Repair of DNA double-strand breaks leaves heritable molecular scars that might shape aging trajectories and cancer risk
Hossein Salari
*

,
Chun-Long Chen
*
*Correspondence to:
Hossein Salari, Institut Curie, PSL Research University, CNRS UMR3244, Dynamics of Genetic Information, Sorbonne Université, 75005 Paris, France.
E-mail: hossein.salari@curie.fr
Chun-Long Chen, Institut Curie, PSL Research University, CNRS UMR3244, Dynamics of Genetic Information, Sorbonne Université, 75005 Paris, France. E-mail: chunlong.chen@curie.fr
Chun-Long Chen, Institut Curie, PSL Research University, CNRS UMR3244, Dynamics of Genetic Information, Sorbonne Université, 75005 Paris, France. E-mail: chunlong.chen@curie.fr
Ageing Cancer Res Treat. 2026;3:202609. 10.70401/acrt.2026.0018
Received: March 13, 2026Accepted: April 15, 2026Published: April 16, 2026
This article belongs to the Special lssue Replication Stress Responses and Genome Stability: Mechanisms, Regulation, and Disease Implications
Abstract
Aging tissues accumulate DNA damage, while genome instability is a defining feature of cancer. Despite this shared foundation, DNA damage is still largely viewed as a transient lesion that is either faithfully repaired or converted into a mutation. New evidence challenges this binary view, indicating that DNA double-strand breaks (DSBs), even when accurately repaired at the sequence level, can leave durable and heritable alterations in chromatin organization and gene regulation. Accordingly, DSB repair restores DNA integrity but does not necessarily re-establish the original regulatory architecture. The biological consequences of such post-repair regulatory memory remain largely underappreciated, progressively contributing to age-associated tissue dysfunction while simultaneously creating permissive states for malignant transformation and therapy resistance. In this commentary, we argue that reframing DNA damage as a source of heritable regulatory change, rather than solely as a mutational event, reshapes our understanding of aging trajectories and cancer risk.
Keywords
DNA repair, three-dimensional genome structure, double-strand break, cancer, aging
1. Introduction
Aging is accompanied by the progressive accumulation of DNA damage across tissues, a phenomenon well documented in both proliferative and post-mitotic cells[1,2]. In parallel, genome instability represents a defining hallmark of cancer, driving mutational burden, clonal evolution, and tumor heterogeneity[3]. Within the prevailing framework, DNA damage is understood both as a byproduct of metabolic and replicative stress during aging and as a causal driver of functional decline and malignant transformation, primarily through mutation accumulation, checkpoint activation, cellular senescence, or apoptosis[4]. Thus, its biological impact has largely been interpreted through the lens of genomic sequence alteration or irreversible cellular outcomes.
Clinically, this paradigm has justified the widespread use of DNA-damaging agents, including chemotherapy and radiotherapy, as cornerstones for cancer treatment[5]. However, such approaches also expose aging tissues to repeated genotoxic stress, often with long-term consequences that extend beyond acute toxicity[6]. Emerging evidence now suggests that DNA breaks, even when efficiently repaired, can induce durable and heritable molecular alterations[7]. These persistent changes may contribute to age-associated tissue dysfunction while simultaneously reshaping cellular states in ways that influence cancer initiation, progression, and therapeutic response.
2. Repair of DNA Breaks as Sources of Heritable Change
Three-dimensional (3D) genome organization is intimately linked to gene regulation, with higher-order chromatin structures such as topologically associating domains (TADs) constraining enhancer–promoter communication and stabilizing transcriptional programs[8,9]. Disruption of local chromatin architecture, through boundary weakening, domain fusion, or altered loop formation, can therefore lead to aberrant gene expression in the surrounding genomic neighborhood[9].
DNA double-strand breaks (DSBs) are now recognized as potent inducers of such structural reorganization. The process of DSB repair is accompanied by chromatin remodeling and transcriptional silencing[10,11]. Acute and localized chromatin remodeling at break sites is a well-established feature of the DNA damage response and facilitates the recruitment and activity of repair factors[12,13]. Strikingly, emerging evidence suggests that these alterations may not be entirely transient (Figure 1a,b). A recent study by Bantele and colleagues suggests that DSB–induced changes in gene transcription inhibition and 3D genome organization can persist beyond the completion of repair, implying that the restoration of DNA sequence integrity does not necessarily equate to a full recovery of regulatory architecture[7].
