Targeting cellular senescence: a promising anticancer strategy
Lei Wang

Yajun Duan
Xiaoyu Ma
Lida Peng
Minxian Qian
*
,
Zhongyuan Wang
*
*Correspondence to:
Minxian Qian, State Key Laboratory of Natural Medicines, Department of Life Science and Technology, China Pharmaceutical University, Nanjing 211198, Jiangsu, China.
E-mail: qmx@cpu.edu.cn
Zhongyuan Wang, State Key Laboratory of Natural Medicines, Department of Life Science and Technology, China Pharmaceutical University, Nanjing 211198, Jiangsu, China. E-mail: wangzhongyuan@cpu.edu.cn
Zhongyuan Wang, State Key Laboratory of Natural Medicines, Department of Life Science and Technology, China Pharmaceutical University, Nanjing 211198, Jiangsu, China. E-mail: wangzhongyuan@cpu.edu.cn
Ageing Cancer Res Treat. 2026;3:202601. 10.70401/acrt.2026.0017
Received: January 09, 2026Accepted: April 01, 2026Published: April 03, 2026
This article belongs to the Special lssue Aging and the Tumor Microenvironment
Abstract
Cellular senescence results in a stable growth arrest of cells that is elicited by endogenous or exogenous stresses. Senescent cells release a broad spectrum of proinflammatory factors, a collective signature known as the senescence-associated secretory phenotype. Senescence modulates tumor initiation and progression via a context-dependent dual role, acting as both a tumor suppressor and a promoter. Triggering cellular senescence restricts cancer progression and enhances therapy outcomes, however, the accumulation of senescent cells drives tumor progression, recurrence, and metastasis. Thus, selective targeting of senescent cells holds the potential to develop novel therapeutic approaches to combat cancer. In this review, we delineate the regulatory mechanisms controlling cellular senescence in tumors. We also summarize the emerging senescence-targeting agents and their utility in anticancer intervention. Given the complex role of senescent cells in cancer, a comprehensive understanding of these processes will create new opportunities for effective anticancer therapy.
Keywords
Cellular senescence, senescence-associated secretory phenotype, senolytics, senomorphics, anticancer therapy
1. Introduction
Cellular senescence is defined as a durable state of cell cycle arrest, distinguished by nuclear and morphological changes, shifted metabolic fluxes, altered transcriptomic profiles, dysregulated autophagic activity, large-scale chromatin remodeling[1], and induction of the senescence-associated secretory phenotype (SASP)[2]. Senescence can be triggered by various stressors, including telomere attrition, persistent DNA damage, oncogene activation, oxidative stress or other cell extrinsic stimuli[3]. While cellular senescence exerts a beneficial physiological function in supporting development and blocking fibrosis and tumorigenesis, aberrant accumulation of senescent cells drives multiple age-associated comorbidities, like cardiovascular disorders and neurodegenerative disorders, and accelerates tumor progression through shaping a pro-inflammatory niche[4].
Given the context-dependent and paradoxical roles of cellular senescence in tumorigenesis and tumor progression, developing approaches to effectively harness cellular senescence’s tumor-suppressive effects while mitigating its pro-tumorigenic outcomes to improve anticancer therapy stands as an emerging and promising direction for senescence-targeted cancer therapeutics. For example, the “One-two Punch” therapeutic approach is engineered to first elicit tumor cell senescence, and subsequently target the newly formed senescent tumor cells through senotherapies[5]. Current senotherapies are developed to target senescent cells via two distinct avenues: senolytics, which selectively eradicate senescent cells, and senomorphics, which aim to attenuate the pro-inflammatory and pro-pathogenic SASP. This review outlines the regulatory mechanisms of tumor-associated cellular senescence and summarizes the emerging senescence-based therapies for cancer treatment.
2. Inducers of Cell Senescence in Tumors
Cellular senescence can be triggered by multiple distinct stimuli, where telomere shortening induces replicative senescence (RS), oncogene activation drives oncogene-induced senescence (OIS), and anticancer therapies elicit therapy-induced senescence (TIS).
Senescent cells accumulate within the tumor microenvironment (TME) throughout cancer initiation, progression, and in response to therapeutic interventions. Despite evading RS, tumor cells retain susceptibility to OIS and TIS. In the context of cancer initiation, the aberrant activation of oncogenes (e.g., RAS or BRAF) or biallelic loss of tumor suppressors (e.g., PTEN) promotes unconstrained cell growth and proliferation[6]. Aberrant hyperproliferation of cells induces DNA replication stress and activates the DNA damage response (DDR). Then, the DDR activates the p53-mediated signaling network, which leads to the upregulation of p16INK4a and p21CIP1, thereby triggering cellular senescence and cell cycle arrest[7]. This process acts as an intrinsic barrier to constrain early tumor progression.
Anticancer interventions can drive both malignant cells and neighboring normal cells into TIS. Genotoxic chemotherapeutic drugs, such as alkylating agents (e.g., cisplatin[8,9], temozolomide[10,11], and cyclophosphamide[8]), topoisomerase inhibitors (e.g., etoposide[12-14] and doxorubicin[15,16]), antimicrotubule agents (e.g., paclitaxel[17,18], docetaxel[19] and vincristine[20]), and antimetabolites (e.g., gemcitabine[21,22] and methotrexate[23]), and radiotherapy (exposure to γ-irradiation or X-ray)[24,25], achieve their anticancer effects by either inhibiting mitosis or inducing DNA damage. Beyond their direct cytotoxic effects on cancer cells, these therapeutic modalities simultaneously trigger cellular senescence. These anticancer therapies elicit DNA damage and initiate DDR signaling cascades mediated by the ATM/CHK2 and ATR/CHK1 axes, which, in turn, drive p53 activation[26]. The severity of treatment-induced DNA damage and the status of p53 are key determinants of cellular fate, with low-dose chemotherapy driving senescence and high-dose chemotherapy favoring apoptotic clearance of damaged cells[27].
