothelial cells, adipocytes, hepatocytes, smooth muscle cells & megakaryocytes/platelets[188] | Inhibition of t-PA & u-PA → inhibition of conversion of plasminogen to plasmin[188] | ↑ [175] |
Va/VIIIa/IXa/Xa: activated factor V/VIII/IX/X; PAI-1: plasminogen activator inhibitor-1; t-PA: tissue plasminogen activator; u-PA: urokinase-type plasminogen activator; vWF: von Willebrand factor; ↑: increased; ↓: decreased; →: caused.
Besides alterations in coagulation factors, aging has been associated with concurrent impairment of counterbalancing anticoagulant mechanisms. Several studies have reported age- and gender-dependent effects on Proteins C and S. However, the age-related increases in coagulation factors generally surpass those in anticoagulant proteins, particularly in men, hence disrupting coagulation balance[167-174]. Likewise, increased PAI-1 levels have been linked to aging and age-related diseases, which in turn correlate with increased risk of thrombosis, atherosclerosis, and progression of cardiovascular disease[175]. These cumulative biomarker changes thus underlie the age-related shift in hemostatic balance toward thrombosis: upregulated fibrinogen, FV, FVIII, and vWF drive thrombin generation, impaired anticoagulant activity limits negative regulation, and enhanced PAI-1 restricts fibrinolysis. Such convergence of prothrombotic alterations can explain the high incidence of arterial and venous thrombotic events in aging populations.
4.2 Micro/nano-plastics
Exposome, the sum of individual’s environmental exposures[189], leaves biological fingerprints[190]. It accelerates biological aging[191], with chemical pollutants exerting deleterious effects in older people[192]. Micro-and-nanoplastics (MNPs) are an emerging component of chemical pollution, and their contributions to aging[193] and cardiovascular diseases are increasingly recognized[12,194]. Two lines of evidence support that MNPs accumulate in the human body, potentially influencing aging processes. The first comes from a predictive model quantifying lifetime accumulation in terms of particles and mass, especially for smaller particles absorbed via the gut, and anticipates accumulation throughout lifetime[195]. The second provides empirical evidence of MNPs presence in brain, kidneys and liver, showing that temporal increases in environmental MNPs concentrations rather than individual age per se, appear to be a key driver of the recent rise in body burden[196].
Patients with MNPs detected in their atheroma exhibited a significantly higher risk of MACE compared to those without detectable MNPs, with the amount of polyethylene correlating with the expression levels of several inflammatory markers (IL-18, IL-1β, IL-6, TNF-α)[197]. Moreover, MNPs were detected in 80% of thrombi surgically retrieved from 30 patients (median age 65.2 years) with ischemic stroke, myocardial infarction, or deep vein thrombosis[198]. Interestingly, MNP concentration in the arterial system was similar to that in the venous system[198]. Mechanistic insights from in vitro models demonstrate that polystyrene latex nanoparticles can bridge non-activated platelets, facilitating physical tethering and agglomeration, and induce concentration-dependent, GPIIb/IIIa-mediated platelet aggregation and secondary granule secretion[199]. Acute exposure to MNPs may destabilize thrombi[200], and depending on their sizes, MNPs can activate the intrinsic pathway by acting as a scaffold for Factor XII, kallikrein, and high-molecular-weight kininogen, as well as the extrinsic pathway[201].
4.3 Air pollution
According to the Global Burden of Disease 2019 study, ambient air pollution accounted for over 4 million deaths worldwide[202], with 64% occurring in individuals aged 65 years or older[192]. Air pollution, particularly exposure to fine particulate matter (PM2.5), is now a major cause of premature cardiovascular death[203]. In 1,934,453 individuals with high-risk conditions (mean age 77 years), PM2.5 exposure was associated with a significant increase in VTE, transient ischemic attack, ischemic stroke and myocardial infarction[204]. Long-term exposure to elevated PM2.5 and NO2 levels has been similarly linked to a higher incidence of stroke in American adults aged 65 years and older[205]. Furthermore, both short- and long-term exposure to PM2.5 have been related to substantially elevated risk of deep vein thrombosis in older adults (mean age 79 years), whereas only long-term exposure confers a great increase in pulmonary embolism risk in those aged ≥ 65 years[206]. Nevertheless, uncertainty remains concerning the net impact of air pollution on VTE incidence[207].
Air pollution triggers cardiovascular events through multiple interrelated mechanisms. Plaque vulnerability is promoted by increased circulating matrix metalloproteinases coupled with reduced levels of their tissue inhibitors. Increased thrombogenicity is reflected by shortened prothrombin time and elevated concentrations of soluble CD40 ligand, soluble P-selectin, PAI-1, vWF, tissue plasminogen activator, thrombin, and fibrinogen/fibrin degradation products. Systemic inflammation further amplifies vascular injury, as indicated by elevated IL-1β and CRP[208-210]. In addition, exposure to PM2.5 induces platelet activation and aggregation, mediated by oxidative stress and stimulation of the MAPK signaling pathway[211]. Overall, air pollution enhances thrombogenicity through multiple mechanisms, including platelet activation and aggregation, coagulation cascade activation[212], suppressed fibrinolysis, oxidative stress, thrombin generation, systemic inflammation, and plaque vulnerability, all of which contribute to an elevated risk of cardiovascular events.
4.4 Sirtuins (SIRTs)
SIRTs are NAD+-dependent deacetylases that modulate metabolism, inflammation, and aging[213,214]. Among them, SIRT1, SIRT3, and SIRT6 demonstrate antithrombotic actions, whereas SIRT5 can be prothrombotic by impairing fibrinolysis. SIRT7 contributes to vascular homeostasis in endothelial and smooth muscle cells, although its direct role in thrombosis remains incompletely defined[215].
Pharmacological inhibition of SIRT1 amplifies TF expression and activity in human endothelial cells by increasing p65 binding to nuclear factor-kappa B (NF-κB), and exacerbates thrombus formation in mice[216]. SIRT1 also regulates platelet reactivity in venous beds, particularly within the pulmonary circulation[217]. SIRT1 activators in wild-type or SIRT1-overexpressing mice decrease pulmonary thrombus formation induced by arachidonic acid or platelet-activating factor. This effect is mediated by SIRT1-dependent proteasomal and lysosomal degradation of the platelet-activating factor receptor expression on platelets[217]. This was confirmed in human platelets, where SIRT1 activation reduced aggregation in vitro, and in vivo[218].
SIRT3, a mitochondrial enzyme, fine-tunes cellular oxidative stress by activating SOD2[219]. SIRT3-/- mice exhibit impaired control of oxidative stress due to reduced SOD2 levels and subsequently develop faster and more stable clot formation following lipopolysaccharide exposure[220]. In patients with acute coronary thrombosis during STEMI, lower leukocyte expression of SIRT3 and SOD2 has been reported, underscoring SIRT3 as a potential therapeutic target in the acute phase of coronary disease.
SIRT6 is another deacetylase that inhibits NF-κB, the pro-inflammatory pathway leading to TF expression[221]. In human aortic endothelial cells, SIRT6 suppression increases TF expression and activates pro-inflammatory pathways such as TNF-α, cleaved polymerase 1, VCAM-1, and COX-2. Consistently, endothelial-specific Sirt6 depletion in mice expresses higher TF levels and promotes arterial occlusion[222], while SIRT6-/- mice present enhanced thrombin-induced platelet activation and aggregation[223].
In contrast, SIRT5 may function as a regulator of fibrinolysis. Loss of SIRT5 in endothelial cells increases fibrinolytic activity by lowering plasma and tissue concentrations of PAI-1[224]. SIRT5 expression is markedly elevated in patients with acute coronary syndrome (ACS) relative to non-ACS controls, indicating a potential association between SIRT5-mediated pathways and thrombotic risk[224]. Together, these findings position SIRTs as critical modulators of thrombosis, and highlight their promising therapeutic targets in age-related cardiovascular disease.
4.5 Amyloid-beta 1-40
Aβ1-40, a cleavage product of amyloid precursor protein implicated in Alzheimer’s disease, is increasingly recognized for its role in atherothrombotic processes[13]. Elevated plasma levels of Aβ1-40 are associated with chronic coronary syndromes (CCS), ACS, and early subclinical stages of cardiovascular disease[1,225-228]. In postmenopausal women, higher Aβ1-40 levels correlate with aortic stiffness, a surrogate marker of arterial aging[227,229].
Mechanistically, Aβ1-40 promotes endothelial dysfunction by inducing ROS production, inflammatory cytokines (IL-1β, IL-6), reduced NO production, and increased adhesion molecules like VCAM-1[230]. In addition, Aβ1-40 favors thrombosis in preclinical and human studies. In vitro and in vivo Alzheimer’s disease models demonstrated that fibrin clots formed in the presence of Aβ exhibit structural abnormalities and are resistant to breakdown. Consistently, fibrinogen has been detected within amyloid-positive blood vessels in both mouse and human Alzheimer’s disease tissues[231]. Aβ1-40 directly promotes platelet activation through multiple pathways. It binds to PAR1, leading to activation of thromboxane signaling[230]. Aβ1-40 plasma levels are associated with plaque echolucency, an ultrasonographic marker of plaque instability, in patients without clinically overt atherosclerotic cardiovascular disease (ASCVD). Also, increased biomarker’s levels correlate with histological features of lower plaque calcification and lower incidence of plaques lacking high-risk features in severely stenotic plaques from patients undergoing endarterectomy[232]. Notably, Aβ1-40 is associated with antiphospholipid syndrome, a systemic autoimmune disorder with often devastating outcomes, characterized by arterial or venous thrombosis[233]. High sensitive-CRP levels are the only independent determinant of Aβ1-40, and elevated Aβ1-40 plasma levels are independently associated with recurrent arterial events, providing further discriminative value beyond the Global AntiPhospholipid Syndrome Score[234]. These results stress the potential role of this biomarker in thrombo-inflammation.
5. Thrombosis Treatment and Clinical Implications
5.1 Current antithrombotic guidelines
Antiplatelet and anticoagulant agents remain the cornerstone of thrombosis management. In elderly patients, adherence to guidelines for selection, dosage modification, and shortened combination therapy optimizes overall benefit, whereas fibrinolysis is reserved for urgent, time-sensitive situations.
Aspirin irreversibly inhibits cyclooxygenase 1, thus preventing Thromboxane-mediated platelet activation and aggregation. As monotherapy, it is effective for secondary prevention[235], but its use in primary prevention among adults ≥ 70 years without established ASCVD, is unfavorable due to significantly higher risk of major hemorrhage[236].
Dual antiplatelet therapy (DAPT) is recommended for patients with ACS and should be maintained for a guideline-specified, risk-adjusted duration[237]. Among older adults (≥ 75 years) with CCS undergoing percutaneous coronary intervention (PCI), clopidogrel 75 mg daily remains the preferred P2Y12 inhibitor in combination with aspirin, for DAPT. For ACS, clopidogrel serves as an alternative to prasugrel 5 mg, which showed similar efficacy and safety[238,239], but no net clinical benefits were observed in clinical trials[240,241]. Furthermore, although ticagrelor was superior to clopidogrel in the PLATO trial, a subgroup analysis indicated its similar effects in ACS patients ≥ 75 years compared with younger patients[242]. However, these results were not confirmed in the POPular AGE trial, which showed that clopidogrel is a safer and equally effective alternative to ticagrelor or prasugrel in patients ≥ 70 years with NSTEMI[243]. Data from the SWEDEHEART registry, evaluating ACS patients discharged on DAPT (aspirin plus clopidogrel or ticagrelor), further suggested that ticagrelor may increase bleeding and mortality in older patients despite similar ischemic outcomes[244]. Meta-analysis supports these findings by showing that potent P2Y12 inhibitors (prasugrel, ticagrelor) reduce cardiovascular death but increase major bleeding without improving overall MACE or all-cause mortality compared to clopidogrel[245]. Notably, genetic variability in the P2Y12 receptor pathway, such as CYP2C19 loss-of-function variants, is associated with poor clopidogrel activation and increased thrombotic risk. Genetic screening for CYP2C19 can help identify poor metabolizers, and guide selection of more potent P2Y12 inhibitors (ticagrelor or prasugrel), which are not influenced by CYP2C19 status and can reduce ischemic events at the expense of increased bleeding[246]. The American Heart Association endorses CYP2C19 genetic testing to optimize antiplatelet therapy in ACS or PCI[247], supporting precision medicine approaches that balance efficacy and safety[248]. On the other hand, the ESC 2024 guidelines do not recommend routine genotype testing for patients with CCS and STEMI, except in the context of high-risk PCI[249].
Residual platelet reactivity complicates therapy in the elderly. Patients over 65 with a history of myocardial infarction[250] or ischemic stroke[159] frequently demonstrate high residual platelet reactivity after stenting, placing them at greater risk for recurrent events, including stent thrombosis[251-254]. Prospective data show platelet reactivity rises with age, partly attributable to reduced cytochrome P450 metabolism[255]. Interestingly, higher platelet reactivity may occur despite adequate P2Y12 pharmacologic response, pointing to intrinsic platelet hyperactivity rather than drug failure[256]. In this setting, switching from clopidogrel to prasugrel enhances platelet inhibition and overcomes residual platelet reactivity in most patients[257]. On the contrary, the observational SENIOR-PLATELET study indicated that patients over 75 years with CAD and PCI history had greater residual platelet reactivity than younger individuals, and that neither doubling clopidogrel to 150 mg nor using prasugrel 10 mg fully eliminates this age-related difference[258]. Interestingly, residual platelet reactivity is also observed in older patients treated with ticagrelor-based DAPT[259]. The presence of comorbidities further complicates thrombotic risk management. Elderly patients with type 2 diabetes are particularly vulnerable, as diabetes amplifies platelet hyperactivity and impairs clopidogrel metabolism[260].
5.2 Atrial fibrillation & anticoagulants in the elderly
Older adults, particularly those with dementia or frailty, are less likely to be prescribed anticoagulants, even though their higher risk of thromboembolic stroke compared with younger patients with atrial fibrillation[261]. This underutilization largely reflects concerns regarding their increased bleeding susceptibility. Aging also affects drug metabolism, including changes in gastrointestinal absorption, hepatic and renal clearance, and pharmacokinetic or pharmacodynamic responses. These alterations, combined with polypharmacy and potential drug interactions, further complicate anticoagulant therapy in the elderly.