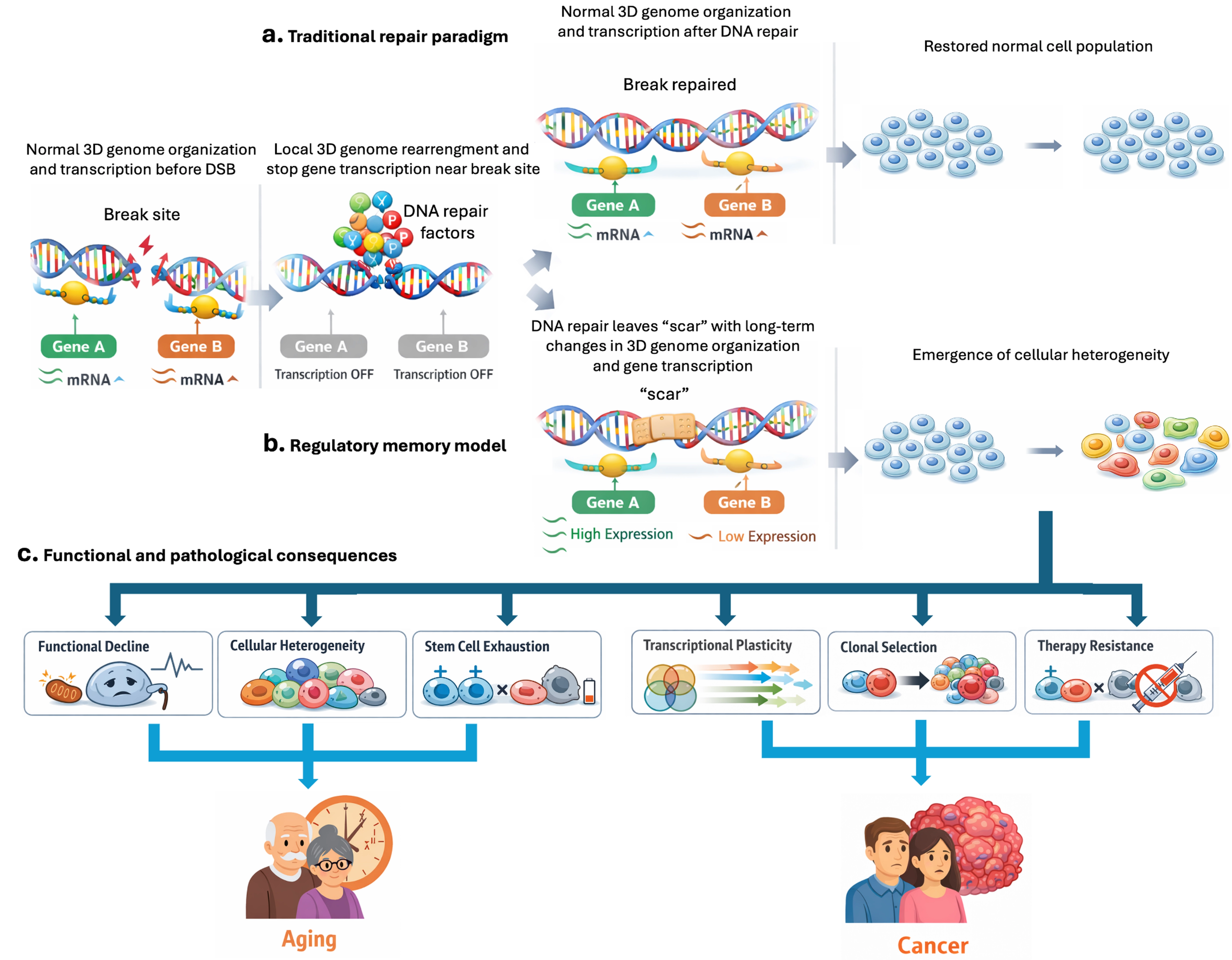
Figure 1. Conceptual model of DSB repair and its long-term consequences for aging and cancer. (a) Traditional view of DSB repair, in which DNA lesions are accurately repaired, and cellular function is fully restored to its pre-damage state; (b) Revised model of DSB repair outcomes, proposing that repaired breaks can leave persistent molecular “scars,” resulting in long-lasting perturbations of 3D genome organization and gene transcription. These regulatory memory alterations may propagate across cell divisions, promoting cellular dysfunction and increased heterogeneity; (c) Cumulative damage-induced architectural alterations underlie key hallmarks of aging and cancer, including functional decline, transcriptional plasticity, clonal selection, stem cell exhaustion, and therapy resistance. DSB: double-strand break; 3D: three-dimensional.