In addition, various targeted therapies can also induce cellular senescence. Cancer cells commonly rely on hyperactivated CDK4/6 signaling to support their proliferation. Notably, CDK4/6 inhibitors (e.g., palbociclib[28-31], abemaciclib[32,33], and ribociclib[34-36]), clinically used for cancer treatment, induce G1-phase cell cycle arrest and cellular senescence by blocking Rb phosphorylation and thereby inhibiting E2F transcriptional activity[37]. In addition, Aurora kinases (AURK) and polo-like kinase (PLK) are essential serine/threonine kinases required for proper spindle assembly and mitotic progression[38,39]. These kinases are frequently overexpressed in multiple tumors. AURK inhibitors such as alisertib, barasertib, danusertib, and tozasertib have been identified as potent inducers of cellular senescence in cancer cells[5,40,41]. The PLK1 inhibitor BI2536 enhances the effect of paclitaxel on breast cancer cells[42], and the PLK4 inhibitor CFI-400945 induces senescence in hepatocellular carcinoma, through the activation of the cyclic GMP–AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway[43]. Poly (ADP-ribose) polymerase (PARP) plays a crucial role in regulating DNA repair and genomic stability, and its inhibitors (e.g., veliparib and olaparib) induce senescence in breast cancer cells, acute myeloid leukemia cells, ovarian, and prostate cancer cells[44-47]. Mitogen-activated protein kinase kinase (MEK) constitutes a key component of the RAS-RAF-MEK-ERK signaling pathway, with alterations in its structure and expression levels being closely associated with tumorigenesis. BRAF inhibitor vemurafenib and MEK inhibitor trametinib demonstrate significant therapeutic efficacy in melanoma whilst inducing cancer cell senescence[48].
3. Characteristics of Senescent Cells
Senescent cells exhibit unique phenotypic features that are associated with irreversible cell cycle arrest and alterations of morphology, molecules, and metabolism, and they also display significant heterogeneity.
Upon the onset of cellular senescence, DDR signaling drives p53 activation. Accordingly, p53 upregulates the expression of its target genes p16INK4a and p21CIP1, which, in turn, inhibit cyclin-CDK complexes, impede division, and mediate senescent cell cycle arrest at the G1 or G2/M phase[49,50]. In addition, p16INK4a and p21CIP1 can be induced in a p53-independent manner. For example, peroxisome proliferator-activated receptor γ (PPARγ) promotes p16INK4a expression[51], while transforming growth factor-β (TGF-β) upregulates p21CIP1 expression[52]. Notably, p21CIP1 plays a critical role in driving senescence entry and initiating cell cycle arrest; conversely, p16INK4a is upregulated in later senescence stages and functions to sustain long-term cell cycle arrest in senescent cells[53,54].
Another hallmark of senescent cells is pronounced cellular morphological remodeling, encompassing cellular enlargement, flattening, and enhanced dispersion[55]. Following senescence induction, cells accumulate cofilin-1 and hyperphosphorylated microtubule-associated protein tau, driving the formation of stiffened cytoskeletal structures[56]. Furthermore, impaired lysosome function results in the accumulation of cytoplasmic particles in senescent cells[57]. Additionally, senescent cells also exhibit expanded nuclei, a structural alteration that is tightly coupled to senescence-associated extensive chromatin reorganization and nuclear lamina remodeling[58]. Senescence-associated heterochromatic foci (SAHF) and senescence-associated distension of satellites (SADS) are two specific DNA structures accumulated in senescent cells. SAHF is characterized by enrichment of repressive epigenetic modifications, particularly the canonical heterochromatic histone marks H3K9me3, which inhibit the expression of cell cycle-related genes[59]. Unlike the condensed heterochromatic architecture of SAHF, SADS are defined as the decondensation of pericentromeric satellite sequences[60]. Moreover, senescence induces autophagic degradation of the nuclear lamina structural component lamin B1 and promotes the formation of DNA damage marker γH2AX-positive cytoplasmic chromatin fragments (CCF) and SAHF[61].
Cellular senescence is also accompanied by extensive epigenetic modifications, such as global DNA hypomethylation, mainly in heterochromatin, and moderate hypermethylation in euchromatin, that silences cell cycle-related genes[59]. Moreover, histone modifications, including acetylation, methylation, and phosphorylation, mediated by epigenetic regulators, can reshape chromatin accessibility to modulate gene expression. For example, the diminished expression of histone deacetylase SIRT1 in senescent cells removes the epigenetic constraint on SASP gene loci, leading to their transcriptional activation[62]. These dynamic, reversible epigenetic mechanisms highlight the core function of epigenetic regulation in orchestrating the cellular senescence program.
Importantly, senescent cells display a suite of reprogrammed metabolic features: elevated glycolytic flux[63], accumulated intracellular lipids[64], and lipofuscin[65], altered amino acid metabolism[66], and disrupted redox homeostasis[67]. Notably, β-galactosidase activity is increased during the process of cellular senescence, and senescence-associated β-galactosidase (SA-β-gal) activity has been widely used to identify senescent cells in culture and tissues[68].
In addition, senescent cells secrete pro-inflammatory cytokines, chemokines, and extracellular matrix proteases, collectively categorized as SASP[2]. The SASP primarily recruits immune cells to eliminate senescent cells[69-72]. Paradoxically, when senescent cells evade immune clearance, persistent secretion of the SASP, like IL-6 and IL-8, induces chronic inflammation and creates a growth-stimulatory and immunosuppressive niche to support tumor development and enhance therapeutic resistance[9,73]. The Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-κB)[74], GATA4[75], and cGAS-STING[76] pathways play critical roles in the regulation of SASP transcription.
Furthermore, cellular senescence is not a uniform or static process but instead exhibits pronounced heterogeneity, driven by the combined effects of multiple factors, including variations in inducing stimuli (such as DNA damage, oxidative stress, and telomere attrition), intrinsic cellular properties (e.g., lineage and differentiation state), and microenvironmental cues[77]. Consistent with this, high-resolution studies based on single-cell transcriptomics have demonstrated that, even under the same inducing conditions, senescent cells display substantial differences in gene expression patterns, metabolic states, and SASP profiles. This variability exists not only across different cell types (such as fibroblasts, epithelial cells, and immune cells), but also within the same cell population, indicating that senescence is not driven by a single pathway but instead involves multiple molecular trajectories and distinct state branches[78]. Overall, the heterogeneity of cellular senescence represents a fundamental biological feature with important implications for aging-related diseases and the development of targeted therapeutic strategies.
