Cytoplasmic chromatin fragments: Divergent roles in senescence and cancer
Weifang Xiang
1

,
Sirui Wang
2
,
Ying Pan
3
,
Xianling Cong
4
,
Jun Lu
2
,
Yu Zhang
1,*
*Correspondence to:
Yu Zhang, Key Laboratory of Molecular Epigenetics of Ministry of Education (MOE), Northeast Normal University, Changchun 130024, Jilin, China.
E-mail: zhangy288@nenu.edu.cn
Ageing Cancer Res Treat. 2026;3:202523. 10.70401/acrt.2026.0016
Received: December 07, 2025Accepted: March 02, 2026Published: March 05, 2026
Abstract
Cytoplasmic chromatin fragments (CCFs) are structures formed by nuclear chromatin leaked into the cytoplasm in response to cellular senescence, stress, or tumorigenesis, primarily due to genomic instability and nuclear envelope rupture. These cytoplasmic DNA fragments are recognized by cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS) and activate the cGAS–STING pathway, which promotes activation of IRF3 and NF-κB, and induces expression of type I interferons and pro-inflammatory cytokines, thereby driving the senescence-associated secretory phenotype (SASP). CCFs are not only a hallmark of cellular senescence but also a critical signaling hub that links DNA damage to chronic inflammation via SASP factors like IL-6 and IL-8, reinforcing senescence through autocrine and paracrine loops. In cancer, CCFs play distinct roles at different stages: in early-stage tumors, they induce cell cycle arrest and enhance immune surveillance, thereby suppressing tumor initiation; whereas in advanced tumors, persistent CCFs chronically activate the cGAS–STING–NF-κB signaling axis, promoting epithelial–mesenchymal transition, angiogenesis, metastasis, and immune evasion. Notably, CCFs formation is heterogeneous and regulated by key factors such as p53, 53BP1, and Lamin B1. Therefore, targeting the CCFs–cGAS–STING pathway and its upstream regulators, including mitochondrial function, autophagy, and epigenetic modifications, offers a promising strategy to alleviate aging-related diseases and improve cancer therapy by suppressing SASP and blocking tumor progression. This review summarizes the mechanisms of CCFs biogenesis, their complex roles in aging and cancer, and emerging therapeutic approaches aimed at this axis, offering insights for both basic research and clinical translation.
Keywords
Cytoplasmic chromatin fragments, cGAS-STING axis, senescence-associated secretory phenotype, DNA damage, senescence, cancer
1. Introduction
Cytoplasmic chromatin fragments (CCFs) are chromatin structures that originate in the nucleus and aberrantly accumulate in the cytoplasm in response to cellular stress, senescence, or tumorigenesis. Unlike canonical nuclear chromatin, CCFs lack an intact nuclear envelope and, when enriched, can serve as specific epigenetic and DNA damage markers, such as γH2AX and H3K27me3. The formation of CCFs is not a random event, but is actively regulated by multiple mechanisms, including persistent DNA damage-induced genomic instability, disruption of the nuclear lamina (particularly through downregulation or degradation of Lamin B1), and dysregulation of cell cycle control[1-4], as well as structural failures such as nuclear envelope rupture or chromosome missegregation during mitosis that lead to micronucleus formation[5-7].
Once in the cytoplasm, CCFs are recognized by innate immune DNA sensor cGAS. Upon binding to double-stranded DNA (dsDNA), cGAS catalyzes the synthesis of 2′3′- cyclic guanosine monophosphate–adenosine monophosphate (cGAMP), which activates the adaptor protein STING (stimulator of interferon genes), and subsequently triggers downstream signaling cascades involving TBK1, IRF3, and NF-κB, ultimately driving expression of the senescence-associated secretory phenotype (SASP), a complex cocktail of pro-inflammatory cytokines (e.g., IL-6, IL-8), chemokines, growth factors, and matrix metalloproteinases[2,8-11]. The biological consequences of the SASP are profoundly context-dependent. The SASP not only promotes tissue repair and immune clearance[12-14], but also contributes to chronic inflammation[15,16], tissue degeneration[17,18], and remodeling of the tumor microenvironment[19-22]. In cancer, CCFs can act as tumor-suppressive signals by inducing senescence[23], but may also be hijacked by cancer cells to promote metastasis and immune evasion[9,24].
In this review, we systematically summarize current knowledge of the molecular mechanisms underlying the biogenesis of CCFs, associated signaling networks, and their divergent roles in aging and cancer. Furthermore, we discuss emerging strategies to modulate the CCFs–cGAS–STING axis, including STING agonists for immunotherapy, cGAS inhibitors for inflammatory diseases, and senomorphic agents to suppress CCFs formation, as promising avenues for precision medicine against age-related disorders and oncological diseases[25-27].
2. Characteristics of CCFs
CCFs are specialized extranuclear chromatin structures that accumulate in the cytoplasm in response to cellular senescence and oncogene activation, and typically consist of dsDNA and carry specific histone modification markers[1,2,4], yet lack nuclear envelope components such as Lamin B1 or Lamin A/C[28,29]. CCFs are not membrane-bound and are frequently found co-localized with autophagy markers (e.g., LC3) or cytosolic DNA sensors (e.g., cGAS)[1,30]. The presence of CCFs in the cytoplasm potently activates innate immune responses, particularly the cGAS–STING pathway, thereby inducing the production of hallmark pro-inflammatory cytokines associated with the SASP[8,31]. Collectively, these characteristics position CCFs as a critical link between nuclear dysfunction and chronic inflammation, aging, and cancer progression.
2.1 Mechanisms underlying the formation of CCFs
The formation of CCFs results from the aberrant leakage of nuclear chromatin into the cytoplasm, primarily driven by multiple mechanisms, including loss of nuclear envelope integrity[28], micronucleus rupture[32], and mitochondrial dysfunction[33,34]. In senescent cells, CCFs formation is primarily driven by persistent DNA damage, telomere shortening, and mitochondrial dysfunction, which trigger nuclear lamina disassembly and heterochromatin release[2,3,34,35]. In contrast, in cancer cells, CCFs predominantly arise from replication stress, mitotic errors, or defective nuclear envelope reassembly after mitosis, and are often closely associated with chromosomal instability[2,9,36]. Although CCFs in senescent and cancer cells are morphologically similar, their underlying triggers and regulatory contexts differ[2,8,9,37].
2.1.1 Loss of nuclear envelope integrity
CCFs are observed in various human cellular senescence models, which are induced by mechanisms such as DNA damage[38], oncogene activation[39], and replicative senescence[5]. In senescent cells, the formation of CCFs is associated with loss of nuclear envelope integrity[29]. Nuclear envelope stability is dependent, in part, on the nuclear lamina, a meshwork structure composed of Lamin A, B1, B2, and C, which provides structural support to the nuclear membrane[40]. Reduced expression of Lamin B1 is a well-established hallmark of cellular senescence[41-44], while overexpression has been shown to delay the senescence process[45]. Knockdown of Lamin B1 can induce the formation of foci-like structures resembling CCFs[28]. Moreover, the formation of CCFs is reportedly associated with autophagic degradation of Lamin B1[1,4]. These findings suggest that the loss of Lamin B1 may be an upstream event in the formation of CCFs. However, some studies have found that in senescent fibroblasts, formation of CCFs is subject to the interaction between Lamin B1 and LC3 in the nucleus, followed by the transport of Lamin B1-containing CCFs to the cytoplasm for autophagic degradation, suggesting that CCFs act as carriers that mediate the degradation of Lamin B1[1] (Figure 1).