When it comes to anticoagulant therapy for atrial fibrillation, two principal treatment categories are available: Vitamin K antagonists (VKAs) and Direct Oral Anticoagulants (DOACs). Several factors limit the use of VKAs, especially in the elderly, including their narrow therapeutic index, the need for complex dose adjustments to maintain therapeutic levels, frequent international normalized ratio monitoring, prolonged pharmacodynamic effects, and numerous food and drug interactions[262]. Yet, current guidelines do not recommend switching from VKA treatment (within stable achievement of therapeutic targets) to a DOAC in frail older patients, as DOACs have been associated with more bleeding complications compared with continuing VKA treatment without a corresponding reduction in thromboembolic events in atrial fibrillation[263,264]. The primary barrier to DOAC initiation in the elderly is their perceived high bleeding risk or prior bleeding, even though the annual incidence of major bleeding remains relatively low[265-267]. Apixaban, in specific, has been associated with reduced stroke and overall mortality, lower bleeding risk and fewer adverse events in elderly and frail patients[266,268]. Dabigatran and rivaroxaban, when compared with warfarin, have demonstrated lower event rates in older but non-frail patients[268]. Individualized risk modification may also incorporate selective assessment of DOAC plasma levels. DOACs are administered at fixed doses based on clinical characteristics such as age, renal function, and body weight, and routine measurement of plasma levels is not recommended except in specific clinical scenarios, such as severe bleeding or thromboembolic events despite apparent DOAC compliance[269]. However, recent evidence illustrates considerable interindividual variability in DOAC concentrations which are linked to acute thromboembolic and bleeding events[270]. As such, further strategies are required to optimize DOAC use in elderly patients at elevated risk for bleeding and stroke. Equally important is the development of alternative stroke-prevention methods for frail patients with prohibitive bleeding risk.
5.3 Novel antiplatelets/anticoagulants
To mitigate residual thrombotic and bleeding risk in the elderly, novel antiplatelet treatments are designed to target not only receptors but also intracellular signaling pathways[271]. These regimens aim to modulate key processes in platelet adhesion, activation and aggregation[15]. Revacept, a GPIV fusion protein, does not increase bleeding risk in patients with stable ischemic heart disease undergoing PCI[272]. In a phase II trial, it reduced the combined safety and efficacy endpoint (incidence of cerebrovascular and coronary ischaemic events, and bleeding complications) in patients with symptomatic carotid stenosis[273]. Glenzocimab, a humanized antibody fragment, safely inhibits collagen-induced platelet aggregation, but demonstrates no significant clinical benefit in stroke[274]. The phase 1b/2a ACTIMIS study showed that glenzocimab 1000 mg, added to the thrombolytic alteplase with or without thrombectomy, was well tolerated and may reduce intracranial hemorrhage and mortality in acute ischemic stroke[275]. Therefore, its role as an add-on therapy is still under consideration (NCT05070260). In contrast, interventions targeting the GPIb–GPV–GPIX–vWF axis seems to increase bleeding risk[276]. The principal platelet activation receptors are P2Y12 and PAR-1/PAR-4. Vorapaxar, a PAR-1 inhibitor approved for patients with prior myocardial infarction or peripheral artery disease[277,278], is limited by safety concerns in ACS[279] and older patients[280]. Various PAR-1 inhibitors are under investigation. Selatogrel, a promising P2Y12 receptor antagonist, has shown superior efficacy and safety compared to ticagrelor in animal models[281], and is currently being tested in patients discharged after acute myocardial infarction (NCT04957719). Next-generation GPIIb/IIIa strategies include conformation-specific single-chain variable fragment antibodies that selectively bind the activated integrin[282], small molecules targeting integrin outside-in signaling (such as the myristoylated ExE peptide motif, mP6[283]), and conformation-specific single-chain variable fragment engineered to incorporate enzymes like CD39, an ADP-hydrolyzing ectonuclease that localizes antithrombotic activity directly at the thrombus site[284]. Multiple preclinical and clinical trials examine the role of intracellular enzymes, such as protein disulfide isomerase involved in GPIIb/IIIa activation[285] and fibrin synthesis[286], and kinases like phosphoinositide 3-kinase a driver of thrombus propagation[287].
Elderly individuals are at increased risk for both thrombosis and bleeding, highlighting the need for specialized antithrombotic treatments in this population. Factor XI, which is elevated in the elderly[184], activates factor IX to enhance the formation of the intrinsic tenase complex (factors IXa and VIIIa), ultimately promoting thrombin production[183]. Notably, individuals with XI deficiency have lower risk of stroke[288] and deep vein thrombosis[289]. Several novel treatments targeting FXI have been developed, such as antisense oligonucleotides, monoclonal antibodies, and small molecules inhibitors[290]. FXI inhibitors significantly reduce both bleeding and thromboembolic risk compared to enoxaparin. However, when compared with DOACs, they are associated with a marked decrease in bleeding events, albeit not thromboembolic events[291]. Additionally, relative to heparin, FXI inhibitors exhibited a lower risk for thromboembolism, as well as major or clinically relevant non-major bleeding compared to low molecular weight heparin[292]. In a phase 3 trial involving patients with atrial fibrillation, asundexian, a small-molecule inhibitor of FXI, was associated with reduced bleeding risk but higher stroke risk compared with apixaban[293].
5.4 Dual antithrombotic therapy
Advancing age is characterized by a substantial comorbidity burden, and elderly patients often exhibit a fragile balance between the competing risks of bleeding and thrombosis. Although antithrombotic therapy is generally indicated in the elderly with cardiovascular disease due to their increased thrombotic risk, a holistic evaluation with systematic bleeding risk assessment is required[237,263]. Among patients ≥ 75 years receiving antiplatelet agents, the long-term risk of major, particularly fatal bleeding seems higher than in younger individuals[294]. Current guidelines recommend discontinuing DAPT after 1-3 months following PCI in ACS patients with high bleeding risk, but not high ischemic risk, and continuing with single antiplatelet therapy[237]. Several clinical trials have demonstrated that monotherapy after 1-3 months following PCI in elderly patients or those at high bleeding risk, was associated with a lower incidence of clinically relevant bleeding, without a significant increase in ischemic cardiovascular events[295-298]. However, the STOPDAPT-2-ACS trial, demonstrated that patients receiving clopidogrel monotherapy for less than 2 months, compared to standard 12 months of DAPT, showed noninferiority for cardiovascular or bleeding events, indicating the need for further clinical investigation[299].
For patients requiring dual pathway inhibition, which means concomitant inhibition of platelets and coagulation, such as patients with atrial fibrillation undergoing PCI, current guidelines support early cessation of aspirin (< 1 week), followed by oral anticoagulant (OAC) and clopidogrel for up to 6 months in patients who are not at high ischemic risk, and then monotherapy with OAC[300]. OACs are usually preferred over VKA. Patients with nonvalvular atrial fibrillation who had undergone PCI, low-dose rivaroxaban plus a P2Y12 inhibitor for 12 months, or very-low-dose rivaroxaban plus DAPT for up to 12 months, demonstrated lower rates of clinically significant bleeding compared with VKA plus DAPT therapy[301]. Low-dose rivaroxaban also appears effective in elderly patients with peripheral artery disease after lower extremity revascularization, where bleeding and ischemic risks simultaneously increase. The VOYAGER PAD trial showed that in this patient group, low-dose rivaroxaban plus aspirin provided overall benefits without increasing major bleeding[302]. In patients with stable CAD, evidence from the COMPASS trial indicates that adding low-dose rivaroxaban to aspirin reduces major cardiovascular events[303,304]. Accordingly, in the context of long-term antithrombotic therapy, the 2024 ESC CCS guidelines provide a Class IIa recommendation for this regimen as an alternative to prolonged DAPT in patients with high ischemic but not high bleeding risk. Nonetheless, bleeding remains a key limitation, and no universally accepted tool exists to guide patient selection. Moreover, most COMPASS participants were enrolled remotely from recent ACS or revascularisation, limiting the applicability of these findings to other CAD subgroups. Antithrombotic therapies are generally beneficial for elderly patients with cardiovascular disease or atrial fibrillation, but systematic bleeding risk assessment and careful consideration of individual patient characteristics are essential for optimizing treatment strategies.
5.5 Anti-inflammatory drugs
The role of inflammation in atherothrombosis is well established and has been tested in landmark clinical trials. In the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS), stable post-MI patients treated with 150 mg canakinumab experienced a 15% lower risk of the primary endpoint compared to placebo[305]. Interestingly, the investigators noted that statin-treated patients with residual inflammatory risk, defined as a baseline hs-CRP ≥ 2 mg/L, experienced future event rates comparable to or higher than those with residual LDL cholesterol-driven risk. Notably, in a subgroup analysis of the CANTOS population, only patients with TET2 mutation demonstrated a lower cardiovascular risk with canakinumab, while no benefit was observed in patients without clonal hematopoiesis or with non-TET2-related mutations. However, canakinumab was also associated with a higher incidence of fatal infection and sepsis. Its high cost, infection risk, and lack of mortality benefit have limited its adoption for atherothrombosis prevention in clinical practice[306]. In contrast, colchicine has emerged as a more accessible anti-inflammatory option. Colchicine has been indicated for patients with established CAD[307,308] in the context of secondary prevention, with Class IIa recommendation[300]. A recent meta-analysis supports its efficacy in reducing MACE among patients with stable CAD[309].
5.6 Gerotherapeutics
Current clinical management of thrombosis in the elderly remains mainly event-driven or guided by population-based risk scores (e.g., CHA2DS2-VASc). While aging-related biology is not yet routinely incorporated, several medications targeting the hallmarks of aging and aiming to directly or indirectly treat thrombosis in the elderly, are under investigation (Table 3).
Table 3. Gerotherapeutics and thrombosis.
| Medication | Function/Target | Treatment result | % of change in treatment result | Model | Type of Thrombosis |
| Gerotherapeutic drugs that have been shown to affect thrombosis in mammals | |||||
| Rapamycin[38] | Inhibition of mTORC1 signaling, inducing aging-related platelet hyperactivation | ↓ experimental DVT in ↓ the average size of MKs, PF4 production, aIIbb3 activation, and platelet aggregation | - | Mice | Venous |
| ROS scavenger NAC[38] | ROS production induces activation of mTORC1 in MKs andplatelets | ↓ venous thrombus size | - | Mice | Venous |
| Torin 1[83] | mTOR specific inhibitor | ↓ platelet aggregation | - | Platelets from young (18-45 years old) and aged (> 65 years old) human donors | Venous |
| EHT 1864[83] | Specific Rac1 inhibitor ↑ TXA2 generation, and platelet hyperreactivity | ↓ platelet aggregation | - | Platelets from young (18-45 years old) and aged (> 65 years old) human donors | Venous |
| Canakinumab[305] | Monoclonal antibody for interleukin-1β | ↓ MACE | Primary end point: ↓ 15% risk than placebo Secondary end point: ↓ 17% risk than placebo,* & Hs-CRP: ↓ 26% in 50-mg dose of canakinumab group, ↓ 37% in the 150-mg group, ↓ 41% in the 300-mg group | Human randomized control trial, not FDA-approved | Arterial |
| Colchicine[307] | Suppresses inflammasome | ↓ MACE | Ischemic cardiovascular events: 5.5% of the patients in the colchicinegroup, 7.1% of the placebo group (p = 0.02) | Human randomized control trial, FDA-approved | Arterial |
| DNase1[310] | Reduction of plasma cfDNA burden | Reduction of age-associated increases in thrombin generation in mice and humans, Decrease in the development of experimental venous thrombi | ↓ 31% and 26% in ETP in young WT and young Gpx1 Tg mice, respectively, and ↓ 43% and 39% in aged WT and aged Gpx1 Tg mice, respectively&↓ 43% in ETP in human plasma samples for young subjects and ↓ 52% for middleaged/older subjects | Mouse model: C57BL6J mice and littermates of glutathione peroxidase1 transgenic and wildtype miceat young (4 month) and old (20 month) ages&Human model: Plasma samples from healthy young (1839 years) or middle-aged/older (50-72 years) humans | Venous |
| Gerotherapeutic drugs that may act on thrombosis events by affecting vascular wall resilience | |||||
| NLRP3 inhibitor MCC950[121] | TET2-deficient hematopoietic cells generate a pool of macrophages with enhanced pro-atherogenic activities, which increase NLRP3 inflammasome-mediated interleukin-1β secretion | Increased atheroprotective activity in chimeric mice reconstituted with TET2-deficient cells | ~↓ 50% in atherosclerotic plaque size in mice transplanted with 10% Tet2-/- and 90% Tet2+/+ cells | Mice | Arterial/Atherosclerosis |
| Trehalose[311] | Autophagy-enhancing agent | Preservation of autophagy markers expression and endothelium-dependent dilatation by reducing oxidative stress, inflammatory cytokine expression and increasing NO bioavailability | - | Mice and cultured human umbilical vein endothelial cells | Vascular endothelial function |
| Trehalose[312] | Autophagy-enhancing agent | Improvement of resistance artery and endothelium-dependent dilatation sensitivity to NO | ↑ 30% FBFACh (marker of resistance artery endothelial function)& ↑ 30% FBFSNP (marker of Endothelium-independent dilation) | Human randomized control trial, not FDA-approved | Vascular endothelial function |
| SIRT1[313] | Regulator from the sirtuin family of anti-ageing proteins, expressed in the vasculature, regulating vascular tone and vasodilatation | Attenuation of COX-2 hyper-activation and upregulation of sGCβ1 in smooth muscle cells, protecting from age-induced vasoconstrictor responses | - | Mice | Vascular endothelial function |
| MitoQ[314] | Mitochondria-targeted antioxidant reducing mitochondrial reactive oxygen species | Improvement of brachial artery flow-mediated dilation, reduction of aortic stiffness and plasma oxidized-LDL | ↑ 42% FMD, ↓ 13% oxidized LDL | Human randomized control trial, not FDA-approved | Vascular endothelial function |
| Senolytic treatment (Dasatinib + Quercetin)[315] | Reduction of senescentcell burden, improvement of vasomotor functionby increasing NO bioavailability | Reduction in senescent cell burden, aortic calcification and osteogenic signaling, improvement of basal NO signaling in medial and intimal regions of atherosclerotic vessels | - | Mice | Vascular endothelial function |
NAC: N-acetyl-L-cysteine; MTORC1: mechanistic target of rapamycin complex 1; DVT: deep vein thrombosis; MK: megakaryocytes; MPV: mean platelet volume; PF4: platelet factor 4; ROS: reactive oxygen species; hs-CRP: high-sensitivity C-reactive protein; ETP: endogenous thrombin potential; WT: wild type; Gpx1 Tg: glutathione peroxidase-1 transgenic; TET2: ten-eleven translocation 2; NO: nitric oxide; FBFACh: forearm blood flow to brachial artery infusion of acetylcholine; FBFSNP: FBF to sodium nitroprusside; SIRT1: sirtuin1; sGC: soluble guanylyl cyclase; *: Patients in the 150-mg dose of canakinumab group, but not the other doses (50 mg, 300 mg), met the prespecified multiplicity-adjusted threshold for statistical significance for the primary and secondary endpoints; COX-2: cyclooxygenase 2; MACE: major adverse cardiovascular events; ↑: increased; ↓: decreased; →: caused.
