From aging to cancer: Genomic instability as a unifying driver and therapeutic nexus
Daijiang Xiong
1,2

,
Lin Cheng
3
,
Yahui Zhu
3,*
,
Li Gu
1,2,*
*Correspondence to:
Yahui Zhu, School of Medicine, Chongqing University, Chongqing 400030, China.
E-mail: zhuyahui861106@foxmail.com
Li Gu, Clinical Laboratory Medicine Research Center, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China. E-mail: ligu@scu.edu.cn
Li Gu, Clinical Laboratory Medicine Research Center, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China. E-mail: ligu@scu.edu.cn
Ageing Cancer Res Treat. 2026;3:202524. 10.70401/acrt.2026.0019
Received: December 09, 2025Accepted: April 24, 2026Published: April 28, 2026
This article belongs to the Special lssue Genomic Instability and Telomeres in Aging and Cancer
This manuscript is made available in its unedited form to allow early access to the
reported findings. Further editing will be completed before final publication. As
such,
the content may include errors, and standard legal disclaimers are applicable.
Abstract
Genomic instability (GI), characterized by the progressive failure of mechanisms that maintain genome integrity, serves as a fundamental link between aging and cancer at the molecular level. It not only drives the aging process but also promotes tumorigenesis through multiple pathways: on one hand, GI can induce cellular senescence and create a pro-inflammatory and tissue remodeling microenvironment via the senescence-associated secretory phenotype; on the other hand, GI can bypass senescence, directly facilitating tumor progression through mechanisms such as aneuploidy, the expansion of pre-malignant clones, and chronic inflammation mediated by DNA damage-associated molecular patterns. The decline in physiological functions accompanying aging and the increased risk of cancer are closely associated with the accumulation of GI, while aging itself may exert anti-cancer effects through irreversible cell cycle arrest in specific contexts. Therefore, a thorough investigation of GI’s dual role in aging and cancer can help reveal the shared biological basis of both processes and provide new strategies for the precise prevention and treatment of age-related tumors.
Keywords
Aging, cancer, genomic instability, SASP, DNA damage
1. Introduction
As global population aging intensifies, the prevention and control of age-related diseases, particularly cancer, has become a significant public health challenge. This conceptual tension, where aging both restrains and fuels cancer, was presciently framed by Campisi as ‘rival demons’ over two decades ago, establishing the foundational paradox that continues to drive mechanistic inquiry today[1]. Genomic instability (GI), the heightened propensity for structural and numerical changes in the genome across the cellular lifespan, is primarily driven by cumulative exposure to endogenous metabolic stressors and environmental insults[2]. It is a
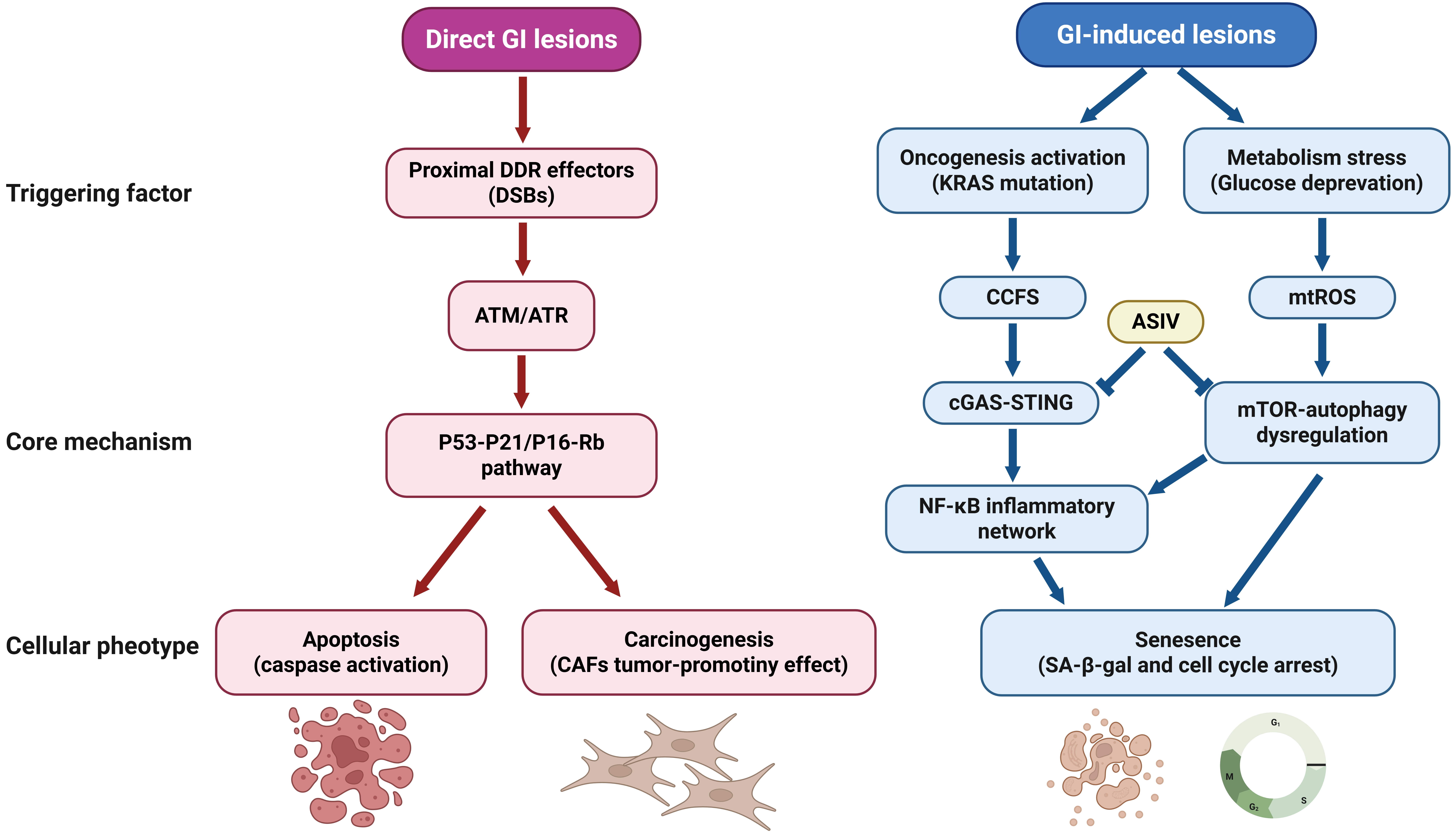
Figure 1. Genomic instability lesions directly engage specific signaling nodes. DNA double-strand breaks→ATM/ATR→p53/p21; Cytoplasmic chromatin fragments→
Table 1. Four interdependent axes of GI: Molecular lesions, biomarkers, and druggable nodes.
| Mechanism Axis | Core Molecular Lesion (GI Manifestation) | Key Biomarkers & Detection Methods | Druggable Nodes & Therapeutic Strategies | Clinical Translation Evidence |
| DNA Damage & Repair Failure | Persistent DSBs (γH2AX foci) | γH2AX IHC/IF | PARP inhibitors (Olaparib) | PARPi approved for BRCA-mutant cancers (HR-GI) |