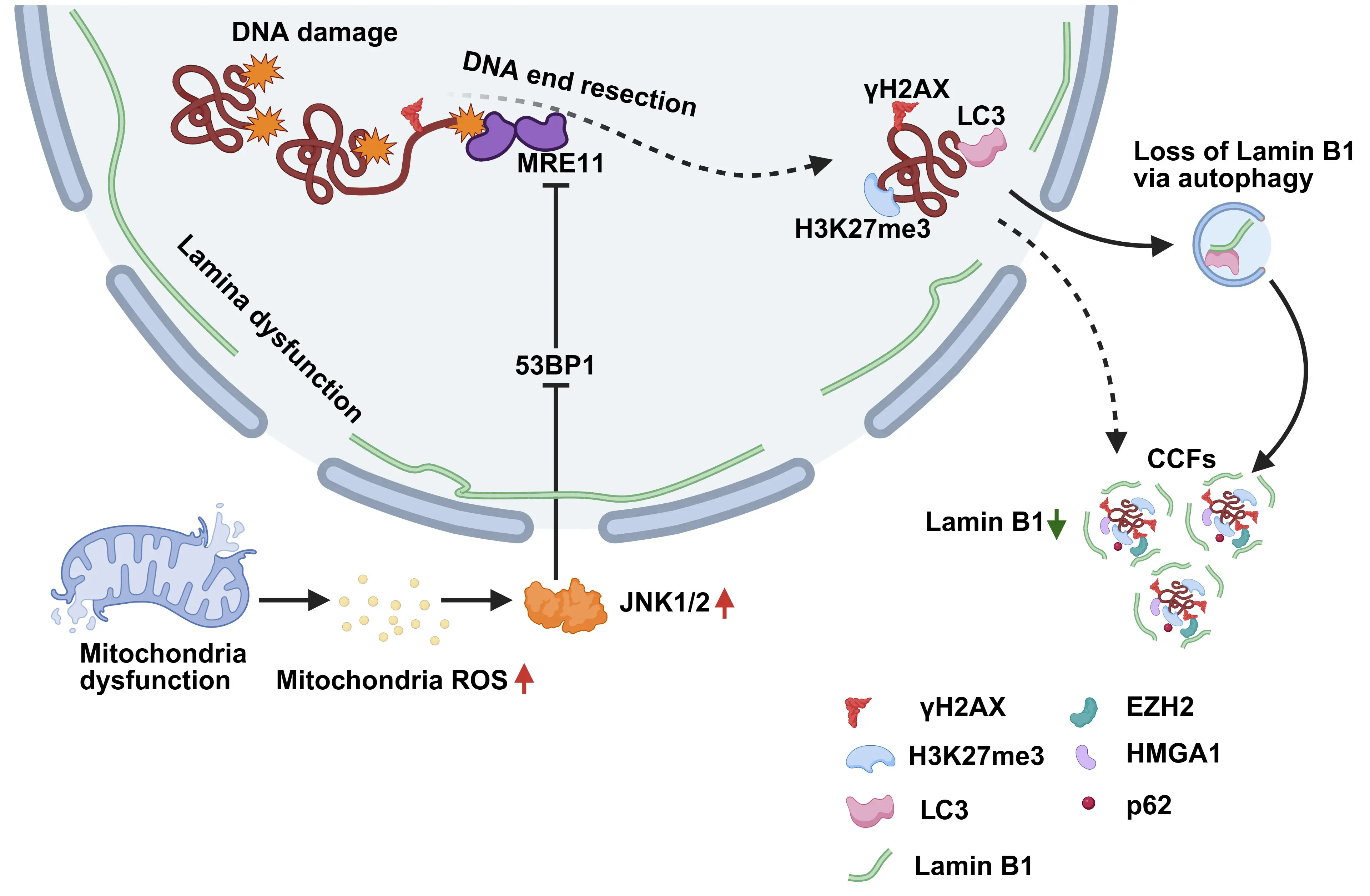
Figure 1. Proposed mechanism of CCFs formation. Loss of Lamin B1 through nuclear autophagy and other mechanisms compromises nuclear envelope integrity and promotes the release of chromatin fragments. Mitochondrial ROS activate JNK1/2 signaling, which further impairs lamina function and increases nuclear fragility. In the nucleus, γH2AX-marked dsDNA breaks undergo MRE11-dependent end resection to drive chromatin fragmentation. The tumor suppressor 53BP1 restricts this process by inhibiting excessive resection, thereby limiting the formation of CCFs. Created in BioRender.com. CCFs: cytoplasmic chromatin fragments; ROS: reactive oxygen species; dsDNA: double-stranded DNA.
2.1.2 Micronucleus rupture
During mitosis of cancer cells, due to abnormal chromosome segregation (e.g., lagging chromosomes or chromosomal bridge breakage), some chromosomes or chromosomal fragments fail to properly incorporate into the main nucleus and are instead encapsulated separately to form micronuclei[32]. Due to the lack of the complete nuclear envelope and key nuclear lamina proteins, such as Lamin B1, micronuclei are highly unstable and prone to rupture, thus releasing the internal chromatin into the cytoplasm, forming CCFs[9].
2.1.3 Decline in lysosomal degradative function
Lysosomes are not only the cell’s “degradation and recycling center” but also multifunctional signaling hubs involved in processes such as nutrient sensing[46, 47], autophagy, organelle communication, and immunity[46-51]. The decline in lysosomal function is closely associated with the accumulation of cytoplasmic chromatin. DNase2, a key nuclease within lysosomes, is responsible for the degradation of DNA fragments that enter the cytoplasm. During the aging process, the decline in lysosomal function may lead to abnormal activity or mislocalization of DNase2. Furthermore, the expression levels of both DNase2 and TREX1 (a cytosolic 3′→5′ DNA exonuclease that degrades free cytoplasmic DNA to maintain genomic homeostasis and prevent aberrant innate immune activation) are significantly downregulated, resulting in the failure to effectively degrade CCFs, thereby leading to subsequent accumulation in the cytoplasm[52].
The decline in lysosomal function directly promotes the accumulation of CCFs and ensures a chronic inflammatory response by impairing DNA clearance, thereby representing a key mechanism driving cellular senescence and associated pathological processes.
2.1.4 Mitochondrial dysfunction
Mitochondria play crucial roles in energy production[53], cell signaling[54], differentiation[55], cell death[56], and aging in eukaryotic cells[57]. The roles of mitochondria in cellular senescence are widely associated with an important function as generators of stochastic molecular damage mediated by reactive oxygen species (ROS)[58-60]. In senescent human fibroblasts (IMR90 cells) induced by ionizing radiation, the expression levels of nucleus-encoded genes associated with mitochondrial oxidative phosphorylation are significantly downregulated, leading to mitochondrial dysfunction and the substantial accumulation of ROS, which further activates the JNK signaling pathway[61]. Activated JNK interacts with the nuclear protein 53BP1, inhibiting the end resection process of double-stranded DNA breaks (DSBs), thereby impairing DNA repair, ultimately leading to aberrant chromatin fragmentation and the formation of CCFs and subsequent release into the cytoplasm[33,34] (Figure 1).
2.2 Regulating factors of CCFs formation
The generation and accumulation of CCFs are tightly regulated by a network of molecular markers, which can serve as positive or negative indicators of their presence or functional state (Table 1). These markers reflect underlying cellular conditions such as genomic instability, nuclear integrity, chromatin dynamics, and DNA damage responses (DDRs).
Table 1. Molecular markers of CCFs.
| Molecular Markers | Formation Pathways | Downstream Pathway and Clearance Pathways | |
| Positive molecular markers | dsDNA[1,10,52] | Loss of nuclear membrane integrity | cGAS-STING activation[2,3] |
| γH2AX[2,29,63] | |||
| H3K27me3[1,2,4] | |||
| cGAS[3,30,64] | Autophagy | ||
| EZH2[67,69] | Lysosomal degradation[52] | ||
| HMGA1[69] | |||
| p62[29] | Decreased lysosomaldegradationmtROS | ||
| Negative molecular markers | H3K9ac[5,29] | ||
| H3K27ac[29] | Exosomal secretion[62] | ||
| Lamin B1[28,69] | |||
| Lamin A/C[4,29] | |||
| 53BP1[29,34] | |||
| p53[77,78] | |||
CCFs: cytoplasmic chromatin fragments; dsDNA: double-stranded DNA; cGAS: cyclic guanosine monophosphate–adenosine monophosphate synthase; EZH2: enhancer of zeste homolog 2; mtROS: mitochondrial reactive oxygen species.
2.2.1 Positive molecular markers of CCFs
Several positive molecular markers either drive or indicate the generation of CCFs. As a fundamental component of CCFs, dsDNA in the cytoplasm typically reflects the breakdown of the nuclear envelope or pathological release of chromatin from the nucleus[1,62]. As a classic marker of dsDNA, γH2AX signals early chromatin damage linked to the initial formation of CCFs[2,29,63]. Certain repressive histone modifications, including H3K9me3 and H3K27me3, promote heterochromatin compaction and are thought to increase chromatin fragility, thereby facilitating fragmentation and subsequent leakage into the cytosol[1,2,4]. The cytosolic DNA sensor cGAS recognizes CCFs-derived dsDNA to activate the STING pathway and trigger inflammatory responses[3,30,64]. Notably, while mammalian cGAS typically suppresses homologous recombination (HR) and promotes genomic instability[65], naked mole-rat cGAS exhibits an opposite function due to four specific amino acid substitutions[66,67]. These four amino acid substitutions reduce cGAS ubiquitination, prolong its retention on damaged chromatin, and thereby enhance HR repair by promoting the interaction between RAD50 and FANCI, ultimately delaying both cellular and organismal aging[66]. Introducing these key mutations into human cGAS can reverse pro-aging effects, highlighting the potential role as a target for anti-aging interventions. Enhancer of zeste homolog 2 (EZH2), which catalyzes the H3K27me3 modification, promotes heterochromatic silencing and indirectly contributes to chromatin destabilization and generation of CCFs, and has been shown to act synergistically with HMGA1[68,69]. The autophagy adaptor protein p62 may also be involved in the intracellular trafficking or degradation of CCFs through selective autophagy, although further investigations are required to elucidate the exact regulatory function contributing to the formation of CCFs[29].
2.2.2 Negative molecular markers of CCFs
The formation of CCFs is counteracted by several negative regulatory factors that maintain chromatin and nuclear integrity. Histone acetylations, such as H3K9ac and H3K27ac, are associated with open, transcriptionally active chromatin states and contribute to chromatin stability, thereby suppressing the generation of CCFs[5,29]. Structural components of the nuclear lamina, including Lamin B1 and Lamin A/C, are essential for nuclear envelope integrity, as their downregulation or loss leads to nuclear rupture and aberrant chromatin leakage into the cytoplasm[4,28,29,70]. The DNA repair protein 53BP1 further restricts CCFs formation by promoting non-homologous end joining (NHEJ)-mediated DSBs repair and inhibiting excessive DNA end resection during HR[71-73], indicating that dysregulated DNA end processing is a key driver of CCFs formation[74-76]. Among these factors, p53 acts as a central regulator that effectively suppresses CCFs formation by coordinating the DDRs, stabilizing chromatin, and preserving nuclear envelope integrity[77,78]. In contrast, p53 inactivation leads to DNA damage accumulation, nuclear structural disruption, and chromatin leakage into the cytoplasm, thereby promoting CCFs accumulation[52,79-81]. Importantly, CCFs formation is not a universal feature of senescence but rather a heterogeneous event influenced by the cellular molecular context and the balance between pro- and anti-CCFs regulators[82]. In senescent cells, the NHEJ and HR pathways are often impaired[77,78]. Under these conditions, the release of CCFs may serve as an alternative mechanism to remove unrepaired chromatin fragments, thereby alleviating nuclear genomic instability. Ultimately, CCFs levels are governed by a dynamic equilibrium between their generation, driven by DNA damage, nuclear envelope breakdown, and epigenetic alterations, and their clearance, potentially mediated by autophagy or other degradation pathways. Disruption of this homeostasis, due to defective DNA repair, compromised nuclear integrity, or impaired clearance mechanisms, is closely linked to aging, chronic inflammation, and cancer progression.