In their study, the authors strategically targeted the MYC gene locus, a master regulator of cell proliferation and a prominent oncogene[14], whose expression is highly sensitive to 3D genome organization[15]. MYC resides within a structurally constrained regulatory domain in which enhancer–promoter communication depends on intact TAD architecture, making it an ideal system to test whether DSB repair perturbs higher-order chromatin structure. Using a combination of 3D DNA fluorescence in situ hybridization (DNA FISH), region capture Micro-C for high-resolution contact mapping, and RNA fluorescence in situ hybridization (RNA FISH) to quantify transcription at the single-cell level, they showed that a single DSB induces persistent local reorganization of chromatin contacts accompanied by stable transcriptional repression. Notably, similar architectural and transcriptional defects were observed when breaks were introduced elsewhere within the same TAD but outside the MYC coding sequence, indicating that the effect is not gene-specific. Comparable long-term impairments were further confirmed at the independent MCM2 locus, demonstrating that post-repair chromatin dysfunction is not restricted to MYC but reflects a more general consequence of DSB repair in structurally sensitive genomic regions.
Importantly, alterations in chromatin contacts, domain insulation, and nuclear positioning were shown to persist across multiple cell divisions. These observations support a model in which transient DNA damage events can be converted into stable, heritable regulatory states. Sustained reorganization of chromatin topology at repaired break sites may therefore stabilize altered transcriptional programs and bias subsequent cell fate decisions.
Although much of the current evidence derives from experimentally induced DSBs, endogenous DNA damage occurs frequently during normal cellular physiology. Mammalian cells are estimated to experience on the order of tens of DSBs per cell cycle, many arising from replication-associated stress and transcription–replication conflicts[16,17], as well as from exogenous exposures such as tobacco smoke and ionizing radiation[18]. It is therefore plausible that naturally occurring DNA breaks, even when efficiently repaired, leave persistent molecular “scars” that accumulate over time, progressively reshaping 3D genome organization and cellular function.
3. Why This Matters for Aging and Cancer
Aging and cancer are traditionally viewed as divergent outcomes of genomic instability, with aging characterized by functional decline and cancer by uncontrolled proliferation[2,3]. Yet, both processes are accompanied by profound and reproducible alterations in chromosome architecture and epigenetic landscapes, including changes in chromatin accessibility, histone modifications, and 3D genome organization[19-21]. These observations suggest that genome instability affects not only DNA sequence integrity but also the regulatory frameworks that govern cellular identity and behavior.
If DNA breaks, even after being efficiently repaired, indeed induce heritable changes in chromatin organization and transcriptional states, then the long-term biological impact of genotoxic stress may extend far beyond the immediate repair response. This view shifts attention from irreversible mutations to reversible, but persistent, regulatory alterations. Persistent architectural and epigenetic alterations could act as a form of cellular memory, progressively reshaping gene regulatory programs across cell divisions (Figure 1c). In aging tissues, the accumulation of such damage-induced memories may contribute to loss of tissue homeostasis, increased cellular heterogeneity, and impaired regenerative capacity[2,22]. In parallel, the same processes may create permissive states for malignant transformation by promoting transcriptional plasticity, stress tolerance, and clonal selection of cells adapted to chronic genomic instability[3,19].
From this perspective, aging and cancer may represent distinct phenotypic outcomes of a shared underlying process: the progressive retention of heritable regulatory changes initiated by DNA damage. Understanding how DNA break–induced memories are established, propagated, and regulated is therefore essential for explaining why aging is a major risk factor for cancer and for developing strategies that maintain tissue function while minimizing oncogenic potential.
4. Future Directions
Despite the conceptual appeal of repair of DNA break–induced regulatory memory, it is important to emphasize that the current evidence remains limited and largely derives from a small number of recent studies, most notably Bantele et al.[7]. While these findings provide compelling proof-of-principle, they do not yet establish the generality of this phenomenon across genomic loci, cell types, or physiological conditions. At present, it remains unclear to what extent damage-induced regulatory memory represents a widespread and robust feature of genome biology versus a context-dependent outcome observed under specific experimental settings. Accordingly, the framework proposed here should be viewed as a working model that requires systematic validation and refinement through broader experimental investigation.
