Inhibition of PAI-1 shifts cardiomyocyte fate from senescence toward apoptosis and mitigates doxorubicin-induced cardiotoxicity
*Correspondence to:
Junichi Sadoshima, Department of Cell Biology and Molecular Medicine, Cardiovascular Research Institute, Rutgers-New Jersey Medical School, 185 South Orange Ave, Newark, NJ 07103, USA.
E-mail: sadoshju@njms.rutgers.edu
Geromedicine. 2026;2:202521. 10.70401/Geromedicine.2026.0018
Received: December 09, 2025Accepted: March 12, 2026Published: March 17, 2026
Abstract
Aims: Doxorubicin (Dox) is an effective chemotherapeutic agent, but its clinical use is limited by cardiotoxicity. Cellular senescence contributes to Dox-induced cardiac dysfunction; however, the underlying molecular mechanism mediating the effect of senescence remains poorly understood. This study aimed to identify senescence-associated factors secreted from cardiomyocytes in Dox-treated hearts and define their functional significance in Dox-induced cardiotoxicity.
Methods: Mice with cardiomyocyte-specific expression of the endoplasmic reticulum BioID secretome profiling system were used to identify Dox-induced secreted factors. Functional analyses were performed in neonatal rat ventricular myocytes (NRVMs). The effects of plasminogen activator inhibitor-1 (PAI-1) inhibition were evaluated in Dox-treated mice by assessing senescence markers, apoptotic responses, and cardiac structure and function. p21High-tdTomato reporter mice were used to examine the fate of senescent cardiomyocytes in vivo.
Results: PAI-1 was identified as a major component of the senescence-associated secretory phenotype and was robustly upregulated in Dox-treated cardiomyocytes. In NRVMs, PAI-1 promoted senescence and maintained the senescent phenotype, in part by conferring resistance to apoptosis. Pharmacological inhibition of PAI-1 reduced senescence markers, enhanced apoptotic responses, and preserved cardiac structure and function in Dox-treated mice. Fate mapping analyses with p21High-tdTomato mice revealed that PAI-1 inhibition decreased the number of p21High senescent cardiomyocytes in Dox-treated hearts. Notably, PAI-1 inhibition did not attenuate Dox cytotoxicity in EO771 murine breast cancer cells.
Conclusion: PAI-1 is a key mediator of Dox-induced cardiac dysfunction. PAI-1 inhibition shifts the fate of cardiomyocytes from senescence toward apoptosis and preserves cardiac structure and function without compromising the antitumor function of Dox, highlighting PAI-1 as a potential therapeutic target for chemotherapy-associated cardiotoxicity.
Keywords
Doxorubicin, PAI-1, BioID, TM5275, senescence
1. Introduction
Doxorubicin (Dox), an anthracycline chemotherapeutic agent widely used in treating hematological and solid tumors, remains a cornerstone of modern cancer therapy. However, its clinical application is significantly limited by its cumulative and dose-dependent cardiotoxicity[1]. Cardiac dysfunction may manifest during treatment or emerge years after therapy, contributing substantially to long-term morbidity and non-cancer mortality in cancer survivors. Despite extensive studies implicating oxidative stress, mitochondrial dysfunction, and DNA damage in Dox-induced cardiomyopathy, effective strategies to prevent or reverse this complication remain elusive[2].
Emerging evidence suggests that cellular senescence plays a pivotal role in driving Dox-induced cardiotoxicity[3,4]. Senescence in cardiomyocytes, characterized by telomere damage and the secretion of pro-inflammatory and pro-fibrotic factors, collectively known as the senescence-associated secretory phenotype (SASP), has been shown to disrupt myocardial structure and function[5,6]. However, the precise molecular mediators that orchestrate the establishment, maintenance, and paracrine effects of cardiomyocyte senescence under Dox stress remain incompletely understood. Autocrine and paracrine factors produced through the SASP are both cell-type and stimulus-specific. To our knowledge, the SASP factors specifically produced by and secreted from cardiomyocytes are poorly understood[7]. To address this question, we used cardiac-specific endoplasmic reticulum (ER)-BioID (cER-BioID) mice, in which a biotin conjugation enzyme is expressed on the endoplasmic reticulum membrane[8]. In these mice, proteins destined to either the plasma membrane or secretion are biotinylated in the presence of biotin. The expression of the ER-BioID system in a
PAI-1, a member of the serine protease inhibitor (serpin) superfamily, has been implicated as a SASP component in non-cardiac tissue, where it contributes to senescence and tissue remodeling[9-11]. Whether PAI-1 plays a similar role in the heart, particularly in response to genotoxic chemotherapy, remains unknown. Furthermore, the origin of PAI-1 in the serum and whether it is produced in senescent cells are unknown.
In this study, we aimed to define the role of PAI-1 in Dox-induced cardiac senescence and dysfunction. By employing the cER-BioID system in both in vivo and in vitro models of Dox-induced cardiomyopathy and pharmacological PAI-1 inhibition, we investigated whether PAI-1 serves as a key mediator of cellular senescence in Dox-induced cardiomyopathy. We further explored whether targeting PAI-1 alters the balance between senescence and apoptosis in cardiomyocytes and improves cardiac function. These findings may offer a novel therapeutic strategy to prevent or reverse Dox-induced cardiotoxicity.
2. Materials and Methods
2.1 Animals
C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). cER-BioID mice were generated by crossing
2.1.1 Dox administration to mice
Phosphate-buffered saline (PBS)-dissolved Dox (Sigma) was administered to mice via intraperitoneal injection at a dose of 5 mg/kg once a week for 2 consecutive weeks[13,14]. Mice injected with PBS were used as controls.
2.1.2 Biotin administration to mice
For in vivo secretome labeling, mice were administered a biotin solution via intraperitoneal and subcutaneous injections. Each mouse received 500 μL of a 2 mg/mL biotin solution (Sigma-Aldrich) once a day for five consecutive days[8]. In parallel, the mice were fed a biotin-supplemented diet during the same period. Serum samples were collected by terminal bleeding, and 50 μL of serum from each mouse was incubated with 20 μL of streptavidin-conjugated beads at 4 ℃ for 2 hours. After washing, the biotinylated proteins were eluted in 2× sodium dodecyl sulfate (SDS) loading buffer and analyzed by mass spectrometry.
2.1.3 ABT-263 (Navitoclax) administration to mice
ABT-263 (Navitoclax, Selleckchem), dissolved in ethanol/polyethylene glycol 400/Phosal 50 PG at a ratio of 10:30:60, was administered to mice via oral gavage at a dose of 50 mg/kg body weight per day for five consecutive days[15]. Control mice received the vehicle solution only.