3. CCFs Activate the cGAS-STING Pathway
DNA in the cytoplasm is typically recognized as a danger signal of pathogen invasion and can be detected by intracellular DNA sensors, thereby activating innate immune responses[67,83,84]. Among these, cGAS serves as a key cytosolic DNA sensor[85]. Upon binding to DNA, cGAS catalyzes the synthesis of adenosine triphosphate and guanosine triphosphate into the second messenger cGAMP[19,86,87], which binds to the adaptor protein STING, located on the endoplasmic reticulum membrane, thereby inducing conformational changes and activating downstream signaling pathways[88]. Activated STING recruits and activates TBK1, which subsequently phosphorylates the transcription factors IRF3 and NF-κB (p65)[25,89]. Phosphorylated IRF3 and NF-κB translocate to the nucleus and induce transcription of type I interferons and NF-κB-dependent pro-inflammatory cytokines, respectively, thereby initiating innate immune responses against pathogens and driving the SASP (Figure 2)[8,31,90-93].
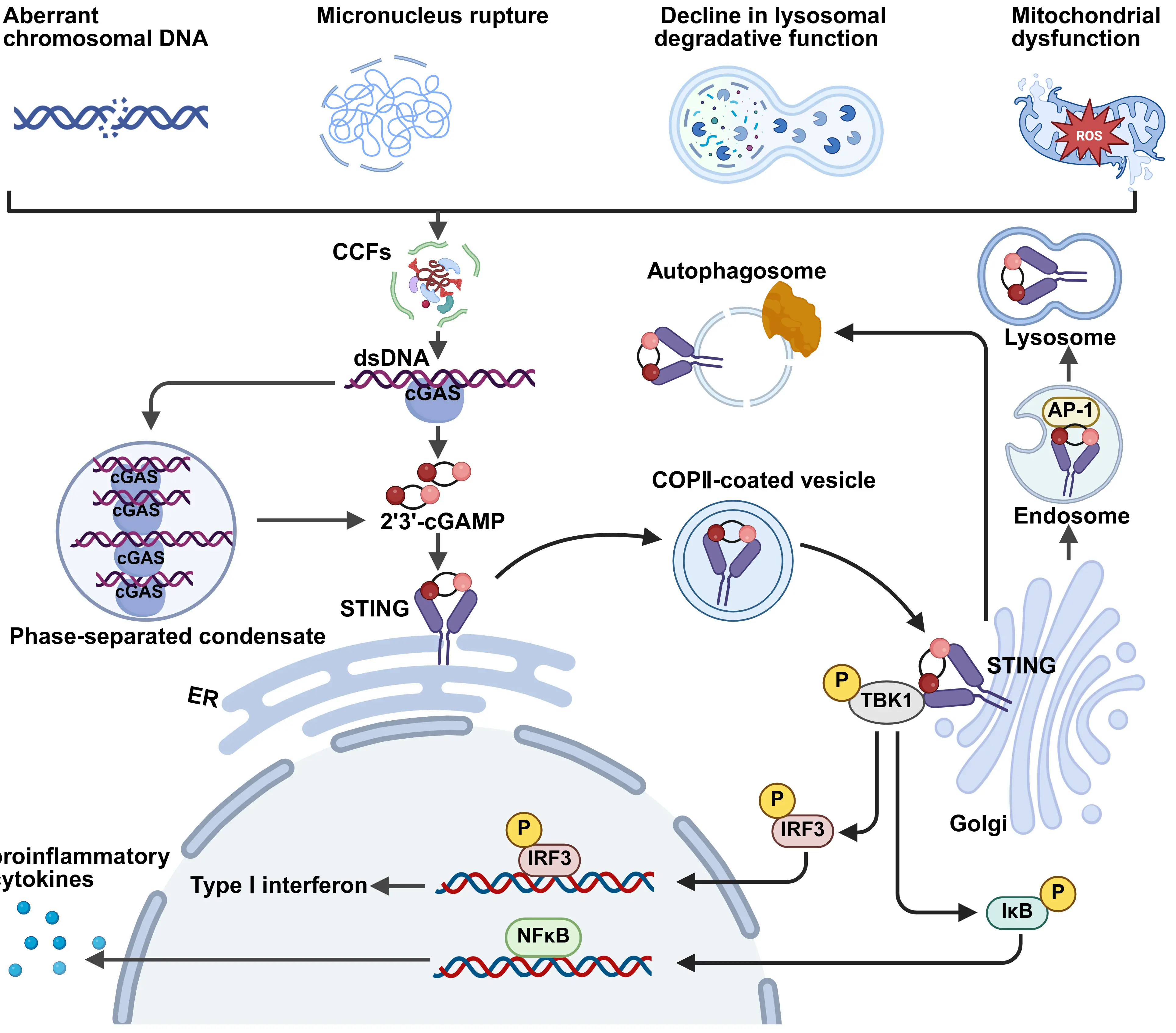
Figure 2. Pathways of CCFs formation and downstream signaling. In the cytoplasm, dsDNA derived from extranuclear chromatin, cytosolic micronuclei, mitochondrial DNA, and aberrant chromosomal DNA can activate cGAS. Activated cGAS synthesizes the second messenger cGAMP, which binds to STING on the endoplasmic reticulum, triggering conformational change. STING then recruits TBK1 to phosphorylate IRF3, promoting IRF3 dimerization and nuclear translocation, leading to the transcription of genes encoding type I interferons. Simultaneously, STING activates the NF-κB pathway (p50/p65), inducing the expression of pro-inflammatory cytokines. Created in BioRender.com. CCFs: cytoplasmic chromatin fragments; dsDNA: double-stranded DNA; cGAS: cyclic guanosine monophosphate–adenosine monophosphate synthase; EZH2: enhancer of zeste homolog 2; cGAMP: cyclic guanosine monophosphate–adenosine monophosphate.
4. The Role of CCFs in Cellular Senescence
CCFs are released into the cytoplasm during cellular senescence, and have recently been recognized as a key molecular link connecting genomic instability to chronic inflammation[2,3,16,94].CCFs are not merely structural markers of cellular senescence, but also serve as a core signaling source driving the SASP and systemic chronic low-grade inflammation, playing a significant role in various age-related diseases[35,37,95-98] (Figure 3).
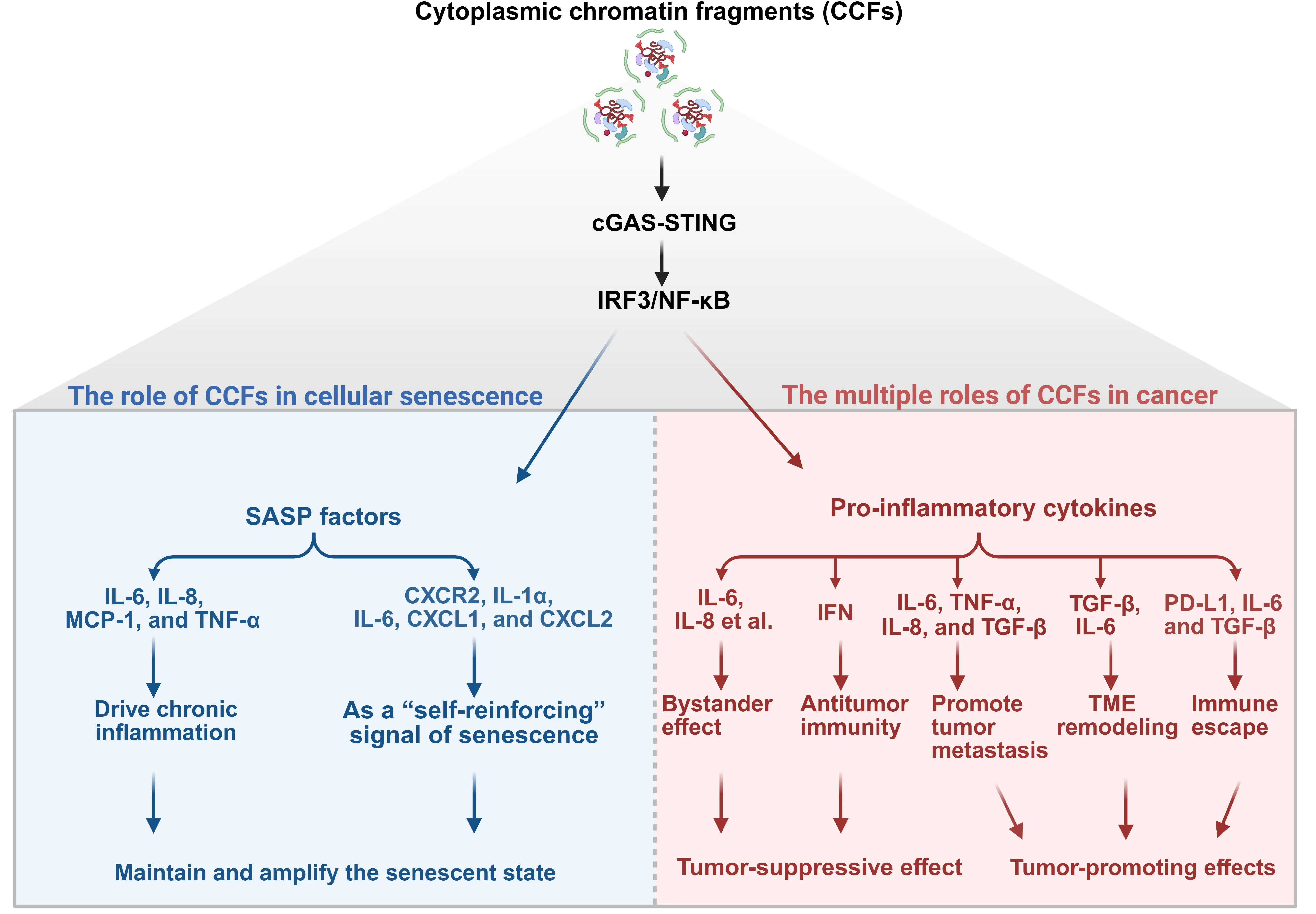
Figure 3. The dual roles of CCFs in cellular senescence and cancer. CCFs are released from the nucleus into the cytoplasm under conditions of genomic stress and can activate the cGAS–STING pathway, leading to IRF3 and NF-κB activation and subsequent secretion of the SASP and pro-inflammatory cytokines. In senescent cells, CCFs act as a “self-reinforcing” signal that drives chronic inflammation and stabilizes the senescent state through SASP-mediated autocrine and paracrine signaling. This contributes to tumor suppression by maintaining growth arrest and promoting immune clearance. In contrast, in cancer cells, CCFs exert dual and context-dependent effects: they can elicit antitumor immunity via bystander effects or immune activation, but more commonly promote tumor progression through sustained SASP production, which fuels metastasis, TME remodeling, and immune escape. Thus, while sharing a common upstream mechanism (cGAS–STING), CCFs play divergent roles, tumor-suppressive in senescence and tumor-promoting in cancer, highlighting their complex biological significance. Created in BioRender.com. CCFs: cytoplasmic chromatin fragments; cGAS: cyclic guanosine monophosphate–adenosine monophosphate synthase; TME: tumor microenvironment; SASP: senescence-associated secretory phenotype.