Taken together, it is important to emphasize that, to date, due to the highly heterogeneous senescent cells, the concurrent use of multiple biomarkers remains necessary for reliable detection of cellular senescence (Table 1).
Table 1. Main biomarkers commonly used to identify senescent cancer cells.
| Characteristics | Markers | References |
| Cell cycle arrest | p16INK4a, p21CIP1, p53, etc. | [79-81] |
| Morphological changes | SAHF, SADS, lamin B1, etc. | [80,82,83] |
| Metabolic features | SA-β-gal, lipofuscin, etc. | [65,84] |
| DNA damage | γH2AX, etc. | [85] |
| SASP | IL-6, IL-8, etc. | [86,87] |
SAHF: senescence-associated heterochromatic foci; SADS: senescence-associated distension of satellites; SA-β-gal: senescence-associated β-galactosidase; γH2AX: phosphorylated histone H2AX (gamma-H2AX); SASP: senescence-associated secretory phenotype.
4. Paradoxical Dual Roles of Cellular Senescence in Cancer
Cellular senescence plays a dual, context-dependent role in tumorigenesis, exerting both tumor-suppressive and tumor-promoting effects.
As mentioned above, oncogenic activation that drives cancer initiation induces cell cycle arrest, a state designated as OIS. Both OIS and TIS act as potent tumor-suppressive barriers that inhibit tumorigenesis via triggering cell cycle arrest and promoting the immune elimination of preneoplastic cells[4,88]. On one hand, the SASP secreted by senescent cells can potentiate cellular senescence through an autocrine signaling axis. Specifically, during TIS in tumor cells, canonical NF-κB signaling is activated to drive the expression of two core SASP cytokines, IL-6 and IL-8, which then act autocrinally to boost p21 and p16 expression and thereby accelerate the establishment of irreversible senescence arrest[71]. On the other hand, the SASP recruits various immune cells to eliminate potential cancerous cells. In ER+ breast cancer, SASP induced by the Kv11.1 channel activates CD4+ Th1 cells and memory T cells, leading to tumor necrosis factor alpha (TNF-α)-mediated elimination of senescent tumor cells[69]. Senescent liver carcinoma cells produce pro-immunogenic SASP, including IL-1β and TNF-α, that either drive M1 polarization of macrophages or recruit NK cells and T cells, collectively favoring the subsequent elimination of these senescent tumor cells[70,72,89].
Although senescence induction serves as an effective antitumor strategy, persistent senescent cells in the TME foster a pro-tumorigenic niche that drives tumor progression, malignant aggressiveness, and resistance to anticancer therapies. The secretion of IL-6, IL-8, IL-1α, IL-1β, and vascular endothelial growth factor (VEGF) by senescent cells facilitates angiogenesis and tumorigenesis. Additionally, the elevated expression of IL-6[90], CCL-18[91], CXCR2-binding chemokines[92], or IL-33[93] triggers the recruitment of immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs) and Tregs, or upregulates immune checkpoint proteins including PD-L1[94] to establish robust immunosuppressive microenvironments, thereby promoting tumor growth and progression. Furthermore, senescent cancer cells induce epithelial-mesenchymal transition (EMT) and promote tumor invasion via cathepsin B, matrix metalloproteases (MMPs), IL-6, and various growth factors[73,95]. Beyond paracrine regulation, senescent cancer cells can overcome cell cycle arrest and re-enter proliferation to drive tumor recurrence and chemotherapy resistance[96,97].
Cellular senescence is not limited to tumor cells but also occurs across multiple stromal compartments within the TME, including cancer-associated fibroblasts (CAFs), tumor-associated endothelial cells, and tissue-specific mesenchymal populations[98]. These senescent stromal cells promote tumor progression through the SASP, which modulates extracellular matrix remodeling, inflammation, and intercellular communication[99]. Senescence in CAFs leads to robust SASP production, promoting tumor cell proliferation, invasion, metabolic reprogramming, therapeutic resistance, and immunosuppression[100]. Similarly, senescent endothelial cells contribute to tumor progression by altering vascular function and secreting pro-angiogenic and pro-metastatic factors, facilitating angiogenesis and metastatic dissemination[101]. In the liver, senescent hepatic stellate cells (HSCs) promote the progression of non-alcoholic steatohepatitis to hepatocellular carcinoma by secreting IL-1β[102]. In this context, targeting stromal senescence may suppress tumor-promoting niche formation more broadly than approaches focused solely on tumor cells. However, stromal senescence is not uniformly detrimental; some senescent stromal populations may transiently support tissue repair or limit injury, and their indiscriminate clearance may result in adverse outcomes, such as fibrosis[103]. Therefore, cell type–specific and context-dependent strategies are essential to selectively modulate stromal senescence while preserving its beneficial functions.
Building on the pivotal role of senescent stromal cells in shaping the TME, these cells also extensively remodel the extracellular matrix (ECM), which serves as a critical but frequently overlooked factor driving tumor progression[104]. Senescent cells drive matrix degradation, aberrant collagen deposition, and altered crosslinking dynamics via the secretion of MMPs, pro-inflammatory cytokines, growth factors, and extracellular vesicles, leading to marked alterations in ECM architecture and mechanics, including increased stiffness and fiber realignment, thereby disrupting tissue homeostasis and establishing a tumor-permissive microenvironment[104-106]. Mechanistically, these effects are mediated by the activation of mechanotransduction pathways, such as integrin–FAK–YAP/TAZ signaling, enhancing tumor cell proliferation, survival, and EMT[107,108]. Moreover, remodeled ECM structures provide physical tracks that facilitate tumor cell invasion and metastasis, while simultaneously impairing immune cell infiltration and fostering an immunosuppressive microenvironment[109,110]. These ECM changes also impede effective drug delivery, and thus contribute to therapeutic resistance[111,112]. Importantly, senescence-associated ECM remodeling may further exacerbate these pro-tumorigenic consequences, suggesting the existence of a feed-forward loop between cell senescence, matrix dysfunction, and tumor progression[104,108,113]. Together, these findings indicate that senescence-associated ECM remodeling is a crucial driver of tumor initiation and progression.