2.1.4 TM5275 administration to mice
TM5275 (MedChemExpress LLC, NJ), suspended in 0.5% carboxymethyl cellulose sodium salt solution, was administered by oral gavage at a dose of 10 mg/kg body weight per day for five days per cycle[16]. Control mice received the vehicle solution only.
2.1.5 Tamoxifen administration to mice
p21High-ER-BioID and cp21-tdTomato mice were fed a tamoxifen-containing diet for two weeks, after which they were switched to a regular diet.
2.2 Single cell RNA sequencing
Dox (5 mg/kg/week) was administered intraperitoneally twice. Control mice were injected with PBS. One week after the final injection, adult mouse heart cells were isolated from both PBS-treated (n = 3) and Dox-treated (n = 3) mice according to previously published protocols[17]. Freshly isolated cardiomyocytes and non-myocytes were pooled into PBS and Dox treatment groups and immediately fixed using Evercode Fixation v2 kits (Parse Biosciences), following the manufacturer’s instructions. Cells were processed to target a total of 10,000 cells across two sublibraries using the Evercode WT Mini v2 kit (Parse Biosciences). Barcoding and library generation were performed according to the manufacturer’s protocols. For data processing, sequencing reads were aligned using the ParseBioscience processing pipeline with default settings set to the GRCm38 mouse genome. Downstream analysis was conducted using the Seurat package in R, following standard procedures. Briefly, aligned count matrices were imported into an AnnData object and subjected to quality control filtering to exclude low-quality cells based on thresholds for gene counts, UMI counts, and mitochondrial gene content. The data were normalized to 10,000 counts per cell and log transformed. Highly variable genes were selected using sc.pp.highly_variable_genes, excluding mitochondrial and ribosomal genes. The data were scaled after regressing out total counts. Principal component analysis was performed on the variable genes. Cell-cell relationships were captured using a neighborhood graph (sc.pp.neighbors), and clusters were identified using the Leiden algorithm (sc.tl.leiden). UMAP was used for
2.3 Primary culture of neonatal rat ventricular cardiomyocytes (NRVMs)
Primary cultures of ventricular cardiomyocytes were prepared from 1-day-old Charles River Laboratories rats and maintained in culture as described previously[18]. A cardiomyocyte-rich fraction was obtained by centrifugation, through a discontinuous Percoll gradient as described.
2.4 SA-β-galactosidase (SA-β-gal) assay
NRVMs were seeded in 12-well plates. After 24 hours, the medium was replaced with fresh medium and the cells were treated with Dox (100 nM) and/or TM5275 (10 μM). After 72 hours, β-galactosidase activity was assessed using the Senescence β-Galactosidase Staining Kit (Cell Signaling Technology) according to the manufacturer’s instructions. Briefly, the cells were washed once with 1× PBS and fixed with fixative solution at room temperature for 15 minutes. The cells were then washed with 1× PBS and stained with
2.5 Cell viability assay
EO771 murine breast cancer cells were seeded in 96-well plates at a density of 2 × 103 cells per well. Cells were treated with
2.6 Echocardiography
Echocardiography was performed using a high-resolution Micro-Ultrasound system (Vevo 3100, FUJIFILM Visual-Sonic Inc., Toronto, Canada). Two-dimensional guided M-mode measurements of left ventricular internal diameter were obtained from at least three beats and then averaged. Left ventricular end-diastolic dimension (LVEDD) was measured at the time of the apparent maximal left ventricular diastolic dimension, and left ventricular end-systolic dimension (LVESD) was measured at the time of the most anterior systolic excursion of the posterior wall. Left ventricular ejection fraction (LVEF) was calculated using the following formula:
2.7 Immunoblotting analysis
Cardiac tissue homogenates were prepared from the left ventricle apex. Cell lysates were prepared from primary cultures of rat ventricular myocytes. Both the homogenates and the cell lysates were prepared in a radioimmunoprecipitation assay (RIPA) buffer containing 150 mM NaCl, 1% Triton-X 100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate, and 50 mM Tris (pH 8.0), and supplemented with a protease inhibitor cocktail (Sigma), 5 mM NaF, and 1 mM sodium orthovanadate. The following antibodies were used: phospho-H2AX (Cell Signaling Technology (CST), #9718), GAPDH (CST, #2118), PAI-1 (CST, #2753), cleaved caspase-3 (CST, #9664), p21 (Santa Cruz Biotechnology, #SC-1661), p53 (Bioss, #8687R), p16 (Invitrogen, #MA5-17142), ATM (CST, #2837), pATM (CST, #4526) and secondary antibodies (anti-rabbit or anti-mouse horseradish peroxidase-conjugated antibodies, CST, #7074 and #7076).
2.8 Pull-down assays
Streptavidin–agarose (Thermo Fisher Scientific) was added to 50 μL of serum and gently rotated at 4 °C for 2 hours. The streptavidin–agarose was then washed three times with PBS and eluted with 2× sample buffer. The eluates were subjected to
2.9 Mass spectrometry analyses
Streptavidin–agarose (Thermo Fisher Scientific) was added to 50 μL of serum and gently rotated at 4 °C for 2 hours. The streptavidin–agarose was then washed three times with PBS and eluted with 2× sample buffer, and the eluates were subjected to
2.10 TUNEL assays
NRVMs were fixed in PBS containing 4% paraformaldehyde. Staining was performed using the In situ Cell Death Detection kit (Roche) as described[19]. Nuclear density was determined by manual counting of DAPI-stained nuclei in six fields for each animal using the
2.11 SDS-PAGE and Western blotting
Heart homogenates and cardiomyocyte lysates for SDS-PAGE and immunoblotting analyses were prepared in RIPA lysis buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate, 1 mM EDTA, 1 mM sodium orthovanadate, 1 mM sodium fluoride, and 1× Halt Protease inhibitor cocktail (Thermo Fisher Scientific). After determining the protein concentrations by BCA assay, equal amounts of protein were loaded on an SDS-PAGE gel with 4× Laemmli sample buffer (200 mM
2.12 Gene silencing via small interfering RNAs (siRNA) transfection
siRNAs were transfected into cells using Lipofectamine RNAiMAX (Thermo Fisher Scientific). Pre-designed Silencer Select siRNA targeting rat Serpine1 (s128154) and non-targeting control siRNA (siControl, #4390843) were purchased from Thermo Fisher Scientific. Lipofectamine RNAiMAX (Thermo Fisher Scientific) was first diluted in Opti-MEM medium (Thermo Fisher Scientific) and subsequently mixed with each siRNA. The mixture was incubated for 15 minutes at room temperature to allow complex formation. The resulting Lipofectamine–siRNA complexes were added to cardiomyocyte cultures, followed by gentle agitation of the mixture. After 24 hours, the transfection medium was replaced with serum-free DMEM/F-12 containing penicillin/streptomycin. Cells were subsequently cultured under standard conditions for downstream analyses.