4.1 CCFs promote the SASP and drive chronic inflammation
CCFs are the primary source of cGAS-STING activation in senescent cells[83,88,99]. CCFs activate the cGAS–STING pathway and its downstream targets IRF3 and NF-κB to induce type I interferons and pro-inflammatory gene expression. However, in senescent cells, this pathway exhibits a unique “signaling bias”[100-102]: although the cGAS-STING pathway is strongly activated, the type I interferon response is significantly suppressed, while the NF-κB-mediated pro-inflammatory pathway is persistently activated[103]. A key mechanism underlying this phenomenon is that persistent activation of the p38-MAPK pathway during senescence suppresses the transcriptional activity of IRF3, thereby blocking the production of type I interferons[104-106]. Therefore, CCFs primarily drive expression of the classical SASP factors IL-6, IL-8, MCP-1, and TNF-α[107,108].
4.2 As a “self-reinforcing” signal of senescence
Once generated, CCFs activate the cGAS–STING signaling pathway, driving SASP secretion and thereby exacerbating DNA damage[109,110]. This creates a self-reinforcing positive feedback loop that maintains and consolidates the senescent state. Senescent cells not only secrete CXCR2 ligands, such as IL-8, but also upregulate CXCR2’s own expression, establishing an autocrine “self-amplifying loop” that enhances the stability and persistence of the SASP[111,112]. Furthermore, multiple key components of the SASP, including IGFBP-7, PAI-1, IL-6, and CXCR2-binding chemokines such as IL-8 and GROα, have been shown to induce cellular senescence.[111,113-115]. For example, during replicative senescence of human fibroblasts, sustained secretion of the SASP factors IL-1α, IL-6, CXCL1, and CXCL2 can activate the NF-κB signaling pathway through autocrine or paracrine mechanisms. In turn, activation of NF-κB promotes the expression of these inflammatory factors, creating a positive feedback loop that further consolidates cell cycle arrest, rendering senescence difficult to reverse[116-119].
5. The Multiple Roles of CCFs in Cancer
CCFs are now widely recognized as key regulatory molecules in cancer biology[2,29]. CCFs primarily originate from the pronounced genomic instability characteristic of tumor cells, and their abnormal accumulation in the cytoplasm is closely associated with cellular senescence, chronic inflammation, and DDRs[90,120]. During tumor progression, the spatial organization of chromatin undergoes significant alterations, with a marked increase in fractal dimension, reflecting heightened structural complexity and decondensation of higher-order chromatin architecture[121,122]. This structural disorganization not only exacerbates genomic instability but is also positively correlated with the invasive capacity and metastatic potential of tumor cells, suggesting that CCFs may play a critical role in tumor evolution[123,124]. However, rather than exerting a uniform effect, CCFs play a dual, context-dependent role in cancer, either acting as tumor-suppressive agents by promoting senescence and immune surveillance or functioning as tumor-promoting drivers by fueling chronic inflammation, metastasis, and therapy resistance (Figure 3).
5.1 Tumor-suppressive effect: senescence induction of cancer cells
In early tumorigenesis or therapy-induced stress, CCFs can serve as critical “danger signals” to activate a series of tumor-suppressive pathways, including cellular senescence, DDRs, and innate immune surveillance[2,32,90,125]. In the early stages of tumorigenesis, aberrant activation of oncogenes (such as RAS and MYC) can trigger replication stress and DDRs, leading to the release of CCFs[126]. These CCFs can initiate cellular senescence programs to block abnormal proliferation. Meanwhile, senescent cells secrete factors associated with the SASP (such as IL-6 and IL-8), which, through signaling pathways such as IL-1α and CXCR2, not only reinforce their own senescent state but also propagate senescence signals to neighboring cells, mediating a “bystander effect”[127,128], expanding growth suppression at the tissue level and blocking tumor progression[129]. DNA released by tumor cells activates the cGAS-STING pathway, triggering type I interferon responses, which promote activation of dendritic cells, thereby initiating CD8⁺ T cell-mediated anti-tumor immune responses[125,130-132]. These findings indicate that, in the early stages of tumorigenesis, CCFs are not merely byproducts, but rather critical tumor-suppressive signaling molecules. By activating the cGAS-STING pathway, CCFs establish a natural line of defense against malignant transformation.
5.2 Tumor-promoting effects
CCFs not only reflect genomic instability in tumor cells but also actively promote tumor progression. On one hand, they persistently activate NF-κB via the cGAS–STING pathway, inducing secretion of pro-inflammatory cytokines such as IL-6, TNF-α, and IL-8, to create an inflammatory microenvironment that favors invasion and metastasis[133-137]. They also cooperate with TGF-β to promote epithelial–mesenchymal transition (EMT)[138-140] and activate cancer-associated fibroblasts (CAFs), thereby remodeling the tumor microenvironment to support metastasis[141]. On the other hand, under chronic stimulation, the CCFs–cGAS–STING signaling shifts from anti-tumor immune activation to immune suppression, upregulating PD-L1, recruiting myeloid-derived suppressor cells (MDSCs), and synergizing with TGF-β and the IL-6/STAT3 pathway to establish a highly immunosuppressive microenvironment that enables tumor immune evasion[142]. Thus, CCFs play a dual tumor-promoting role in both metastasis and immune escape, and their downstream signaling networks represent promising targets for novel anti-metastatic and immunotherapeutic strategies.
5.2.1 CCFs promote tumor metastasis via cGAS-STING signaling
CCFs play a critical role in promoting tumor metastasis. CCFs-induced sustained activation of NF-κB, together with TBK1-mediated IRF3 phosphorylation, drives pro-metastatic inflammatory signaling. Sustained activation of NF-κB is particularly critical, driving robust expression of the pro-inflammatory cytokines IL-6, TNF-α, and IL-8, thereby creating a pro-inflammatory microenvironment that promotes tumor cell invasion and metastasis. IL-6 not only enhances the invasive capacity of tumor cells through the IL-6-STAT3 signaling axis[133-137], but also upregulates factors such as TGF-β to induce EMT, thereby promoting the metastatic process[138-140]. Furthermore, IL-6 and IL-8 act synergistically to induce fibroblast-like morphology of MCF-7 cells, increase CD44 expression, and enhance migratory capacity, self-renewal, and multilineage differentiation potential, further highlighting the central role of inflammatory cytokines in driving the acquisition of malignant phenotypes[143]. Notably, cellular crosstalk within the tumor microenvironment also contributes to this regulation. For example, IL-6 secreted by myofibroblasts can activate STAT3 in lung cancer cells and promote TGF-β expression, while TGF-β produced by tumor cells, in turn, stimulates myofibroblasts to release IL-6, forming a positive feedback loop that cooperatively promotes tumor growth[144]. Therefore, targeting the CCFs-cGAS-STING-NF-κB-inflammatory cytokine axis may provide a novel therapeutic strategy to inhibit tumor metastasis.
5.2.2 CCFs mediate remodeling of the tumor microenvironment to promote metastasis
As a transcription factor, NF-κB directly binds to the promoter region of TGF-β, driving its upregulation[145]. TGF-β is a key activator of CAFs[141]. As a major cellular component of the stromal microenvironment in various cancers, CAFs promote tumor progression by enhancing cancer cell proliferation, invasion, and metastasis[146,147]. The pro-tumorigenic activity of CAFs has been reported in cancers of the prostate[148], breast[149,150], pancreas[151], and colon/rectum[152]. Moreover, other microenvironmental signals can also induce tumor cells to secrete pro-inflammatory cytokines, such as IL-6[153]. Activation of IL-6 signaling in tumor cells promotes tumor metastasis through multiple mechanisms. First, IL-6 activates the STAT3 pathway, upregulates the expression of MMP-2, promotes extracellular matrix degradation, and enhances the invasive capacity of tumor cells[154,155]. Second, the IL-6/STAT3 axis induces expression of VEGF and bFGF, driving tumor angiogenesis and providing essential nutritional support for tumor growth and metastasis[142]. In summary, CCFs activate the cGAS-STING-NF-κB signaling pathway to upregulate the expression of TGF-β and IL-6, which not only promotes CAFs activation and remodeling of the tumor microenvironment to drive tumor progression, but also activates the IL-6/STAT3 axis in tumor cells, further enhancing invasiveness, angiogenic capacity, and metastatic potential, collectively forming a coordinated molecular network that promotes tumor metastasis.