One key pathways leading to platelet aggregation and thrombosis in the elderly involves the activation of mTORC1 in megakaryocytes and platelets, induced by increased ROS. mTORC1 activation, in turn, enhances bone marrow megakaryocytes size, mean platelet volume and platelet activation. Administration of rapamycin, an mTORC1 inhibitor, appears to significantly reduce susceptibility to experimental deep vein thrombosis in aged mice, whereas treatment with a ROS scavenger, N-acetyl-L-cysteine, decreases megakaryocytes size, mean platelet volume, platelet activation, and thrombus size[38]. Similarly, it was shown that aging alters mTOR phosphorylation, enhancing Rac1 and p38 activation, which increases thromboxane production, platelet hyperactivity, and thrombosis. Inhibition of mTOR by Torin 1 and Rac1 by EHT 1864 reverses platelet hyperactivity[83]. Aging also induces increases in circulating cell‐free DNA, contributing to a prothrombotic state. Treatment with DNase 1, likely through hydrolyzing cell‐free DNA, reduces thrombin generation and protects against venous thrombosis during aging[310].
Other emerging medications may indirectly mitigate thrombosis in the elderly. TET2-deficient macrophages exhibit an increase in NLRP3 inflammasome-mediated IL-1β secretion. Administration of an NLRP3 inhibitor, MCC950, has been shown to enhance atheroprotective activity[121]. Aging also contributes to endothelial dysfunction through impaired autophagy. Treatment with trehalose, an autophagy-enhancing agent, preserves arterial endothelial function by reducing oxidative stress and inflammation and increasing NO bioavailability via an autophagy-dependent mechanism[311,312]. Vascular aging is further exacerbated by loss of function of SIRT1, a longevity regulator with atheroprotective properties, leading to downregulation of soluble guanylyl cyclase (sGC) and upregulation of COX-2 in arteries. Increased endothelial SIRT1 expression appears to prevent vascular aging by upregulating sGCβ1 in smooth muscle cells, thus maintaining vasodilator responses[313]. Another key mechanism of age-induced vascular dysfunction is excessive superoxide-related oxidative stress, mainly originating from the mitochondria. Supplementation with MitoQ, a mitochondrial-targeted antioxidant, improves markers of endothelial function, including brachial artery flow-mediated dilation and aortic stiffness, both in preclinical and clinical studies, and emerges as a new potential therapeutic agent[314]. Aging is also known to increase senescent cell burden. Preclinical evidence suggests that chronic administration of senolytic treatment improves vasomotor function by increasing NO bioavailability, while reducing aortic calcification and osteogenic signaling[315]. The effectiveness and safety of novel therapeutic agents targeting the aging hallmarks, with the potential to lower the thrombotic risk in age-related morbidity and mortality, require further investigated.
6. Future Directions
6.1 Multi-omics: A new era in thrombosis
As clinical practice moves beyond a “one-size-fits-all” approach, multi-omics technologies are laying the groundwork for precision medicine in thrombosis[316,317]. Multi-omics approaches (i.e., genomics, epigenomics, transcriptomics, proteomics, metabolomics, and epitranscriptomics) offer solutions to key challenges, such as high residual platelet reactivity, optimal DAPT duration, and identification of novel therapeutic targets[318,319]. Recently, the concept of “immunothrombolysis” has emerged, challenging the view of thromboinflammation as purely detrimental[320]. In stroke patients, integrative multi-omics using scRNA-seq and CITE-seq revealed a pro-resolving monocyte-neutrophil axis. NR4A1hi non-classical monocytes, activated by coagulation, recruited neutrophils via CXCL8-CXCR1/2 signaling. Within thrombi, neutrophils adopted a hypoxia-driven thrombus-resolving urokinase receptor (PLAUR)+ phenotype, regulated by HIF1α, thereby promoting thrombus resolution[320]. Another systems-level study applied Mendelian randomization and network analysis across genomics, proteomics, transcriptomics, metabolomics, and immunomics to explore causal links between cytokines and thrombotic disease traits. This approach identified 20 independent cytokine-thrombotic disease associations, revealing shared and autonomous mechanisms governing inflammatory-thrombotic interactions in arterial and venous thrombosis. By integrating diverse omic approaches with advanced in vivo and in vitro experimental systems, computational biology, and multiplex high-dimensional multi-color cytometry, these systems-based approach provide novel insights into dynamic cellular programs and intercellular communication, ultimately advancing the resolution of thrombotic disease.
6.2 Artificial intelligence and thrombosis in elderly
Artificial intelligence and machine learning models are emerging as promising tools for optimizing treatment adherence, anticoagulant dosing, and adverse event prediction[321]. Several machine learning models have been developed to predict the optimal dosage of warfarin treatment, both at hospital discharge and during long-term maintenance therapy using international normalised ratio prediction tools[322,323]. These tools exhibit robust predictive value, often exceeding physicians’ estimation, though external validation is still limited. Interestingly, machine learning models have also been used to identify drug interactions in warfarin-treated patients with stable international normalized ratio. Random forest models accurately detected known drug interactions, such as β-lactamase-resistant penicillins, antithrombotic agents, class III antiarrhythmics, opioids, glucocorticoids and triazole derivatives, and identified previously unrecognized interactions with antipropulsives, which appear to increase international normalised ratio levels[324]. Moreover, emerging artificial intelligence models predict the risk of major bleeding, stroke/systemic embolism, and all-cause mortality in patients with atrial fibrillation, leveraging serial prothrombin time and international normalized ratio during the first 30 days of VKA treatment. These models demonstrate predictive accuracy, although several limitations remain to be addressed[325]. Similarly, machine learning models have been developed to predict bleeding and vascular events risk in patients receiving rivaroxaban or dabigatran, offering the potential of more individualized treatment options[326,327].
7. Conclusion
Aging is a biological process that reshapes the vascular landscape and shifts the delicate hemostasis balance toward thrombosis. The thrombotic risk in older adults is not merely an inevitable consequence of chronological age, but the cumulative impact of molecular and cellular alterations across the vascular wall, blood cells, and immune system. Indeed, elevated epigenetic age is associated with a prothrombotic hemostatic profile[328]. These alterations, compounded by environmental exposures, chronic inflammation, and clonal hematopoiesis, create a prothrombotic milieu that challenges current prevention and treatment strategy.
Most antithrombotic therapies remain event-driven, largely ignoring the upstream biology of aging. Given that aging itself is the strongest risk factor for arterial and venous thrombosis, this disparity is particularly striking. The advance of geroscience-guided interventions (e.g., mTOR inhibitors, senolytics, autophagy enhancers, mitochondrial antioxidants) offer the potential to address the underlying causes of vascular aging rather than its effects. Yet, these promising strategies are predominantly confined to preclinical research, creating a substantial translational gap.
In the near future, risk prediction and therapeutic personalization may be revolutionized by incorporating biological age metrics, multi-omics profiling, and artificial intelligence into clinical practice. When paired with innovative gerotherapeutics, these strategies hold the potential to reduce thrombotic disease burden in aging populations. The transition from reactive to proactive approaches that maintain vascular health throughout life is ultimately the challenge.
Acknowledgments
We acknowledge the use of large language models to identify improvements in the writing style. BioRender.com was used to create the figures.
Authors contribution
Stellos K: Conceptualization, data curation, supervision, writing-review & editing.
Zervas G: Data curation, visualization, writing-original draft, writing-review & editing.
Konstantaki C: Data curation, writing-original draft, writing-review & editing.
Stamatelopoulos K: Data curation, supervision, writing-review & editing.
Tual-Chalot S: Data curation, supervision, visualization, writing-review & editing.
Sigl M: Writing-review & editing.
Conflicts of interest
Konstantinos Stellos is an Editorial Board Member of Geromedicine. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Simon Tual-Chalot is supported by grants from the British Heart Foundation grant (PG/23/11093) and the Royal Society (RG\R1\241197). Konstantinos Stellos is supported by the German Research Foundation (DFG) (CRC1366 C07, project no. 394046768), the German Centre for Cardiovascular Research (DZHK), the Health + Life Science Alliance Heidelberg Mannheim GmbH and the Helmholtz-Institute for Translational AngioCardioScience (HI-TAC) of the Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC) at the Heidelberg University.
Copyright
© The Author(s) 2025.
References
-
1. Liberale L, Tual-Chalot S, Sedej S, Ministrini S, Georgiopoulos G, Grunewald M, et al. Roadmap for alleviating the manifestations of ageing in the cardiovascular system. Nat Rev Cardiol. 2025;22(8):577-605.[DOI]
-
2. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell. 2023;186(2):243-278.[DOI]
-
3. Herzog MJ, Müller P, Lechner K, Stiebler M, Arndt P, Kunz M, et al. Arterial stiffness and vascular aging: Mechanisms, prevention, and therapy. Sig Transduct Target Ther. 2025;10(1):282.[DOI]
-
4. Ding Y, Zuo Y, Zhang B, Fan Y, Xu G, Cheng Z, et al. Comprehensive human proteome profiles across a 50-year lifespan reveal aging trajectories and signatures. Cell. 2025;188(20):5763-5784.e5726.[DOI]
-
5. Wendelboe AM, Raskob GE. Global burden of thrombosis. Circ Res. 2016;118(9):1340-1347.[DOI]
-
6. Spray L, Richardson G, Booth LK, Haendeler J, Altschmied J, Bromage DI, et al. How to measure and model cardiovascular aging. Cardiovasc Res. 2025;121(10):1489-1508.[DOI]
-
7. Previtali E, Bucciarelli P, Passamonti SM, Martinelli I. Risk factors for venous and arterial thrombosis. Blood Transfus. 2011;9(2):120-138.[DOI]
-
8. Spray L, Richardson G, Haendeler J, Altschmied J, Rumampouw V, Wallis SB, et al. Cardiovascular inflammaging: Mechanisms, consequences, and therapeutic perspectives. Cell Rep Med. 2025;6(9):102264.[DOI]
-
9. Stark K, Massberg S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol. 2021;18(9):666-682.[DOI]
-
10. Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, et al. Gut microbial metabolite tmao enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165(1):111-124.[DOI]
-
11. Hou K, Wu ZX, Chen XY, Wang JQ, Zhang D, Xiao C, et al. Microbiota in health and diseases. Signal Transduct Target Ther. 2022;7(1):135.[DOI]
-
12. Prattichizzo F, Ceriello A, Pellegrini V, La Grotta R, Graciotti L, Olivieri F, et al. Micro-nanoplastics and cardiovascular diseases: Evidence and perspectives. Eur Heart J. 2024;45(38):4099-4110.[DOI]
-
13. Aivalioti E, Georgiopoulos G, Tual-Chalot S, Bampatsias D, Delialis D, Sopova K, et al. Amyloid-beta metabolism in age-related neurocardiovascular diseases. Eur Heart J. 2025;46(3):250-272.[DOI]
-
14. Młynarska E, Czarnik W, Fularski P, Hajdys J, Majchrowicz G, Stabrawa M, et al. From atherosclerotic plaque to myocardial infarction—The leading cause of coronary artery occlusion. Int J Mol Sci. 2024;25(13):7295.[DOI]
-
15. McFadyen JD, Schaff M, Peter K. Current and future antiplatelet therapies: Emphasis on preserving haemostasis. Nat Rev Cardiol. 2018;15(3):181-191.[DOI]
-
16. Fahed AC, Jang IK. Plaque erosion and acute coronary syndromes: Phenotype, molecular characteristics and future directions. Nat Rev Cardiol. 2021;18(10):724-734.[DOI]
-
17. Yao M, Ma J, Wu D, Fang C, Wang Z, Guo T, et al. Neutrophil extracellular traps mediate deep vein thrombosis: From mechanism to therapy. Front Immunol. 2023;14:1198952.[DOI]
-
18. Navarro S, Stegner D, Nieswandt B, Heemskerk JWM, Kuijpers MJE. Temporal roles of platelet and coagulation pathways in collagen- and tissue factor-induced thrombus formation. Int J Mol Sci. 2022;23(1):358.[DOI]
-
19. Ruggeri ZM. Old concepts and new developments in the study of platelet aggregation. J Clin Invest. 2000;105(6):699-701.[DOI]
-
20. ten Cate H, Guzik TJ, Eikelboom J, Spronk HMH. Pleiotropic actions of factor xa inhibition in cardiovascular prevention: Mechanistic insights and implications for anti-thrombotic treatment. Cardiovasc Res. 2021;117(9):2030-2044.[DOI]
-