2.13 Immunofluorescence analyses
Mouse hearts were harvested, fixed with 10% formalin and embedded in wax, and cross-sections (5 μm thick) were prepared. Neonatal cardiomyocytes were cultured on coverslips and fixed with 4% paraformaldehyde. An overnight incubation with specific antibodies against phospho-H2AX, IL-6 (Thermo Fisher Scientific, #P620), RFP (Rockland, #600-401-379), and cTNT (Invitrogen,
2.14 Cell death detection
NRVMs were seeded in 4-well chamber slides and cultured overnight. The medium was then replaced with fresh medium and the cells were treated with Dox (100 nM) and/or TM5275 (25 μM) for 3 hours. Apoptosis and necrosis were detected using the Cell MeterTM Apoptotic and Necrotic Detection kit (AAT Bioquest, Inc., Sunnyvale, CA) according to the manufacturer’s protocol. The cells were observed and analyzed in a blinded manner using fluorescent microscopy.
2.15 Statistical analyses
All data are expressed as the mean ± SEM. All statistical analyses were performed using an unpaired Student’s t-test or one-way ANOVA followed by a post hoc Bonferroni-Dunn’s comparison test for multiple group comparisons, or two-way ANOVA followed by Sidak’s multiple comparison test. A value of P < 0.05 was considered significant.
3. Results
3.1 Dox treatment enhances cardiomyocyte-derived PAI-1 secretion in cER-BioID mice
To characterize the cardiomyocyte-specific secretome during the acute phase of Dox-induced cardiomyopathy, we generated
Our previous study demonstrated that repeated administration of Dox (5 mg/kg/week, four times) induces cardiac dysfunction and cardiomyocyte senescence in mice[14]. To capture the cardiomyocyte-specific secretome associated with Dox-induced cardiac dysfunction, cER-BioID mice were treated with Dox (5 mg/kg) or PBS on Day 0 and on Day 7, the point at which LVEF typically begins to decline[14]. We confirmed that Dox treatment induced cardiac dysfunction in cER-BioID mice similar to that in wild type mice as reported previously[14] (Figure S2B). Biotin supplementation was administered via intraperitoneal (i.p.) injections and biotin-enriched chow for five consecutive days beginning after the second Dox dose (on Day 8, 500 μL of 2 mg/mL biotin) (Figure S1D). Serum samples were then collected on Day 14, and biotinylated proteins were isolated using streptavidin-conjugated beads and analyzed via mass spectrometry (MS). Proteins upregulated in the Dox + biotin group relative to both the PBS + biotin and the PBS without biotin groups were identified as candidate cardiomyocyte-specific Dox-induced secreted factors (Figure S3A,B). Of these proteins, two molecules in the serine protease inhibitor superfamily (serpins), namely PAI-1 (Serpine1) and alpha-1-antitrypsin 1-5 (Serpina1e), were the most elevated (Figure S3A,B). The Search Tool for the Retrieval of Interacting Genes/Proteins network analysis highlighted that serpins have functional interactions with other proteins identified in the screen, including Murinoglobulin-1, Haptoglobin and Leucine-rich HEV glycoprotein (Figure S3C). Serpins have diverse functions and are involved in blood coagulation, fibrinolysis, programmed cell death, development and inflammation[9]. PAI-1 mediates Dox-induced senescence in alveolar type II cells and cultured cardiomyocytes[10,11]. However, the involvement of PAI-1 in Dox-induced cardiomyopathy in vivo, particularly its function as a SASP factor, the cell type that produces PAI-1, and its functional significance are unknown. Given the known ability of PAI-1 to induce tissue remodeling[20], we focused on PAI-1 in this study and hypothesized that it is secreted from senescent cardiomyocytes in response to Dox treatment.