5.2.3 CCFs promote immune escape
CCFs also serve as a key signaling molecule to initiate immune evasion. Recognition of CCFs by cGAS and transient activation of the STING pathway induce the production of type I interferons and other pro-inflammatory factors, thereby initiating an anti-tumor immune response[156-158]. However, when this signaling is chronically activated by persistently present CCFs, the STING pathway shifts to become a “driver” of immune suppression. On one hand, this shift promotes the expression of the immune checkpoint molecule PD-L1 by activating the RelA/NF-κB signaling pathway, thereby directly aiding tumor cells to evade surveillance and killing by cytotoxic T lymphocytes[159]. On the other hand, the sustained activation of the STING signaling pathway synergizes with pro-inflammatory cytokines such as IL-6 to further amplify the immunosuppressive effect through the JAK/STAT3 pathway[160]. This process not only upregulates inhibitory molecules like PD-L1 but also promotes the recruitment of myeloid-derived suppressor cells, collectively constructing a local immunosuppressive microenvironment that suppresses the activity of cytotoxic T lymphocytes, thereby driving tumor progression and immune evasion[161,162]. In addition, TGF-β, another core immunosuppressive factor, plays a significant role in this process[163-165] by bidirectionally regulating either the generation of immunosuppressive regulatory T cells in the presence of cytokines such as IL-2[166,167], or inducing the generation of pro-inflammatory Th17 cells in the presence of inflammatory cytokines such as IL-6[168,169]. Meanwhile, TGF-β can also inhibit the functions of NK cells, T cells, and B cells, and polarize macrophages and neutrophils towards an immunosuppressive phenotype, thereby comprehensively suppressing anti-tumor immune responses[170]. These mechanisms collectively form a complex network intertwined with multiple pathways such as STING, NF-κB, IL-6, and TGF-β, enabling tumor cells to efficiently evade immune system attacks[165].
In summary, CCFs can trigger anti-tumor immune responses at the initial stage by activating signaling pathways such as STING. However, under conditions of chronic and persistent activation, CCFs become a key factor in driving the formation of an immunosuppressive microenvironment. Interconnected and synergistic with multiple signaling pathways, including NF-κB, IL-6, and TGF-β, CCFs not only upregulate the expression of immune checkpoint molecules but also promote the expansion and infiltration of immunosuppressive cells, thereby comprehensively suppressing the functions of anti-tumor immune cells, such as effector T cells. This complex regulatory network reveals the underlying mechanisms by which tumor cells evade immune surveillance, providing a crucial theoretical foundation for the development of novel immunotherapeutic strategies targeting CCFs and related downstream signaling pathways. Future interventions targeting key nodes within this network have the potential to reverse the tumor immunosuppressive microenvironment, enhance the efficacy of current immunotherapies, and offer new breakthroughs in cancer treatment.
6. Therapeutic Strategies
In terms of therapeutic potential, targeting cellular senescence-related pathways offers a novel strategy for intervening in age-related diseases. The CCFs-SASP pathway, as a newly identified mechanism in senescence research, provides a key target for suppressing SASP-driven inflammation and tissue damage[171-173]. Senostatics are agents that inhibit the paraclockwise spread of senescence by blocking paracrine signaling, such as through NF-κB inhibition or antioxidant activity, thereby preventing the bystander effect without killing senescent cells[174,175]. Senomorphics, on the other hand, suppress the secretion or activity of harmful SASP components to alleviate inflammation and tissue damage, also without inducing cell death[176]. While both strategies are non-senolytic, senostatics specifically aim to halt the propagation of senescence, whereas senomorphics focus on modulating the senescent phenotype. Inhibiting SASP signaling through “senomorphics” or “senostatics”, rather than directly eliminating senescent cells, represents a promising alternative therapeutic approach (Figure 4)[177-179].
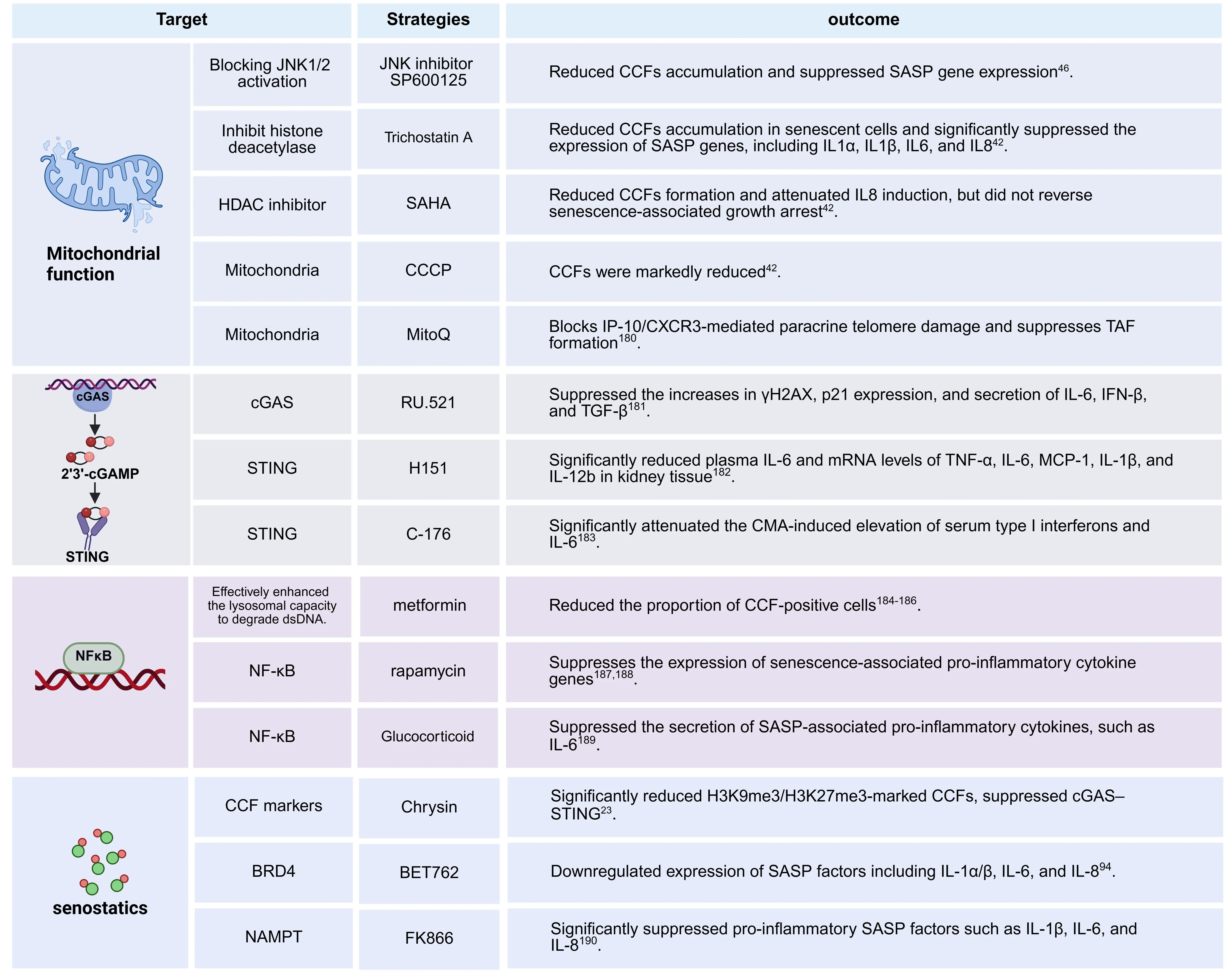
Figure 4. Therapeutic targets of CCFs and CCFs-related pathways[23,42,46,94,180-190]. Histone deacetylase inhibitors (e.g., TSA, SAHA) reduce mtROS and prevent the formation of CCFs by enhancing mitochondrial function. JNK inhibitors (e.g., SP600125) block the mtROS-JNK-Lamin B1 axis to stabilize the nucleus. cGAS-STING inhibitors (e.g., H151, RU.521) suppress DNA sensor activation after the release of CCFs. NF-κB modulators (e.g., rapamycin, glucocorticoids, metformin) inhibit the activities of transcription factors that drive the SASP. senostatic agents (e.g., chrysin, BET 762, and FK866) broadly suppress the SASP without eliminating senescent cells. Created in BioRender.com. CCFs: cytoplasmic chromatin fragments; cGAS: cyclic guanosine monophosphate–adenosine monophosphate synthase; SASP: senescence-associated secretory phenotype; mtROS: mitochondrial reactive oxygen species.
6.1 Strategies targeting mitochondrial function
Improving mitochondrial homeostasis is a key strategy for inhibiting CCFs formation. Mitochondrial dysfunction leads to excessive ROS accumulation, activates the JNK pathway, and impairs DNA repair, thereby promoting chromatin fragmentation and CCFs generation. Treatment with JNK inhibitors (e.g., SP600125), low-dose HDAC inhibitors (e.g., trichostatin A, SAHA)[34], or mitochondrial-targeted antioxidants (e.g., MitoQ) effectively reduces CCFs accumulation and suppresses SASP factor expression[180]. Moreover, the mitochondrial uncoupling agent CCCP has been shown to significantly reduce CCFs levels, underscoring that preserving mitochondrial integrity can mitigate aging-associated inflammation at its source.