21. Ren D, Fedorova J, Davitt K, Van Le TN, Griffin JH, Liaw PC, et al. Activated protein c strengthens cardiac tolerance to ischemic insults in aging. Circ Res. 2022;130(2):252-272.[DOI]
-
22. Khan F, Tritschler T, Kahn SR, Rodger MA. Venous thromboembolism. The Lancet. 2021;398(10294):64-77.[DOI]
-
23. Wolberg AS, Rosendaal FR, Weitz JI, Jaffer IH, Agnelli G, Baglin T, et al. Venous thrombosis. Nat Rev Dis Primers. 2015;1(1):15006.[DOI]
-
24. Zifkos K, Dubois C, Schäfer K. Extracellular vesicles and thrombosis: Update on the clinical and experimental evidence. Int J Mol Sci. 2021;22(17):9317.[DOI]
-
25. Grover SP, Mackman N. Tissue factor: an essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol. 2018;38(4):709-725.[DOI]
-
26. Undas A, Brummel-Ziedins KE, Mann KG. Statins and blood coagulation. Arterioscler Thromb Vasc Biol. 2005;25(2):287-294.[DOI]
-
27. Van Bruggen S, Martinod K. The coming of age of neutrophil extracellular traps in thrombosis: Where are we now and where are we headed? Immunol Rev. 2023;314(1):376-398.[DOI]
-
28. Abdellatif M, Rainer PP, Sedej S, Kroemer G. Hallmarks of cardiovascular ageing. Nat Rev Cardiol. 2023;20(11):754-777.[DOI]
-
29. Morgan RG, Walker AE, Trott DW, Machin DR, Henson GD, Reihl KD, et al. Induced Trf2 deletion leads to aging vascular phenotype in mice associated with arterial telomere uncapping, senescence signaling, and oxidative stress. J Mol Cell Cardiol. 2019;127:74-82.[DOI]
-
30. Durik M, Kavousi M, van der Pluijm I, Isaacs A, Cheng C, Verdonk K, et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation. 2012;126(4):468-478.[DOI]
-
31. Ezzat D, Uddin MM, Xue L, Pershad Y, Zhang S, Collins JM, et al. Clonal hematopoiesis and cardiovascular outcomes in older women. J Am Coll Cardiol. 2025;86(15):1093-1106.[DOI]
-
32. Zhang L, Yang P, Chen J, Chen Z, Liu Z, Feng G, et al. Cd44 connects autophagy decline and ageing in the vascular endothelium. Nat Commun. 2023;14(1):5524.[DOI]
-
33. Xu F, Zhang C, Zou Z, Fan EKY, Chen L, Li Y, et al. Aging-related atg5 defect impairs neutrophil extracellular traps formation. Immunology. 2017;151(4):417-432.[DOI]
-
34. Lan Y, Li YJ, Li DJ, Li P, Wang JY, Diao YP, et al. Long noncoding RNA MEG3 prevents vascular endothelial cell senescence by impairing miR-128-dependent Girdin downregulation. Am J Physiol Cell Physiol. 2019;316(6):C830-C843.[DOI]
-
35. Hofmann P, Sommer J, Theodorou K, Kirchhof L, Fischer A, Li Y, et al. Long non-coding RNA H19 regulates endothelial cell aging via inhibition of STAT3 signalling. Cardiovasc Res. 2019;115(1):230-242.[DOI]
-
36. Hu J, Leisegang MS, Looso M, Drekolia M-K, Wittig J, Mettner J, et al. Disrupted binding of cystathionine γ-lyase to p53 promotes endothelial senescence. Circ Res. 2023;133(10):842-857.[DOI]
-
37. Wang Q, Wu S, Zhu H, Ding Y, Dai X, Ouyang C, et al. Deletion of PRKAA triggers mitochondrial fission by inhibiting the autophagy-dependent degradation of DNM1L. Autophagy. 2017;13(2):404-422.[DOI]
-
38. Yang J, Zhou X, Fan X, Xiao M, Yang D, Liang B, et al. mTORC1 promotes aging-related venous thrombosis in mice via elevation of platelet volume and activation. Blood. 2016;128(5):615-624.[DOI]
-
39. Pastorek M, Konečná B, Janko J, Janovičová Ľ, Podracká Ľ, Záhumenský J, et al. Mitochondria-induced formation of neutrophil extracellular traps is enhanced in the elderly via Toll-like receptor 9. J Leukoc Biol. 2023;114(6):651-665.[DOI]
-
40. Silva GC, Abbas M, Khemais-Benkhiat S, Burban M, Ribeiro TP, Toti F, et al. Replicative senescence promotes prothrombotic responses in endothelial cells: Role of NADPH oxidase- and cyclooxygenase-derived oxidative stress. Exp Gerontol. 2017;93:7-15.[DOI]
-
41. Ghosh AK, Rai R, Park KE, Eren M, Miyata T, Wilsbacher LD, et al. A small molecule inhibitor of PAI-1 protects against doxorubicin-induced cellular senescence. Oncotarget. 2016;7(45):72443.[DOI]
-
42. Alavi P, Yousef Abdualla R, Brown D, Mojiri A, Nagendran J, Lewis J, et al. Aging is associated with organ-specific alterations in the level and expression pattern of von willebrand factor. Arterioscler Thromb Vasc Biol. 2023;43(11):2183-2196.[DOI]
-
43. Spyridopoulos I, Martin-Ruiz C, Hilkens C, Yadegarfar ME, Isaacs J, Jagger C, et al. CMV seropositivity and T-cell senescence predict increased cardiovascular mortality in octogenarians: Results from the Newcastle 85+ study. Aging Cell. 2016;15(2):389-392.[DOI]
-
44. Bergström I, Backteman K, Lundberg A, Ernerudh J, Jonasson L. Persistent accumulation of interferon-γ-producing cd8+cd56+ T cells in blood from patients with coronary artery disease. Atherosclerosis. 2012;224(2):515-520.[DOI]
-
45. Gardner SE, Humphry M, Bennett MR, Clarke MCH. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1α–dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol. 2015;35(9):1963-1974.[DOI]
-
46. Corica F, Allegra A, Ientile R, Buemi M, Corsonello A, D’Angelo R, et al. Reduced intraplatelet magnesium concentrations in elderly patients with non-insulin dependent diabetes mellitus (NIDDM). Arch Gerontol Geriatr. 1997;25(3):255-262.[DOI]
-
47. Gleerup G, Winther K. The effect of ageing on human platelet sensitivity to serotonin. Eur J Clin Invest. 1988;18(5):504-506.[DOI]
-
48. Liberale L, Holy EW, Akhmedov A, Bonetti NR, Nietlispach F, Matter CM, et al. Interleukin-1β mediates arterial thrombus formation via NET-associated tissue factor. J Clin Med. 2019;8(12):2072.[DOI]
-
49. Jia X, Buckley L, Sun C, Al Rifai M, Yu B, Nambi V, et al. Association of interleukin-6 and interleukin-18 with cardiovascular disease in older adults: Atherosclerosis risk in communities study. Eur J Prev Cardiol. 2023;30(16):1731-1740.[DOI]
-
50. Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357(24):2482-2494.[DOI]
-
51. Cadroy Y, Diquélou A, Dupouy D, Bossavy JP, Sakariassen KS, Sié P, et al. The thrombomodulin/protein C/protein S anticoagulant pathway modulates the thrombogenic properties of the normal resting and stimulated endothelium. Arterioscler Thromb Vasc Biol. 1997;17(3):520-527.[DOI]
-
52. Wang M, Hao H, Leeper NJ, Zhu L. Thrombotic regulation from the endothelial cell perspectives. Arterioscler Thromb Vasc Biol. 2018;38(6):e90-e95.[DOI]
-
53. Olgasi C, Assanelli S, Cucci A, Follenzi A. Hemostasis and endothelial functionality: The double face of coagulation factors. Haematologica. 2024;109(7):2041.[DOI]
-
54. Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118(4):620-636.[DOI]
-
55. Ungvari Z, Tarantini S, Kiss T, Wren JD, Giles CB, Griffin CT, et al. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol. 2018;15(9):555-565.[DOI]
-
56. Russo I, Barale C, Melchionda E, Penna C, Pagliaro P. Platelets and cardioprotection: The role of nitric oxide and carbon oxide. Int J Mol Sci. 2023;24(7):6107.[DOI]
-
57. Yang Y, Loscalzo J. Regulation of tissue factor expression in human microvascular endothelial cells by nitric oxide. Circulation. 2000;101(18):2144-2148.[DOI]
-
58. Sakamoto T, Ishibashi T, Sakamoto N, Sugimoto K, Egashira K, Ohkawara H, et al. Endogenous NO blockade enhances tissue factor expression via increased Ca2+ influx through MCP-1 in endothelial cells by monocyte adhesion. Arterioscler Thromb Vasc Biol. 2005;25(9):2005-2011.[DOI]
-
59. Gries A, Bode C, Peter K, Herr A, Böhrer H, Motsch J, et al. Inhaled nitric oxide inhibits human platelet aggregation, P-selectin expression, and fibrinogen binding in vitro and in vivo. Circulation. 1998;97(15):1481-1487.[DOI]
-
60. Tejero J, Shiva S, Gladwin MT. Sources of vascular nitric oxide and reactive oxygen species and their regulation. Physiol Rev. 2019;99(1):311-379.[DOI]
-
61. Nieto-Torres JL, Hansen M. Macroautophagy and aging: The impact of cellular recycling on health and longevity. Mol Aspects Med. 2021;82:101020.[DOI]
-
62. Torisu T, Torisu K, Lee IH, Liu J, Malide D, Combs CA, et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med. 2013;19(10):1281-1287.[DOI]
-
63. Coppola R, Mari D, Lattuada A, Franceschi C. Von Willebrand factor in Italian centenarians. Haematologica. 2003;88(1):39-43.[PubMed]
-
64. Conlan MG, Folsom AR, Finch A, Davis CE, Sorlie P, Marcucci G, et al. Associations of factor VIII and von Willebrand factor with age, race, sex, and risk factors for atherosclerosis. Thromb Haemost. 1993;70(03):380-385.[DOI]
-
65. Tong ZQ, Chen SC, Zhang FT. Effects of stimulation of the rabbit caudate nucleus on units responsive to noxious stimuli in the parafascicular nucleus of the thalamus. Acta Physiologica Sinica. 1985;37(2):128-136.[PubMed]
-
66. Grootaert MOJ, Moulis M, Roth L, Martinet W, Vindis C, Bennett MR, et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res. 2018;114(4):622-634.[DOI]
-
67. Flynn PD, Byrne CD, Baglin TP, Weissberg PL, Bennett MR. Thrombin generation by apoptotic vascular smooth muscle cells. Blood. 1997;89(12):4378-4384.[DOI]
-
68. Clarke MCH, Talib S, Figg NL, Bennett MR. Vascular smooth muscle cell apoptosis induces interleukin-1—directed inflammation: Effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ Res. 2010;106(2):363-372.[DOI]
-
69. Wang L, Miller C, Swarthout RF, Rao M, Mackman N, Taubman MB. Vascular smooth muscle-derived tissue factor is critical for arterial thrombosis after ferric chloride-induced injury. Blood. 2009;113(3):705-713.[DOI]
-
70. Tull SP, Anderson SI, Hughan SC, Watson SP, Nash GB, Rainger GE. Cellular pathology of atherosclerosis: Smooth muscle cells promote adhesion of platelets to cocultured endothelial cells. Circ Res. 2006;98(1):98-104.[DOI]
-
71. Wise SG, Waterhouse A, Michael P, Ng MKC. Extracellular matrix molecules facilitating vascular biointegration. J Funct Biomater. 2012;3(3):569-587.[DOI]
-
72. Habibi A, Ruf W, Schurgers L. Protease-activated receptors in vascular smooth muscle cells: A bridge between thrombo-inflammation and vascular remodelling. Cell Commun Signal. 2025;23(1):57.[DOI]
-
73. DeRoo E, Zhou T, Yang H, Stranz A, Henke P, Liu B. A vein wall cell atlas of murine venous thrombosis determined by single-cell RNA sequencing. Commun Biol. 2023;6(1):130.[DOI]
-
74. Grover A, Sanjuan-Pla A, Thongjuea S, Carrelha J, Giustacchini A, Gambardella A, et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat Commun. 2016;7(1):11075.[DOI]
-
75. Vázquez-Santiago M, Ziyatdinov A, Pujol-Moix N, Brunel H, Morera A, Soria JM, et al. Age and gender effects on 15 platelet phenotypes in a Spanish population. Comput Biol Med. 2016;69:226-233.[DOI]
-
76. Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447(7145):725-729.[DOI]
-
77. Kasjanovová D, Baláẑ V. Age-related changes in human platelet function in vitro. Mech Ageing Dev. 1986;37(2):175-182.[DOI]
-
78. Gnanenthiran SR, Pennings GJ, Reddel CJ, Campbell H, Kockx M, Hamilton JR, et al. Identification of a distinct platelet phenotype in the elderly: ADP hypersensitivity coexists with platelet PAR (protease-activated receptor)-1 and PAR-4-mediated thrombin resistance. Arterioscler Thromb Vasc Biol. 2022;42(8):960-972.[DOI]
-
79. Poscablo DM, Worthington AK, Smith-Berdan S, Rommel MGE, Manso BA, Adili R, et al. An age-progressive platelet differentiation path from hematopoietic stem cells causes exacerbated thrombosis. Cell. 2024;187(12):3090-3107.[DOI]
-
80. Davizon-Castillo P, McMahon B, Aguila S, Bark D, Ashworth K, Allawzi A, et al. Tnf-α—driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood. 2019;134(9):727-740.[DOI]
-
81. Fender AC, Rauch BH, Geisler T, Schrör K. Protease-activated receptor PAR-4: An inducible switch between thrombosis and vascular inflammation? Thromb Haemost. 2017;117(11):2013-2025.[DOI]
-
82. Sonkar VK, Eustes AS, Ahmed A, Jensen M, Solanki MV, Swamy J, et al. Endogenous SOD2 (superoxide dismutase) regulates platelet-dependent thrombin generation and thrombosis during aging. Arterioscler Thromb Vasc Biol. 2023;43(1):79-91.[DOI]
-
83. Portier I, Manne BK, Kosaka Y, Tolley ND, Denorme F, Babur Ö, et al. Aging-related alterations in mechanistic target of rapamycin signaling promote platelet hyperreactivity and thrombosis. J Thromb Haemost. 2024;22(9):2576-2588.[DOI]
-
84. Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U.S.A. 1990;87(23):9383-9387.[DOI]
-
85. Morrow JD. Quantification of isoprostanes as indices of oxidant stress and the risk of atherosclerosis in humans. Arterioscler Thromb Vasc Biol. 2005;25(2):279-286.[DOI]
-
86. Praticò D, Smyth EM, Violi F, FitzGerald GA. Local amplification of platelet function by 8-epi prostaglandin F2α is not mediated by thromboxane receptor isoforms. J Biol Chem. 1996;271(25):14916-14924.[DOI]
-
87. Davì G, Guagnano MT, Ciabattoni G, Basili S, Falco A, Marinopiccoli M, et al. Platelet activation in obese women role of inflammation and oxidant stress. JAMA. 2002;288(16):2008-2014.[DOI]
-
88. Davı̀ G, Ciabattoni G, Consoli A, Mezzetti A, Falco A, Santarone S, et al. In vivo formation of 8-iso-prostaglandin F2α and platelet activation in diabetes mellitus. Circulation. 1999;99(2):224-229.[DOI]
-
89. John T, Kretsch K, Maurer FM, Recktenwald SM, Kaestner L, Wagner C. Viscosity and density measurements on the cytosol of human red blood cells. Biophys J. 2025;124(16):2668-2676.[DOI]