| Replication fork collapse | RAD51 foci assay | ATR inhibitors (Ceralasertib) | NCT045: cGAS inhibitors + PD-1 for CCF-high tumors (ref Clinical Translation section) | |
| MSI | MSI testing (PCR/NGS) | MMR restoration (mRNA therapy) | ||
| Telomere Attrition | Critically short telomeres (qFISH) | Telomere length (Flow-FISH, qPCR) | Telomerase activators (TA-65) | Telomere attrition predicts glioma risk |
| Telomere fusion-derived dicentric chromosomes | TERT promoter mutations (NGS) | Shelterin stabilizers (TRF2 mimetics) | TERT promoter mutations are diagnostic in glioblastoma (WHO CNS5) | |
| Chromothripsis at shelterin regions | BFB cycle inhibitors (ATRi) | |||
| Epigenetic Dysregulation | Global hypomethylation (LINE-1) | Epigenetic clocks (GrimAge, DNAmTL) | DNMT inhibitors (Azacitidine) | GrimAge acceleration predicts colorectal cancer risk |
| Promoter hypermethylation (CDKN2A, MLH1) | Methylation arrays (EPIC) | EZH2 inhibitors (Tazemetostat) | EZH2-mutant lymphomas sensitive to JAK/STAT inhibition | |
| H3K27ac gain at | ChIP-seq for histone marks | BET inhibitors (JQ1) | ||
| Mitochondrial-ROS Axis | mtDNA deletions | mtDNA/nDNA ratio (qPCR) | Mitophagy inducers (Urolithin A) | Betaine inhibits TBK1→suppresses SASP |
| 8-oxoG accumulation (HPLC-MS) | Urinary 8-oxoG (ELISA) | ROS scavengers (MitoQ) | mtDNA mutations correlate with SBS1/SBS5 mutational signatures | |
| Cardiolipin peroxidation (C11-BODIPY assay) | Serum TFAM (ELISA) | TBK1 inhibitors (Amlexanox) |
DDR: DNA damage response; SASP: senescence-associated secretory phenotype; GI: genomic instability; CCFs: cytoplasmic chromatin fragments; cGAS-STING: cyclic
With epidemiological data showing increased GI in tumors of elderly patients, including SCNAs and the accumulation of somatic mutations[8], this association is particularly evident in specific tumor types such as gliomas and endometrial cancer[8]. The altered microenvironment of aging tissues provides critical conditions for tumorigenesis: on one hand, aging tissues tend to form cell clones with normal histological appearance but altered genotypes[12]; on the other hand, aging tissues provide a permissive milieu for the clonal expansion of somatically mutated cells, often with histologically normal architecture but acquired driver mutations
Therefore, GI is the molecular core of the “central paradox” connecting aging and cancer. As an upstream substrate of genetic instability, it accumulates during the aging process, directly driving cellular functional decline while simultaneously providing a genetic basis for tumorigenesis. Understanding its mechanisms and targeted interventions is fundamentally significant for addressing the challenges of age-related cancers. To systematically dissect this unifying driver, we classify GI into four interrelated mechanistic axes: DNA damage and repair failure, telomere attrition, epigenetic dysregulation, and mitochondrial-reactive oxygen species (ROS) dysfunction, each with distinct molecular lesions, quantifiable biomarkers, druggable nodes, and emerging clinical translation evidence. This framework is summarized in Table 1, which provides a mechanistic roadmap linking specific GI manifestations to actionable therapeutic strategies and validated biomarker applications in elderly cancer patients. Throughout this review, ‘genomic instability’ is operationally defined, not as a synonym for cellular phenotypes (e.g., senescence), but as the quantifiable, upstream accumulation of DNA lesions, chromosomal aberrations, and retrotransposon activation resulting from failed genome maintenance. All discussed hallmarks (senescence, SASP, inflammaging) are contextual effectors of GI, not its synonyms”.