6.2 Strategies targeting the cGAS-STING pathway
Inhibiting the CCFs–cGAS–STING pathway can effectively suppress IRF3 and NF-κB activation, thereby alleviating SASP. Several small-molecule inhibitors are currently under investigation. For example, the cGAS inhibitor RU.521 reduces the secretion of IL-6, IFN-β, and TGF-β[181], whereas STING inhibitors such as H151 and C-176 significantly lower serum levels of type I interferons and IL-6[182,183]. These inhibitors directly interrupt the inflammatory signaling cascade triggered by CCFs, offering precise targets for controlling chronic inflammation.
6.3 Strategies targeting the NF-κB signaling pathway
NF-κB represents a critical downstream therapeutic target to inhibit pro-inflammatory gene expression. Various drugs suppress the SASP by inhibiting NF-κB activity. For example, metformin enhances lysosomal clearance of cytoplasmic DNA, thereby reducing the proportion of CCFs-positive cells[184-186]. Rapamycin downregulates SASP-related factors by inhibiting the mTOR–NF-κB axis, while glucocorticoids directly suppress the secretion of inflammatory mediators such as IL-6[181,187-189]. Although these strategies do not prevent CCFs formation, they effectively suppress its inflammatory output and mitigate tissue damage.
6.4 Senostatic strategy targeting the characteristics of senescent cells
Unlike senolytic approaches that eliminate senescent cells, senostatic strategies aim to suppress their deleterious functions without inducing cell death. For instance, chrysin reduces H3K9me3- and H3K27me3-marked CCFs and suppresses the cGAS–STING pathway[23]; the BET inhibitor BET762 markedly downregulates IL-1α, IL-1β, IL-6, and IL-8[94]; and the NAMPT inhibitor FK866 potently inhibits multiple SASP factors[190]. These strategies suppress the secretory phenotype of senescent cells while preserving their potential physiological functions, thereby effectively mitigating aging-associated pathological processes. In mouse models, whether by blocking the induction of SASP upstream, eliminating senescent cells at the source, or directly suppressing SASP itself, the procarcinogenic process mediated by the “deoxycholic acid (DCA)-SASP axis” can all be effectively inhibited, thereby preventing the development of obesity-associated hepatocellular carcinoma[24,191]. In summary, intervention strategies targeting the key nodes can not only effectively suppress SASP secretion and alleviate aging-related pathological phenotypes, but also provide a solid theoretical foundation and potential therapeutic targets for developing novel therapies against aging and age-related diseases[192].
7. Perspective
In summary, targeting the formation of CCFs and their associated signaling pathways holds significant promise for anti-aging and anticancer interventions, yet numerous critical scientific questions remain unresolved.
(1) Mechanisms of CCFs formation:
CCFs are strongly enriched with the DNA damage marker γH2AX but lack the DNA repair protein 53BP1, which normally colocalizes with γH2AX[34,74,193]. Multiple studies have clearly established 53BP1 as a negative regulator of CCFs formation, suggesting that DSBs and their subsequent processing play a central role in CCFs biogenesis. It has been shown that 53BP1 restricts CCFs generation by inhibiting nucleolytic resection of DSBs ends. Accordingly, pharmacological inhibition of MRE11, a key nuclease that initiates DSBs end resection[194], with the small-molecule inhibitor mirin significantly suppresses CCFs formation[34]. However, it remains unclear whether other key factors involved in DSBs end resection or homologous recombination repair, such as CtIP, EXO1, and BRCA1, also contribute to the regulation of CCFs biogenesis. Furthermore, whether 53BP1 influences CCFs formation through additional mechanisms beyond its canonical role warrants further investigation.
(2) Diversity of DDR–SASP signaling pathways
Although CCFs can promote the SASP by activating the cGAS–STING pathway, the DDR can also trigger inflammatory signaling through CCFs-independent mechanisms. For example, ATM can phosphorylate NEMO, a key regulator of the NF-κB pathway, thereby directly activating NF-κB[195-197]. Therefore, future studies should employ genetic or pharmacological approaches to specifically block CCFs formation, enabling a clear distinction between CCFs-dependent and CCFs-independent components of SASP.
Moreover, it remains an open question whether different senescence triggers preferentially activate distinct SASP signaling routes. For instance, do stimuli such as ionizing radiation, oncogene activation, and replication stress exhibit distinct biases toward the ATM–NF-κB axis versus the CCFs–cGAS–STING pathway? Elucidating these context-specific signaling preferences will deepen our understanding of SASP heterogeneity and its functional implications across diverse pathological settings.
(3) Non-Canonical functions of STING during cellular senescence
Notably, in multiple cellular senescence models, the proportion of CCFs-positive cells is significantly increased, accompanied by activation of the cGAS–STING pathway, yet the protein expression level of STING itself is markedly downregulated[198]. This seemingly paradoxical observation raises new questions: does STING have non-canonical functions in the context of senescence beyond its classical role in inflammatory signaling? For instance, could reduced STING expression perturb endoplasmic reticulum calcium homeostasis, thereby promoting cytosolic Ca2+ release and contributing to other senescence-associated processes[199]? These questions remain to be thoroughly investigated.
(4) Mitochondria–Nucleus crosstalk in CCFs formation and inflammatory signaling
Meanwhile, mitochondrial dysfunction also plays a critical role in CCFs formation. Existing studies have shown that mtROS can promote CCFs generation by activating the stress-activated kinases JNK1/2, which in turn impair 53BP1 function[34]. However, it remains unclear how mtROS precisely regulates chromatin stability in the nucleus. Are specific mitochondrial metabolites, lipid messengers, or retrograde signaling molecules directly involved in chromatin remodeling or disruption of nuclear envelope integrity? Addressing these questions will advance the emerging field of mitochondria–epigenome crosstalk. In addition, mitochondrial DNA (mtDNA) itself can activate the cGAS–STING pathway[200], further complicating the cytosolic nucleic acid sensing network in senescence. CCFs and mtDNA may therefore cooperatively or competitively modulate inflammatory responses.
(5) Biological significance of CCFs
Finally, a fundamental question remains unresolved: are CCFs merely passive byproducts of failed DNA repair in senescent cells, or do they represent functional structures with active biological significance? For instance, could CCFs act as a “safety valve” by sequestering irreparably damaged chromatin into the cytoplasm to prevent the spread of genomic instability within the nucleus? Alternatively, might they promote the clearance of senescent cells by activating immune surveillance mechanisms? Addressing these questions will not only deepen our understanding of the nature of cellular senescence but also provide a theoretical foundation for developing novel anti-aging or anticancer strategies targeting the CCFs pathway.
Cellular senescence plays a dual role in cancer: on one hand, it acts as a crucial natural tumor-suppressive mechanism, whereby oncogene activation or chemotherapy can induce senescence of tumor cells, inhibiting proliferation and promoting immune clearance, thereby contributing to therapeutic efficacy[201-204]. However, on the other hand, senescence also poses significant risks. Due to frequent defects in cell cycle checkpoints of cancer cells, therapy-induced senescence is often unstable, making it easier for tumor cells to escape from this state[205-208]. These “escapee” cells may acquire stem cell-like properties through epigenetic reprogramming, exhibiting enhanced tumorigenic and metastatic potential[209]. Moreover, therapy can also induce senescence of normal cells within the tumor microenvironment, and the secreted SASP-related factors can promote EMT[210,211], proliferation[212], and invasion[104,213] of residual cancer cells through bystander effects, thereby creating a microenvironment that favors tumor recurrence and metastasis[214]. Therefore, although helping in short-term tumor control, the induction of senescence may promote tumor progression and systemic aging in the long term. Future approaches need to carefully weigh these benefits and risks, and explore combination strategies with senolytics or senomorphics to eliminate senescent cells or suppress the SASP, to improve treatment safety and long-term efficacy.
Acknowledgements
The authors declare that AI tools were used solely for language polishing during the manuscript preparation process. During the language polishing process, we utilized the following AI tools: Youdao (11.2.14), Tongyi Qianwen (Qwen3.5-Plus), and DeepSeek (1.7.8). All research content, including study design, data analysis, interpretations, figures, and tables, is original and was not generated using AI tools.
Authors contribution
Xiang W: Writing-original draft.
Wang S, Pan Y, Cong X, Lu J: Investigation, writing-review & editing.
Zhang Y: Conceptualization, supervision, writing-original draft.
Conflicts of interest
The authors declare no competing interests.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This study was supported by the grants from the National Natural Science Foundation of China (grant numbers: 32371216, 32271207,82571794), the Development and Reform Commission of Jilin Province (grant numbers: 2024C013-1), and the Scientific Research Innovation Capability Support Project for Young Faculty (grant number: SRICSPYF-ZY2025126).
Copyright
© The Author(s) 2026.