-
90. Martínez de Toda I, Vida C, Garrido A, De la Fuente M. Redox parameters as markers of the rate of aging and predictors of life span. J Gerontol A Biol Sci Med Sci. 2020;75(4):613-620.[DOI]
-
91. Mohanty J, Nagababu E, Rifkind JM. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front Physiol. 2014;5:84.[DOI]
-
92. MacKinney A, Woska E, Spasojevic I, Batinic-Haberle I, Zennadi R. Disrupting the vicious cycle created by NOX activation in sickle erythrocytes exposed to hypoxia/reoxygenation prevents adhesion and vasoocclusion. Redox Biol. 2019;25:101097.[DOI]
-
93. Whelihan MF, Zachary V, Orfeo T, Mann KG. Prothrombin activation in blood coagulation: The erythrocyte contribution to thrombin generation. Blood. 2012;120(18):3837-3845.[DOI]
-
94. Semeraro F, Ammollo CT, Esmon NL, Esmon CT. Histones induce phosphatidylserine exposure and a procoagulant phenotype in human red blood cells. J Thromb Haemost. 2014;12(10):1697-1702.[DOI]
-
95. Nader E, Romana M, Guillot N, Fort R, Stauffer E, Lemonne N, et al. Association between nitric oxide, oxidative stress, eryptosis, red blood cell microparticles, and vascular function in sickle cell anemia. Front Immunol. 2020;11:551441.[DOI]
-
96. Eduard JvB, Marianne CLS, René JB, Rienk N, Augueste S, Frederiek FvD, et al. Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica. 2009;94(11):1513-1519.[DOI]
-
97. Camus SM, De Moraes JA, Bonnin P, Abbyad P, Le Jeune S, Lionnet F, et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood. 2015;125(24):3805-3814.[DOI]
-
98. Setty BNY, Betal SG, Zhang J, Stuart MJ. Heme induces endothelial tissue factor expression: Potential role in hemostatic activation in patients with hemolytic anemia. J Thromb Haemost. 2008;6(12):2202-2209.[DOI]
-
99. Annarapu GK, Nolfi-Donegan D, Reynolds M, Wang Y, Kohut L, Zuckerbraun B, et al. Heme stimulates platelet mitochondrial oxidant production to induce targeted granule secretion. Redox Biol. 2021;48:102205.[DOI]
-
100. Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, Frenette PS. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood. 2014;123(24):3818-3827.[DOI]
-
101. Thomas AM, Gerogianni A, McAdam MB, Fløisand Y, Lau C, Espevik T, et al. Complement component C5 and TLR molecule CD14 mediate heme-induced thromboinflammation in human blood. J Immunol. 2019;203(6):1571-1578.[DOI]
-
102. Pernow J, Mahdi A, Yang J, Zhou Z. Red blood cell dysfunction: A new player in cardiovascular disease. Cardiovasc Res. 2019;115(11):1596-1605.[DOI]
-
103. Somogyi V, Peto K, Deak A, Tanczos B, Nemeth N. Effects of aging and gender on micro-rheology of blood in 3 to 18 months old male and female Wistar (Crl: WI) rats. Biorheology. 2017;54(5-6):127-140.[DOI]
-
104. Turpin C, Catan A, Guerin-Dubourg A, Debussche X, Bravo SB, Álvarez E, et al. Enhanced oxidative stress and damage in glycated erythrocytes. PLoS One. 2020;15(7):e0235335.[DOI]
-
105. Zhou Z, Mahdi A, Tratsiakovich Y, Zahorán S, Kövamees O, Nordin F, et al. Erythrocytes from patients with type 2 diabetes induce endothelial dysfunction via arginase I. J Am Coll Cardiol. 2018;72(7):769-780.[DOI]
-
106. Mahdi A, Tengbom J, Alvarsson M, Wernly B, Zhou Z, Pernow J. Red blood cell peroxynitrite causes endothelial dysfunction in type 2 diabetes mellitus via arginase. Cells. 2020;9(7):1712.[DOI]
-
107. Mazrouei S, Sharifpanah F, Caldwell RW, Franz M, Shatanawi A, Muessig J, et al. Regulation of MAP kinase-mediated endothelial dysfunction in hyperglycemia via arginase I and eNOS dysregulation. Biochim Biophys Acta Mol Cell Res. 2019;1866(9):1398-1411.[DOI]
-
108. Chandra S, Fulton DJR, Caldwell RB, Caldwell RW, Toque HA. Hyperglycemia-impaired aortic vasorelaxation mediated through arginase elevation: Role of stress kinase pathways. Eur J Pharmacol. 2019;844:26-37.[DOI]
-
109. Turpin C, Catan A, Meilhac O, Bourdon E, Canonne-Hergaux F, Rondeau P. Erythrocytes: Central actors in multiple scenes of atherosclerosis. Int J Mol Sci. 2021;22(11):5843.[DOI]
-
110. Grossin N, Wautier MP, Wautier JL. Red blood cell adhesion in diabetes mellitus is mediated by advanced glycation end product receptor and is modulated by nitric oxide. Biorheology. 2009;46(1):63-72.[DOI]
-
111. Deischinger C, Dervic E, Nopp S, Kaleta M, Klimek P, Kautzky-Willer A. Diabetes mellitus is associated with a higher relative risk for venous thromboembolism in females than in males. Diabetes Res Clin Pract. 2022;194:110190.[DOI]
-
112. Gagnon DR, Zhang T-J, Brand FN, Kannel WB. Hematocrit and the risk of cardiovascular disease—the framingham study: A 34-year follow-up. Am Heart J. 1994;127(3):674-682.[DOI]
-
113. Sigrid KB, Ellisiv BM, Inger N, Tom W, John-Bjarne H. Hematocrit and risk of venous thromboembolism in a general population. The tromsø study. Haematologica. 2010;95(2):270-275.[DOI]
-
114. Mittelbrunn M, Kroemer G. Hallmarks of T cell aging. Nat Immunol. 2021;22(6):687-698.[DOI]
-
115. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488-2498.[DOI]
-
116. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111-121.[DOI]
-
117. Bhattacharya R, Zekavat SM, Haessler J, Fornage M, Raffield L, Uddin MM, et al. Clonal hematopoiesis is associated with higher risk of stroke. Stroke. 2022;53(3):788-797.[DOI]
-
118. Jaiswal S, Libby P. Clonal haematopoiesis: Connecting ageing and inflammation in cardiovascular disease. Nat Rev Cardiol. 2020;17(3):137-144.[DOI]
-
119. Murphy AJ, Dragoljevic D, Natarajan P, Wang N. Hematopoiesis of indeterminate potential and atherothrombotic risk. Thromb Haemost. 2022;122(09):1435-1442.[DOI]
-
120. Tremblay D, Kremyanskaya M, Mascarenhas J, Hoffman R. Diagnosis and treatment of polycythemia vera: A review. JAMA. 2025;333(2):153-160.[DOI]
-
121. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355(6327):842-847.[DOI]
-
122. Saadatagah S, Kim RB, Sukumar S, Uddin MM, Folsom AR, Cushman M, et al. Clonal hematopoiesis of indeterminate potential and incidence of venous thromboembolism in older adults. J Thromb Haemost. 2025;23(7):2235-2241.[DOI]
-
123. Arends CM, Liman TG, Strzelecka PM, Kufner A, Löwe P, Huo S, et al. Associations of clonal hematopoiesis with recurrent vascular events and death in patients with incident ischemic stroke. Blood. 2023;141(7):787-799.[DOI]
-
124. Liu Q, Smedby KE, Xue H, Wästerlid T, Li J, Fang F, et al. Clonal hematopoiesis of indeterminate potential and the risk of pulmonary embolism: An observational study. EClinicalMedicine. 2024;74:102753.[DOI]
-
125. Yokokawa T, Misaka T, Kimishima Y, Wada K, Minakawa K, Kaneshiro T, et al. Clonal hematopoiesis and JAK2V617F mutations in patients with cardiovascular disease. J Am Coll Cardiol CardioOnc. 2021;3(1):134-136.[DOI]
-
126. Zhang Y, Zhao Y, Liu Y, Zhang M, Zhang J. New advances in the role of JAK2 V617F mutation in myeloproliferative neoplasms. Cancer. 2024;130(24):4229-4240.[DOI]
-
127. Byun JH, Jung J, Woo J. Clonal hematopoiesis and thromboembolic diseases: A review. J Thromb Haemost. 2025;23(9):2697-2709.[DOI]
-
128. Rossi M, Meggendorfer M, Zampini M, Tettamanti M, Riva E, Travaglino E, et al. Clinical relevance of clonal hematopoiesis in persons aged ≥ 80 years. Blood. 2021;138(21):2093-2105.[DOI]
-
129. Zon RL, Sekar A, Clapham K, Oren O, Niroula A, Bick AG, et al. Jak2-mutant clonal hematopoiesis is associated with venous thromboembolism. Blood. 2024;144(20):2149-2154.[DOI]
-
130. Moliterno AR, Kaizer H, Reeves BN. Jak2v617f allele burden in polycythemia vera: Burden of proof. Blood. 2023;141(16):1934-1942.[DOI]
-
131. Schuermans A, Honigberg MC. Clonal haematopoiesis in cardiovascular disease: Prognostic role and novel therapeutic target. Nat Rev Cardiol. 2025;22(11):845-856.[DOI]
-
132. Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592(7853):296-301.[DOI]
-
133. Edelmann B, Gupta N, Schnoeder TM, Oelschlegel AM, Shahzad K, Goldschmidt J, et al. JAK2-V617F promotes venous thrombosis through β1/β2 integrin activation. J Clin Invest. 2018;128(10):4359-4371.[DOI]
-
134. Liu W, Pircher J, Schuermans A, Ul Ain Q, Zhang Z, Honigberg MC, et al. Jak2v617f clonal hematopoiesis promotes arterial thrombosis via platelet activation and cross talk. Blood. 2024;143(15):1539-1550.[DOI]
-
135. Molinaro R, Sellar RS, Vromman A, Sausen G, Folco E, Sukhova GK, et al. The clonal hematopoiesis mutation Jak2V617F aggravates endothelial injury and thrombosis in arteries with erosion-like intimas. Int J Cardiol. 2024;409:132184.[DOI]
-
136. Wang S, Luo X, Hu S, Zhao C, Sun Q, Zeng M, et al. Plaque erosion risk and JAK2 V617F variant. Eur Heart J. 2025;46(23):2205-2219.[DOI]
-
137. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm‐aging: An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908(1):244-254.[DOI]
-
138. Liberale L, Montecucco F, Tardif JC, Libby P, Camici GG. Inflamm-ageing: The role of inflammation in age-dependent cardiovascular disease. Eur Heart J. 2020;41(31):2974-2982.[DOI]
-
139. Ferrucci L, Fabbri E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15(9):505-522.[DOI]
-
140. Beaulieu LM, Lin E, Mick E, Koupenova M, Weinberg EO, Kramer CD, et al. Interleukin 1 receptor 1 and interleukin 1β regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arterioscler Thromb Vasc Biol. 2014;34(3):552-564.[DOI]
-
141. Morton AC, Rothman AMK, Greenwood JP, Gunn J, Chase A, Clarke B, et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA Heart Study. Eur Heart J. 2015;36(6):377-384.[DOI]
-
142. Ministrini S, Puspitasari YM, Han J, Kirmes K, Niederberger R, Bengs S, et al. Direct il-6 inhibition prevents arterial thrombosis by reducing collagen-mediated platelets activation. Eur Heart J. 2024;45(Supplement_1):ehae666.3881.[DOI]
-
143. Papadopoulos A, Palaiopanos K, Björkbacka H, Peters A, de Lemos JA, Seshadri S, et al. Circulating interleukin-6 levels and incident ischemic stroke: A systematic review and meta-analysis of prospective studies. Neurology. 2022;98(10):e1002-e1012.[DOI]
-
144. Libby P. Targeting inflammatory pathways in cardiovascular disease: The inflammasome, interleukin-1, interleukin-6 and beyond. Cells. 2021;10(4):951.[DOI]
-
145. Vaiserman A, Romanenko M, Piven L, Moseiko V, Lushchak O, Kryzhanovska N, et al. Differences in the gut firmicutes to bacteroidetes ratio across age groups in healthy ukrainian population. BMC Microbiol. 2020;20(1):221.[DOI]
-
146. Güemes-González AM, Arriaga-Pizano LA, Chacón-Salinas R, Wong-Baeza I, Ferat-Osorio E. Regulation of intestinal permeability in health and disease: Possible therapeutic applications. Arch Med Res. 2026;57(3):103321.[DOI]
-
147. Wu J, Ren W, Chen L, Lou Y, Liu C, Huang Y, et al. Age-related changes in the composition of intestinal microbiota in elderly chinese individuals. Gerontology. 2022;68(9):976-988.[DOI]
-
148. Khuu MP, Paeslack N, Dremova O, Benakis C, Kiouptsi K, Reinhardt C. The gut microbiota in thrombosis. Nat Rev Cardiol. 2025;22(2):121-137.[DOI]
-
149. Nemet I, Saha PP, Gupta N, Zhu W, Romano KA, Skye SM, et al. A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell. 2020;180(5):862-877.e822.[DOI]
-
150. Jäckel S, Kiouptsi K, Lillich M, Hendrikx T, Khandagale A, Kollar B, et al. Gut microbiota regulate hepatic von willebrand factor synthesis and arterial thrombus formation via toll-like receptor-2. Blood. 2017;130(4):542-553.[DOI]
-
151. Pieterse E, Rother N, Yanginlar C, Hilbrands LB, Van der Vlag J. Neutrophils discriminate between lipopolysaccharides of different bacterial sources and selectively release neutrophil extracellular traps. Front Immunoly. 2016;7:484.[DOI]
-
152. Heianza Y, Ma W, Manson JE, Rexrode KM, Qi L. Gut microbiota metabolites and risk of major adverse cardiovascular disease events and death: A systematic review and meta-analysis of prospective studies. J Am Heart Assoc. 2017;6(7):e004947.[DOI]
-
153. Lei D, Yu W, Liu Y, Jiang Y, Li X, Lv J, et al. Trimethylamine N-Oxide (TMAO) inducing endothelial injury: Uplc-MS/MS-based quantification and the activation of cathepsin B-mediated NLRP3 inflammasome. Molecules. 2023;28(9):3817.[DOI]
-
154. Rath S, Rox K, Kleine Bardenhorst S, Schminke U, Dörr M, Mayerle J, et al. Higher trimethylamine-N-oxide plasma levels with increasing age are mediated by diet and trimethylamine-forming bacteria. mSystems. 2021;6(5):10-128.[DOI]
-
155. Netea Mihai G, Quintin J, van der Meer Jos WM. Trained immunity: A memory for innate host defense. Cell Host Microbe. 2011;9(5):355-361.[DOI]
-
156. Gritsenko A, Green JP, Brough D, Lopez-Castejon G. Mechanisms of NLRP3 priming in inflammaging and age related diseases. Cytokine Growth Factor Rev. 2020;55:15-25.[DOI]
-
157. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. 2018;172(1):162-175.e114.[DOI]
-
158. Flores-Gomez D, Bekkering S, Netea MG, Riksen NP. Trained immunity in atherosclerotic cardiovascular disease. Arterioscler Thromb Vasc Biol. 2021;41(1):62-69.[DOI]
-
159. Li S, Peng Y, Wang X, Qian Y, Xiang P, Wade SW, Guo H, Lopez JA, Herzog CA, Handelsman Y. Cardiovascular events and death after myocardial infarction or ischemic stroke in an older Medicare population. Clin Cardiol. 2019;42(3):391-399.[DOI]
