Microglial autophagy and other LC3-dependent pathways in neurodegeneration
Jennifer E. Palmer
1,2

,
David C. Rubinsztein
1,2,*
*Correspondence to:
David C. Rubinsztein, Cambridge Institute for Medical Research, University of Cambridge, Cambridge CB2 OXY, UK; UK Dementia Research Institute, Cambridge Biomedical Campus, Cambridge CB2 0XY, UK.
E-mail: dcr1000@cam.ac.uk
Geromedicine. 2026;2:202525. 10.70401/Geromedicine.2026.0022
Received: December 23, 2025Accepted: April 29, 2026Published: April 30, 2026
This manuscript is made available in its unedited form to allow early access to the
reported findings. Further editing will be completed before final publication. As
such,
the content may include errors, and standard legal disclaimers are applicable.
Abstract
Autophagy is a conserved cellular clearance pathway that supports homeostasis by removing damaged or superfluous intracellular components. Within microglia, autophagy is emerging as a regulator of key processes that modify neurodegeneration, including phagocytosis, cytokine secretion, and senescence. Many studies that have examined the effect of disrupted autophagy on microglial functions have used genetic knockouts of the machinery required to conjugate microtubule-associated light chain 3 (LC3) to the autophagic membrane. However, much of this molecular machinery is also required for a set of distinct but related pathways known as the conjugation of ATG8s to single membranes (CASM). CASM includes processes of particular importance in microglia, such as LC3-associated phagocytosis and LC3-associated endocytosis. It is thus not clear which of the effects of the disruption of LC3 conjugation in microglia are attributable to the loss of autophagy or the loss of CASM function. In this review, we describe the mechanisms of autophagy and CASM and highlight the effects of the loss of these pathways on key microglial processes relevant to brain ageing and neurodegenerative diseases. We discuss recent literature that has revealed the effects of ageing and neurodegeneration on microglial autophagy, and the effects of microglial autophagy and/or CASM disruption on key microglial functions such as phagocytosis, cytokine secretion, and senescence. Finally, we discuss the potential therapeutic implications of these findings for neurodegeneration and highlight key unanswered questions for future research.
Keywords
Autophagy, macroautophagy, microglia, ageing, neurodegeneration, LC3-associated phagocytosis, LC3-associated endocytosis, conjugation of ATG8s to single membranes
1. Introduction
Neurodegenerative diseases are a group of progressive brain diseases that share key characteristics such as the accumulation of aggregation-prone proteins, neuroinflammation, and neuronal cell death[1]. Ageing is the biggest risk factor for neurodegenerative diseases, and many of the hallmarks of ageing are also observed in neurodegeneration, including loss of proteostasis, senescence, and inflammation[2]. Neurodegenerative diseases are becoming increasingly prevalent as global societies age, placing a growing burden on societal and healthcare systems[3].
Microglia, the resident phagocytic immune cells of the brain, are one of the major modifiers of neurodegeneration through their protective and detrimental effects on neuronal and glial function. Microglia have key roles in the maintenance of brain health, including the removal of cellular debris and protein deposits, and regulate myelination[4,5]. However, during ageing and neurodegenerative diseases, microglia transition to a variety of hyperactivated or senescent states that exacerbate neurodegeneration through the release of neuronally-damaging proinflammatory cytokines, excessive phagocytic removal of synapses, and demyelination[4,5].
The maintenance of normal microglial function depends on critical homeostatic pathways, including autophagy. Autophagy is a conserved cellular recycling pathway that delivers cytoplasmic components to the lysosome for degradation[6]. In macroautophagy, this occurs by the capture of autophagy substrates within double-membraned autophagosomes, which then fuse with lysosomes to enable the clearance and recycling of the substrates. A defining molecular event in macroautophagy is the conjugation of the small protein microtubule-associated protein 1 light chain 3 (LC3) to the autophagic membrane[7]. The disruption of this conjugation event through genetic knockouts of the core molecular machinery abrogates autophagy and is commonly used as a tool to study the effects of autophagy loss-of-function. As discussed in this review, such approaches have been used to show that microglial-specific autophagy loss results in microglial senescence and proinflammatory cytokine secretion from microglia that exacerbates neurodegeneration, highlighting the importance of microglial autophagy in preventing neurodegeneration.
However, LC3 can also be conjugated to single membranes in a variety of pathways collectively known as the conjugation of ATG8s to single membranes (CASM), and the knockout of the LC3 conjugation machinery also abrogates these pathways[8,9]. CASM includes processes of particular importance in microglia, including LC3-associated phagocytosis (LAP), where LC3 is conjugated to the phagosome membrane, and LC3-associated endocytosis (LANDO), where LC3 is conjugated to the endosome membrane[10]. As discussed in this review, recent literature also indicates that the loss of these pathways in microglia exacerbates neurodegeneration.
In the following sections, we define the mechanisms and molecular regulation of autophagy (Section 2) and CASM (Section 3) pathways, and note which components of the molecular machinery are shared by both pathways or are specific to one pathway. We then describe how microglia modulate neurodegeneration, including brief descriptions of the key characteristics and roles of microglia in ageing and neurodegenerative diseases (Section 4). We consider the effects of ageing and neurodegeneration-associated processes on microglial autophagy and CASM (Section 5) and how microglial autophagy and/or CASM disruption affects key microglial functions that modify neurodegeneration (Section 6). We draw on the literature in other cell types to discuss the potential mechanisms through which autophagy and CASM alter microglia cytokine secretion, senescence, and phagocytosis (Section 7). Finally, we discuss the potential therapeutic implications of these findings (Section 8) and highlight key unanswered questions for future research (Section 9).
2. Autophagy
Autophagy is a conserved cellular clearance pathway that removes and recycles superfluous or damaged cellular components by delivering them to the lysosome, wherein they are degraded by acid hydrolysis, and their molecular subunits (such as amino acids) are recycled to the cytoplasm. There are three main autophagy pathways, which differ in their mechanism of delivering the autophagy substrates to the lysosome; these are macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy
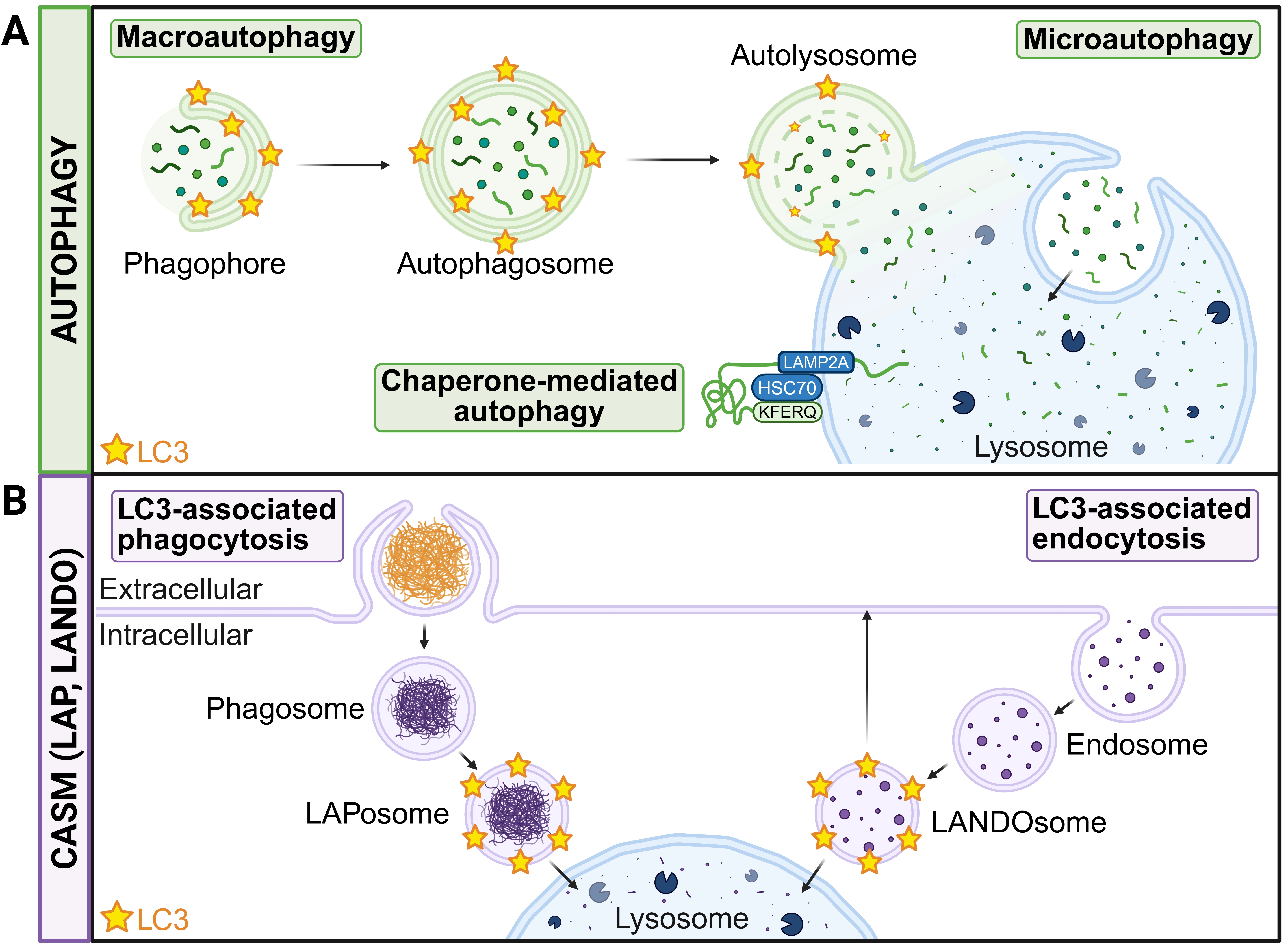
Figure 1. Autophagy and CASM pathways in microglia. (A) Autophagy includes macroautophagy (which proceeds through the conjugation of LC3 to the phagophore double membrane), CMA (which involves the direct lysosomal translocation of KFERQ motif-containing proteins dependent on HSC70 and LAMP2A) and microautophagy (in which the lysosomal membrane invaginates to enable the direct uptake of solutes); (B) CASM describes the conjugation of LC3 to single membranes. For example, in LAP, LC3 is conjugated to a phagosome to form a LAPosome, and in LANDO, LC3 is conjugated to an endosome to form a LANDOsome. Created in BioRender.com. CASM: conjugation of ATG8s to single membranes; LC3: light chain 3; CMA: chaperone-mediated autophagy; HSC70: heat shock cognate protein 70; LAMP2A: lysosome-associated membrane protein 2A; LAP: LC3-associated phagocytosis; LANDO: LC3-associated endocytosis.