γH2AX/CCFs: Guiding clinical trials of cGAS-STING agonists (such as ADU-S100) combined with PD-1 inhibitors (NCT045);
Breakage-fusion-bridge (BFB) loops/telomere fusions: Indicating MYC/EGFR amplification, suggesting a risk of resistance to CDK4/6 inhibitors;
SBS1/SBS5: Used to identify oxidative damage-driven hepatocellular carcinoma (HCC), guiding antioxidant interventions or PARPi combination strategies;
Replication fork collapse: The irreversible structural disintegration of a stalled replication fork under conditions of replication stress (such as DNA damage, nucleotide depletion, topological stress, etc.) due to the failure of protective mechanisms. This leads to the breaking of newly synthesized DNA strands, excessive accumulation of single-stranded DNA, and often initiates the pathological process of DSBs.
2. Core Mechanisms Linking GI-Driven Senescence
The accumulation of unrepaired DNA damage and the progressive failure of DNA repair machinery represent the most direct manifestations of GI and serve as primary triggers of cellular senescence. At its core, aging reflects a systemic imbalance between macromolecular damage and maintenance capacity, with genomic instability acting as both cause and consequence in this
3. Accumulation of DNA Damage and Dysfunction of Repair Systems
The accumulation of DNA damage and the failure of the repair system are the most direct initiating events and core phenotypes of gastrointestinal diseases: endogenous metabolic stress and continuous external damage impact the genome, while repair capacity declines with age, leading to the accumulation of DSBs, replication fork collapse, and other lesions, which directly trigger aging and provide mutation substrates for carcinogenesis. Replicative stress is a major source of GI, and defects in prelamin A processing caused by mutations in the lamin A/C or zinc metallopeptidase STE24 genes can lead to premature aging[19]. Deficiencies in repair mechanisms, such as mismatch repair (MMR) deficiency, further exacerbate mutation accumulation, creating a vicious cycle[20,21]. This instability manifests as chromosomal breaks, translocations, or aneuploidy, directly driving cellular senescence. DNA damage can also activate key proteins like p53 through ataxia telangiectasia mutated (ATM)/ataxia telangiectasia and Rad3-related (ATR) kinases, triggering downstream DDR signaling to the mitochondria, inducing mitochondrial dysfunction, and exacerbating DNA damage, ultimately inducing senescence[22,23]. More severe damage leads to the release of CCFs into the cytoplasm, carrying DNA damage markers γH2AX, and activating inflammatory responses through the cGAS-STING pathway, reinforcing the senescence
4. Telomere Attrition
Telomere shortening is a specific form and accelerator of GI: it not only serves as the “mitotic clock” of replicative senescence, but also induces large-scale chromosomal instability (CIN) through mechanisms such as telomere fusion and the BFB cycle, escalating localized damage into genomic chaos. Telomeres, nucleoprotein structures at chromosome ends, protect against recognition as DSBs and prevent end-to-end fusions. With each round of cell division, telomeres progressively shorten due to the end-replication problem and oxidative damage. The ‘end-replication problem’ arises because DNA polymerases cannot fully replicate the 3′ ends of linear chromosomes, resulting in progressive telomere shortening with each cell division[29,30]. When telomeres reach a critically short length, they lose their protective capping function and are recognized as persistent DNA damage, thereby activating the
5. Epigenetic Dysregulation