-
160. Dong Z, Hou L, Luo W, Pan LH, Li X, Tan HP, et al. Myocardial infarction drives trained immunity of monocytes, accelerating atherosclerosis. Eur Heart J. 2024;45(9):669-684.[DOI]
-
161. Rehill AM, McCluskey S, Ledwith AE, Ryan TA, Ünlü B, Leon G, et al. Trained immunity causes myeloid cell hypercoagulability. Sci Adv. 2025;11(10):eads0105.[DOI]
-
162. Konieczyńska M, Natorska J, Undas A. Thrombosis and aging: fibrin clot properties and oxidative stress. Antioxid Redox Signal. 2024;41(4-6):233-254.[DOI]
-
163. Favaloro EJ, Soltani S, McDonald J, Grezchnik E, Easton L. Cross-laboratory audit of normal reference ranges and assessment of abo blood group, gender and age on detected levels of plasma coagulation factors. Blood Coagul Fibrinolysis. 2005;16(8):597-605.[DOI]
-
164. Favaloro EJ, Soltani S, McDonald J, Grezchnik E, Easton L, Favaloro JWC. Reassessment of abo blood group, sex, and age on laboratory parameters used to diagnose von willebrand disorder: Potential influence on the diagnosis vs the potential association with risk of thrombosis. Am J Clin Pathol. 2005;124(6):910-917.[DOI]
-
165. van Paridon PC, Panova-Noeva M, van Oerle R, Schulz A, Hermanns IM, Prochaska JH, et al. Thrombin generation in cardiovascular disease and mortality—results from the Gutenberg Health Study. Haematologica. 2020;105(9):2327-2334.[DOI]
-
166. Costanzo S, De Laat B, Di Castelnuovo A, Van Der Vorm L, De Curtis A, Cerletti C, et al. Age-and sex-dependency of thrombin generation parameters in the general Italian population: The Moli-sani study. Front Cardiovasc Med. 2025;12:1528871.[DOI]
-
167. Mari D, Mannucci P, Coppola R, Bottasso B, Bauer K, Rosenberg R. Hypercoagulability in centenarians: The paradox of successful aging. Blood. 1995;85(11):3144-3149.[DOI]
-
168. Mennen LI, Witteman JC, den Breeijen JH, Schouten EG, de Jong PT, Hofman A, et al. The association of dietary fat and fiber with coagulation factor VII in the elderly: The Rotterdam Study. Am J Clin Nutr. 1997;65(3):732-736.[DOI]
-
169. Dolan G, Neal K, Cooper P, Brown P, Preston FE. Protein C, antithrombin III and plasminogen: Effect of age, sex and blood group. Br J Haematol. 1994;86(4):798-803.[DOI]
-
170. Favaloro EJ, Soltani S, McDonald J, Grezchnik E, Easton L. Laboratory identification of familial thrombophilia: do the pitfalls exceed the benefits? A reassessment of ABO-blood group, gender, age, and other laboratory parameters on the potential influence on a diagnosis of protein C, protein S, and antithrombin deficiency and the potential high risk of a false positive diagnosis. Lab Hematol. 2005;11(3):174-184.[PubMed]
-
171. Lowe GD, Rumley A, Woodward M, Morrison CE, Philippou H, Lane DA, et al. Epidemiology of coagulation factors, inhibitors and activation markers: The Third Glasgow MONICA survey. I. Br J Haematol. 1997;97(4):775-784.[DOI]
-
172. Stout RW, Crawford VLS, McDermott MJ, Rocks MJ, Morris TCM. Seasonal changes in haemostatic factors in young and elderly subjects. Age Ageing. 1996;25(3):256-258.[DOI]
-
173. Cadroy Y, Daviaud P, Saivin S, Sie P, Boneu B. Distribution of 16 hemostatic laboratory variables assayed in 100 blood donors. Nouv Rev Fr Hematol. 1990;32(4):259-264.[PubMed]
-
174. Dykes AC, Walker ID, McMahon AD, Islam SIAM, Tait RC. A study of protein s antigen levels in 3788 healthy volunteers: Influence of age, sex and hormone use, and estimate for prevalence of deficiency state. Br J Haematol. 2001;113(3):636-641.[DOI]
-
175. Yamamoto K, Takeshita K, Kojima T, Takamatsu J, Saito H. Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: Implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc Res. 2005;66(2):276-285.[DOI]
-
176. Rui V, Richard JF, Alessandro C, Marguerite N-A. Fibrin(ogen) in human disease: Both friend and foe. Haematologica. 2020;105(2):284-296.[DOI]
-
177. Rui V, Richard JF, Alessandro C, Marguerite NA. Fibrin(ogen) in human disease: Both friend and foe. Haematologica. 2020;105(2):284-296.[DOI]
-
178. Munsch G, Mohapatra AK, van Hylckama Vlieg A, Kleber ME, Martinez-Perez A, Le NQ, et al. A multiomics approach reveals novel regulators of plasma factor V levels, highlighting CLEC4M as a clearance receptor. Blood. 2025;146(5):628-637.[DOI]
-
179. Hayakawa M, Sakata A, Hayakawa H, Matsumoto H, Hiramoto T, Kashiwakura Y, et al. Characterization and visualization of murine coagulation factor VIII-producing cells in vivo. Sci Rep. 2021;11(1):14824.[DOI]
-
180. Pan J, Dinh TT, Rajaraman A, Lee M, Scholz A, Czupalla CJ, et al. Patterns of expression of factor VIII and von Willebrand factor by endothelial cell subsets in vivo. Blood. 2016;128(1):104-109.[DOI]
-
181. Camire RM. Blood coagulation factor x: Molecular biology, inherited disease, and engineered therapeutics. J Thromb Thrombolysis. 2021;52(2):383-390.[DOI]
-
182. Gailani D, Sun M-F, Sun Y. A comparison of murine and human factor XI. Blood. 1997;90(3):1055-1064.[DOI]
-
183. Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010;115(13):2569-2577.[DOI]
-
184. Wang H, Rosendaal FR, Cushman M, van Hylckama Vlieg A. Procoagulant factor levels and risk of venous thrombosis in the elderly. J Thromb Haemost. 2021;19(1):186-193.[DOI]
-
185. Mojzisch A, Brehm MA. The manifold cellular functions of von Willebrand factor. Cells. 2021;10(9):2351.[DOI]
-
186. Adams R, Coleman R, Stanton T. Performance of chromogenic protein C (PC) testing. In: Favaloro EJ, Gosselin RC, editors. Methods in Molecular Biology; New York: Springer; 2023. p. 225-232.[DOI]
-
187. Rezende SM, Simmonds RE, Lane DA. Coagulation, inflammation, and apoptosis: Different roles for protein S and the protein S-C4b binding protein complex. Blood. 2004;103(4):1192-1201.[DOI]
-
188. Morrow GB, Mutch NJ. Past, present, and future perspectives of plasminogen activator inhibitor 1 (PAI-1). Semin Thromb Hemost 2023; 49(03): 305-313.[DOI]
-
189. Wild CP. Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Biomarkers Prev. 2005;14(8):1847-1850.[DOI]
-
190. Münzel T, Sørensen M, Hahad O, Nieuwenhuijsen M, Daiber A. The contribution of the exposome to the burden of cardiovascular disease. Nat Rev Cardiol. 2023;20(10):651-669.[DOI]
-
191. Pandics T, Major D, Fazekas-Pongor V, Szarvas Z, Peterfi A, Mukli P, et al. Exposome and unhealthy aging: Environmental drivers from air pollution to occupational exposures. Geroscience. 2023;45(6):3381-3408.[DOI]
-
192. Landrigan PJ, Fuller R, Acosta NJR, Adeyi O, Arnold R, Basu N, et al. The Lancet Commission on pollution and health. Lancet. 2018;391(10119):462-512.[DOI]
-
193. Mahmud F, Sarker DB, Jocelyn JA, Sang QXA. Molecular and cellular effects of microplastics and nanoplastics: Focus on inflammation and senescence. Cells. 2024;13(21):1788.[DOI]
-
194. Zhang T, Liao Y, Ling J, Zhang J, Zhang D, Yin X, et al. Tiny trouble: Microplastics, nanoplastics, and their heartfelt impact on cardiovascular health. Cardiovasc Res. 2025;121(7):992-1010.[DOI]
-
195. Mohamed Nor NH, Kooi M, Diepens NJ, Koelmans AA. Lifetime accumulation of microplastic in children and adults. Environ Sci Technol. 2021;55(8):5084-5096.[DOI]
-
196. Nihart AJ, Garcia MA, El Hayek E, Liu R, Olewine M, Kingston JD, et al. Bioaccumulation of microplastics in decedent human brains. Nat Med. 2025;31(4):1114-1119.[DOI]
-
197. Marfella R, Prattichizzo F, Sardu C, Fulgenzi G, Graciotti L, Spadoni T, D’Onofrio N, Scisciola L, La Grotta R, Frigé C, Pellegrini V. Microplastics and nanoplastics in atheromas and cardiovascular events. N Engl J Med. 2024;390(10):900-910.[DOI]
-
198. Wang T, Yi Z, Liu X, Cai Y, Huang X, Fang J, et al. Multimodal detection and analysis of microplastics in human thrombi from multiple anatomically distinct sites. eBioMedicine. 2024;103:105118.[DOI]
-
199. Smyth E, Solomon A, Vydyanath A, Luther PK, Pitchford S, Tetley TD, et al. Induction and enhancement of platelet aggregation in vitro and in vivo by model polystyrene nanoparticles. Nanotoxicology. 2015;9(3):356-364.[DOI]
-
200. Chen L, Zheng Y, Liu Y, Tian P, Yu L, Bai L, et al. Microfluidic-based in vitro thrombosis model for studying microplastics toxicity. Lab Chip. 2022;22(7):1344-1353.[DOI]
-
201. Christodoulides A, Hall A, Alves NJ. Exploring microplastic impact on whole blood clotting dynamics utilizing thromboelastography. Front Public Health. 2023;11:1215817.[DOI]
-
202. Murray CJL, Aravkin AY, Zheng P, Abbafati C, Abbas KM, Abbasi-Kangevari M, et al. Global burden of 87 risk factors in 204 countries and territories, 1990-2019: A systematic analysis for the global burden of disease study 2019. Lancet. 2020;396(10258):1223-1249.[DOI]
-
203. Al-Kindi SG, Brook RD, Biswal S, Rajagopalan S. Environmental determinants of cardiovascular disease: Lessons learned from air pollution. Nat Rev Cardiol. 2020;17(10):656-672.[DOI]
-
204. Nethery RC, Josey K, Gandhi P, Kim JH, Visaria A, Bates B, et al. Air pollution and cardiovascular and thromboembolic events in older adults with high-risk conditions. Am J Epidemiol. 2023;192(8):1358-1370.[DOI]
-
205. Jin T, Di Q, Réquia WJ, Danesh Yazdi M, Castro E, Ma T, et al. Associations between long-term air pollution exposure and the incidence of cardiovascular diseases among american older adults. Environ Int. 2022;170:107594.[DOI]
-
206. Kloog I, Zanobetti A, Nordio F, Coull BA, Baccarelli AA, Schwartz J. Effects of airborne fine particles (PM2.5) on deep vein thrombosis admissions in the northeastern united states. J Thromb Haemost. 2015;13(5):768-774.[DOI]
-
207. Swan D, Turner R, Franchini M, Mannucci PM, Thachil J. Air pollution and venous thromboembolism: Current knowledge and future perspectives. Lancet Haematol. 2025;12(1):e68-e82.[DOI]
-
208. Xu H, Wang T, Liu S, Brook RD, Feng B, Zhao Q, et al. Extreme levels of air pollution associated with changes in biomarkers of atherosclerotic plaque vulnerability and thrombogenicity in healthy adults: The Beijing AIRCHD Study. Circ Res. 2019;124(5):e30-e43.[DOI]
-
209. Wang K, Wang W, Lei L, Lan Y, Liu Q, Ren L, et al. Association between short-term exposure to ambient air pollution and biomarkers of coagulation: A systematic review and meta-analysis. Environ Res. 2022;215:114210.[DOI]
-
210. Bonzini M, Tripodi A, Artoni A, Tarantini L, Marinelli B, Bertazzi PA, et al. Effects of inhalable particulate matter on blood coagulation. J Thromb Haemost. 2010;8(4):662-668.[DOI]
-
211. Xie Y, Fan D, Wu B, Guo J, Wang G, Yu L, et al. PM2.5 promotes platelet activation and thrombosis via ROS/MAPKs pathway-mediated mitochondrial dysfunction. Environ Res. 2025;283:122116.[DOI]
-
212. Kilinç E, Schulz H, Kuiper GJ, Spronk HMH, ten Cate H, Upadhyay S, et al. The procoagulant effects of fine particulate matter in vivo. Part Fibre Toxicol. 2011;8(1):12.[DOI]
-
213. Grootaert MOJ, Bennett MR. Sirtuins in atherosclerosis: Guardians of healthspan and therapeutic targets. Nat Rev Cardiol. 2022;19(10):668-683.[DOI]
-
214. Ding YN, Wang HY, Chen XF, Tang X, Chen HZ. Roles of sirtuins in cardiovascular diseases: Mechanisms and therapeutics. Circ Res. 2025;136(5):524-550.[DOI]
-
215. Winnik S, Auwerx J, Sinclair DA, Matter CM. Protective effects of sirtuins in cardiovascular diseases: From bench to bedside. Eur Heart J. 2015;36(48):3404-3412.[DOI]
-
216. Breitenstein A, Stein S, Holy EW, Camici GG, Lohmann C, Akhmedov A, et al. Sirt1 inhibition promotes in vivo arterial thrombosis and tissue factor expression in stimulated cells. Cardiovasc Res. 2011;89(2):464-472.[DOI]
-
217. Kim YH, Bae JU, Kim IS, Chang CL, Oh SO, Kim CD. SIRT1 prevents pulmonary thrombus formation induced by arachidonic acid via downregulation of PAF receptor expression in platelets. Platelets. 2016;27(8):735-742.[DOI]
-
218. Yao X, Chen W, Liu J, Liu H, Zhan JY, Guan S, et al. Deep vein thrombosis is modulated by inflammation regulated via sirtuin 1/NF-κB signalling pathway in a rat model. Thromb Haemost. 2019;119(03):421-430.[DOI]
-
219. He J, Liu X, Su C, Wu F, Sun J, Zhang J, et al. Inhibition of mitochondrial oxidative damage improves reendothelialization capacity of endothelial progenitor cells via SIRT3 (Sirtuin 3)-enhanced SOD2 (Superoxide Dismutase 2) deacetylation in hypertension. Arterioscler Thromb Vasc Biol. 2019;39(8):1682-1698.[DOI]
-
220. Gaul DS, Weber J, van Tits LJ, Sluka S, Pasterk L, Reiner MF, et al. Loss of Sirt3 accelerates arterial thrombosis by increasing formation of neutrophil extracellular traps and plasma tissue factor activity. Cardiovasc Res. 2018;114(8):1178-1188.[DOI]
-
221. Mackman N, Brand K, Edgington TS. Lipopolysaccharide-mediated transcriptional activation of the human tissue factor gene in THP-1 monocytic cells requires both activator protein 1 and nuclear factor kappa b binding sites. J Exp Med. 1991;174(6):1517-1526.[DOI]
-
222. Gaul DS, Calatayud N, Pahla J, Bonetti NR, Wang YJ, Weber J, et al. Endothelial SIRT6 deficiency promotes arterial thrombosis in mice. J Mol Cell Cardiol. 2023;174:56-62.[DOI]
-
223. Qi Z, Hu L, Zhang J, Yang W, Liu X, Jia D, et al. PCSK9 (Proprotein Convertase Subtilisin/Kexin 9) enhances platelet activation, thrombosis, and myocardial infarct expansion by binding to platelet CD36. Circulation. 2021;143(1):45-61.[DOI]