2.1 Macroautophagy
Macroautophagy is uniquely defined by the formation of double-membraned autophagosomes positive for the autophagic marker protein LC3[6,7]. Cargoes are captured by finger-like membrane protrusions known as phagophores, which form from recycling endosomes proximal to the ER[11,12]. Phagophore extension is facilitated by lipid transport from the ER, dependent on the ATG2-ATG9A complex[13]. Phagophores are then closed by the endosomal sorting complex required for transport (ESCRT) machinery and subsequently released from the recycling endosome compartment by dynamin 2 (DNM2)-dependent scission[14]. The released autophagosomes are retrogradely trafficked on microtubules and undergo Soluble NSF Attachment Protein Receptor-dependent fusion with lysosomes to enable the acid hydrolysis of the autophagic substrates[15].
Autophagy is upregulated in response to a variety of cellular stresses, most notably amino acid or glucose starvation[16]. A basal level of autophagy also continually occurs and is important for cytoplasmic quality control, nutrient recycling, and development, among many other functions[17]. Low nutrient availability results in the inhibition of the mammalian target of rapamycin complex 1 (mTORC1) kinase complex, and low energy availability results in the activation of the AMP-activated protein kinase (AMPK)[16]. These proteins signal to the Unc-51-like autophagy-activating kinase 1 (ULK1), which is inhibited by mTORC1 and activated by AMPK, and thus, cellular starvation results in the activation of ULK1[18]. ULK1 phosphorylates components of the autophagy-specific,
Macroautophagy can be further divided into bulk and selective pathways. Bulk macroautophagy captures a pool of cytoplasmic components in phagophores, resulting in their degradation and recycling. In selective autophagy, specific cargoes are targeted for autophagic degradation, such as mitochondria (mitophagy), invading bacteria (xenophagy), and aggregated proteins (aggrephagy), among many others. Selective autophagy utilises specific autophagy receptors, including (but certainly not limited to) p62, OPTN, TAX1BP1, and NDP52[24]. These selective autophagy receptors contain LC3-interacting regions and are often (but not always) activated in ubiquitination-dependent mechanisms[24]. Clustering of these receptors leads to the recruitment and activation of the ULK1 complex, initiating phagophore formation[24].
2.2 CMA
CMA and microautophagy involve the direct lysosomal uptake of autophagic cargoes and are independent of the formation of the LC3-conjugated autophagic double membrane that is characteristic of macroautophagy. The substrate proteins of CMA contain KFERQ motifs, which are recognised by the heat shock cognate protein 70 (HSC70). The protein is unfolded into a linear polypeptide and then imported into the lysosome dependent on the lysosome-associated membrane protein 2A (LAMP2A)[25]. This restricts the substrates of CMA to KFERQ-containing soluble proteins, which includes approximately 40% of the proteins in the mammalian proteome[26]. CMA is independent of LC3 conjugation and may be upregulated as a compensatory mechanism in response to macroautophagy impairment[27].
2.3 Microautophagy
In microautophagy, the lysosomal membrane invaginates to enable the direct uptake of soluble cargoes. The substrates of microautophagy include soluble proteins, organelle-derived vesicles, and dissolved solutes[28]. Like macroautophagy, microautophagy can be bulk or selective[28], although selective microautophagy has been studied mostly in yeast with comparatively few mammalian studies. In mammalian cells, selective microautophagy can be initiated by the binding of HSC70 to KFERQ motif-containing proteins. In contrast to CMA, where HSC70 binds to LAMP2A to import the substrate across the lysosomal membrane, HSC70 in microautophagy is internalised with the substrate during membrane invagination[25]. Microautophagy is dependent on the ESCRT complexes I, II, and III[25], and may also require components of the core autophagy machinery, although direct evidence for this is lacking in mammalian systems[29].
3. CASM Pathways
In addition to the conjugation of LC3 to the autophagic double membrane in macroautophagy, LC3 can also be conjugated to single membranes in a set of processes collectively known as the conjugation of LC3 to single membranes[8,9,30] (Figure 1). CASM includes processes of particular importance in microglia, including LAP and LANDO, which have been implicated in neurodegenerative diseases[10,31,32]. Whilst the alternative terminology of “non-canonical autophagy” has occasionally been used to refer to CASM pathways, we discourage the use of this term to reduce ambiguity and confusion, as non-canonical autophagy is also used to refer to macroautophagy occurring independent of some components of the core autophagy machinery.
3.1 LAP
Conjugation of LC3 to the phagosome membrane, known as LAP, is an important regulator of phagosome maturation and phagosome-lysosome fusion[30,33]. LC3 conjugation to the phagosome membrane occurs after the phagosome has closed[30,33], and unlike the exclusive conjugation to PE in macroautophagy, LC3 can be conjugated to both PE and phosphatidylserine on phagosomes in LAP[34].
LAP has mostly been studied in the context of phagosome alkalinisation in response to bacterial infection. Alkalinisation promotes the assembly of the vacuolar ATPase (v-ATPase), which recruits ATG16L1, leading to LC3 conjugation[35-38]. This recruitment of ATG16L1 is dependent on its v-ATPase-interacting WD40 domain and independent of its interaction partner in macroautophagy, WIPI2[38,39]. The initiation mechanisms for the microglial LAP of sterile phagocytic substrates are currently poorly understood, although they appear to also be dependent on the ATG16L1 WD40 domain[40].
3.2 LANDO
Conjugation of LC3 to endosomes, known as LANDO, regulates the recycling of endocytosed phagocytic receptors in microglia[31]. LANDO is comparatively understudied; however, like LAP, it is induced in response to altered pH and osmotic imbalance[41], and appears to use the same machinery as LAP[31]. LANDO has been implicated in the recycling of the phagocytic receptors TLR4, TREM2, and CD36 in microglia[31].
As it involves LC3 conjugation, the molecular machinery of CASM overlaps with that of macroautophagy (Figure 2). For example, the loss of ATG5 or ATG7, which are commonly used to abrogate autophagy in microglia (Table 1), prevents LC3 conjugation, disrupting both macroautophagy and CASM. This shared molecular machinery can make it challenging to disentangle which effects are due to the loss of autophagy or the loss of CASM. An approach to selectively impair autophagy without affecting CASM is to use ULK1 inhibitors, as CASM is independent of ULK1[33,49]. To selectively impair CASM but not affect autophagy, the ATG16L1 WD40 domain can be deleted; this domain is required for CASM but is dispensable for autophagy[38,39,50].