We confirmed that Dox- and biotin-treated cER-BioID mice exhibit significantly elevated serum PAI-1 levels compared to PBS- or
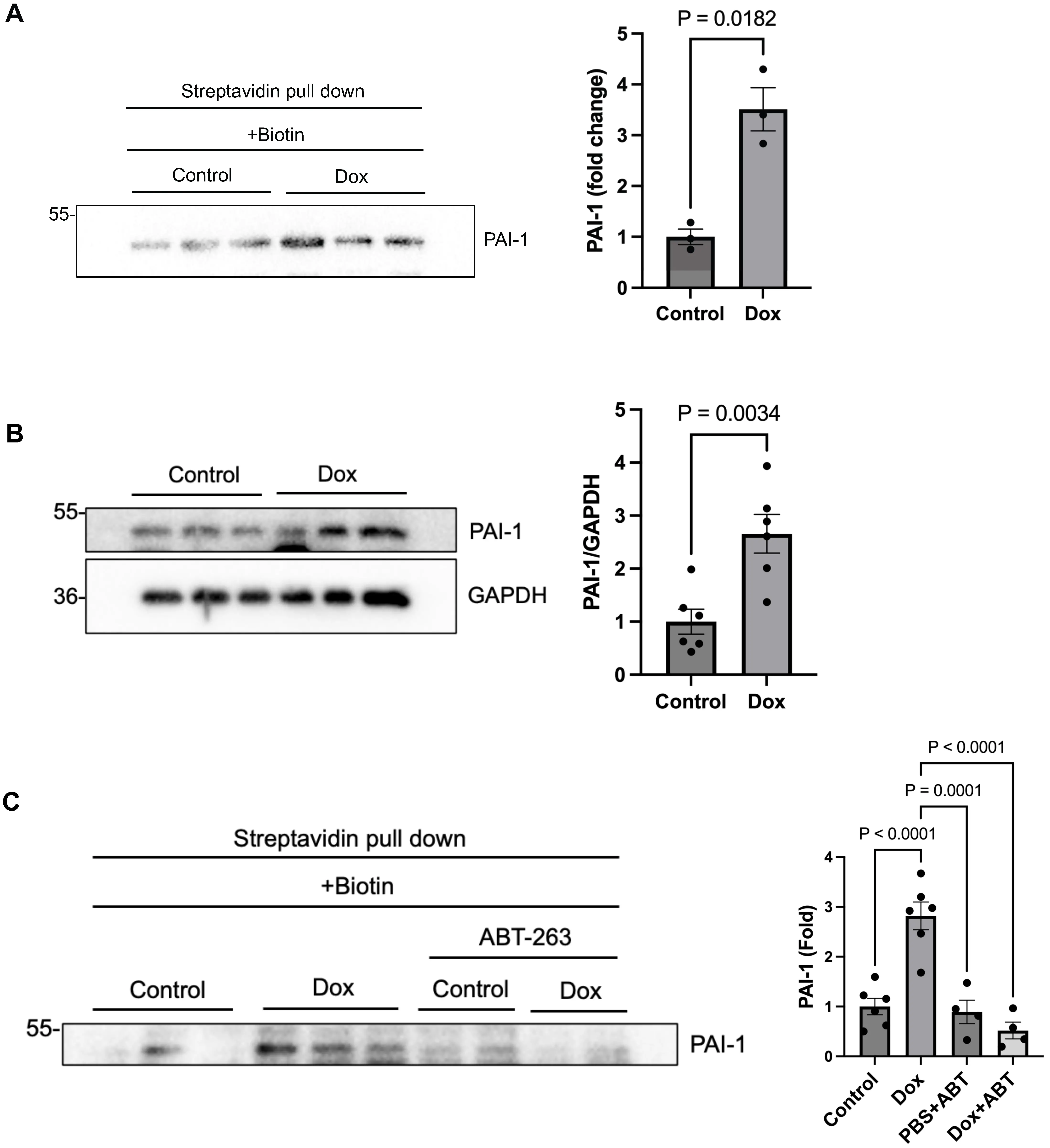
Figure 1. Identification of PAI-1 as a secreted protein induced by Dox using cER-BioID mice. (A) Left: Western blot analysis of PAI-1in serum from
3.2 PAI-1 secreted from senescent cardiomyocytes spreads senescence to neighboring cells
To determine whether Dox directly induces upregulation and secretion of PAI-1 in cardiomyocytes in a cell-autonomous manner, we treated NRVMs with Dox (100 nM for 72 hours) or PBS and evaluated the levels of senescence markers. Dox treatment significantly increased the expression of both PAI-1 (Figure 2A) and senescence markers, including p21 and γH2AX (Figure 2B), indicating that Dox upregulates both senescence and the production of PAI-1 in cardiomyocytes. To confirm that Dox upregulates PAI-1 in cardiomyocytes in the heart in vivo, we conducted single cell RNA sequencing analyses of mouse hearts with or without Dox treatment (Figure S4). mRNA expression of Serpine1 (Pai1) was significantly upregulated in the cardiomyocyte fraction in response to Dox treatment. Interestingly, upregulation of p53 and downregulation of plasminogen activator 1 (Plat) were also observed in the cardiomyocyte fraction.
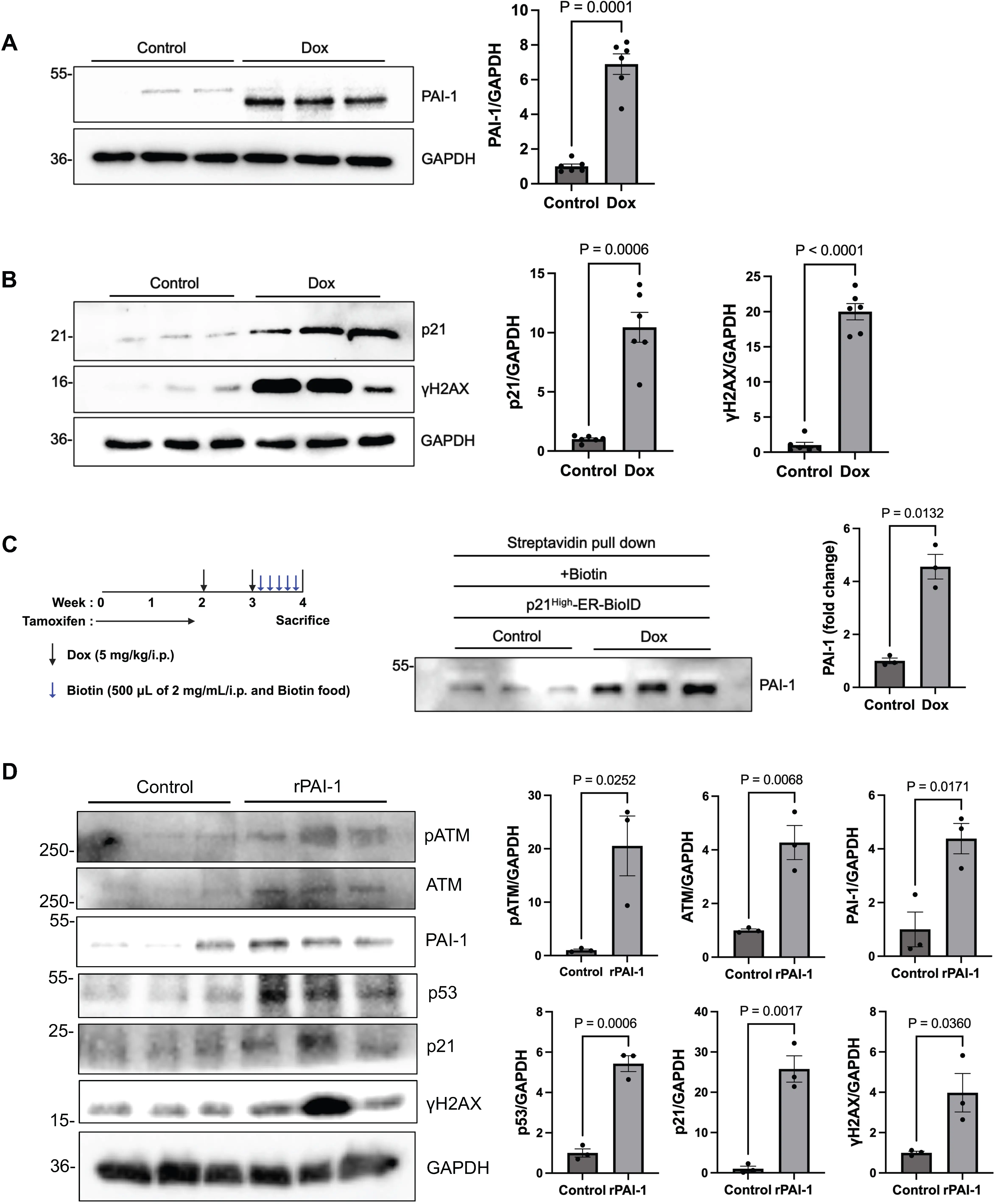
Figure 2. PAI-1 and other cellular senescence markers are elevated in cardiomyocytes exposed to Dox. (A) Left: Western blot analysis of PAI-1 in NRVMs treated with PBS (control) or Dox. Right: Quantification of PAI-1 band intensity; (B) Left: Western blot analysis of p21 and γH2AX in control or Dox-treated NRVMs. Right: Quantification of p21 and γH2AX band intensity; (C) Left: Schematic representation of the tamoxifen, Dox, and biotin administration protocol in p21High-ER-BioID mice. Center: Western blot analysis of PAI-1 in serum from p21High-ER-BioID mice treated with PBS + biotin (control) or Dox + biotin. Right: Quantification of PAI-1 band intensity; (D) NRVMs were treated with
To further investigate whether PAI-1 is specifically secreted by senescent cells, we expressed the ER-BioID system under the control of the p21High promoter, which is known to be activated in a senescent cell-specific manner[12]. To this end, we generated
3.3 Inhibition of PAI-1 attenuates cardiomyocyte senescence and enhances apoptosis signaling
To examine whether PAI-1 plays a functional role in mediating senescence in cardiomyocytes, we performed gene silencing in NRVMs using siRNA targeting PAI-1 (Pai-1si). After 48 hours, cardiomyocytes were treated with Dox for 72 hours to induce senescence. Treatment with Pai-1si downregulated PAI-1 protein levels and key senescence markers, including p53, p21, and p16, in the presence of Dox (Figure 3A), indicating that endogenous PAI-1 contributes to either the establishment or the maintenance of senescence in cardiomyocytes during Dox treatment.