-
224. Liberale L, Akhmedov A, Vlachogiannis NI, Bonetti NR, Nageswaran V, Miranda MX, et al. Sirtuin 5 promotes arterial thrombosis by blunting the fibrinolytic system. Cardiovasc Res. 2021;117(10):2275-2288.[DOI]
-
225. Stamatelopoulos K, Sibbing D, Rallidis LS, Georgiopoulos G, Stakos D, Braun S, et al. Amyloid-beta (1-40) and the risk of death from cardiovascular causes in patients with coronary heart disease. J Am Coll Cardiol. 2015;65(9):904-916.[DOI]
-
226. Amyloid-β (1-40) and mortality in patients with non—ST-segment elevation acute coronary syndrome: A Cohort Study. Ann Intern Med. 2018;168(12):855-865.[DOI]
-
227. Stamatelopoulos K, Pol CJ, Ayers C, Georgiopoulos G, Gatsiou A, Brilakis ES, et al. Amyloid-beta (1-40) peptide and subclinical cardiovascular disease. J Am Coll Cardiol. 2018;72(9):1060-1061.[DOI]
-
228. Laina A, Stellos K, Stamatelopoulos K. Vascular ageing: Underlying mechanisms and clinical implications. Exp Gerontol. 2018;109:16-30.[DOI]
-
229. Lambrinoudaki I, Delialis D, Georgiopoulos G, Tual-Chalot S, Vlachogiannis NI, Patras R, et al. Circulating amyloid beta 1-40 is associated with increased rate of progression of atherosclerosis in menopause: A prospective cohort study. Thromb Haemost. 2021;121(05):650-658.[DOI]
-
230. Stakos DA, Stamatelopoulos K, Bampatsias D, Sachse M, Zormpas E, Vlachogiannis NI, et al. The alzheimer’s disease amyloid-beta hypothesis in cardiovascular aging and disease: JACC Focus Seminar. J Am Coll Cardiol. 2020;75(8):952-967.[DOI]
-
231. Cortes-Canteli M, Paul J, Norris EH, Bronstein R, Ahn HJ, Zamolodchikov D, et al. Fibrinogen and β-amyloid association alters thrombosis and fibrinolysis: A possible contributing factor to Alzheimer’s disease. Neuron. 2010;66(5):695-709.[DOI]
-
232. Delialis D, Georgiopoulos G, Tual-Chalot S, Angelidakis L, Aivalioti E, Mavraganis G, et al. Amyloid beta is associated with carotid wall echolucency and atherosclerotic plaque composition. Sci Rep. 2024;14(1):14944.[DOI]
-
233. Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295-306.[DOI]
-
234. Tektonidou MG, Kravvariti E, Vlachogiannis NI, Georgiopoulos G, Mantzou A, Sfikakis PP, et al. Clinical value of amyloid-beta1-40 as a marker of thrombo-inflammation in antiphospholipid syndrome. Rheumatology. 2021;60(4):1669-1675.[DOI]
-
235. Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324(7329):71-86.[DOI]
-
236. McNeil JJ, Wolfe R, Woods RL, Tonkin AM, Donnan GA, Nelson MR, et al. Effect of aspirin on cardiovascular events and bleeding in the healthy elderly. N Engl J Med. 2018;379(16):1509-1518.[DOI]
-
237. Byrne RA, Rossello X, Coughlan JJ, Barbato E, Berry C, Chieffo A, et al. 2023 ESC guidelines for the management of acute coronary syndromes: Developed by the task force on the management of acute coronary syndromes of the European Society of Cardiology (ESC). Eur Heart J. 2023;44(38):3720-3826.[DOI]
-
238. Savonitto S, Ferri LA, Piatti L, Grosseto D, Piovaccari G, Morici N, et al. Comparison of reduced-dose prasugrel and standard-dose clopidogrel in elderly patients with acute coronary syndromes undergoing early percutaneous revascularization. Circulation. 2018;137(23):2435-2445.[DOI]
-
239. Roe MT, Goodman SG, Ohman EM, Stevens SR, Hochman JS, Gottlieb S, et al. Elderly patients with acute coronary syndromes managed without revascularization. Circulation. 2013;128(8):823-833.[DOI]
-
240. Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001-2015.[DOI]
-
241. Roe MT, Armstrong PW, Fox KAA, White HD, Prabhakaran D, Goodman SG, et al. Prasugrel versus clopidogrel for acute coronary syndromes without revascularization. N Engl J Med. 2012;367(14):1297-1309.[DOI]
-
242. Husted S, James S, Becker RC, Horrow J, Katus H, Storey RF, et al. Ticagrelor versus clopidogrel in elderly patients with acute coronary syndromes: A substudy from the prospective randomized PLATelet inhibition and patient Outcomes (PLATO) trial. Circ Cardiovasc Qual Outcomes. 2012;5(5):680-688.[DOI]
-
243. Gimbel M, Qaderdan K, Willemsen L, Hermanides R, Bergmeijer T, de Vrey E, et al. Clopidogrel versus ticagrelor or prasugrel in patients aged 70 years or older with non-ST-elevation acute coronary syndrome (POPular AGE): The randomised, open-label, non-inferiority trial. Lancet. 2020;395(10233):1374-1381.[DOI]
-
244. Szummer K, Montez-Rath ME, Alfredsson J, Erlinge D, Lindahl B, Hofmann R, et al. Comparison between ticagrelor and clopidogrel in elderly patients with an acute coronary syndrome: Insights from the SWEDEHEART registry. Circulation. 2020;142(18):1700-1708.[DOI]
-
245. Fujisaki T, Kuno T, Ando T, Briasoulis A, Takagi H, Bangalore S. Potent P2Y12 inhibitors versus Clopidogrel in elderly patients with acute coronary syndrome: Systematic review and meta-analysis. Am Heart J. 2021;237:34-44.[DOI]
-
246. Pereira NL, Farkouh ME, So D, Lennon R, Geller N, Mathew V, et al. Effect of genotype-guided oral p2y12 inhibitor selection vs conventional clopidogrel therapy on ischemic outcomes after percutaneous coronary intervention: The tailor-pci randomized clinical trial. JAMA. 2020;324(8):761-771.[DOI]
-
247. Pereira NL, Cresci S, Angiolillo DJ, Batchelor W, Capers Q, Cavallari LH, et al. CYP2C19 genetic testing for oral P2Y12 inhibitor therapy: A scientific statement from the american heart association. Circulation. 2024;150(6):e129-e150.[DOI]
-
248. van den Broek WWA, Azzahhafi J, Chan Pin Yin DRPP, van der Sangen NMR, Sivanesan S, Dijksman LM, et al. Cost-effectiveness of implementing a genotype-guided de-escalation strategy in patients with acute coronary syndrome. Eur Heart J Cardiovasc Pharmacother. 2025;11(3):230-240.[DOI]
-
249. Ingraham BS, Farkouh ME, Lennon RJ, So D, Goodman SG, Geller N, et al. Genetic-guided oral P2Y12 inhibitor selection and cumulative ischemic events after percutaneous coronary intervention. JACC Cardiovasc Interv. 2023;16(7):816-825.[DOI]
-
250. Nair R, Johnson M, Kravitz K, Huded C, Rajeswaran J, Anabila M, et al. Characteristics and outcomes of early recurrent myocardial infarction after acute myocardial infarction. J Am Heart Assoc. 2021;10(16):e019270.[DOI]
-
251. Geisler T, Langer H, Wydymus M, Göhring K, Zürn C, Bigalke B, et al. Low response to clopidogrel is associated with cardiovascular outcome after coronary stent implantation. Eur Heart J. 2006;27(20):2420-2425.[DOI]
-
252. Price MJ, Endemann S, Gollapudi RR, Valencia R, Stinis CT, Levisay JP, et al. Prognostic significance of post-clopidogrel platelet reactivity assessed by a point-of-care assay on thrombotic events after drug-eluting stent implantation. Eur Heart J. 2008;29(8):992-1000.[DOI]
-
253. Patti G, Nusca A, Mangiacapra F, Gatto L, D’Ambrosio A, Sciascio GD. Point-of-care measurement of clopidogrel responsiveness predicts clinical outcome in patients undergoing percutaneous coronary intervention: Results of the ARMYDA-PRO (Antiplatelet therapy for Reduction of MYocardial Damage during Angioplasty-Platelet Reactivity Predicts Outcome) study. J Am Coll Cardiol. 2008;52(14):1128-1133.[DOI]
-
254. Hochholzer W, Trenk D, Bestehorn HP, Fischer B, Valina CM, Ferenc M, et al. Impact of the degree of peri-interventional platelet inhibition after loading with clopidogrel on early clinical outcome of elective coronary stent placement. J Am Coll Cardiol. 2006;48(9):1742-1750.[DOI]
-
255. Gremmel T, Steiner S, Seidinger D, Koppensteiner R, Panzer S, Kopp CW. Adenosine diphosphate-inducible platelet reactivity shows a pronounced age dependency in the initial phase of antiplatelet therapy with clopidogrel. J Thromb Haemost. 2010;8(1):37-42.[DOI]
-
256. Cuisset T, Quilici J, Grosdidier C, Fourcade L, Gaborit B, Pankert M, et al. Comparison of platelet reactivity and clopidogrel response in patients ≤ 75 years versus > 75 years undergoing percutaneous coronary intervention for non-ST-segment elevation acute coronary syndrome. Am J Cardiol. 2011;108(10):1411-1416.[DOI]
-
257. Capranzano P, Tamburino C, Capodanno D, Miccichè E, D’Urso L, Calvi V, et al. Platelet function profiles in the elderly: results of a pharmacodynamic study in patients on clopidogrel therapy and effects of switching to prasugrel 5 mg in patients with high platelet reactivity. Thromb Haemost. 2011;106(12):1149-1157.[DOI]
-
258. Silvain J, Cayla G, Hulot JS, Finzi J, Kerneis M, O’Connor SA, et al. High on-thienopyridine platelet reactivity in elderly coronary patients: The SENIOR-PLATELET study. Eur Heart J. 2012;33(10):1241-1249.[DOI]
-
259. Verdoia M, Pergolini P, Rolla R, Nardin M, Schaffer A, Barbieri L, et al. Advanced age and high‐residual platelet reactivity in patients receiving dual antiplatelet therapy with clopidogrel or ticagrelor. J Thromb Haemost. 2016;14(1):57-64.[DOI]
-
260. Angiolillo DJ, Jakubowski JA, Ferreiro JL, Tello-Montoliu A, Rollini F, Franchi F, et al. Impaired responsiveness to the platelet P2Y12 receptor antagonist clopidogrel in patients with type 2 diabetes and coronary artery disease. J Am Coll Cardiol. 2014;64(10):1005-1014.[DOI]
-
261. Ko D, Lin KJ, Bessette LG, Lee SB, Walkey AJ, Cheng S, et al. Trends in use of oral anticoagulants in older adults with newly diagnosed atrial fibrillation, 2010-2020. JAMA Netw Open. 2022;5(11):e2242964.[DOI]
-
262. Bernacki GM, Becker RC. Oral anticoagulants in older adults with atrial fibrillation. J Thromb Thrombolysis. 2013;36(4):403-415.[DOI]
-
263. Van Gelder IC, Rienstra M, Bunting KV, Casado-Arroyo R, Caso V, Crijns HJGM, et al. 2024 ESC Guidelines for the management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS) Developed by the task force for the management of atrial fibrillation of the European Society of Cardiology (ESC), with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Endorsed by the European Stroke Organisation (ESO). Eur Heart J. 2024;45(36):3314-3414.[DOI]
-
264. Joosten LPT, van Doorn S, van de Ven PM, Köhlen BTG, Nierman MC, Koek HL, et al. Safety of switching from a vitamin k antagonist to a non-vitamin K antagonist oral anticoagulant in frail older patients with atrial fibrillation: Results of the FRAIL-AF randomized controlled trial. Circulation. 2024;149(4):279-289.[DOI]
-
265. Sandhu RK, Seiler A, Johnson CJ, Bunch TJ, Deering TF, Deneke T, et al. Heart rhythm society atrial fibrillation centers of excellence study: A survey analysis of stakeholder practices, needs, and barriers. Heart Rhythm. 2022;19(6):1039-1048.[DOI]
-
266. Halvorsen S, Atar D, Yang H, De Caterina R, Erol C, Garcia D, et al. Efficacy and safety of apixaban compared with warfarin according to age for stroke prevention in atrial fibrillation: Observations from the ARISTOTLE trial. Eur Heart J. 2014;35(28):1864-1872.[DOI]
-
267. Halperin JL, Hankey GJ, Wojdyla DM, Piccini JP, Lokhnygina Y, Patel MR, et al. Efficacy and safety of rivaroxaban compared with warfarin among elderly patients with nonvalvular atrial fibrillation in the Rivaroxaban Once Daily, Oral, Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET AF). Circulation. 2014;130(2):138-146.[DOI]
-
268. Frailty and clinical outcomes of direct oral anticoagulants versus warfarin in older adults with atrial fibrillation: A cohort study. Ann Intern Med. 2021;174(9):1214-1223.[DOI]
-
269. Suwa M, Nohara Y, Morii I, Kino M. Safety and efficacy re-evaluation of edoxaban and rivaroxaban dosing with plasma concentration monitoring in non-valvular atrial fibrillation: With observations of on-label and off-label dosing. Circ Rep. 2023;5(3):80-89.[DOI]
-
270. Godino C, Mazza R, Gaspardone C, Minerva A, Sena R, Cozzani G, et al. Plasma levels measurement of the 4 direct oral anticoagulants in patients with atrial fibrillation at the time of acute thromboembolic and bleeding events. J Thromb Haemost. 2025;23(11):3515-3526.[DOI]
-
271. Gawaz M, Geisler T, Borst O. Current concepts and novel targets for antiplatelet therapy. Nat Rev Cardiol. 2023;20(9):583-599.[DOI]
-
272. Mayer K, Hein-Rothweiler R, Schüpke S, Janisch M, Bernlochner I, Ndrepepa G, et al. Efficacy and safety of revacept, a novel lesion-directed competitive antagonist to platelet glycoprotein VI, in patients undergoing elective percutaneous coronary intervention for stable ischemic heart disease: The randomized, double-blind, placebo-controlled ISAR-PLASTER phase 2 trial. JAMA Cardiol. 2021;6(7):753-761.[DOI]
-
273. Uphaus T, Richards T, Weimar C, Neugebauer H, Poli S, Weissenborn K, et al. Revacept, an inhibitor of platelet adhesion in symptomatic carotid stenosis: A multicenter randomized phase II trial. Stroke. 2022;53(9):2718-2729.[DOI]
-
274. Wichaiyo S, Parichatikanond W, Rattanavipanon W. Glenzocimab: A GPVI (Glycoprotein VI)-targeted potential antiplatelet agent for the treatment of acute ischemic stroke. Stroke. 2022;53(11):3506-3513.[DOI]
-
275. Mazighi M, Köhrmann M, Lemmens R, Lyrer PA, Molina CA, Richard S, et al. Safety and efficacy of platelet glycoprotein VI inhibition in acute ischaemic stroke (ACTIMIS): A randomised, double-blind, placebo-controlled, phase 1b/2a trial. Lancet Neurol. 2024;23(2):157-167.[DOI]