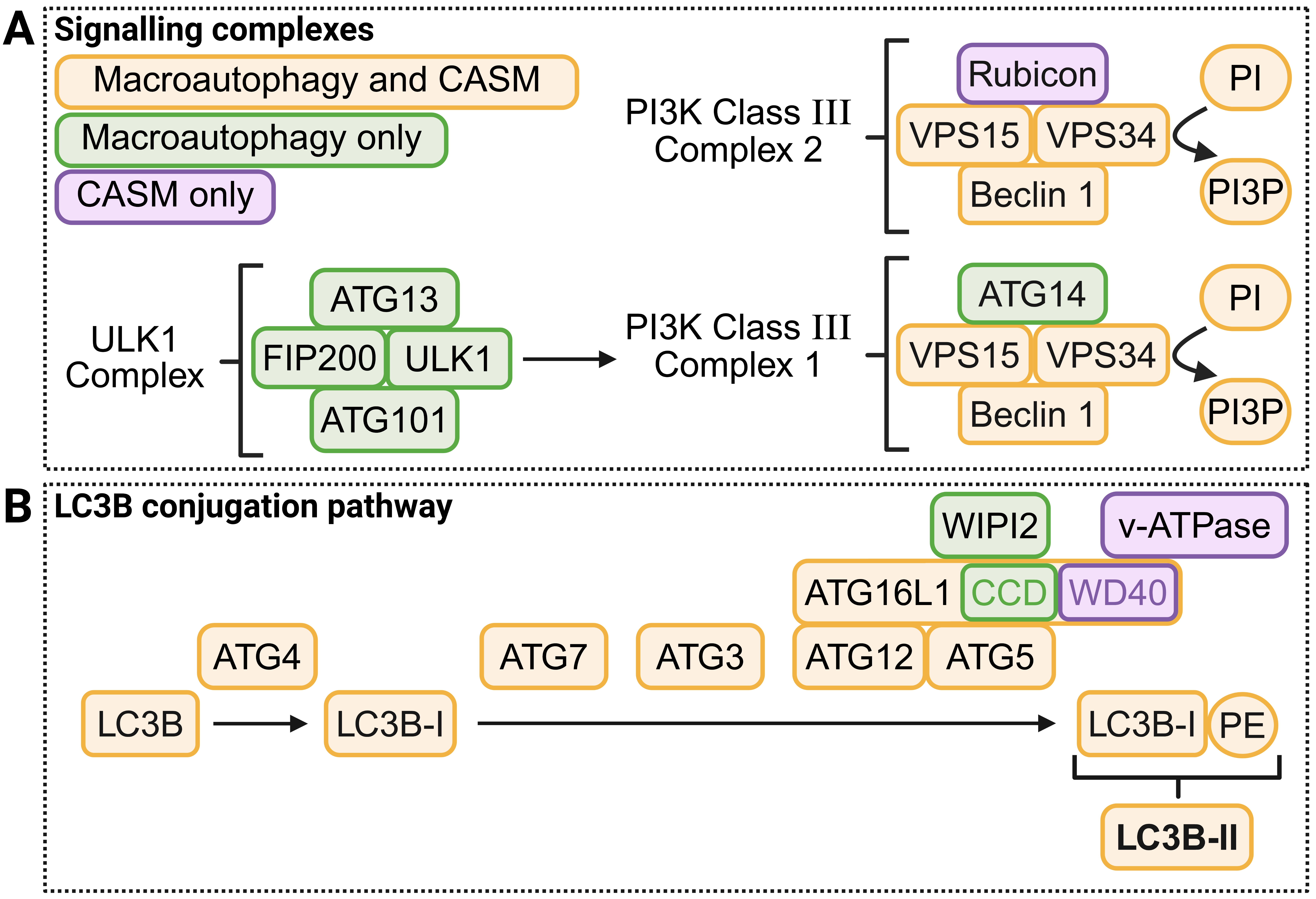
Figure 2. Molecular regulation of autophagy and CASM. Proteins and small molecules involved in key upstream signalling complexes (A) and the LC3B conjugation pathway (B) are shown and coloured according to the pathway(s) they function in. Molecular machinery that is shared by both pathways is shown in yellow, those specific to macroautophagy are shown in green, and those specific to CASM are shown in purple. Within ATG16L1, the CCD (autophagy) and WD40 (v-ATPase) domains are coloured independently. Created in BioRender.com. CASM: conjugation of ATG8s to single membranes; CCD: coiled-coil domain; v-ATPase: vacuolar ATPase.
Table 1. Effects of neurodegenerative disease-associated proteins on microglial autophagy.
| Disease | Protein | Effect on microglial autophagy-lysosomal function | Reference |
| AD | BIN1 | BIN1 upregulation resulted in an accumulation of autophagosomes due to the inhibition of the release of newly formed autophagosomes from the recycling endosome. | Palmer et al.[42] |
| AD | PSEN1 | Phospho-deficient PSEN1 mutations resulted in an accumulation of autophagosomes and lysosomal alkalinisation. | Ledo et al.[43] |
| AD | TREM2 | TREM2 loss-of-function mutations and knockout resulted in mTOR inhibition, AMPK activation, and autophagy upregulation. | Ulland et al.[44] |
| ALS | C9orf72 | C9orf72 patient iPSC-derived microglia had decreased autophagosome formation. | Banerjee et al.[45] |
| FTD | Progranulin | Progranulin knockout resulted in microglial lysosome dysfunction. | Wu et al.[46] |
| Niemann-Pick type A | Acid sphingomyelinase | Acid sphingomyelinase knockout resulted in microglial lysosome dysfunction. | Gabandé-Rodríguez et al.[47] |
| PD | LRRK2 | LRRK2 gain-of-function mutations resulted in decreased TFEB/TFE3/MITF-mediated expression of autophagy-lysosomal pathway genes and lysosome dysfunction. | Yadavalli & Ferguson[48] |
AD: Alzheimer’s disease; PSEN1: presenilin 1; mTOR: mammalian target of rapamycin; AMPK: AMP-activated protein kinase; ALS: amyotrophic lateral sclerosis; iPSC: induced pluripotent stem cell; FTD: frontotemporal dementia; PD: Parkinson’s disease; TFEB: transcription factor EB; MITF: microphthalmia-associated transcription factor.
4. Microglia in Ageing and Neurodegeneration
Microglia are the resident phagocytic immune cells of the brain, which have key functions in the maintenance of brain function but can also exacerbate brain ageing and neurodegeneration[4,5]. Genetic risk for many neurodegenerative diseases is enriched in microglia-specific enhancers, such as in BIN1, PICALM, INPP5D, TREM2, and RABEP1 in Alzheimer’s disease (AD), TMEM163 in Parkinson’s disease (PD), and CLECL1 and MCM9 in multiple sclerosis[51,52], implicating microglia in the pathogenesis of neurodegenerative diseases. Furthermore, the genetic variation associated with longevity is enriched in a homeostatic microglial gene expression network[53], implicating microglia in the modulation of brain ageing.
A common observation in ageing and neurodegenerative diseases is the loss of microglia with homeostatic gene expression signatures, and an increase in the abundance of highly inflammatory, dystrophic, or senescent microglia[54-56]. Importantly, both the loss of protective microglial functions, and the detrimental effects of increased numbers of hyperactivated and dystrophic microglia can contribute to pathological processes occurring during ageing and neurodegenerative diseases.
4.1 Microglial activation in ageing and neurodegenerative diseases
Ageing and neurodegenerative diseases are associated with an increase in microglial activation and neuroinflammation[4,55,57-59]. This heightened microglial activation occurs in response to damage-associated patterns resulting from extracellular protein aggregates and dying neurons, and microglial activation closely correlates with, and propagates with, markers of neuropathology such as tau[60]. Acute, regulated microglial activation can be protective by enhancing the phagocytic clearance of debris and plaques[4,61]. A protective, activated microglial state known as disease-associated microglia (DAM) is observed in AD, which restricts neurodegeneration by enhancing the phagocytic clearance of amyloid-β (Aβ) plaques[56,61]. Importantly, the transition to the DAM state is dependent on TREM2[61], and loss of function mutations in TREM2 increase the risk of developing AD[62,63], highlighting the importance of microglial activation in preventing neurodegeneration.
However, sustained microglial activation and hyperactivation exacerbate neurodegeneration through a variety of mechanisms. This includes the release of proinflammatory cytokines that impair neuronal and glial function, including impairing neuronal
4.2 Microglial dystrophy and senescence in ageing and neurodegenerative diseases
Prolonged microglial activation and processes occurring during ageing and neurodegenerative diseases can also lead to microglial dysfunction, which is typically characterised by microglial senescence[59,70,75-77]. Key characteristics of microglial senescence include reduced proliferation, increased oxidative damage, lipid droplet accumulation, and lipofuscin accumulation[78]. Senescence impairs microglial phagocytic clearance[78], which could be a contributing factor to the decline in microglial phagocytosis with age[79-82]. Importantly, senescent microglia release a senescence-associated secretory phenotype (SASP)[83], which includes proinflammatory cytokines such as TNFα and CCL3 that exacerbate neurodegeneration[78]. Therefore, microglial senescence actively worsens neuropathology, particularly through the SASP.
This results in a detrimental positive feedback loop, whereby neurodegeneration results in heightened microglial activation and eventually microglial dystrophy and senescence, which exacerbates neurodegeneration through the increased release of proinflammatory cytokines and impaired clearance of plaques and debris. It is thus unsurprising that microglia have been suggested as key drivers of the progression of neurodegeneration. For example, Lau, Ramer and Tremblay[84] predicted that the accumulation of senescent glia (particularly microglia) drives the transition to AD, and Simons, Levin and Dichgans[85] hypothesised that the transition to dementia occurs once the homeostasis of the glial, immune and vascular systems are disrupted, and highlighted the transition from DAM to dystrophic, senescent microglia as a key contributor to dementia progression.