5. Strategies to Target Senescent Cells for Cancer Therapy
Notably, lingering senescent cells in the TME after the aforementioned senescence-inducing therapies have detrimental effects on tumor progression, metastasis, and drug resistance by SASP secretion and senescence reversibility. Consequently, targeting senescent cells, either by directly eliminating them (senolytics)[114,115] and/or regulating the SASP (senomorphics)[116], emerges as a promising strategy to improve therapeutic outcomes and alleviate the adverse consequences (Figure 1).
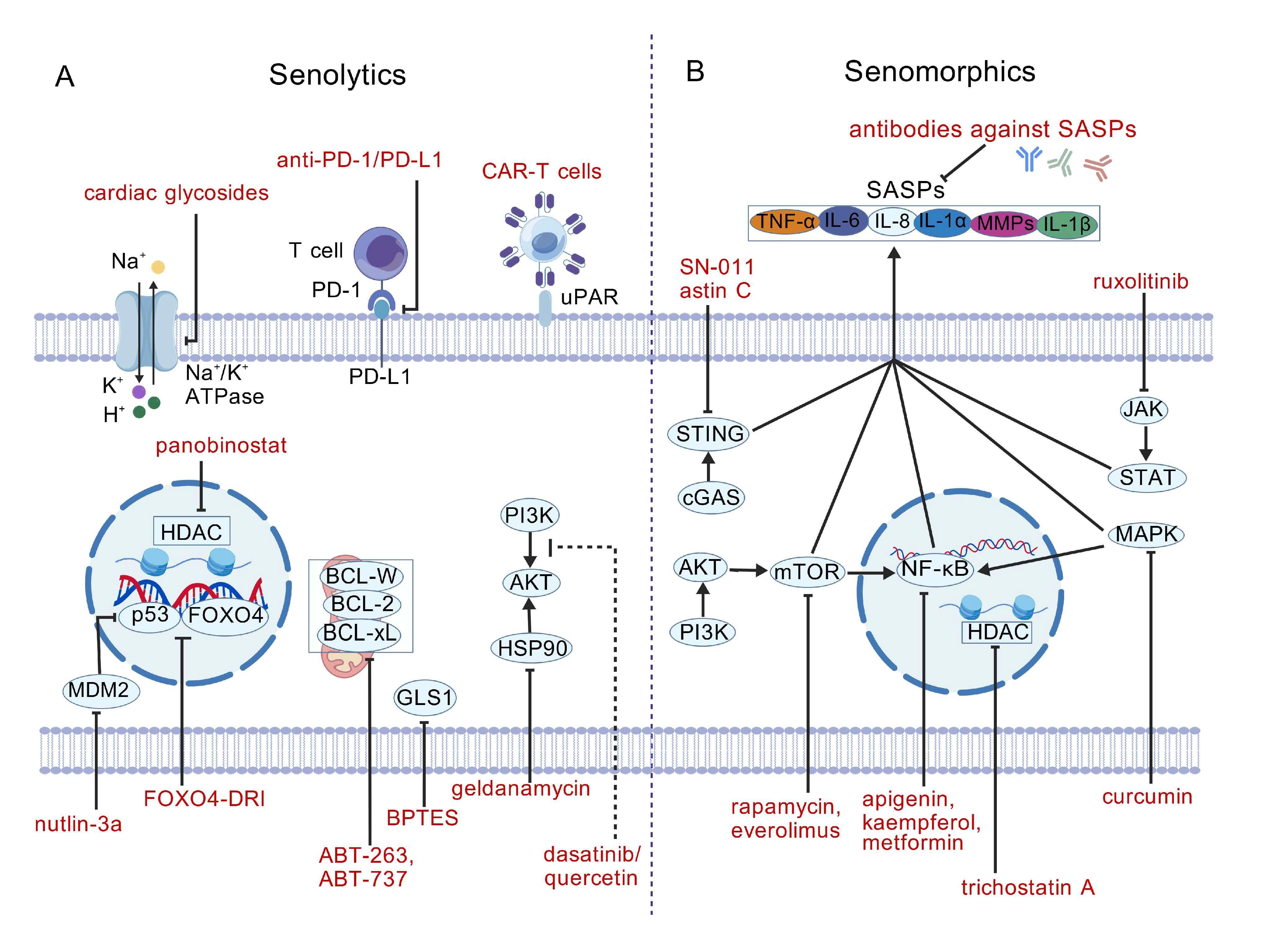
Figure 1. Targeting senescent cells through senolytics and senomorphics. (A) Senolytics induce apoptosis in senescent cells by targeting various molecules or pathways, which include MDM2 inhibitors (nutlin-3a), BCL-2 family member inhibitors (ABT-263 and ABT-737), PI3K/AKT inhibitors (dasatinib + quercetin), GLS1 inhibitors (BPTES), HSP90 inhibitors (geldanamycin), Na+/K+ ATPase inhibitors (cardiac glycosides) and FOXO4-p53 inhibitors (FOXO4-D-retro-inverso (DRI) peptides). Moreover, immune-mediated clearance of senescent cells can be potentiated via anti-PD-1/PD-L1 antibody therapy or CAR-T cells; (B) Senomorphics, including NF-κB pathway inhibitors (apigenin, kaempferol, and metformin), MAPK pathway inhibitors (curcumin), PI3K/AKT/mTOR pathway inhibitors (rapamycin and everolimus), cGAS/STING pathway inhibitors (SN-011 and astin C), JAK/STAT pathway inhibitors (ruxolitinib), and inducers of SASP reprogramming (HDAC inhibitors such as trichostatin A) suppress the release of SASP factors by senescent cells. In addition, antibodies against SASP components (e.g., IL-6, IL-8, IL-1α, IL-1β, TNF-α, and MMPs) antagonize the biological effects of SASP factors. Created with BioGDP.com[117]. MDM2: mouse double minute 2 homolog; BCL-2: B-cell lymphoma 2; GLS1: glutaminase1; CAR-T: chimeric antigen receptor T cells; MAPK: mitogen-activated protein kinases; cGAS: cyclic GMP-AMP synthase; STING: stimulator of interferon genes; SASP: senescence-associated secretory phenotype; MMPs: matrix metalloproteases; DRI: D-retro-inverso (peptides); CAR-T cells: chimeric antigen receptor T cells; NF-κB: Nuclear Factor kappa-light-chain-enhancer of activated B cells; JAK/STAT: Janus kinase/signal transducers and activators of transcription; PI3K/AKT/mTOR: phosphatidylinositol 3-kinase/protein kinase-B/mammalian target of rapamycin; HDAC: histone deacetylase.