References
-
1. Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527:105-109.[DOI]
-
2. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402-406.[DOI]
-
3. Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke N, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19:1061-1070.[DOI]
-
4. Miller KN, Victorelli SG, Salmonowicz H, Dasgupta N, Liu T, Passos JF, et al. Cytoplasmic DNA: Sources, sensing, and role in aging and disease. Cell. 2021;184(22):5506-5526.[DOI]
-
7. Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, et al. Chromothripsis from DNA damage inmicronuclei. Nature. 2015;522:179-184.[DOI]
-
9. MacKenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461-465.[DOI]
-
10. Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A. 2017;114(23):E4612-E4620.[DOI]
-
11. Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498:332-337.[DOI]
-
12. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu Rev Pathol Mech Dis. 2010;5:99-118.[DOI]
-
15. Islam MT, Tuday E, Allen S, Kim J, Trott DW, Holland WL, et al. Senolytic drugs, dasatinib and quercetin, attenuate adipose tissue inflammation, and ameliorate metabolic function in old age. Aging Cell. 2023;22(2):e13767.[DOI]
-
16. Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. Cellular senescence: Aging, cancer, and injury. Physiol Rev. 2019;99(2):1047-1078.[DOI]
-
17. Hoenicke L, Zender L. Immune surveillance of senescent cells: Biological significance in cancer- and non-cancer pathologies. Carcinogenesis. 2012;33(6):1123-1126.[DOI]
-
18. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134(4):657-667.[DOI]
-
20. Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016;7:11762.[DOI]
-
22. Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7(6):513-520.[DOI]
-
25. Wang Y, Luo J, Alu A, Han X, Wei Y, Wei X. cGAS-STING pathway in cancer biotherapy. Mol Cancer. 2020;19(1):136.[DOI]
-
27. Kong X, Zuo H, Huang HD, Zhang Q, Chen J, He C, et al. STING as an emerging therapeutic target for drug discovery: Perspectives from the global patent landscape. J Adv Res. 2023;44:119-133.[DOI]
-
28. Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell. 2013;154(1):47-60.[DOI]
-
29. Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, et al. Lysosome-mediated processing of chromatin in senescence. J Cell Biol. 2013;202(1):129-143.[DOI]
-
31. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339(6121):826-830.[DOI]
-
34. Vizioli MG, Liu T, Miller KN, Robertson NA, Gilroy K, Lagnado AB, et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020;34(5-6):428-445.[DOI]
-
36. Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, et al. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016;352(6283):353-358.[DOI]
-
38. Lan YY, Londoño D, Bouley R, Rooney MS, Hacohen N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. 2014;9(1):180-192.[DOI]
-
39. Han X, Chen H, Gong H, Tang X, Huang N, Xu W, et al. Autolysosomal degradation of cytosolic chromatin fragments antagonizes oxidative stress–induced senescence. J Biol Chem. 2020;295(14):4451-4463.[DOI]
-
40. Kovacs MT, Vallette M, Wiertsema P, Dingli F, Loew D, de Freitas Nader GP, et al. DNA damage induces nuclear envelope rupture through ATR-mediated phosphorylation of lamin A/C. Mol Cell. 2023;83(20):3659-3668.e10.[DOI]
-
41. Garvalov BK, Muhammad S, Dobreva G. Lamin B1 in cancer and aging. Aging. 2019;11(18):7336-7338.[DOI]
-
42. Gao H, Nepovimova E, Heger Z, Valko M, Wu Q, Kuca K, et al. Role of hypoxia in cellular senescence. Pharmacol Res. 2023;194:106841.[DOI]
-
44. Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. MBoC. 2012;23(11):2066-2075.[DOI]
-
46. Bonifacino JS, Neefjes J. Moving and positioning the endolysosomal system. Curr Opin Cell Biol. 2017;47:1-8.[DOI]
-
47. Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354-358.[DOI]
-
48. Andrews NW, Almeida PE, Corrotte M. Damage control: Cellular mechanisms of plasma membrane repair. Trends Cell Biol. 2014;24(12):734-742.[DOI]
-
49. Chapel A, Kieffer-Jaquinod S, Sagné C, Verdon Q, Ivaldi C, Mellal M, et al. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol Cell Proteom. 2013;12(6):1572-1588.[DOI]
-
52. Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, Wakita M, et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun. 2018;9:1249.[DOI]
-
53. Schatz G. The protein import machinery of mitochondria. Protein Sci. 1993;2(2):141-146.[DOI]
-
54. Chandel NS. Mitochondria as signaling organelles. BMC Biol. 2014;12(1):34.[DOI]
-
55. Khacho M, Slack RS. Mitochondrial activity in the regulation of stem cell self-renewal and differentiation. Curr Opin Cell Biol. 2017;49:1-8.[DOI]
-
58. Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Investig. 2018;128(4):1238-1246.[DOI]
-
59. Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5(5):e110.[DOI]
-
60. Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. Mitochondria in cell senescence: Is mitophagy the weakest link? EBioMedicine. 2017;21:7-13.[DOI]
-
62. Takahashi A, Okada R, Nagao K, Kawamata Y, Hanyu A, Yoshimoto S, et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun. 2017;8:15287.[DOI]
-
63. Zhao B, Liu P, Fukumoto T, Nacarelli T, Fatkhutdinov N, Wu S, et al. Topoisomerase 1 cleavage complex enables pattern recognition and inflammation during senescence. Nat Commun. 2020;11:908.[DOI]
-
69. Duan D, Shang M, Han Y, Liu J, Liu J, Kong SH, et al. EZH2–CCF–cGAS axis promotes breast cancer metastasis. Int J Mol Sci. 2022;23(3):1788.[DOI]
-
71. Scully R, Panday A, Elango R, Willis NA. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 2019;20:698-714.[DOI]
-
72. Pâques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks inSaccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63(2):349-404.[DOI]
-
74. Mirman Z, de Lange T. 53BP1: A DSB escort. Genes Dev. 2020;34(1-2):7-23.[DOI]
-
75. Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243-254.[DOI]
-
76. Fink LS, Roell M, Caiazza E, Lerner C, Stamato T, Hrelia S, et al. 53BP1 contributes to a robust genomic stability in human fibroblasts. Aging. 2011;3(9):836-845.[DOI]
-
77. Seluanov A, Mittelman D, Pereira-Smith OM, Wilson JH, Gorbunova V. DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc Natl Acad Sci U S A. 2004;101(20):7624-7629.[DOI]
-
80. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):e301.[DOI]
-
81. Wiley CD, Schaum N, Alimirah F, Lopez-Dominguez JA, Orjalo AV, Scott G, et al. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci Rep. 2018;8:2410.[DOI]
-
82. Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, et al. Integration of interferon-α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424(6948):516-523.[DOI]
-
83. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788-792.[DOI]
-
84. Barber GN. STING: Infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760-770.[DOI]
-
86. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380-384.[DOI]
-
88. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674-678.[DOI]
-
89. Buss H, Dörrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. Constitutive and Interleukin-1-inducible Phosphorylation of p65 NF-κB at Serine 536 Is Mediated by Multiple Protein Kinases Including IκB Kinase (IKK)-α, IKKβ, IKKϵ, TRAF Family Member-associated (TANK)-binding Kinase 1 (TBK1), and an Unknown Kinase and Couples p65 to TATA-binding Protein-associated Factor II31-mediated Interleukin-8 Transcription. J Biol Chem. 2004;279(53):55633-55643.