5. Effects of Ageing and Neurodegeneration on Microglial Autophagy
Microglial autophagy and neurodegeneration have a bidirectional association, wherein ageing and neurodegeneration affect microglial autophagy, and microglial autophagy affects key microglial functions that modulate processes of ageing and neurodegeneration. In this section, we consider the effects of ageing and neurodegenerative diseases on microglial autophagy. In the following section, we consider the effects of microglial autophagy and/or CASM disruption on microglial function, ageing and neurodegeneration.
5.1 Effects of ageing on microglial autophagy
Autophagic function declines with age in most cell types and autophagy compromise is a hallmark of ageing[86]. Furthermore, autophagy disruption shortens lifespan, whereas autophagy upregulation extends lifespan, in a variety of model organisms[86-89]. This suggests that autophagy has a causal role in ageing. Potential contributors to the decreased autophagy in aged organisms include increased mTORC1 activity[90], the reduced expression of autophagy proteins such as ATG5, ATG12, LC3B, Beclin 1, and p62[90], and impaired lysosomal function[91], among other factors. One cause for decreased autophagy in the ageing mammalian brain is increased SORBS3 expression, which compromises YAP/TAZ signalling[92].
There are few studies directly examining the effect of ageing on microglial autophagy. Microglial senescence, which increases during ageing, is associated with decreased autophagy[93]. This suggests that, like many cell types, microglial autophagy function declines during ageing.
Additionally, ageing is associated with an increased abundance of a protective population of microglia known as
5.2 Effects of neurodegenerative diseases on microglial autophagy
Impaired autophagic flux is a conserved characteristic of many neurodegenerative diseases[91,95]. Whilst comparatively few studies have investigated autophagic function specifically in microglia, the existing studies indicate that neurodegenerative diseases are associated with alterations in microglial autophagy. For example, postmortem brains from human AD patients show an accumulation of LC3-positive vesicles in microglia[96], which could indicate either enhanced autophagosome formation or impaired autophagic clearance. Microglia in mouse models of AD have an accumulation of LC3 and autophagic substrates such as p62 and ubiquitin, particularly in the microglia surrounding Aβ plaques[97], which suggests that microglial autophagic clearance may be impaired in AD. Microglia from adult mice overexpressing α-synuclein in neurons (but not in microglia) show decreased LC3B-II and increased p62, suggesting microglial autophagy impairment in response to neuronal α-synuclein pathology[98]. Microglia also appear to capture
Impaired microglial autophagy in neurodegenerative diseases can be due to a combination of factors such as disease-associated genetic variants in autophagic and lysosomal proteins, changes in cellular signalling pathways and/or nutrients that regulate autophagy, and the inhibition of autophagy by neurodegenerative disease-associated aggregation-prone proteins[91,95], along with indirect, non-cell-autonomous effects arising from altered microglial state due to neuronal death and neurodegenerative pathology.
Many of the neurodegenerative disease-associated proteins that regulate autophagy are also expressed in microglia, and so they likely also regulate microglial autophagy, although this has not generally been experimentally confirmed. Table 1 lists the studies that have directly examined the effect of neurodegeneration-associated proteins and mutations on microglial autophagy. For example, mutations in presenilin 1, which are known to impair autophagosome maturation and clearance, result in an accumulation of autophagosomes in microglia in mouse models of AD[43]. Similarly, impaired microglial lysosome function, which impairs microglial autophagic flux, has been demonstrated with frontotemporal dementia-associated mutations in progranulin[46] and in Niemann-Pick type A microglia[47]. Microglia with amyotrophic lateral sclerosis C9orf72 mutations have decreased autophagosome formation, impaired phagocytosis, and increased secretion of pro-inflammatory cytokines[45], although it is unclear whether these are causally related. The PD-associated LRRK2 protein inhibits the TFEB-mediated expression of autophagic and lysosomal genes in microglia, impairing microglial autophagy[48].
Importantly, the specific impairment of microglial autophagy may affect the risk of neurodegeneration. For example, AD
TREM2 is a major regulator of microglial function and activation, which is required for the transition to the protective DAM state[61]. TREM2 AD-associated loss-of-function mutations or knockout results in decreased mTOR activity, an increased number of
6. Effects of Microglial Autophagy and CASM on Ageing and Neurodegeneration
Most publications that have tested the effect of microglial autophagy disruption on key microglial processes involved in ageing or neurodegeneration have used genetic knockouts of core components of the LC3 conjugation machinery, such as ATG5 or ATG7
Table 2. Effects of autophagy and/or CASM disruption in microglia.
| Protein | Process(es) | Effects | References |
| ATG5 | Macroautophagy; CASM | Microglial-specific Atg5 deletion or depletion in wild-type mice resulted in an age-dependent PD-like phenotype, with the loss of dopamine neurons, increased α-synuclein, increased proinflammatory cytokines, and defects in motor coordination. | Cheng et al.[102] |
| Microglial-specific Atg5 deletion impaired the transition to DAM in the 5xFAD mouse model of AD. | Walter et al.[101] | ||
| Microglial-specific Atg5 deletion exacerbated neurodegeneration in the MPTP-treated mouse model of PD, with impaired motor coordination, reduced dopaminergic neurons, and increased proinflammatory cytokines. | Qin et al.[103] | ||
| Microglial-specific inducible Atg5 deletion resulted in decreased demyelination at 10 days post demyelinating insult, but exacerbated demyelination at 28 days post insult, in mice. Proinflammatory cytokine production was decreased. | Zhou et al.[104] | ||
| Microglial-specific Atg5 deletion exacerbated neurodegeneration in the 5xFAD mouse model of AD, possibly linked to impaired recycling of phagocytic receptors such as TLR4, TREM2 and CD36. | Heckmann et al.[31] | ||
| Glial knockdown of Atg5 impaired the clearance of axonal debris in a Drosophila model of traumatic brain injury, possibly due to impaired LAP. | Szabó et al.[40] | ||
| ATG7 | Macroautophagy; CASM | Microglial-specific Atg7 deletion exacerbated neurodegeneration in the 5xFAD mouse model of AD, with reduced microglia around amyloid plaques, increased amyloid and tau pathology, increased microglial senescence and proinflammatory cytokine secretion. | Choi et al.[97] |
| Microglial-specific Atg7 deletion increased inflammation and microglial lipid accumulation in wild-type and PS19 tauopathy mice. | Xu et al.[105] | ||
| Microglial-specific Atg7 deletion exacerbated neurodegeneration in mice overexpressing α-synuclein. | Choi et al.[99] | ||
| Microglial-specific Atg7 deletion resulted in increased neuronal damage and proinflammatory cytokine secretion in mice injected with Aβ. | Cho et al.[106] | ||
| Microglial-specific Atg7 knockout decreased synaptic pruning during brain development in mice, altering dendritic spine density and social behaviour. | Kim et al.[107] | ||
| Microglial-specific Atg7 deletion impaired recovery in a mouse model of autoimmune encephalitis. | Berglund et al.[108] | ||
| ATG14 | Macroautophagy | Microglial-specific Atg14 deletion resulted in a senescence-associated secretory profile in mice. | Choi et al.[97] |
| Microglial-specific Atg14 deletion exacerbated neurodegeneration in mice overexpressing α-synuclein. | Choi et al.[99] | ||
| Glial knockdown of Atg14 did not affect the clearance of axonal debris in a Drosophila model of traumatic brain injury (whereas knockdown of Atg5, Atg8, or Atg16 impaired it). | Szabó et al.[40] | ||
| ATG16L1 | Macroautophagy; CASM NB: the WD40 domain is required for CASM but dispensable for macroautophagy | Ubiquitous deletion of the ATG16L1 WD40 domain resulted in neurodegeneration, including the accumulation of Aβ and phosphorylated tau pathology, in aged wild-type mice. This may be linked to reduced recycling of phagocytic receptors such as TREM2, CD36 and TLR4 in microglia. | Heckmann et al.[32] |
| Glial knockdown of Atg16 impaired the clearance of axonal debris in a Drosophila model of traumatic brain injury, possibly due to impaired LAP. | Szabó et al.[40] | ||
| Beclin 1 | Macroautophagy; CASM; endocytosis | Microglial-specific Beclin 1 heterozygous deletion resulted in increased proinflammatory cytokine secretion in APP/PS1 AD model mice. | Houtman et al.[109] |
| Beclin 1 depletion in BV-2 microglia impaired phagocytosis and the recycling of phagocytic receptors. | Lucin et al.[110] | ||
| FIP200 | Macroautophagy | Microglial-specific Fip200 deletion did not significantly affect neurodegeneration in the 5xFAD mouse model of AD (whereas Atg5 or Rubicon deletion exacerbated it). | Heckmann et al.[31] |