Several key challenges must be addressed to establish repair of DNA break–induced regulatory memory as a unifying framework for aging and cancer. A central question is whether DSB-induced regulatory memory exhibits locus specificity. Are certain genomic regions, such as highly transcribed genes, super-enhancer–associated loci, TAD boundaries, or structurally constrained regulatory domains, more susceptible to persistent architectural and transcriptional alterations? Addressing this will require systematic mapping of break-induced chromatin and 3D genome changes across diverse genomic contexts, cell types, and tissue environments. In particular, studies in primary cells and in vivo models will be essential to determine the frequency, stability, and functional impact of these alterations under physiological conditions, and to assess their broader biological relevance.
Second, the molecular mechanisms that underlie the persistence of damage-induced regulatory changes remain poorly defined. A major unresolved question is how transient DNA lesions are converted into stable, heritable chromatin states. Does regulatory memory arise from incomplete restoration of DNA and histone modifications following repair, or from altered stability and reassembly of 3D genome organizers such as CCCTC-binding factor (CTCF) and cohesin? Alternatively, persistent effects may reflect changes in chromatin loop stabilization, domain insulation strength, nuclear repositioning, or repair-coupled chromatin assembly pathways. Dissecting the relative contribution of these mechanisms will be essential to define the structural and epigenetic basis of chromatin memory. Identifying factors that either reinforce or actively erase damage-induced memory states may further uncover new therapeutic points of intervention.
Beyond DSBs, it will also be important to determine whether other stress responses that perturb transcription and 3D genome organization, such as replication fork stalling and transcription–replication conflicts[23], can similarly generate long-lasting epigenetic memory. If so, regulatory memory may represent a broader principle of genome stress responses rather than a DSB-specific phenomenon.
Third, the implications of damage-induced regulatory memory extend beyond aging-related regenerative decline and increased cellular heterogeneity, potentially conferring transcriptional plasticity and selective advantages that facilitate tumor initiation and clonal expansion. Persistent alterations in chromatin architecture may create epigenetic states that bias cell fate decisions, promote adaptive stress responses, and enable the emergence of competitively advantaged clones. These considerations are particularly relevant for genome-editing strategies such as Clustered Regularly-Interspaced Short Palindromic Repeats (CRISPR)–Cas9, where therapeutic DSBs could inadvertently induce long-term chromatin and transcriptional perturbations at target sites or neighboring loci, underscoring the need to evaluate their long-term physiological consequences.
Integrating single-cell multi-omics, lineage-tracing strategies, and high-resolution 3D genome profiling will therefore be essential to directly link damage-induced architectural changes to clonal dynamics, cell fate trajectories, and functional outcomes during aging and tumor evolution. Such approaches may elucidate how cellular heterogeneity gradually develops and how adaptive yet dysfunctional transcriptional states become selectively stabilized over time.
Finally, a deeper understanding of DNA damage memory has important clinical implications. Defining how genotoxic therapies reshape chromatin states in aging tissues may help explain long-term toxicities, secondary malignancies, and variable treatment responses. Ultimately, strategies that preserve genome organization and regulatory integrity, rather than simply minimizing mutations, may offer new avenues to mitigate age-associated decline while limiting oncogenic risk.
Acknowledgements
ChatGPT (OpenAI) was used to assist with language editing of the manuscript, including improvements to clarity, readability, and grammar. In addition, it was used to assist in figure editing. All authors reviewed and revised the manuscript and take full responsibility for the integrity, accuracy, and originality of the final content.
Authors contribution
Salari H, Chen CL: Conceptualization, writing-original draft, writing review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
C.L.C.’s team was supported by the ATIP-Avenir programme from the Centre National de la Recherche Scientifique (CNRS) and the Institut National de la Santé et de la Recherche Médicale (INSERM) (Grant No. 18CT014-00), Plan Cancer, the Agence Nationale de la Recherche (ANR) (ReDeFINe, Grant No. 19-CE12-0016-02; TELOCHROM, Grant No. 19-CE12-0020-02; SMART, Grant No. 21-CE12-0033-02; ReSPoND, Grant No. 23-CE12-0020-02; DetGap, Grant No. 25-CE12-4452-03), the Institut National du Cancer (INCa) (PLBIO19-076; INCa-AMED ASPIRE, Grant No. 2025-016), and the Impulscience programme of the Bettencourt Schueller Foundation.
Copyright
© The Author(s) 2026.