-
276. Varon D, Spectre G. Antiplatelet agents. Berlin: Springer; 2012.
-
277. Magnani G, Bonaca MP, Braunwald E, Dalby AJ, Fox KAA, Murphy SA, et al. Efficacy and safety of vorapaxar as approved for clinical use in the united states. J Am Heart Assoc. 2015;4(3):e001505.[DOI]
-
278. Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366(15):1404-1413.[DOI]
-
279. Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, Van de Werf F, et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012;366(1):20-33.[DOI]
-
280. Morrow DA, Alberts MJ, Mohr JP, Ameriso SF, Bonaca MP, Goto S, et al. Efficacy and safety of vorapaxar in patients with prior ischemic stroke. Stroke. 2013;44(3):691-698.[DOI]
-
281. Rey M, Kramberg M, Hess P, Morrison K, Ernst R, Haag F, et al. The reversible P2Y12 antagonist ACT-246475 causes significantly less blood loss than ticagrelor at equivalent antithrombotic efficacy in rat. Pharmacol Res Perspect. 2017;5(5):e00338.[DOI]
-
282. Schwarz M, Meade G, Stoll P, Ylanne J, Bassler N, Chen YC, et al. Conformation-specific blockade of the integrin GPIIb/IIIa: A novel antiplatelet strategy that selectively targets activated platelets. Circ Res. 2006;99(1):25-33.[DOI]
-
283. Shen B, Zhao X, O’Brien KA, Stojanovic-Terpo A, Delaney MK, Kim K, et al. A directional switch of integrin signalling and a new anti-thrombotic strategy. Nature. 2013;503(7474):131-135.[DOI]
-
284. Hohmann JD, Wang X, Krajewski S, Selan C, Haller CA, Straub A, et al. Delayed targeting of CD39 to activated platelet GPIIb/IIIa via a single-chain antibody: Breaking the link between antithrombotic potency and bleeding? Blood. 2013;121(16):3067-3075.[DOI]
-
285. Leader A, Mor-Cohen R, Ram R, Sheptovitsky V, Seligsohn U, Rosenberg N, et al. The role of protein disulfide isomerase in the post-ligation phase of β3 integrin-dependent cell adhesion. Thromb Res. 2015;136(6):1259-1265.[DOI]
-
286. Cho J, Furie BC, Coughlin SR, Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest. 2008;118(3):1123-1131.[DOI]
-
287. Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, et al. Pi 3-kinase p110β: A new target for antithrombotic therapy. Nat Med. 2005;11(5):507-514.[DOI]
-
288. Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111(8):4113-4117.[DOI]
-
289. Salomon O, Steinberg DM, Zucker M, Varon D, Zivelin A, Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb Haemost. 2011;105(02):269-273.[DOI]
-
290. Feng D, Wang J. Factor XI inhibitors are the novel promising anticoagulants in the treatment of age related thrombotic disease. Front Cardiovasc Med. 2025;12:1498826.[DOI]
-
291. Ali AE, Awad MK, Ali K, Abouzid MR, Ahmed MH, Mazroua MS. Factor xi as a new target for prevention of thromboembolism in cardiovascular disease: A meta-analysis of randomized controlled trials. J Thromb Thrombolysis. 2025;58(1):1-14.[DOI]
-
292. Presume J, Ferreira J, Ribeiras R, Mendes M. Achieving higher efficacy without compromising safety with factor XI inhibitors versus low molecular weight heparin for the prevention of venous thromboembolism in major orthopedic surgery—Systematic review and meta-analysis. J Thromb Haemost. 2022;20(12):2930-2938.[DOI]
-
293. Capodanno D, Alexander JH, Bahit MC, Eikelboom JW, Gibson CM, Goodman SG, et al. Factor XI inhibitors for the prevention and treatment of venous and arterial thromboembolism. Nat Rev Cardiol. 2025;22(11):896-912.[DOI]
-
294. Li L, Geraghty OC, Mehta Z, Rothwell PM. Age-specific risks, severity, time course, and outcome of bleeding on long-term antiplatelet treatment after vascular events: A population-based cohort study. Lancet. 2017;390(10093):490-499.[DOI]
-
295. Mehran R, Baber U, Sharma SK, Cohen DJ, Angiolillo DJ, Briguori C, et al. Ticagrelor with or without aspirin in high-risk patients after PCI. N Engl J Med. 2019;381(21):2032-2042.[DOI]
-
296. Tomaniak M, Chichareon P, Modolo R, Takahashi K, Chang CC, Kogame N, et al. Ticagrelor monotherapy beyond one month after PCI in ACS or stable CAD in elderly patients: a pre-specified analysis of the GLOBAL LEADERS trial. EuroIntervention. 2020;15(18):e1605-e1614.[DOI]
-
297. Valgimigli M, Frigoli E, Heg D, Tijssen J, Jüni P, Vranckx P, et al. Dual antiplatelet therapy after PCI in patients at high bleeding risk. N Engl J Med. 2021;385(18):1643-1655.[DOI]
-
298. Costa F, Montalto C, Branca M, Hong SJ, Watanabe H, Franzone A, et al. Dual antiplatelet therapy duration after percutaneous coronary intervention in high bleeding risk: A meta-analysis of randomized trials. Eur Heart J. 2023;44(11):954-968.[DOI]
-
299. Watanabe H, Morimoto T, Natsuaki M, Yamamoto K, Obayashi Y, Ogita M, et al. Comparison of clopidogrel monotherapy after 1 to 2 months of dual antiplatelet therapy with 12 months of dual antiplatelet therapy in patients with acute coronary syndrome: The STOPDAPT-2 ACS randomized clinical trial. JAMA Cardiol. 2022;7(4):407-417.[DOI]
-
300. Vrints C, Andreotti F, Koskinas KC, Rossello X, Adamo M, Ainslie J, et al. 2024 ESC guidelines for the management of chronic coronary syndromes: developed by the task force for the management of chronic coronary syndromes of the European Society of Cardiology (ESC) endorsed by the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J. 2024;45(36):3415-3537.[DOI]
-
301. Gibson CM, Mehran R, Bode C, Halperin J, Verheugt FW, Wildgoose P, et al. Prevention of bleeding in patients with atrial fibrillation undergoing PCI. N Engl J Med. 2016;375(25):2423-2434.[DOI]
-
302. Krantz MJ, Debus SE, Hsia J, Patel MR, Anand SS, Nehler MR, et al. Low-dose rivaroxaban plus aspirin in older patients with peripheral artery disease undergoing acute limb revascularization: Insights from the VOYAGER PAD trial. Eur Heart J. 2021;42(39):4040-4048.[DOI]
-
303. Branch KRH, Probstfield JL, Bosch J, Bhatt DL, Maggioni AP, Muehlhofer E, et al. Total events and net clinical benefit of rivaroxaban and aspirin in patients with chronic coronary or peripheral artery disease: The COMPASS. Am Heart J. 2023;258:60-68.[DOI]
-
304. Connolly SJ, Eikelboom JW, Bosch J, Dagenais G, Dyal L, Lanas F, et al. Rivaroxaban with or without aspirin in patients with stable coronary artery disease: An international, randomised, double-blind, placebo-controlled trial. Lancet. 2018;391(10117):205-218.[DOI]
-
305. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119-1131.[DOI]
-
306. Svensson EC, Madar A, Campbell CD, He Y, Sultan M, Healey ML, et al. TET2-driven clonal hematopoiesis and response to canakinumab: An exploratory analysis of the CANTOS randomized clinical trial. JAMA Cardiol. 2022;7(5):521-528.[DOI]
-
307. Tardif J-C, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. 2019;381(26):2497-2505.[DOI]
-
308. Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. 2020;383(19):1838-1847.[DOI]
-
309. Samuel M, Berry C, Dubé MP, Koenig W, López-Sendón J, Maggioni AP, et al. Long-term trials of colchicine for secondary prevention of vascular events: A meta-analysis. Eur Heart J. 2025;46(26):2552-2563.[DOI]
-
310. Kumar R, Sonkar VK, Swamy J, Ahmed A, Sharathkumar AA, Pierce GL, et al. Dnase 1 protects from increased thrombin generation and venous thrombosis during aging: Cross-sectional study in mice and humans. J Am Heart Assoc. 2022;11(2):e021188.[DOI]
-
311. LaRocca TJ, Henson GD, Thorburn A, Sindler AL, Pierce GL, Seals DR. Translational evidence that impaired autophagy contributes to arterial ageing. J Physiol. 2012;590(14):3305-3316.[DOI]
-
312. Kaplon RE, Hill SD, Bispham NZ, Santos-Parker JR, Nowlan MJ, Snyder LL, et al. Oral trehalose supplementation improves resistance artery endothelial function in healthy middle-aged and older adults. Aging. 2016;8(6):1167-1183.[DOI]
-
313. Guo Y, Xu C, Man AWC, Bai B, Luo C, Huang Y, et al. Endothelial SIRT1 prevents age-induced impairment of vasodilator responses by enhancing the expression and activity of soluble guanylyl cyclase in smooth muscle cells. Cardiovasc Res. 2019;115(3):678-690.[DOI]
-
314. Rossman MJ, Santos-Parker JR, Steward CAC, Bispham NZ, Cuevas LM, Rosenberg HL, et al. Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension. 2018;71(6):1056-1063.[DOI]
-
315. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15(5):973-977.[DOI]
-
316. Wang RS, Maron BA, Loscalzo J. Multiomics network medicine approaches to precision medicine and therapeutics in cardiovascular diseases. Arterioscler Thromb Vasc Biol. 2023;43(4):493-503.[DOI]
-
317. Nordestgaard LT, Wolford BN, de Gonzalo-Calvo D, Sopić M, Devaux Y, Matic L, et al. Multiomics in atherosclerotic cardiovascular disease. Atherosclerosis. 2025;408:120414.[DOI]
-
318. Solari FA, Krahn D, Swieringa F, Verhelst S, Rassaf T, Tasdogan A, et al. Multi-omics approaches to study platelet mechanisms. Curr Opin Chem Biol. 2023;73:102253.[DOI]
-
319. Doran S, Arif M, Lam S, Bayraktar A, Turkez H, Uhlen M, et al. Multi-omics approaches for revealing the complexity of cardiovascular disease. Brief Bioinform. 2021;22(5):bbab061.[DOI]
-
320. Pekayvaz K, Kilani B, Joppich M, Eivers L, Brambs S, Knottenberg V, et al. Immunothrombolytic monocyte-neutrophil axes dominate the single-cell landscape of human thrombosis and correlate with thrombus resolution. Immunity. 2025;58(5):1343-1358.[DOI]
-
321. Costa F, Gomez Doblas JJ, Díaz Expósito A, Adamo M, D’Ascenzo F, Kołtowski L, et al. Artificial intelligence in cardiovascular pharmacotherapy: Applications and perspectives. Eur Heart J. 2025;46(37):3616-3627.[DOI]
-
322. Choi H, Kang HJ, Ahn I, Gwon H, Kim Y, Seo H, et al. Machine learning models to predict the warfarin discharge dosage using clinical information of inpatients from South Kore. Sci Rep. 2023;13(1):22461.[DOI]
-
323. Lee H, Kim HJ, Chang HW, Kim DJ, Mo J, Kim JE. Development of a system to support warfarin dose decisions using deep neural networks. Sci Rep. 2021;11(1):14745.[DOI]
-
324. Hansen PW, Clemmensen L, Sehested TSG, Fosbøl EL, Torp-Pedersen C, Køber L, et al. Identifying drug-drug interactions by data mining: A pilot study of warfarin-associated drug interactions. Circ Cardiovasc Qual Outcomes. 2016;9(6):621-628.[DOI]
-
325. Goto S, Goto S, Pieper KS, Bassand JP, Camm AJ, Fitzmaurice DA, et al. New artificial intelligence prediction model using serial prothrombin time international normalized ratio measurements in atrial fibrillation patients on vitamin K antagonists: GARFIELD-AF. Eur Heart J Cardiovasc Pharmacother. 2020;6(5):301-309.[DOI]
-
326. Chen C, Yin C, Wang Y, Zeng J, Wang S, Bao Y, et al. Xgboost-based machine learning test improves the accuracy of hemorrhage prediction among geriatric patients with long-term administration of rivaroxaban. BMC Geriatr. 2023;23(1):418.[DOI]
-
327. Huang YC, Cheng YC, Jhou MJ, Chen M, Lu CJ. Integrated machine learning decision tree model for risk evaluation in patients with non-valvular atrial fibrillation when taking different doses of dabigatran. Int J Environ Res Public Health. 2023;20(3):2359.[DOI]
-
328. Ward-Caviness CK, Huffman JE, Everett K, Germain M, van Dongen J, Hill WD, et al. DNA methylation age is associated with an altered hemostatic profile in a multiethnic meta-analysis. Blood. 2018;132(17):1842-1850.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Zervas G, Konstantaki C, Sigl M, Stamatelopoulos K, Tual-Chalot S, Stellos K. Vascular aging as a driver of thrombosis in older adults: From mechanisms to gerotherapeutics. Geromedicine. 2026;2:202513. https://doi.org/10.70401/Geromedicine.2025.0010
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Zervas G, Konstantaki C, Sigl M, Stamatelopoulos K, Tual-Chalot S, Stellos K. Vascular aging as a driver of thrombosis in older adults: From mechanisms to gerotherapeutics. Geromedicine. 2026;2:202513. https://doi.org/10.70401/Geromedicine.2025.0010
copy
Share Link
copy