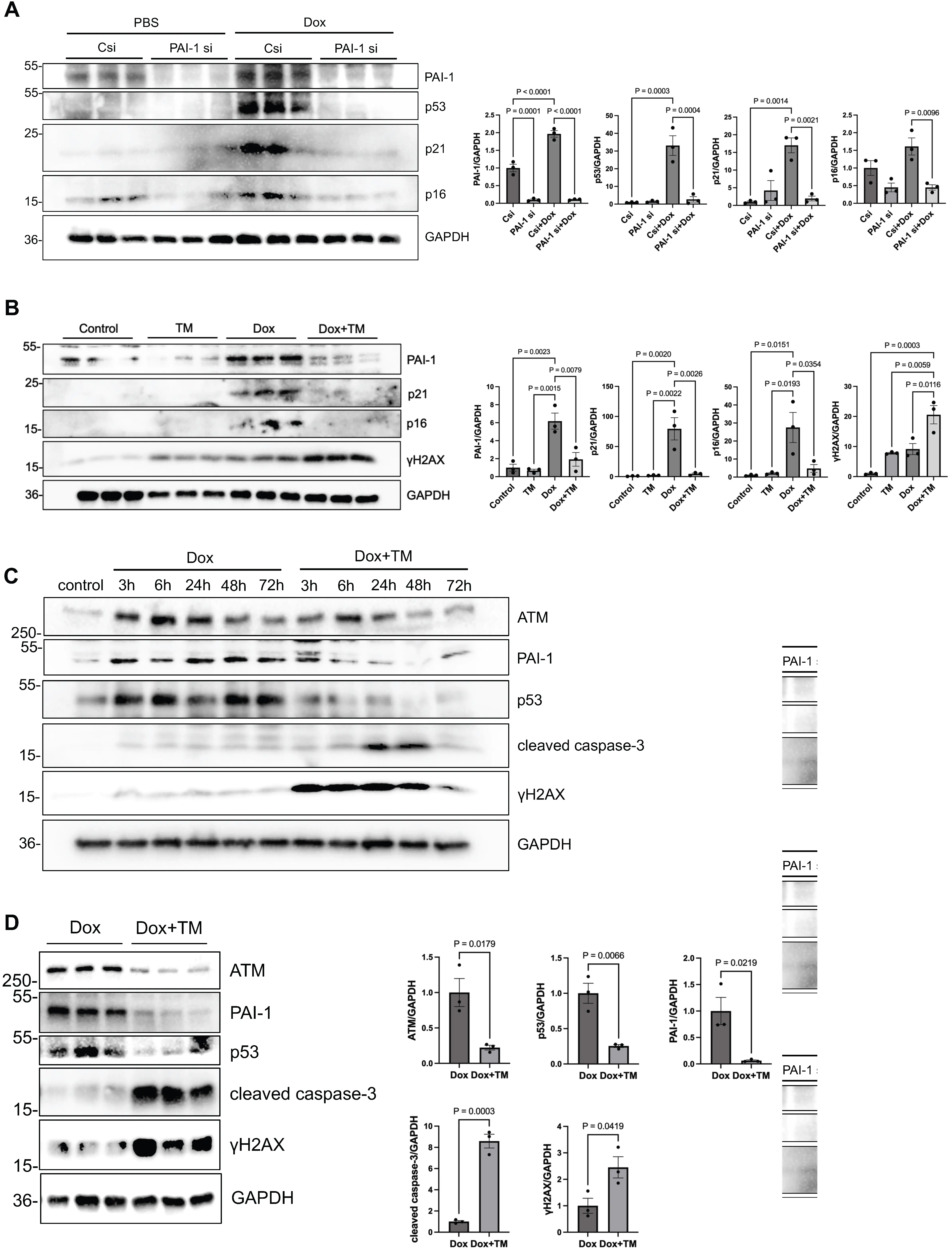
Figure 3. Doxorubicin-induced cardiomyocyte senescence involves PAI-1 and is attenuated by TM5275. (A) Left: Western blot analysis of PAI-1, p53, p21, and p16 in NRVMs treated with PBS (control), Csi, or PAI-1 si, with or without Dox (100 nM). Right: Quantification of band intensity for each protein; (B) NRVMs were treated with Dox (100 nM) and/or TM5275 (25 μM) for 72 hours. Left: Western blot analysis of PAI-1, p21, p16, and γH2AX in NRVMs treated with vehicle (control), TM5275, Dox, or Dox + TM5275.
We next assessed the effect of pharmacological inhibition of PAI-1 on Dox-induced senescence. NRVMs were treated with Dox in the presence or absence of TM5275, a selective PAI-1 inhibitor[24]. Although Dox treatment for 72 hours increased the protein levels of PAI-1, p21, and p16, co-treatment with TM5275 suppressed the upregulation of both PAI-1 and the senescence markers, with the exception of γH2AX (Figure 3B).
To directly assess cellular senescence, we performed SA-β-gal staining in NRVMs. Dox treatment significantly increased the proportion of SA-β-gal-positive cells, whereas PAI-1 inhibition with TM5275 markedly reduced the SA-β-gal positivity (Figure S6A,B), confirming attenuation of Dox-induced senescence. Importantly, to determine whether PAI-1 inhibition interferes with the antitumor efficacy of Dox, we assessed Dox-induced cytotoxicity in EO771 breast cancer cells. TM5275 did not affect Dox-induced cytotoxicity in EO771 cells (Figure S6C), indicating that PAI-1 inhibition does not compromise the anticancer effects of Dox.