| Knockdown of Atg17/Fip200 in glia did not affect the clearance of axonal debris in a Drosophila model of traumatic brain injury (whereas knockdown of Atg5, Atg8, or Atg16 impaired it). | Szabó et al.[40] | ||
| LC3 | Macroautophagy; CASM | Map1lc3B depletion in mouse primary microglia resulted in increased proinflammatory cytokines in response to Aβ. | Cho et al.[106] |
| Knockdown of Atg8A (Drosophila homologue of LC3) in glia impaired the clearance of axonal debris in a Drosophila model of traumatic brain injury, possibly due to impaired LAP. | Szabó et al.[40] | ||
| Rubicon | CASM; endocytosis; negative regulator of autophagy | Microglial-specific Rubicon deletion exacerbated neurodegeneration in the 5xFAD mouse model of AD, possibly linked to impaired recycling of phagocytic receptors such as TLR4, TREM2 and CD36. | Heckmann et al.[31] |
| Glial knockdown of Rubicon impaired the clearance of axonal debris in a Drosophila model of traumatic brain injury, possibly due to impaired LAP. | Szabó et al.[40] | ||
| Syntaxin 17 | Macroautophagy | Glial knockdown of Syntaxin 17 did not affect the clearance of axonal debris in a Drosophila model of traumatic brain injury (whereas knockdown of Atg5, Atg8, or Atg16 impaired it). | Szabó et al.[40] |
| ULK1 | Macroautophagy | Microglial-specific Ulk1 knockout resulted in the loss of a protective, | Berglund et al.[94] |
| Microglial-specific Ulk1 knockout did not affect the recovery from autoimmune encephalitis (whereas Atg7 knockout impaired it). | Berglund et al.[108] | ||
| Glial knockdown of Atg1 (the Drosophila homologue of ULK1) or Atg13 (a component of the Drosophila Atg1 complex) did not affect the clearance of axonal debris in a Drosophila model of traumatic brain injury (whereas knockdown of Atg5, Atg8, or Atg16 impaired it). | Szabó et al.[40] | ||
| VPS34 | Macroautophagy; CASM; endocytosis | Glial knockdown of Vps34 impaired the clearance of axonal debris in a Drosophila model of traumatic brain injury, possibly due to impaired LAP. | Szabó et al.[40] |
CASM: conjugation of ATG8s to single membranes; ATG: autophagy-related gene; PD: Parkinson’s disease; DAM: disease-associated microglia; AD: Alzheimer’s disease;
However, as LC3 conjugation is required not just for autophagy but also for CASM processes such as LAP and LANDO, it is not evident whether the observed effects are due to the loss of autophagy and/or CASM functions. Some publications have made microglial deletions of ATG14, which abrogates autophagy but not CASM, and observed similar exacerbation of neuroinflammation and neurodegeneration[97,99]. This suggests that the disruption of microglial autophagy is sufficient to recapitulate the effects of microglial LC3 conjugation disruption. However, this is challenged by a study wherein the microglial-specific deletion of FIP200, which impairs autophagy but not CASM, did not affect neurodegeneration in a mouse model of AD. In that same study, the microglial-specific deletion of ATG5 or Rubicon did exacerbate neurodegeneration[31], which could suggest that the observed effects of ATG5 disruption were due to the loss of CASM and not autophagy. No other studies have investigated the specific effect of microglial CASM disruption on autophagy, however, the ubiquitous deletion of the WD40 domain of ATG16L1 (which abrogates CASM without affecting
7. Potential Mechanisms Linking Autophagy and CASM to Microglial Function
As described in the previous section, the loss of autophagy and/or CASM function in microglia is associated with increased secretion of proinflammatory cytokines, microglial senescence, and reduced microglial phagocytic clearance, which exacerbates neurodegeneration. The mechanisms linking autophagy and/or CASM dysfunction to these effects on microglial processes are currently poorly understood. However, studies in other cells suggest possible mechanisms through which autophagy and/or CASM can regulate cytokine production, senescence, and phagocytic clearance. It is also worth noting that in vivo, non-cell-autonomous effects may contribute to the proinflammatory effect of autophagy inhibition, particularly when extensive cell death occurs and results in microglial activation. However, as the increased proinflammatory cytokines, senescence, and impaired phagocytosis can be observed when autophagy and/or CASM are specifically disrupted in microglia, we have chosen to focus on potential
7.1 Autophagy, CASM, and proinflammatory cytokine secretion
Autophagy degrades many immune signalling molecules to restrain inflammation. For example, the NLRP3 and AIM2 inflammasomes are degraded by autophagy through a ubiquitination-dependent mechanism[111-113], decreasing inflammasome activation and the subsequent production of proinflammatory cytokines. Many cytokine receptors, pattern recognition receptors, and their adaptors and downstream signalling proteins are autophagy substrates, including pro-IL-1β[114], the TLR3/4 adaptor TRIF[115,116], and the NF-κB regulator BCL10[117]. Therefore, autophagy disruption in microglia would be expected to result in an increased abundance of these immune adaptors, which likely contributes to the increased cytokine signalling, increased cytokine production, and an overall neuroinflammatory secretory profile observed upon microglial autophagy disruption. In neurons, the CCL3/4/5 receptor CCR5 is degraded by autophagy, dependent on its internalisation and trafficking to the recycling endosome[64]. As CCR5 inhibits autophagy, its activation by microglial-secreted CCL3/4/5 results in reduced degradation of CCR5, resulting in a positive feedback loop that enhances CCR5 activation[64].
Autophagy, particularly mitophagy, is a major regulator of inflammatory signalling pathways through the removal of damaged mitochondria. For example, one of the major proinflammatory signalling cascades involves the release of mitochondrial DNA (mtDNA) into the cytoplasm, which is sensed by the cGAS-STING pathway to promote inflammation[118]. Damaged mitochondria also generate reactive oxygen species (ROS), which activates the NLRP3 inflammasome[119,120]. Mitophagy, through its removal of damaged mitochondria, prevents this mtDNA release and ROS generation, reducing cGAS-STING and inflammasome activation[121-123]. Consequently, impaired mitophagy results in an accumulation of damaged mitochondria, increased mitochondria-derived ROS, and increased cytosolic mtDNA, driving inflammation[119,120]. Additionally, inflammasome activation induces mitochondrial damage and inhibits mitophagy[124], potentially leading to a positive feedback loop that drives further neuroinflammation.
Additionally, to the removal of damaged mitochondria through mitophagy, autophagy also regulates inflammation through the degradation of cytosolic mtDNA, cGAS, and STING. For example, autophagy degrades cytosolic mtDNA through a TFAM-dependent selective autophagy pathway[125], cGAS is degraded by autophagy through a ubiquitination-dependent mechanism[126], and STING is degraded by autophagy through a p62-dependent mechanism[127,128]. STING activation also upregulates autophagy through enhancing the activity of the PI3K-C1[128], which may potentially act as a homeostatic mechanism to shut down proinflammatory signalling.
The association between CASM and inflammatory signalling is less established. However, LAP deficiency results in heightened
7.2 Autophagy, CASM, and senescence
Whilst the mechanisms leading to microglial senescence are poorly understood, the production of the SASP is dependent on the transcription factor GATA4, which is degraded by p62-dependent selective autophagy[130]. Impaired autophagy during senescence results in GATA4 stabilisation, activating NF-κB and initiating the SASP[130]. The cGAS-STING pathway, which is inhibited by autophagy, also promotes senescence[131,132].
A characteristic associated with microglial dystrophy and senescence is the accumulation of lipid droplets in microglia, which is observed in ageing, neurodegeneration, and demyelination[78,104,133-135]. Lipid-droplet containing microglia have defective phagocytosis, elevated ROS, and secrete pro-inflammatory cytokines, exacerbating neurodegeneration in a non-cell-autonomous manner[78,133,135-137]. Importantly, autophagy impairment is associated with the accumulation of lipid droplets[105,104] and lipofuscin[97] in microglia, suggesting that autophagy impairment may be related to lipid droplet accumulation. Similarly, mutation or depletion of the neurodegeneration-associated endo-lysosomal proteins progranulin[78], PICALM[136], and NPC1[137], are also associated with microglial lipid droplet accumulation. The association between impaired autophagy and lipid droplet accumulation could be bidirectional, as lipid droplet accumulation likely alters membrane dynamics and the abundance of lipids that regulate
There are also close interplays between autophagy and the survival, differentiation and activation of immune cells such as B cells and T cells, possibly through the regulation of their metabolism[140-145]. It is unknown whether autophagy has similar effects on microglial metabolism and survival. However, the requirement for the upregulation of autophagy and lysosomal genes in the transition to the protective DAM and ADAM states[61,94,97] suggests that autophagy regulates microglial metabolism and activation.