As the most intensively explored modality of senotherapies, senolytics exert their effects by selectively eliminating senescent cells and can be combined with other therapies for cancer treatment (Figure 1A). Due to the upregulation of anti-apoptotic B-cell lymphoma 2 (BCL-2) family proteins (e.g., BCL-2, BCL-W and BCL-xL), senescent cells acquire resistance to apoptotic cell death[118]. BH3 mimetics ABT-263[47,119,120] and ABT-737[121], that target both BCL-2 and BCL-XL, function as senolytics to induce apoptosis in senescent cells. Furthermore, inhibitors of p53-specific E3 ubiquitin ligase mouse double minute 2 homolog (MDM2), such as nutlin-3a, not only induce p53-dependent senescence but also exert senolytic activity by exacerbating the inherent DDR in senescent cells[122-124]. Induction of p53 nuclear exclusion by FOXO4 inhibition (FOXO4 D-retro-inverso (DRI) isoform peptides or peptidomimetics) also triggers apoptosis[125,126]. Moreover, senescent cells exhibit reduced intracellular pH because of lysosome membrane permeabilization[127]. Treatment with the glutaminase1 (GLS1) inhibitor BPTES blocks the neutralization of intracellular pH via glutaminolysis, thereby removing senescent cells[92]. Similarly, cardiac glycosides, including digoxin and ouabain, inhibit the Na+/K+ exchanger, leading to a rise in intracellular H+ concentrations and subsequent senescent cell death[128]. Additionally, a panel of flavonoids, including quercetin[115], fisetin, and curcumin[129], has also been demonstrated to possess senolytic properties. The combination of the tyrosine kinase inhibitor (TKI) dasatinib and quercetin (D + Q) holds promise for the clearance of senescent cells and improves clinical outcomes in patients with pulmonary fibrosis[115] and chronic kidney disease[130]. Another alternative approach for senescent cell clearance is immune cell-mediated elimination. Senescent cells upregulate immune checkpoint proteins, including PD-L1, to evade T cell surveillance[131]. Accordingly, anti-PD-L1/PD-1 therapy promotes the removal of senescent cells by enhancing T cell-mediated cytotoxicity[132,133]. In addition, chimeric antigen receptor (CAR)-T cells engineered to target the senescence-specific antigen urokinase-type plasminogen activator receptor (uPAR) can effectively ablate senescent cells[134,135]. Notably, despite the promising clinical potential, the senolytic therapies may be associated with certain toxic side effects, including platelet toxicity.
Senomorphics are another class of drugs targeting important properties (e.g., SASP) of senescent cells (Figure 1B). Unlike senolytics, senomorphics do not primarily aim to kill senescent cells; instead, they attenuate or reshape the harmful signaling output of these cells and may be safer in some scenarios. Based on mechanisms of action, senomorphics can be classified into three classes: (1) Inhibit upstream signaling pathways, including the DDR pathway, p38-mitogen-activated protein kinases (MAPK) pathway, Janus kinase (JAK)-signal transducers and activators of transcription (STAT) pathway, phosphatidylinositol 3-kinase (PI3K)-protein kinase-B (AKT)-mammalian target of rapamycin (mTOR) pathway, cGAS-STING pathway, NF-κB, or C/EBPβ signaling to suppress the transcription or secretion of SASP factors (e.g., rapamycin[116], metformin[136] and SN-011[137]); (2) Neutralizing antibodies against SASP components (e.g., IL-8 and IL-6) that interfere with the interaction between SASP factors and their receptors on target cells[138]; (3) Inducers of SASP reprogramming. SASP reprogramming does not globally suppress secretory activity but instead reshapes the composition and functional output of the SASP[139]. Mechanistically, this process involves transcriptional and epigenetic remodeling via chromatin regulators, as well as modulation of key signaling pathways, such as JAK–STAT, NF-κB, and MAPK, thereby shifting the SASP from a pro-inflammatory, tumor-promoting profile to a context-dependent state with altered cytokine and chemokine composition[140]. In contrast to broad SASP inhibition, which may impair beneficial processes such as tissue repair and immune-mediated clearance of senescent cells, SASP reprogramming enables selective modulation of secretory profiles[56]. For example, treating senescent breast cancer cells with the JAK2/STAT3 inhibitor ruxolitinib reduces the secretion of immunosuppressive factors such as IL-6, IL-10 and IL-13, while preserving immunostimulatory SASPs, including CCL5, ICAM-1, and MCP-1, thereby enhancing the immunogenic efficacy of alisertib (Aurora kinase inhibitor)[141]. Importantly, SASP reprogramming can reshape the TME by attenuating chronic inflammation while maintaining or promoting immune surveillance, including the recruitment and activation of cytotoxic immune cells such as NK cells and CD8+ T cells[142]. In cancer therapy, this property is particularly advantageous; for example, combining chemotherapy with JAK2 inhibitors or the CDK4/6 inhibitor palbociclib and the MEK inhibitor trametinib has been shown to reprogram the SASP toward a pro-immune phenotype, enhance immune cell recruitment, and facilitate tumor clearance[143].