We further evaluated the time-dependent effects of PAI-1 inhibition in NRVMs. Dox treatment rapidly upregulated p53 within 3 hours, whereas TM5275 inhibited Dox-induced upregulation of p53. Interestingly, while Dox increased the expression of γH2AX, a marker of DNA double-strand breaks, TM5275 enhanced its elevation, starting as early as after 3 hours of Dox treatment, with attenuation observed at 72 hours. This suggests that DNA damage is promoted in the presence of PAI-1 inhibition. In parallel, cleaved
3.4 PAI-1 inhibition mitigates Dox-induced cardiac dysfunction and remodeling in vivo
To evaluate the cardioprotective effects of PAI-1 inhibition in vivo, wild-type mice were treated with Dox in the presence or absence of TM5275. Dox was injected twice, at weeks 0 and 1 (i.p. injection, 5 mg/kg), in the presence or absence of TM5275 for 2 weeks
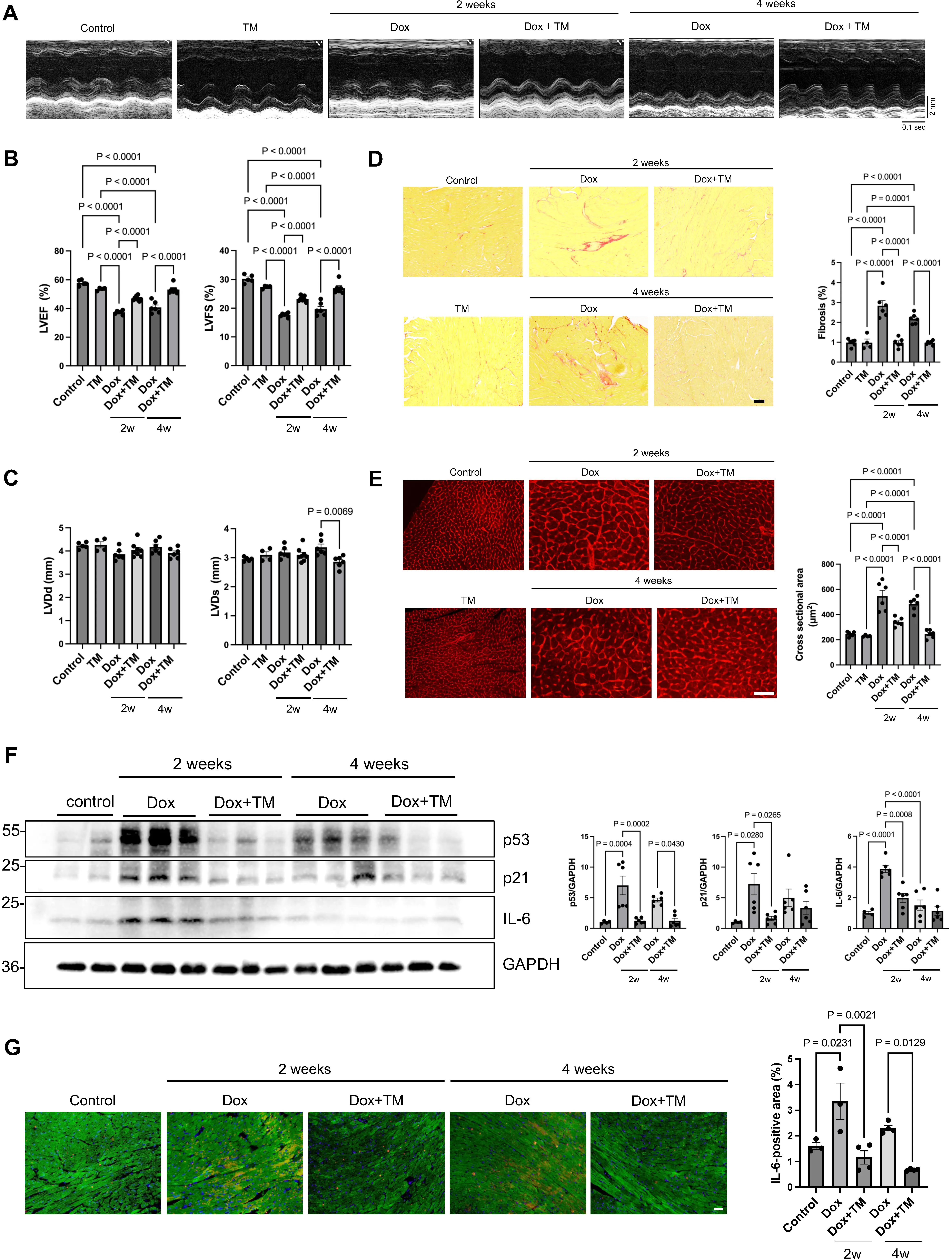
Figure 4. TM5275 improves cardiac function and reduces senescence- and stress-related markers in a mouse model of doxorubicin-induced cardiotoxicity. Wild-type mice were treated with Dox in the presence or absence of TM5275. Dox was injected two times, at weeks 0 and 1 (i.p. injection, 5 mg/kg), in either the presence or absence of TM5275 for 2 weeks (10 mg/kg orally). One group of mice was sacrificed at 2 weeks to evaluate the effect of Dox and TM5275 in the acute phase and another group of mice was sacrificed at 4 weeks to evaluate the chronic effect. (A) Representative M-mode echocardiographic images from control, TM5275, Dox, and Dox + TM5275 mice at
3.5 PAI-1 inhibition may promote apoptosis of cardiomyocytes with persistent DNA damage while reducing overall cell death
We next examined whether PAI-1 inhibition modulates the fate of cardiomyocytes under Dox treatment. Using a Cell MeterTM apoptotic and necrotic multiplexing detection assay, we found that TM5275 did not affect apoptosis or necrosis in NRVMs in the absence of Dox. In Dox-treated cells, TM5275 reduced overall apoptosis and necrosis (Figure 5A), indicating attenuation of net cell death. To determine whether the inhibition of cell death is seen uniformly in every cell type, we performed γH2AX/TUNEL
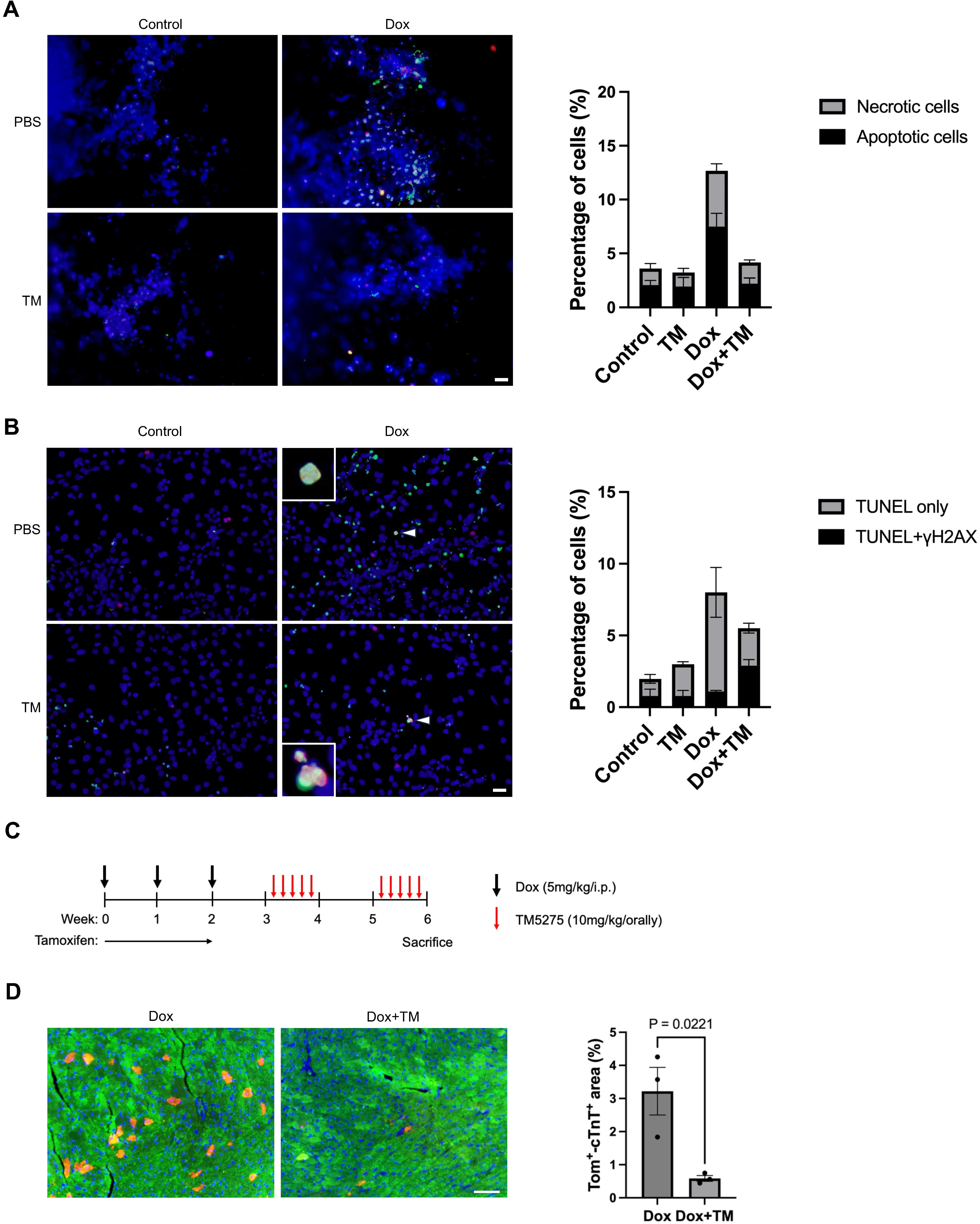
Figure 5. PAI-1 inhibition promotes apoptosis and clearance of senescent cardiomyocytes after exposure to doxorubicin. (A) NRVMs were treated with Dox (100 nM) and/or TM5275 (10 μM) for 3 hours. Left: Representative images of live/dead cell staining showing healthy cells (blue), apoptotic cells (green), and necrotic cells (red) in NRVMs treated with vehicle (control), TM5275, Dox, or Dox + TM5275. Scale bar = 100 μm. Right: Quantification of the proportions of healthy, apoptotic, and necrotic cells in each group; (B) NRVMs were treated with Dox (100 nM) and/or TM5275 (10 μM) for 3 hours. Left: Representative immunofluorescence images of TUNEL (green), γH2AX (red), and DAPI (blue) staining in NRVMs treated with vehicle (control), TM5275, Dox, and Dox + TM5275. Right: Quantification of TUNEL-positive only and double-positive (TUNEL and