7.3 Autophagy, CASM, and phagocytosis
Disruption of autophagy is associated with an increased abundance of phagocytic receptors such as MARCO and MSR1 on macrophages[146]. Interestingly, this increased abundance appears to be due to increased transcription of scavenger receptors due to the accumulation of p62 in autophagy-null cells, rather than a direct effect of autophagy on the degradation of the cytokine
CASM is a key modulator of phagocytosis and the loss of CASM function may contribute to the impaired phagocytosis observed in autophagy-null microglia. LAP regulates the maturation of phagosomes and their lysosomal fusion[30,33]. Consequently, LAP affects both the clearance of phagocytosed particles, and the MHC class II presentation of antigens[147,148], regulating downstream immune signalling. LANDO regulates the recycling of phagocytic receptors such as TREM2, CD36 and TLR4, and LANDO disruption impairs the phagocytic clearance of Aβ due to reduced abundance of these receptors at the plasma membrane[31].
8. Therapeutic Implications of Microglial Autophagy in Ageing and Neurodegenerative Diseases
Autophagy upregulation is a promising strategy to combat ageing and neurodegenerative diseases[149]. In addition to its beneficial effects on neuronal proteostasis, brain autophagy upregulation will likely be beneficial by maintaining microglial function and restraining neuroinflammation. Many in vitro studies of microglia have shown beneficial effects of mTOR inhibitors such as rapamycin (an autophagy-activating immune suppressant) in reducing the production of proinflammatory cytokines. However, mTOR inhibitors also reduce protein synthesis, resulting in decreased cytokine production, so these findings are expected and may be independent of any effects on microglial autophagy. To properly test the potential beneficial effects of microglial autophagy upregulation, mTOR-independent therapeutics will need to be used. Furthermore, there are currently no therapeutic approaches to enhance LAP or LANDO.
Given that microglial autophagy impairment exacerbates neurodegeneration likely through increasing microglial proinflammatory cytokine secretion and microglial senescence, the modulation of these pathways may be beneficial in ageing and neurodegenerative diseases. For example, the removal of senescent microglia using senolytics has been beneficial in animal models of neurodegeneration, including in a microglial autophagy-null model of AD[97,150-152]. The inhibition of the NLRP3 inflammasome has also been successful in animal models of neurodegeneration with impaired microglial autophagy and/or CASM[32,102]. Careful selection of the immune target is required, in order to maintain the beneficial functions of microglia. In this respect, senolytics may be more advantageous, as their treatment could be intermittent to remove senescent microglia and reduce the SASP, whereas the inhibition of neuroinflammation would likely need to be continuous[153]. Another potential therapeutic approach may be to prevent the effect of specific microglial-secreted cytokines on neuronal function, such as the blockade of CCL3/4/5 signalling to CCR5 using maraviroc, which was protective in tauopathy and HD mouse models[64]. However, careful analysis will be required to monitor for any unintended consequences of the removal of senescent cells or modulation of immune signalling.
It is also worth considering that the beneficial effects of anti-Aβ immunotherapies in AD are dependent on microglial activation, including the increased abundance of DAM and microglial phagocytosis of Aβ plaques[154,155]. As microglial autophagy impairment prevents the transition to DAM and reduces phagocytic clearance, these therapies may be less effective in conditions where microglial autophagy is impaired.
9. Future Directions
The relationship between autophagy, CASM, and microglial function is a new and growing field, with promising advances and many unanswered questions. One of the largest unresolved questions is which of the effects of the disruption of LC3 conjugation in microglia (such as when using ATG5 or ATG7 knockout models) are attributable to autophagy loss and which are due to CASM loss. Selective abrogation of autophagy can be achieved by ULK1/2 inhibition or knockout, and selective abrogation of CASM can be achieved using mutation of the ATG16L1 WD40 domain, which is required for CASM but dispensable for autophagy. Rubicon knockout has also been used as a mechanism to impair CASM, however, as Rubicon is also a negative regulator of autophagy, Rubicon depletion results in enhanced autophagy. Furthermore, the role of Rubicon and PI3P production in microglial CASM needs to be clarified, as key papers in this research area have been retracted, including the initial paper proposing a role for Rubicon in LAP. Whilst some subsequent papers have reported a requirement for Rubicon and VPS34 for CASM[31,40], these proteins do not appear to be universally required for CASM[49,156]. It may also be challenging to differentiate the role of Rubicon and PI3P in endosome maturation[157] and in CASM. The ATG16L1ΔWD40 model may be a more reliable model, although a microglial-specific ATG16L1ΔWD40 model needs to be created to ensure that any effects are due to cell-autonomous effects in microglia.
Another challenge for future research is to differentiate changes in autophagy and CASM function resulting directly from neurodegeneration-associated proteins from those arising due to changes in microglial activation and microglial state, which indirectly affect autophagy. Such effects likely contribute to the different effects reported for proteins such as Aβ and α-synuclein on microglial autophagy. It is also possible that different oligomeric states and/or posttranslational modifications of the neurodegeneration-associated proteins may have different effects on microglial autophagy.
Importantly, any effects will need to be studied specifically in microglia, as microglia and macrophages can have disparate responses to the same stimuli. For example, treatment with LPS, which activates both microglia and macrophages, results in autophagy upregulation in macrophages, but autophagy inhibition in microglia[158]. A frequently overlooked aspect of in vivo studies is that ageing and neurodegeneration result in increased infiltration of the brain by macrophages and other monocytes. It will be important to confirm which cell type(s) are involved in any observed effects.
Many studies have employed Cx3cr1-Cre systems to achieve selective knockout of core autophagy genes in microglia. However, Cx3cr1 is also expressed in other myeloid cells, particularly mononuclear phagocytes[159]. Cx3cr1-Cre lines may thus also result in the depletion of target genes in central nervous system border-associated macrophages, including perivascular macrophages, meningeal macrophages, and choroid plexus macrophages[160], making the attribution of any effects specifically to microglia challenging. This may be further confounded by potential neurodevelopmental effects when using constitutive Cre driver systems; inducible Cre-driver systems may be more appropriate for studying effects on ageing and neurodegeneration.
Finally, microglia display significant sexual dimorphism, which may contribute to the differences in the relative risk of neurodegenerative diseases such as AD (higher in females) and PD (higher in males)[161,162]. It is currently unknown whether the relationships between microglial LC3 conjugation-dependent pathways and neurodegeneration are affected by sex. Almost all the existing microglial autophagy and/or CASM studies have not subgrouped their analyses by sex. Interestingly, microglia from female AD mice show greater increases in DAM-associated genes, including lysosomal genes such as CTSD, CD68, and LAMP1[163,164], and female mouse microglia also show a greater increase in mTORC1 activation during ageing than microglia from male mice[164]. However, it is unknown whether this relates to sex differences in microglial autophagy. Future studies should aim to investigate potential sex-specific changes associated with microglial autophagy in ageing and neurodegenerative diseases, with sufficient power for sex-specific analysis and careful experimentation to attempt to deconvolute non-cell-autonomous effects resulting from potentially different extents of neuropathology and microglial state.
10. Conclusion
Emerging evidence indicates that microglial autophagy may be altered by ageing and neurodegenerative diseases. The loss of autophagy and/or CASM function in microglia is a potential exacerbator of neurodegeneration, resulting in increased secretion of proinflammatory cytokines, microglial senescence, and worsened neuronal protein accumulation and degeneration. The transition to the protective DAM and ADAM states is dependent on autophagy, and autophagy also restricts inflammation and senescence by degrading immune effectors such as the inflammasome and cGAS-STING. Autophagy upregulation, the removal of senescent microglia, and/or the modulation of microglial proinflammatory signalling may be promising therapeutic strategies for neurodegeneration. A critical area for future work is to differentiate which of the effects of microglial LC3 conjugation loss are attributable to autophagy or CASM disruption. Overall, autophagy and other LC3-dependent pathways affect microglial functions to modulate brain ageing and neurodegenerative diseases.
Acknowledgements
We thank our colleagues in this research field and regret that, due to space constraints, we were unable to include all relevant literature on this topic.
Authors contribution
Palmer JE, Rubinsztein DC: Writing-original draft, writing review & editing.
Conflicts of interest
David C. Rubinsztein is an Editorial Board Member of Geromedicine. The author has consulted for or is a consultant for Drishti Discoveries, PAQ Therapeutics, MindRank AI, Retro Biosciences, Alexion Pharma International Operations Limited, Carlyle Investment Management LLC, Aladdin Healthcare Technologies Ltd, Nido Biosciences, ProtosBio, and is a co-founder of Acuity Technologies Ltd. The other author declares no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the UK Dementia Research Institute through UK DRI Ltd, principally funded by the Medical Research Council
Copyright
© The Author(s) 2026.