Targeting senescent stromal cells also represents a promising therapeutic strategy in cancer. Therapy-induced senescent stromal cells contribute to tumor relapse and resistance, and their clearance by senolytics, such as BCL-2 family inhibitors (e.g., navitoclax/ABT-263), significantly attenuates these pro-tumorigenic effects and enhances therapeutic efficacy[100,115]. Notably, targeting senescent stromal cells can also improve the response to immunotherapy by relieving T cell suppression within the tumor microenvironment[144]. Emerging strategies further expand the therapeutic landscape; for instance, uPAR-directed chimeric antigen receptor T (CAR-T) cells have been shown to selectively eliminate senescent cells in vivo, including stromal populations, thereby improving disease outcomes[145]. Moreover, engineered nanovesicle-based delivery systems capable of simultaneously targeting tumor cells and senescent stromal cells have demonstrated enhanced antitumor efficacy in preclinical models[30].
Given the important role of senescence-associated ECM remodeling in driving tumor progression, targeting the senescent ECM has become a complementary and prospective strategy to further reprogram the TME. One approach focuses on normalizing ECM structure and mechanics by reducing aberrant matrix deposition and stiffness, such as with collagen-degrading enzymes and the lysyl oxidase (LOX)/lysyl oxidase-like (LOXL) family inhibitors (e.g., β-aminopropionitrile), enhancing drug penetration and therapeutic outcomes[146,147]. Additionally, targeting key mediators of ECM interactions, including MMPs and CD44/HA signaling pathways, may suppress tumor growth, invasion, and metastasis[148,149]. Senescence-targeting therapies such as senolytics and senomorphics can also indirectly restore ECM homeostasis by eliminating senescent cells or attenuating SASP-driven matrix remodeling[150,151]. Combining ECM-targeting strategies with chemotherapy or immunotherapy provides synergistic benefits and enhances drug delivery in a variety of cancers [152-154]. Overall, these findings underscore that reprogramming the senescent ECM represents a multifaceted strategy to reshape the TME and improve therapeutic outcomes.
From a translational perspective, the clinical development of senotherapies remains at an early stage. Although the number of clinical studies has increased in recent years, their application in oncology is still limited[155]. Intermittent senolytic regimens, such as dasatinib plus quercetin (D + Q), have entered small human studies outside oncology and provided proof-of-concept that senescent-cell burden can be reduced in vivo[156,157]. In parallel, navitoclax-based strategies are under active investigation, particularly in combination with chemotherapy, but on-target thrombocytopenia remains a major barrier to their clinical utility[158]. Senomorphics, including the repurposing of metformin, are particularly appealing due to their clinical familiarity; however, robust evidence for their anticancer efficacy and SASP modulation in patients remains limited[136,159]. Emerging immunotherapeutic strategies, such as uPAR-directed CAR-T cells, have shown promising preclinical activity but have yet to reach advanced clinical evaluation[145]. Several agents targeting ECM remodeling, including MMP inhibitors and LOX/LOXL inhibitors, have entered clinical evaluation; however, their efficacy has been constrained by issues such as off-target effects, toxicity, and limited clinical efficacy[160,161]. To date, there are still no large phase III trials establishing senotherapies as standard treatment, and key hurdles include toxicity, optimization of intermittent versus continuous dosing schedules, appropriate patient selection, and the lack of standardized biomarkers to confirm target engagement (Table 2)[158]. Collectively, these limitations highlight the need for further clinical investigation to fully realize the translational potential of senescence-targeting strategies.
Table 2. Senescence-targeting strategies in cancer: clinical development and major translational challenges.
| Agents/strategies | Targets | Clinical status | Challenges |
| D + Q | Multi-target (e.g., tyrosine kinases, PI3K) | Early-phase clinical (Phase I/II, primarily non-oncology)[162] | Limited cancer-specific evidence; optimal intermittent dosing; patient selection |
| Navitoclax | BCL-2/BCL-xL inhibition | Phase I/II trials (often in combination)[163] | Dose-limiting toxicity (thrombocytopenia); therapeutic window; dosing schedule optimization |
| Metformin | AMPK activation; mTOR inhibition; SASP modulation | Ongoing clinical studies[164] | Uncertain efficacy in senescence targeting; lack of specific biomarkers; patient stratification |
| Rapamycin | mTOR pathway inhibition; SASP suppression | Clinical use (non-senescence-specific)[164] | Off-target effects; immunosuppression; unclear senescence-specific benefit |
| Ruxolitinib | JAK/STAT signaling; SASP regulation | preclinical/early clinical[165] | Systemic immunosuppression; limited specificity for senescent cells |
| CAR-T cells | senescent cells | Preclinical | Safety concerns; on-target/off-tumor effects |
| FOXO4-DRI | FOXO-p53 interaction disruption | Preclinical | Delivery challenges; stability; lack of clinical validation |
| Nanoparticle/EV-based delivery systems | Senescence-associated markers (e.g., SA-β-gal, uPAR) | Preclinical | Targeting specificity; scalability; in vivo safety and biodistribution |
| BAPN | LOX/LOXL inhibition | Preclinical | High toxicity; induction of vascular pathologies |
PI3K: phosphatidylinositol 3-kinase; BCL-2: B-cell lymphoma 2; mTOR: mammalian target of rapamycin; SASP: senescence-associated secretory phenotype; JAK: Janus kinase; STAT: signal transducers and activators of transcription; CAR-T: chimeric antigen receptor T; SA-β-gal: senescence-associated β-galactosidase; uPAR: urokinase-type plasminogen activator receptor; LOX: lysyl oxidase; LOXL: lysyl oxidase-like; BAPN: β-aminopropionitrile; FOXO4-DRI: forkhead box O4-D-retro-inverso peptide; EV: extracellular vesicle; BCL-xL: B-cell lymphoma-extra large; AMPK: AMP-activated protein kinase.