-
90. Li T, Chen ZJ. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215(5):1287-1299.[DOI]
-
91. Brzostek-Racine S, Gordon C, Van Scoy S, Reich NC. The DNA damage response induces IFN. J Immunol. 2011;187(10):5336-5345.[DOI]
-
94. Tasdemir N, Banito A, Roe JS, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, et al. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 2016;6(6):612-629.[DOI]
-
95. Birch J, Gil J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020;34(23-24):1565-1576.[DOI]
-
96. Chandra A, Rajawat J. Skeletal aging and osteoporosis: Mechanisms and therapeutics. Int J Mol Sci. 2021;22(7):3553.[DOI]
-
97. Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020;18(1):e3000599.[DOI]
-
98. Wang Y, Wang L, Wen X, Hao D, Zhang N, He G, et al. NF-κB signaling in skin aging. Mech Ageing Dev. 2019;184:111160.[DOI]
-
100. Aguado J, Chaggar HK, Gómez-Inclán C, Shaker MR, Leeson HC, Mackay-Sim A, et al. Inhibition of the cGAS-STING pathway ameliorates the premature senescence hallmarks of Ataxia-Telangiectasia brain organoids. Aging Cell. 2021;20(9):e13468.[DOI]
-
101. Gao M, He Y, Tang H, Chen X, Liu S, Tao Y. cGAS/STING: Novel perspectives of the classic pathway. Mol Biomed. 2020;1(1):7.[DOI]
-
102. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016;167(6):1540-1554.e12.[DOI]
-
103. Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25(20):2125-2136.[DOI]
-
104. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656-660.[DOI]
-
105. Chen Y, Wang L, Jin J, Luan Y, Chen C, Li Y, et al. p38 inhibition provides anti–DNA virus immunity by regulation of USP21 phosphorylation and STING activation. J Exp Med. 2017;214(4):991-1010.[DOI]
-
106. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response‐independent regulator of the senescence‐associated secretory phenotype. EMBO J. 2011;30(8):1536-1548.[DOI]
-
109. Alqahtani S, Alqahtani T, Venkatesan K, Sivadasan D, Ahmed R, Sirag N, et al. SASP modulation for cellular rejuvenation and tissue homeostasis: Therapeutic strategies and molecular insights. Cells. 2025;14(8):608.[DOI]
-
111. Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019-1031.[DOI]
-
113. Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133(6):1006-1018.[DOI]
-
115. Malaquin N, Rodier F. Dynamic and scalable assessment of the senescence-associated secretory phenotype (SASP). In: Cellular senescence and aging. Amsterdam: Elsevier; 2024. p. 181-195. [[DOI]
-
116. Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132(3):363-374.[DOI]
-
118. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978-990.[DOI]
-
119. Gardner SE, Humphry M, Bennett MR, Clarke MCH. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1α–dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol. 2015;35(9):1963-1974.[DOI]
-
121. Metze K, Adam R, Florindo JB. The fractal dimension of chromatin - a potential molecular marker for carcinogenesis, tumor progression and prognosis. Expert Rev Mol Diagn. 2019;19(4):299-312.[DOI]
-
122. Almassalha LM, Tiwari A, Ruhoff PT, Stypula-Cyrus Y, Cherkezyan L, Matsuda H, et al. The global relationship between chromatin physical topology, fractal structure, and gene expression. Sci Rep. 2017;7:41061.[DOI]
-
123. Metze K. Fractal dimension of chromatin and cancer prognosis. Epigenomics. 2010;2(5):601-604.[DOI]
-
124. Bedin V, Adam RL, de Sá BC, Landman G, Metze K. Fractal dimension of chromatin is an independent prognostic factor for survival in melanoma. BMC Cancer. 2010;10:260.[DOI]
-
125. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830-842.[DOI]
-
126. Bergsmedh A, Szeles A, Henriksson M, Bratt A, Folkman MJ, Spetz AL, et al. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc Natl Acad Sci U S A. 2001;98(11):6407-6411.[DOI]
-
128. Braig M, Schmitt CA. Oncogene-induced senescence: Putting the brakes on tumor development. Cancer Res. 2006;66(6):2881-2884.[DOI]
-
129. Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A. 2015;112(50):15408-15413.[DOI]
-
130. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989-2003.[DOI]
-
131. Abe T, Harashima A, Xia T, Konno H, Konno K, Morales A, et al. STING recognition of cytoplasmic DNA instigates cellular defense. Mol Cell. 2013;50(1):5-15.[DOI]
-
132. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515-518.[DOI]
-
134. Ma Z, Sun Q, Zhang C, Zheng Q, Liu Y, Xu H, et al. RHOJ induces epithelial-to-mesenchymal transition by IL-6/STAT3 to promote invasion and metastasis in gastric cancer. Int J Biol Sci. 2023;19(14):4411-4426.[DOI]
-
135. Jia C, Wang G, Wang T, Fu B, Zhang Y, Huang L, et al. Cancer-associated Fibroblasts induce epithelial-mesenchymal transition via the Transglutaminase 2-dependent IL-6/IL6R/STAT3 axis in Hepatocellular Carcinoma. Int J Biol Sci. 2020;16(14):2542-2558.[DOI]
-
137. Rodriguez JA, Huerta-Yepez S, Law IKM, Baay-Guzman GJ, Tirado-Rodriguez B, Hoffman JM, et al. Diminished expression of corticotropin-releasing hormone receptor 2 in human colon cancer promotes tumor growth and epithelial-to-mesenchymal transition via persistent interleukin-6/Stat3 signaling. Cell Mol Gastroenterol Hepatol. 2015;1(6):610-630.[DOI]
-
138. Li Z, Low V, Luga V, Sun J, Earlie E, Parang B, et al. Tumor-produced and aging-associated oncometabolite methylmalonic acid promotes cancer-associated fibroblast activation to drive metastatic progression. Nat Commun. 2022;13:6239.[DOI]
-
141. Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q, et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Sig Transduct Target Ther. 2021;6:218.[DOI]
-
143. Ortiz-Montero P, Londoño-Vallejo A, Vernot JP. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal. 2017;15(1):17.[DOI]
-
144. Shi J, Feng J, Xie J, Mei Z, Shi T, Wang S, et al. Targeted blockade of TGF-β and IL-6/JAK2/STAT3 pathways inhibits lung cancer growth promoted by bone marrow-derived myofibroblasts. Sci Rep. 2017;7:8660.[DOI]
-
145. Li J, Kong X, Jiang S, Liao W, Zhang Z, Song J, et al. miR-627/HMGB1/NF-κB regulatory loop modulates TGF-β1-induced pulmonary fibrosis. J Cell Biochem. 2019;120(3):2983-2993.[DOI]
-
148. Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Cunha GR, Hein P, et al. Carcinoma-associated fibroblasts stimulate tumor progression of initiated human epithelium. Breast Cancer Res. 2000;2(S1):S.19.[DOI]
-
150. Shekhar MP, Werdell J, Santner SJ, Pauley RJ, Tait L. Breast stroma plays a dominant regulatory role in breast epithelial growth and differentiation: Implications for tumor development and progression. Cancer Res. 2001;61(4):1320-1326.
-
151. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68(3):918-926.[DOI]
-
152. Henriksson ML, Edin S, Dahlin AM, Oldenborg PA, Öberg Å, Van Guelpen B, et al. Colorectal cancer cells activate adjacent fibroblasts resulting in FGF1/FGFR3 signaling and increased invasion. Am J Pathol. 2011;178(3):1387-1394.[DOI]
-
154. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860-867.[DOI]
-
156. Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol. 2008;28(16):5014-5026.[DOI]
-
157. Sun W, Li Y, Chen L, Chen H, You F, Zhou X, et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A. 2009;106(21):8653-8658.[DOI]
-
161. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147(6):1393-1404.[DOI]
-
162. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J Biomed Sci. 2019;26(1):78.[DOI]
-
163. Yoshimura A, Wakabayashi Y, Mori T. Cellular and molecular basis for the regulation of inflammation by TGF-β. J Biochem. 2010;147(6):781-792.[DOI]
-
165. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGF-β. Nat Rev Immunol. 2010;10:554-567.[DOI]
-
167. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9(6):632-640.[DOI]
-
168. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGF-β in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179-189.[DOI]
-
170. Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-β controls T helper type 1 cell development through regulation of natural killer cell interferon-γ. Nat Immunol. 2005;6(6):600-607.[DOI]
-
171. He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169(6):1000-1011.[DOI]
-
173. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397-408.[DOI]
-
174. Nelson G, Kucheryavenko O, Wordsworth J, von Zglinicki T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech Ageing Dev. 2018;170:30-36.[DOI]
-
175. Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell. 2012;11(2):345-349.[DOI]
-
178. Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD. The clinical potential of senolytic drugs. J Am Geriatr Soc. 2017;65(10):2297-2301.[DOI]
-
180. Victorelli S, Lagnado A, Halim J, Moore W, Talbot D, Barrett K, et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J. 2019;38(23):EMBJ2019101982.[DOI]
-
187. Wang R, Yu Z, Sunchu B, Shoaf J, Dang I, Zhao S, et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell. 2017;16(3):564-574.[DOI]
-
188. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015;17:1205-1217.[DOI]
-
191. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97-101.[DOI]
-
194. Swift ML, Zhou R, Syed A, Moreau LA, Tomasik B, Tainer JA, et al. Dynamics of the DYNLL1–MRE11 complex regulate DNA end resection and recruitment of Shieldin to DSBs. Nat Struct Mol Biol. 2023;30:1456-1467.[DOI]
-
195. Zhao Y, Simon M, Seluanov A, Gorbunova V. DNA damage and repair in age-related inflammation. Nat Rev Immunol. 2023;23:75-89.[DOI]
-
196. Sandoval C, Nisson K, Fregoso OI. HIV-1 Vpr-induced DNA damage activates NF-κB through ATM-NEMO independent of cell cycle arrest. mBio. 2024;15(10):e00240-e00224.[DOI]
-
197. Zhao J, Zhang L, Lu A, Han Y, Colangelo D, Bukata C, et al. ATM is a key driver of NF-κB-dependent DNA-damage-induced senescence, stem cell dysfunction and aging. Aging. 2020;12(6):4688-4710.[DOI]
-
198. Kong SH, Ma L, Yuan Q, Liu X, Han Y, Xiang W, et al. Inhibition of EZH2 alleviates SAHA-induced senescence-associated secretion phenotype in small cell lung cancer cells. Cell Death Discov. 2023;9:289.[DOI]
-
202. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638-642.[DOI]
-
203. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109(3):335-346.[DOI]
-
204. Goldman JW, Shi P, Reck M, Paz-Ares L, Koustenis A, Hurt KC. Treatment rationale and study design for the JUNIPER study: A randomized phase III study of abemaciclib with best supportive care versus erlotinib with best supportive care in patients with stage IV non-small-cell lung cancer with a detectable KRAS mutation whose disease has progressed after platinum-based chemotherapy. Clin Lung Cancer. 2016;17(1):80-84.
-
208. Chitikova ZV, Gordeev SA, Bykova TV, Zubova SG, Pospelov VA, Pospelova TV. Sustained activation of DNA damage response in irradiated apoptosis-resistant cells induces reversible senescence associated with mTOR downregulation and expression of stem cell markers. Cell Cycle. 2014;13(9):1424-1439.[DOI]
-
211. Cantelli G, Crosas-Molist E, Georgouli M, Sanz-Moreno V. TGFΒ-induced transcription in cancer. Semin Cancer Biol. 2017;42:60-69.[DOI]
-
212. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98(21):12072-12077.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Xiang W, Wang S, Pan Y, Cong X, Lu J, Zhang Y. Cytoplasmic chromatin fragments: Divergent roles in senescence and cancer. Ageing Cancer Res Treat. 2026;3:202523. https://doi.org/10.70401/acrt.2026.0016
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Characteristics of CCFs
- 3. CCFs Activate the cGAS-STING Pathway
- 4. The Role of CCFs in Cellular Senescence
- 5. The Multiple Roles of CCFs in Cancer
- 6. Therapeutic Strategies
- 7. Perspective
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- Copyright
Science Exploration Style
Xiang W, Wang S, Pan Y, Cong X, Lu J, Zhang Y. Cytoplasmic chromatin fragments: Divergent roles in senescence and cancer. Ageing Cancer Res Treat. 2026;3:202523. https://doi.org/10.70401/acrt.2026.0016
copy
Share Link
copy