References
-
1. Wilson DM, Cookson MR, Van Den Bosch L, Zetterberg H, Holtzman DM, Dewachter I. Hallmarks of neurodegenerative diseases. Cell. 2023;186(4):693-714.[DOI]
-
2. Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15(10):565-581.[DOI]
-
4. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21(10):1359-1369.[DOI]
-
5. Prinz M, Jung S, Priller J. Microglia biology: One century of evolving concepts. Cell. 2019;179(2):292-311.[DOI]
-
6. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36(13):1811-1836.[DOI]
-
7. Kabeya Y. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720-5728.[DOI]
-
8. Figueras-Novoa C, Timimi L, Marcassa E, Ulferts R, Beale R. Conjugation of ATG8s to single membranes at a glance. J Cell Sci. 2024;137(15):jcs261031.[DOI]
-
9. Deretic V, Duque T, Trosdal E, Paddar M, Javed R, Akepati P. Membrane atg8ylation in canonical and noncanonical autophagy. J Mol Biol. 2024;436(15):168532.[DOI]
-
10. Peña-Martinez C, Rickman AD, Heckmann BL. Beyond autophagy: LC3-associated phagocytosis and endocytosis. Sci Adv. 2022;8(43):eabn1702.[DOI]
-
11. Puri C, Vicinanza M, Ashkenazi A, Gratian MJ, Zhang Q, Bento CF, et al. The RAB11A-positive compartment is a primary platform for autophagosome assembly mediated by WIPI2 recognition of PI3P-RAB11A. Dev Cell. 2018;45(1):114-131.[DOI]
-
12. Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11(12):1433-1437.[DOI]
-
13. Matoba K, Kotani T, Tsutsumi A, Tsuji T, Mori T, Noshiro D, et al. Author Correction: Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat Struct Mol Biol. 2020;27(12):1209.[DOI]
-
14. Puri C, Manni MM, Vicinanza M, Hilcenko C, Zhu Y, Runwal G, et al. A DNM2 centronuclear myopathy mutation reveals a link between recycling endosome scission and autophagy. Dev Cell. 2020;53(2):154-168.[DOI]
-
15. Zhao YG, Codogno P, Zhang H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat Rev Mol Cell Biol. 2021;22(11):733-750.[DOI]
-
16. Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159(6):1263-1276.[DOI]
-
17. Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, et al. Autophagy in major human diseases. EMBO J. 2021;40(19):e108863.[DOI]
-
18. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132-141.[DOI]
-
19. Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741-750.[DOI]
-
20. Dooley HC, Razi M, Polson HEJ, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell. 2014;55(2):238-252.[DOI]
-
21. Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36(12):2503-2518.[DOI]
-
22. Settembre C, Di Malta C, Polito VA, Arencibia MG, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429-1433.[DOI]
-
23. Takla M, Keshri S, Rubinsztein DC. The post-translational regulation of transcription factor EB (TFEB) in health and disease. EMBO Rep. 2023;24(11):e57574.[DOI]
-
24. Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2023;24(3):167-185.[DOI]
-
25. Tekirdag K, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: Jointed by a chaperone. J Biol Chem. 2018;293(15):5414-5424.[DOI]
-
26. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19(6):365-381.[DOI]
-
27. Kaushik S, Massey AC, Mizushima N, Cuervo AM. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell. 2008;19(5):2179-2192.[DOI]
-
28. Wang L, Klionsky DJ, Shen HM. The emerging mechanisms and functions of microautophagy. Nat Rev Mol Cell Biol. 2023;24(3):186-203.[DOI]
-
29. Sakai Y, Oku M. ATGandESCRTcontrol multiple modes of microautophagy. FEBS Lett. 2024;598(1):48-58.[DOI]
-
30. Sanjuan MA, Dillon CP, Tait SWG, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450(7173):1253-1257.[DOI]
-
31. Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M, et al. LC3-associated endocytosis facilitates β-amyloid clearance and mitigates neurodegeneration in murine Alzheimer’s disease. Cell. 2020;183(6):1733-1734.[DOI]
-
32. Heckmann BL, Teubner BJW, Boada-Romero E, Tummers B, Guy C, Fitzgerald P, et al. Noncanonical function of an autophagy protein prevents spontaneous Alzheimer’s disease. Sci Adv. 2020;6(33):eabb9036.[DOI]
-
33. Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol. 2011;13(11):1335-1343.[DOI]
-
34. Durgan J, Lystad AH, Sloan K, Carlsson SR, Wilson MI, Marcassa E, et al. Non-canonical autophagy drives alternative ATG8 conjugation to phosphatidylserine. Mol Cell. 2021;81(9):2031-2040.[DOI]
-
35. Timimi L, Wrobel AG, Chiduza GN, Maslen SL, Torres-Méndez A, Montaner B, et al. The V-ATPase/ATG16L1 axis is controlled by the V1H subunit. Mol Cell. 2024;84(15):2966-2983.[DOI]
-
36. Hooper KM, Jacquin E, Li T, Goodwin JM, Brumell JH, Durgan J, et al. V-ATPase is a universal regulator of LC3-associated phagocytosis and non-canonical autophagy. J Cell Biol. 2022;221(6):e202105112.[DOI]
-
37. Ulferts R, Marcassa E, Timimi L, Lee LC, Daley A, Montaner B, et al. Subtractive CRISPR screen identifies the ATG16L1/vacuolar ATPase axis as required for non-canonical LC3 lipidation. Cell Rep. 2021;37(4):109899.[DOI]
-
38. Fletcher K, Ulferts R, Jacquin E, Veith T, Gammoh N, Arasteh JM, et al. TheWD40 domain ofATG16L1 is required for its non-canonical role in lipidation ofLC3 at single membranes. EMBO J. 2018;37(4):e97840.[DOI]
-
39. Lystad AH, Carlsson SR, de la Ballina LR, Kauffman KJ, Nag S, Yoshimori T, et al. Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy-related processes. Nat Cell Biol. 2019;21(3):372-383.[DOI]
-
40. Szabó Á, Vincze V, Chhatre AS, Jipa A, Bognár S, Varga KE, et al. LC3-associated phagocytosis promotes glial degradation of axon debris after injury in Drosophila models. Nat Commun. 2023;14:3077.[DOI]
-
41. Florey O, Gammoh N, Kim SE, Jiang X, Overholtzer M. V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy. 2015;11(1):88-99.[DOI]
-
42. Palmer JE, Puri C, O’Rourke RS, Son SM, Sang C, Huang YA, et al. Coordination of autophagosome closure and release by the Alzheimer’s disease-associated protein BIN1. Cell
-
43. Ledo JH, Liebmann T, Zhang R, Chang JC, Azevedo EP, Wong E, et al. Presenilin 1 phosphorylation regulates amyloid-β degradation by microglia. Mol Psychiatry. 2021;26(10):5620-5635.[DOI]
-
44. Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell. 2017;170(4):649-663.[DOI]
-
45. Banerjee P, Mehta AR, Nirujogi RS, Cooper J, James OG, Nanda J, et al. Cell-autonomous immune dysfunction driven by disrupted autophagy in C9orf72-ALS iPSC-derived microglia contributes to neurodegeneration. Sci Adv. 2023;9(16):eabq0651.[DOI]
-
46. Wu Y, Shao W, Todd TW, Tong J, Yue M, Koga S, et al. Microglial lysosome dysfunction contributes to white matter pathology and TDP-43 proteinopathy in GRN-associated FTD. Cell Rep. 2021;36(8):109581.[DOI]
-
47. Gabandé-Rodríguez E, Pérez-Cañamás A, Soto-Huelin B, Mitroi DN, Sánchez-Redondo S, Martínez-Sáez E, et al. Lipid-induced lysosomal damage after demyelination corrupts microglia protective function in lysosomal storage disorders. EMBO J. 2019;38(2):e99553.[DOI]
-
48. Yadavalli N, Ferguson SM. LRRK2 suppresses lysosome degradative activity in macrophages and microglia through MiT-TFE transcription factor inhibition. Proc Natl Acad Sci U S A. 2023;120(31):e2303789120.[DOI]
-
49. Durgan J, Florey O. Many roads lead to CASM: Diverse stimuli of noncanonical autophagy share a unifying molecular mechanism. Sci Adv. 2022;8(43):eabo1274.[DOI]
-
50. Rai S, Arasteh M, Jefferson M, Pearson T, Wang Y, Zhang W, et al. The ATG5-binding and coiled coil domains of ATG16L1 maintain autophagy and tissue homeostasis in mice independently of the WD domain required for LC3-associated phagocytosis. Autophagy. 2019;15(4):599-612.[DOI]
-
51. Bryois J, Calini D, MacNair W, Foo L, Urich E, Ortmann W, et al. Cell-type-specific cis-eQTLs in eight human brain cell types identify novel risk genes for psychiatric and neurological disorders. Nat Neurosci. 2022;25(8):1104-1112.[DOI]
-
52. Askarova A, Yaa RM, Marzi SJ, Nott A. Genetic risk for neurodegenerative conditions is linked to disease-specific microglial pathways. PLoS Genet. 2025;21(4):e1011407.[DOI]
-
53. Graham AC, Bellou E, Harwood JC, Yaman U, Celikag M, Magusali N, et al. Human longevity and Alzheimer’s disease variants act via microglia and oligodendrocyte gene networks. Brain. 2025;148(3):969-984.[DOI]