3.6 TM5275 selectively reduces p21High senescent cardiomyocytes in vivo
To determine whether TM5275 alters the balance between senescence and apoptosis in cardiomyocytes in vivo, we generated
4. Discussion
Using a cardiomyocyte-specific secretome profiling approach with cER-BioID mice, we identified PAI-1 as a key protein secreted by senescent cardiomyocytes following Dox treatment. Functionally, our findings indicate that PAI-1 contributes to Dox-induced cardiotoxicity in vivo by promoting and sustaining cellular senescence in the heart. Mechanistically, our data suggest that PAI-1 promotes senescence development and supports the survival of senescent cardiomyocytes.
Cellular senescence is characterized by the presence of the SASP, which involves the release of pro-inflammatory and
At the functional level, siRNA-mediated knockdown of PAI-1 in vitro attenuated Dox-induced upregulation of senescence markers in cardiomyocytes. Consistent with these findings, pharmacological inhibition of PAI-1 with TM5275 in vivo attenuated Dox-induced upregulation of senescence markers in the heart (e.g., p53 and IL-6) and mitigated Dox-induced cardiomyopathy. These results support the notion that PAI-1 plays a functional role in mediating Dox-induced senescence in cardiomyocytes and consequent pathological cardiac remodeling and dysfunction. Importantly, under the conditions we tested, TM5275 did not attenuate
Although PAI-1 has previously been shown to be involved in Dox-induced senescence in several cell types, including
What is the mechanism through which PAI-1 inhibition promotes apoptotic signaling in senescent cells? A previous study showed that PAI-1 activates cell survival signaling pathways, including Akt and ERK1/2, and inhibits caspase-3[28]. In the present study, we show that recombinant PAI-1 activates ATM, a master regulator of DNA repair, in cultured NRVMs, suggesting that PAI-1 is sufficient to activate DNA repair signaling. Conversely, pharmacological inhibition of PAI-1 promoted γH2AX in the presence of Dox, accompanied by downregulation of ATM. A previous study showed that Serpine2 directly interacts with ATM, thereby stimulating phosphorylation and activation of ATM[29]. Since ATM allows senescent cells to repair damage and avoid apoptosis[30,31], PAI-1 inhibition may promote apoptosis of senescent cells by inhibiting ATM.
It has been shown that p53 plays an important role in mediating transcription of Pai-1 in fibroblasts in response to a carcinogen[32]. We found that Dox-induced upregulation of Pai-1 in cardiomyocytes is accompanied by upregulation of p53. Furthermore, recombinant PAI-1 also upregulates p53. Thus, it is possible that p53 and PAI-1 stimulate one another. Together with the fact that inhibition of PAI-1 attenuates expression of PAI-1 in cardiomyocytes, this suggests that Dox-induced upregulation of PAI-1 initiates a self-amplification mechanism to promote PAI-1 and senescence through p53.