6. Summary
Over the past decade, accumulating studies have revealed the pivotal role of cellular senescence in orchestrating the pathophysiology of age-associated disorders, ranging from neurodegeneration and cardiovascular disease to cancer[166]. Although senotherapies harbor tremendous therapeutic promise, several critical challenges must be addressed: (1) Improving the specificity of senotherapeutic agents to selectively target pathological senescent cells while sparing healthy cells. As a key pathogenic driver, the accumulation of senescent cells in the absence of effective and specific clearance mechanisms contributes to the development or exacerbation of cancer. Moreover, emerging precision delivery systems, such as proteolysis-targeting chimeras (PROTACs), antibody-drug conjugates (ADCs), galacto-nanoparticles, and senolytic CAR-T therapy hold great promise for improving the targeting specificity and therapeutic index of senotherapeutic strategies. For example, sacituzumab govitecan, as a representative ADC, has shown significant clinical antitumor activity in patients with metastatic triple-negative breast cancer and greatly improved their survival[167]. In addition, senescent cells exhibit elevated activity of lysosomal β-galactosidase, providing the rationale for delivering senotherapeutic drugs using galacto‐oligosaccharide encapsulated nanoparticles, which can reduce toxicity and increase selectivity[168]. (2) Overcoming the inherent heterogeneity of senescent cells, a barrier that limits the universal efficacy of single-agent senotherapies. Recent advances in single-cell and spatial multi-omics technologies, coupled with the development of machine learning and artificial intelligence (AI) classifiers, are beginning to resolve this diversity at higher resolution and may improve patient stratification and senotherapy selection in the future[169-171]. (3) Mitigating long-term adverse effects that may arise from prolonged treatment or off-target interactions.
In summary, while significant challenges remain to be addressed, senotherapies are a highly interesting and nascent field with broad translational potential. With the expansion of our knowledge of the molecular and cellular networks regulating senescence, we will refine strategies to address prior challenges and accelerate the translation of senotherapies into anticancer therapies.
Authors contribution
Wang L, Duan Y, Ma X, Peng L: Conceptualization, writing-original draft.
Wang Z, Qian M: Conceptualization, writing-review & editing.
Conflicts of interest
Minxian Qian is a Youth Editorial Board Member of Ageing and Cancer Research & Treatment. Other authors declared that there are no conflicts of interest.
Ethical approval
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by grants from the National Key Research and Development Program of China (Grant No. 2023YFA1801900); the National Natural Science Foundation of China (Grant Nos. 32571473, 82271602); Natural Science Foundation of Zhejiang Province, China (Grant No. LMS25C050001); the Natural Science Foundation of Chongqing, China (Grant No. CSTB2024NSCQ-MSX1152); the Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (Grant No. SKLNMZZ2024JS39); and the Fundamental Research Funds for the Central Universities (Grant No. 2632025TD05).
Copyright
© The Author(s) 2026.
References
-
1. Zhang JW, Zhang D, Yu BP. Senescent cells in cancer therapy: Why and how to remove them. Cancer Lett. 2021;520:68-79.[DOI]
-
8. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109(3):335-346.[DOI]
-
11. Knizhnik AV, Roos WP, Nikolova T, Quiros S, Tomaszowski KH, Christmann M, et al. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS One. 2013;8(1):e55665.[DOI]
-
13. Singh V, Verma S, Fatima F, Samanta SK, Varadwaj PK, Sahoo AK. In silico study of a small bioactive molecule targeting topoisomerase II and P53-MDM2 complex in triple-negative breast cancer. ACS Omega. 2023;8(41):38025-38037.[DOI]
-
24. Macià i Garau M. Radiobiology of stereotactic body radiation therapy (SBRT). Rep Pract Oncol Radiother. 2017;22(2):86-95.[DOI]
-
27. d’Adda di Fagagna F. Living on a break: Cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8(7):512-522.[DOI]
-
34. Guenther LM, Dharia NV, Ross L, Conway A, Robichaud AL, Catlett JL, et al. A combination CDK4/6 and IGF1R inhibitor strategy for Ewing sarcoma. Clin Cancer Res. 2019;25(4):1343-1357.[DOI]
-
41. Cilibrasi C, Guzzi A, Bazzoni R, Riva G, Cadamuro M, Hochegger H, et al. A ploidy increase promotes sensitivity of glioma stem cells to aurora kinases inhibition. J Oncol. 2019;2019:9014045.[DOI]
-
42. Prashanth Kumar BN, Rajput S, Bharti R, Parida S, Mandal M. BI2536–A PLK inhibitor augments paclitaxel efficacy in suppressing tamoxifen induced senescence and resistance in breast cancer cells. Biomed Pharmacother. 2015;74:124-132.[DOI]
-
49. Kumari R, Jat P. Mechanisms of cellular senescence: Cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol. 2021;9:645593.[DOI]
-
62. Hayakawa T, Iwai M, Aoki S, Takimoto K, Maruyama M, Maruyama W, et al. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS One. 2015;10(1):e0116480.[DOI]
-
65. Georgakopoulou EA, Tsimaratou K, Evangelou K, Fernandez Marcos PJ, Zoumpourlis V, Trougakos IP, et al. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging. 2013;5(1):37-50.
-
67. Ngoi NY, Liew AQ, Chong SJF, Davids MS, Clement MV, Pervaiz S. The redox-senescence axis and its therapeutic targeting. Redox Biol. 2021;45:102032.[DOI]
-
68. Feng B, Chu F, Bi A, Huang X, Fang Y, Liu M, et al. Fidelity-oriented fluorescence imaging probes for beta-galactosidase: From accurate diagnosis to precise treatment. Biotechnol Adv. 2023;68:108244.[DOI]
-
69. Saatloo MV, Delisi D, Eskandari N, Krieg C, Gentile S. Kv11.1-dependent senescence activates a lethal immune response via tumor necrosis factor alpha. Neoplasia. 2025;63:101148.[DOI]
-
73. Dalmasso G, Cougnoux A, Faïs T, Bonnin V, Mottet-Auselo B, Nguyen HT, et al. Colibactin-producing Escherichia coli enhance resistance to chemotherapeutic drugs by promoting epithelial to mesenchymal transition and cancer stem cell emergence. Gut Microbes. 2024;16(1):2310215.