-
54. Rexach JE, Cheng Y, Chen L, Polioudakis D, Lin LC, Mitri V, et al. Cross-disorder and disease-specific pathways in dementia revealed by single-cell genomics. Cell. 2024;187(20):5753-5774.[DOI]
-
55. Li X, Li Y, Jin Y, Zhang Y, Wu J, Xu Z, et al. Transcriptional and epigenetic decoding of the microglial aging process. Nat Aging. 2023;3(10):1288-1311.[DOI]
-
56. Martins-Ferreira R, Calafell-Segura J, Leal B, Rodríguez-Ubreva J, Martínez-Saez E, Mereu E, et al. The Human Microglia Atlas (HuMicA) unravels changes in disease-associated microglia subsets across neurodegenerative conditions. Nat Commun. 2025;16:739.[DOI]
-
57. Jin C, Shao Y, Zhang X, Xiang J, Zhang R, Sun Z, et al. A unique type of highly-activated microglia evoking brain inflammation via mif/Cd74 signaling axis in aged mice. Aging Dis. 2021;12(8):2125.[DOI]
-
58. Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007;55(4):412-424.[DOI]
-
59. Millet A, Ledo JH, Tavazoie SF. An exhausted-like microglial population accumulates in aged and APOE4 genotype Alzheimer’s brains. Immunity. 2024;57(1):153-170.[DOI]
-
60. Pascoal TA, Benedet AL, Ashton NJ, Kang MS, Therriault J, Chamoun M, et al. Microglial activation and tau propagate jointly across Braak stages. Nat Med. 2021;27(9):1592-1599.[DOI]
-
61. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276-1290.[DOI]
-
62. Ulrich JD, Ulland TK, Colonna M, Holtzman DM. Elucidating the role of TREM2 in Alzheimer’s disease. Neuron. 2017;94(2):237-248.[DOI]
-
63. Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6(243):e3009093.[DOI]
-
64. Festa BP, Siddiqi FH, Jimenez-Sanchez M, Won H, Rob M, Djajadikerta A, et al. Microglial-to-neuronal CCR5 signaling regulates autophagy in neurodegeneration. Neuron. 2023;111(13):2021-2037.[DOI]
-
65. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712-716.[DOI]
-
66. Wang C, Yue H, Hu Z, Shen Y, Ma J, Li J, et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science. 2020;367(6478):688-694.[DOI]
-
67. Pereira-Iglesias M, Maldonado-Teixido J, Melero A, Piriz J, Galea E, Ransohoff RM, et al. Microglia as hunters or gatherers of brain synapses. Nat Neurosci. 2025;28(1):15-23.[DOI]
-
68. Marzan DE, Brügger-Verdon V, West BL, Liddelow S, Samanta J, Salzer JL. Activated microglia drive demyelination via CSF1R signaling. Glia. 2021;69(6):1583-1604.[DOI]
-
69. Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18(11):1584-1593.[DOI]
-
70. Brelstaff JH, Mason M, Katsinelos T, McEwan WA, Ghetti B, Tolkovsky AM, et al. Microglia become hypofunctional and release metalloproteases and tau seeds when phagocytosing live neurons with P301S tau aggregates. Sci Adv. 2021;7(43):eabg4980.[DOI]
-
71. D’Errico P, Ziegler-Waldkirch S, Aires V, Hoffmann P, Mezö C, Erny D, et al. Microglia contribute to the propagation of Aβ into unaffected brain tissue. Nat Neurosci. 2022;25(1):20-25.[DOI]
-
72. Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature. 2017;552(7685):355-361.[DOI]
-
73. Zhang X, Wang R, Chen H, Jin C, Jin Z, Lu J, et al. Aged microglia promote peripheral T cell infiltration by reprogramming the microenvironment of neurogenic niches. Immun Ageing. 2022;19:34.[DOI]
-
74. Chen X, Firulyova M, Manis M, Herz J, Smirnov I, Aladyeva E, et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023;615(7953):668-677.[DOI]
-
75. Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118(4):475-485.[DOI]
-
76. Streit WJ, Sammons NW, Kuhns AJ, Sparks DL. Dystrophic microglia in the aging human brain. Glia. 2004;45(2):208-212.[DOI]
-
77. Carr L, Mustafa S, Collins-Praino LE. The hallmarks of ageing in microglia. Cell Mol Neurobiol. 2025;45:45.[DOI]
-
78. Marschallinger J, Iram T, Zardeneta M, Lee SE, Lehallier B, Haney MS, et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci. 2020;23(2):194-208.[DOI]
-
79. Gabandé-Rodríguez E, Keane L, Capasso M. Microglial phagocytosis in aging and Alzheimer’s disease. J Neurosci Res. 2020;98(2):284-298.[DOI]
-
80. Floden AM, Combs CK. Microglia demonstrate age-dependent interaction with amyloid-β fibrils. J Alzheimers Dis. 2011;25(2):279-293.[DOI]
-
81. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28(33):8354-8360.[DOI]
-
82. Bliederhaeuser C, Grozdanov V, Speidel A, Zondler L, Ruf WP, Bayer H, et al. Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 2016;131(3):379-391.[DOI]
-
83. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28(6):436-453.[DOI]
-
84. Lau V, Ramer L, Tremblay MÈ. An aging, pathology burden, and glial senescence build-up hypothesis for late onset Alzheimer’s disease. Nat Commun. 2023;14:1670.[DOI]
-
85. Simons M, Levin J, Dichgans M. Tipping points in neurodegeneration. Neuron. 2023;111(19):2954-2968.[DOI]
-
86. Aman Y, Schmauck-Medina T, Hansen M, Morimoto RI, Simon AK, Bjedov I, et al. Autophagy in healthy aging and disease. Nat Aging. 2021;1(8):634-650.[DOI]
-
87. Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: Insights from model organisms. Nat Rev Mol Cell Biol. 2018;19(9):579-593.[DOI]
-
88. Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013;4:2300.[DOI]
-
89. Fernández ÁF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;558(7708):136-140.[DOI]
-
90. Ott C, König J, Höhn A, Jung T, Grune T. Macroautophagy is impaired in old murine brain tissue as well as in senescent human fibroblasts. Redox Biol. 2016;10:266-273.[DOI]
-
91. Nixon RA, Rubinsztein DC. Mechanisms of autophagy-lysosome dysfunction in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2024;25(11):926-946.[DOI]
-
92. Park SJ, Frake RA, Karabiyik C, Son SM, Siddiqi FH, Bento CF, et al. Vinexin contributes to autophagic decline in brain ageing across species. Cell Death Differ. 2022;29(5):1055-1070.[DOI]
-
93. Hong B, Ohtake Y, Itokazu T, Yamashita T. Glial senescence enhances α-synuclein pathology owing to its insufficient clearance caused by autophagy dysfunction. Cell Death Discov. 2024;10:50.[DOI]
-
94. Berglund R, Cheng Y, Piket E, Adzemovic MZ, Zeitelhofer M, Olsson T, et al. The aging mouse CNS is protected by an autophagy-dependent microglia population promoted by IL-34. Nat Commun. 2024;15:383.[DOI]
-
95. Palmer JE, Wilson N, Son SM, Obrocki P, Wrobel L, Rob M, et al. Autophagy, aging, and age-related neurodegeneration. Neuron. 2025;113(1):29-48.[DOI]
-
96. Pomilio C, Gorojod RM, Riudavets M, Vinuesa A, Presa J, Gregosa A, et al. Microglial autophagy is impaired by prolonged exposure to β-amyloid peptides: Evidence from experimental models and Alzheimer’s disease patients. GeroScience. 2020;42(2):613-632.[DOI]
-
97. Choi I, Wang M, Yoo S, Xu P, Seegobin SP, Li X, et al. Autophagy enables microglia to engage amyloid plaques and prevents microglial senescence. Nat Cell Biol. 2023;25(7):963-974.[DOI]
-
98. Tu HY, Yuan BS, Hou XO, Zhang XJ, Pei CS, Ma YT, et al. α-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell. 2021;20(12):e13522.[DOI]
-
99. Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K, et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 2020;11:1386.[DOI]
-
100. Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, et al. Brain cell type–specific enhancer–promoter interactome maps and disease-risk association. Science. 2019;366(6469):1134-1139.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Palmer JE, Rubinsztein DC. Microglial autophagy and other LC3-dependent pathways in neurodegeneration. Geromedicine. 2026;2(3):202525. https://doi.org/10.70401/Geromedicine.2026.0022
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Autophagy
- 3. CASM Pathways
- 4. Microglia in Ageing and Neurodegeneration
- 5. Effects of Ageing and Neurodegeneration on Microglial Autophagy
- 6. Effects of Microglial Autophagy and CASM on Ageing and Neurodegeneration
- 7. Potential Mechanisms Linking Autophagy and CASM to Microglial Function
- 8. Therapeutic Implications of Microglial Autophagy in Ageing and Neurodegenerative Diseases
- 9. Future Directions
- 10. Conclusion
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Palmer JE, Rubinsztein DC. Microglial autophagy and other LC3-dependent pathways in neurodegeneration. Geromedicine. 2026;2(3):202525. https://doi.org/10.70401/Geromedicine.2026.0022
copy
Share Link
copy