Several limitations of this study should be acknowledged. While cER-BioID enables cardiomyocyte-specific secretome profiling, it does not exclude contributions of PAI-1 secreted from other cardiac cell types or from extracardiac sources to Dox-induced cardiomyopathy. Because PAI-1 is produced by multiple cell populations, the cardioprotective effect of TM5275 may not be solely attributable to the inhibition of cardiomyocyte-derived PAI-1. Moreover, our analyses focused on early-phase injury and the use of a single model of cardiotoxicity. Long-term consequences of PAI-1 inhibition, particularly regarding cardiac regeneration and systemic effects, remain unclear. Additionally, while TM5275 is a selective inhibitor, off-target effects cannot be entirely ruled out. Future studies should explore the timing and duration of PAI-1 inhibition, and combination of PAI-1 inhibition and other cardioprotective or senescence-modulating strategies. Investigating the role of PAI-1 in other forms of cardiac stress or in human cardiomyocytes will also be crucial for clinical translation.
5. Conclusion
In summary, our study provides the proof-of-concept for utilizing ER-BioID mice in conjunction with other tools for investigating cellular senescence to identify cell-type-specific SASP factors in an unbiased manner. Our findings position PAI-1 as a critical mediator of Dox-induced cardiac dysfunction in vivo. PAI-1 promotes senescence in cardiomyocytes and supports the persistence of senescent cardiomyocytes by favoring their survival over apoptotic signaling, thereby contributing to maladaptive cardiac remodeling and functional decline. Inhibition of PAI-1 attenuates cellular senescence, enhances apoptotic signaling in cardiomyocytes with persistent DNA damage, and preserves cardiac structure and function without compromising the antitumor efficacy of Dox. These results provide a strong rationale for developing PAI-1-targeted therapies to mitigate chemotherapy-associated cardiac dysfunction by modulating the survival of senescent cardiomyocytes in Dox-induced cardiomyopathy hearts.
Supplementary materials
The supplementary material for this article is available at: Supplementary materials.
Acknowledgements
We thank Liu T, Li H, and Tian Y for their excellent technical assistance.
Authors contribution
Shiheido-Watanabe Y: Methodology, investigation, data curation, formal analysis, writing-original draft.
Sung EA: Resources, methodology, investigation, data curation, formal analysis.
Ivessa A: Methodology, investigation, data curation, formal analysis.
Zhai P: Resources, methodology.
Takada T, Ikeda S, Matsushita M: Investigation, formal analysis.
Zablocki D: Writing-review & editing.
Sadoshima J: Conceptualization, methodology, formal analysis, writing-original draft.
Conflicts of interest
Junichi Sadoshima is an Editorial Board Member of Geromedicine. The other authors declare no conflicts of interest.
Ethical approval
All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Rutgers New Jersey Medical School, The State University of New Jersey. The approval number is 201900133.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
The data that support the findings, including statistical analyses, and reagents used are available from the corresponding author upon request.
Funding
This work was supported in part by the JSPS KAKENHI (Grant No. 22J40087 to Yuka Shiheido-Watanabe), American Heart Association (AHA) Predoctoral Fellowship (Grant No. 915784 to Eun-Ah Sung), Merit Award 20 Merit (Grant No. 35120374 to Junichi Sadoshima), and Transformational Project Award (Grant No. 25TPA1481361 to Junichi Sadoshima), U.S. Public Health Service (Grant No. HL91469,
Copyright
© The Author(s) 2026.
References
-
1. Bhutani V, Varzideh F, Wilson S, Kansakar U, Jankauskas S, Santulli G. Doxorubicin-induced cardiotoxicity: A comprehensive update. J Cardiovasc Dev Dis. 2025;12(6):207.[DOI]
-
6. Zhai P, Sadoshima J. Cardiomyocyte senescence and the potential therapeutic role of senolytics in the heart. J Cardiovasc Aging. 2024;4(2):18.[DOI]
-
8. Liu J, Jang JY, Pirooznia M, Liu S, Finkel T. The secretome mouse provides a genetic platform to delineate tissue-specific in vivo secretion. Proc Natl Acad Sci U S A. 2021;118(3):e2005134118.[DOI]
-
13. Liu Y, Zhang W, Hu T, Ni J, Xu B, Huang W. A doxorubicin-induced murine model of dilated cardiomyopathy in vivo. J Vis Exp. 2020;159:e61158.[DOI]
-
14. Zhai P, Sung EA, Shiheido-Watanabe Y, Takayama K, Tian Y, Sadoshima J. Suppression of autophagy induces senescence in the heart. J Mol Cell Cardiol. 2024;195:83-96.[DOI]
-
19. Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, et al. Mst1 inhibits autophagy by promoting Beclin1-Bcl-2 interaction. Nat Med. 2013;19(11):1478-1488.[DOI]
-
23. Siddiqi S, Sussman MA. Cardiac hegemony of senescence. Curr Transl Geriatr Exp Gerontol Rep. 2013;2(4):247-254.[DOI]
-
29. Zhang J, Wu Q, Zhu L, Xie S, Tu L, Yang Y, et al. SERPINE2/PN-1 regulates the DNA damage response and radioresistance by activating ATM in lung cancer. Cancer Lett. 2022;524:268-283.[DOI]
-
30. Phan LM, Rezaeian AH. ATM: Main features, signaling pathways, and its diverse roles in DNA damage response, tumor suppression, and cancer development. Genes. 2021;12(6):845.[DOI]
-
32. Parra M, Jardí M, Koziczak M, Nagamine Y, Muñoz-Cánoves P. p53 Phosphorylation at serine 15 is required for transcriptional induction of the plasminogen activator inhibitor-1 (PAI-1) gene by the alkylating agent N-methyl-N'-nitro-N-nitrosoguanidine. J Biol Chem. 2001;276(39):36303-36310.
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Shiheido-Watanabe Y, Sung AH, Ivessa A, Zhai P, Takada T, Ikeda S, et al. Inhibition of PAI-1 shifts cardiomyocyte fate from senescence toward apoptosis and mitigates doxorubicin-induced cardiotoxicity. Geromedicine. 2026;2:202521. https://doi.org/10.70401/Geromedicine.2026.0018
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Materials and Methods
- 3. Results
- 4. Discussion
- 5. Conclusion
- Supplementary materials
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Shiheido-Watanabe Y, Sung AH, Ivessa A, Zhai P, Takada T, Ikeda S, et al. Inhibition of PAI-1 shifts cardiomyocyte fate from senescence toward apoptosis and mitigates doxorubicin-induced cardiotoxicity. Geromedicine. 2026;2:202521. https://doi.org/10.70401/Geromedicine.2026.0018
copy
Share Link
copy