References
-
1. Schumacher B, Pothof J, Vijg J, Hoeijmakers JHJ. The central role of DNA damage in the ageing process. Nature. 2021;592(7856):695-703.[DOI]
-
2. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell. 2023;186(2):243-278.[DOI]
-
3. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022;12(1):31-46.[DOI]
-
4. Vijg J. From DNA damage to mutations: All roads lead to aging. Ageing Res Rev. 2021;68:101316.[DOI]
-
5. Moon J, Kitty I, Renata K, Qin S, Zhao F, Kim W. DNA damage and its role in cancer therapeutics. Int J Mol Sci. 2023;24(5):4741.[DOI]
-
6. Wang S, Prizment A, Thyagarajan B, Blaes A. Cancer treatment-induced accelerated aging in cancer survivors: Biology and assessment. Cancers. 2021;13(3):427.[DOI]
-
7. Bantele S, Mordini I, Biran A, Alcaraz N, Zonderland G, Wenger A, et al. Repair of DNA double-strand breaks leaves heritable impairment to genome function. Science. 2025;390(6773):eadk6662.[DOI]
-
8. Zuin J, Roth G, Zhan Y, Cramard J, Redolfi J, Piskadlo E, et al. Nonlinear control of transcription through enhancer–promoter interactions. Nature. 2022;604(7906):571-577.[DOI]
-
9. Yang JH, Hansen AS. Enhancer selectivity in space and time: From enhancer–promoter interactions to promoter activation. Nat Rev Mol Cell Biol. 2024;25(7):574-591.[DOI]
-
10. Machour FE, Ayoub N. Transcriptional regulation at DSBs: Mechanisms and consequences. Trends Genet. 2020;36(12):981-997.[DOI]
-
11. He S, Huang Z, Liu Y, Ha T, Wu B. DNA break induces rapid transcription repression mediated by proteasome-dependent RNAPII removal. Cell Rep. 2024;43(7):114420.[DOI]
-
12. Arnould C, Rocher V, Saur F, Bader AS, Muzzopappa F, Collins S, et al. Chromatin compartmentalization regulates the response to DNA damage. Nature. 2023;623(7985):183-192.[DOI]
-
13. Arnould C, Rocher V, Finoux AL, Clouaire T, Li K, Zhou F, et al. Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature. 2021;590(7847):660-665.[DOI]
-
14. Dhanasekaran R, Deutzmann A, Mahauad-Fernandez WD, Hansen AS, Gouw AM, Felsher DW. The MYC oncogene: The grand orchestrator of cancer growth and immune evasion. Nat Rev Clin Oncol. 2022;19(1):23-36.[DOI]
-
15. Hyle J, Zhang Y, Wright S, Xu B, Shao Y, Easton J, et al. Acute depletion of CTCF directly affects MYC regulation through loss of enhancer–promoter looping. Nucleic Acids Res. 2019;47(13):6699-6713.[DOI]
-
16. Goehring L, Huang TT, Smith DJ. Transcription–replication conflicts as a source of genome instability. Annu Rev Genet. 2023;57:157-179.[DOI]
-
17. Niehrs C, Luke B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat Rev Mol Cell Biol. 2020;21(3):167-178.[DOI]
-
19. Terekhanova NV, Karpova A, Liang WW, Strzalkowski A, Chen S, Li Y, et al. Epigenetic regulation during cancer transitions across 11 tumour types. Nature. 2023;623(7986):432-441.[DOI]
-
20. Wang K, Liu H, Hu Q, Wang L, Liu J, Zheng Z, et al. Epigenetic regulation of aging: Implications for interventions of aging and diseases. Sig Transduct Target Ther. 2022;7:374.[DOI]
-
21. Wu Z, Qu J, Liu GH. Roles of chromatin and genome instability in cellular senescence and their relevance to ageing and related diseases. Nat Rev Mol Cell Biol. 2024;25(12):979-1000.[DOI]
-
22. Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun. 2019;10:963.[DOI]
-
23. Kramarz K, Schirmeisen K, Boucherit V, Ait Saada A, Lovo C, Palancade B, et al. The nuclear pore primes recombination-dependent DNA synthesis at arrested Forks by promoting SUMO removal. Nat Commun. 2020;11:5643.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Salari H, Chen CL. Repair of DNA double-strand breaks leaves heritable molecular scars that might shape aging trajectories and cancer risk. Ageing Cancer Res Treat. 2026;3:202609. https://doi.org/10.70401/acrt.2026.0018
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Repair of DNA Breaks as Sources of Heritable Change
- 3. Why This Matters for Aging and Cancer
- 4. Future Directions
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent to publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Salari H, Chen CL. Repair of DNA double-strand breaks leaves heritable molecular scars that might shape aging trajectories and cancer risk. Ageing Cancer Res Treat. 2026;3:202609. https://doi.org/10.70401/acrt.2026.0018
copy
Share Link
copy