-
79. Safwan-Zaiter H, Wagner N, Wagner KD. P16INK4A: More than a senescence marker. Life. 2022;12(9):1332.[DOI]
-
86. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):e301.[DOI]
-
88. Sun Y, Coppé JP, Lam EW-F. Cellular senescence: The sought or the unwanted? Trends Mol Med. 2018;24(10):871-885.[DOI]
-
92. Bullock K, Richmond A. Suppressing MDSC recruitment to the tumor microenvironment by antagonizing CXCR2 to enhance the efficacy of immunotherapy. Cancers. 2021;13(24):6293.[DOI]
-
95. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):e301.[DOI]
-
101. Ma L, He X, Fu Y, Ge S, Yang Z. Senescent endothelial cells promote liver metastasis of uveal melanoma in single-cell resolution. J Transl Med. 2024;22:605.[DOI]
-
102. Meng SS, Gu HW, Zhang T, Li YS, Tang HB. Gradual deterioration of fatty liver disease to liver cancer via inhibition of AMPK signaling pathways involved in energy-dependent disorders, cellular aging, and chronic inflammation. Front Oncol. 2023;13:1099624.[DOI]
-
103. Lagoumtzi SM, Chondrogianni N. Senolytics and senomorphics: Natural and synthetic therapeutics in the treatment of aging and chronic diseases. Free Radic Biol Med. 2021;171:169-190.[DOI]
-
105. Hawthorne L, Yang J, Zorlutuna P. Aging and the extracellular matrix: A tumor-permissive microenvironment driving cancer progression. Curr Opin Biomed Eng. 2025;36:100618.[DOI]
-
107. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29(6):783-803.[DOI]
-
112. Nia HT, Munn LL, Jain RK. Physical traits of cancer. Science. 2020;370(6516):eaaz0868.[DOI]
-
113. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15(8):978-990.[DOI]
-
122. Harakandi C, Nininahazwe L, Xu H, Liu B, He C, Zheng YC, et al. Recent advances on the intervention sites targeting USP7-MDM2-p53 in cancer therapy. Bioorg Chem. 2021;116:105273.[DOI]
-
123. Natarajan U, Venkatesan T, Dhandayuthapani S, Dondapatti P, Rathinavelu A. Differential mechanisms involved in RG-7388 and Nutlin-3 induced cell death in SJSA-1 osteosarcoma cells. Cell Signal. 2020;75:109742.[DOI]
-
124. Carvajal LA, Ben Neriah D, Senecal A, Benard L, Thiruthuvanathan V, Yatsenko T, et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci Transl Med. 2018;10(436):eaao3003.[DOI]
-
129. Chamcheu JC, Esnault S, Adhami VM, Noll AL, Banang-Mbeumi S, Roy T, et al. Fisetin, a 3, 7, 3', 4'-tetrahydroxyflavone inhibits the PI3K/Akt/mTOR and MAPK pathways and ameliorates psoriasis pathology in 2D and 3D organotypic human inflammatory skin models. Cells. 2019;8(9):1089.
-
133. You L, Wu Q. Cellular senescence in tumor immune escape: Mechanisms, implications, and therapeutic potential. Crit Rev Oncol. 2025;208:104628.[DOI]
-
134. Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020;583(7814):127-132.[DOI]
-
136. Abdelgawad IY, Agostinucci K, Sadaf B, Grant MKO, Zordoky BN. Metformin mitigates SASP secretion and LPS-triggered hyper-inflammation in Doxorubicin-induced senescent endothelial cells. Front Aging. 2023;4:1170434.[DOI]
-
138. Saleh T, Tyutynuk-Massey L, Cudjoe EK Jr, Idowu MO, Landry JW, Gewirtz DA. Non-cell autonomous effects of the senescence-associated secretory phenotype in cancer therapy. Front Oncol. 2018;8:164.[DOI]
-
139. Feng T, Xie F, Lee LMY, Lin Z, Tu Y, Lyu Y, et al. Cellular senescence in cancer: From mechanism paradoxes to precision therapeutics. Mol Cancer. 2025;24(1):213.[DOI]
-
141. Wang Z, Chen Y, Fang H, Xiao K, Wu Z, Xie X, et al. Reprogramming cellular senescence in the tumor microenvironment augments cancer immunotherapy through multifunctional nanocrystals. Sci Adv. 2024;10(44):eadp7022.[DOI]
-
145. Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020;583(7814):127-132.[DOI]
-
150. Yang D, Tian X, Ye Y, Liang Y, Zhao J, Wu T, et al. Identification of GL-V9 as a novel senolytic agent against senescent breast cancer cells. Life Sci. 2021;272:119196.[DOI]
-
151. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu Rev Pathol Mech Dis. 2010;5:99-118.[DOI]
-
153. Zhao Q, He X, Qin X, Liu Y, Jiang H, Wang J, et al. Enhanced therapeutic efficacy of combining losartan and chemo-immunotherapy for triple negative breast cancer. Front Immunol. 2022;13:938439.[DOI]
-
156. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine. 2019;40:554-563.[DOI]
-
157. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. eBioMedicine. 2019;47:446-456.[DOI]
-
162. Saliev T, Singh PB. Senotherapeutics for brain aging management. Neurol Int. 2025;17(12):204.[DOI]
-
168. Guerrero A, Guiho R, Herranz N, Uren A, Withers DJ, Martínez-Barbera JP, et al. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell. 2020;19(4):e13133.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Wang L, Duan Y, Ma X, Peng L, Qian M, Wang Z. Targeting cellular senescence: a promising anticancer strategy. Ageing Cancer Res Treat. 2026;3:202601. https://doi.org/10.70401/acrt.2026.0017
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Inducers of Cell Senescence in Tumors
- 3. Characteristics of Senescent Cells
- 4. Paradoxical Dual Roles of Cellular Senescence in Cancer
- 5. Strategies to Target Senescent Cells for Cancer Therapy
- 6. Summary
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Wang L, Duan Y, Ma X, Peng L, Qian M, Wang Z. Targeting cellular senescence: a promising anticancer strategy. Ageing Cancer Res Treat. 2026;3:202601. https://doi.org/10.70401/acrt.2026.0017
copy
Share Link
copy