Epigenetic dysregulation is a bidirectional amplifier and functional mediator of GI. Aging-associated epigenetic dysregulation, characterized by global DNA hypomethylation (e.g., LINE-1), promoter-specific hypermethylation (e.g., CDKN2A, MLH1), loss of heterochromatin marks (e.g., H3K9me3), and gain of activating marks (e.g., H3K27ac), drives GI and cellular senescence. It impairs genome stability maintenance, reactivates transposable elements, and promotes formation of CCFs. Critically, DNA methylation–based epigenetic clocks (e.g., GrimAge, DNAmTL), which are computational biomarkers derived from DNA methylation levels at age-informative CpG sites, providing a quantitative estimate of biological (as opposed to chronological) age, quantify biological aging and independently predict cancer risk (e.g., glioma, colorectal cancer), even after adjusting for chronological age and lifestyle factors[38-41]. Critically, these age-associated methylation changes are not merely correlative but quantitatively captured by DNA methylation-based epigenetic clocks, robust molecular surrogates of biological aging that outperform chronological age in predicting cancer incidence and therapy-related morbidity[39,40,42]. Notably, chemotherapy itself induces rapid epigenetic aging. Chemotherapy accelerates epigenetic aging (e.g., 2-5 years GrimAge in 6 months), establishing a vicious cycle: aging→epigenetic drift→cancer→therapy→accelerated aging→secondary malignancy[43]. Notably, there is a bidirectional relationship between epigenetic dysregulation and GI. This functional interplay extends beyond stochastic drift to clinically actionable biomarkers: Yu et al. demonstrated that epigenetic aging clocks not only track biological age but also predict cancer risk and therapeutic response, revealing their role as dynamic ‘molecular rheostats’ rather than passive timers[44]. Epigenetic alterations also directly shape the SASP and modulate senescence execution[45-47]. Notably, age-associated hypermethylation preferentially targets Polycomb group target (PCGT) genes, a signature first mapped by Teschendorff et al. in normal tissues and consistently observed in dysplastic lesions
6. Mitochondrial Dysfunction and Oxidative Stress
Mitochondrial dysfunction is a key upstream trigger and downstream amplification circuit of GI. Mitochondrial dysfunction, marked by reduced OXPHOS, membrane depolarization, mtDNA deletions, and cardiolipin peroxidation, elevates ROS production. Excess ROS damages nuclear DNA (e.g., 8-oxoguanine, DSBs), erodes telomeres, and mutates mtDNA, further impairing mitochondrial function and amplifying ROS leakage, a self-reinforcing loop[50,51]. ROS also inhibit TET enzymes, promoting DNA hypermethylation and heterochromatin loss[52]. Mitochondrial damage contributes to CCF generation, activating cGAS-STING and SASP[9,53]. Notably, mtDNA mutations correlate with oxidative mutational signatures (SBS1/SBS5) in cancers like HCC[54-56]. This axis converges with p53 and
In summary, GI underlies the molecular pathology of aging, primarily operating through four interrelated axes: accumulation of DNA damage and repair failure, telomere shortening, epigenetic dysregulation, and mitochondrial dysfunction. Importantly, while the four axes (Figure 2), DNA damage/repair failure, telomere attrition, epigenetic dysregulation, and mitochondrial dysfunction, are interrelated, only the first two represent primary manifestations of GI (i.e., direct physical/genetic lesions: DSBs, telomere fusions, chromothripsis)[2,3,29]. In contrast, epigenetic dysregulation and mitochondrial dysfunction are robustly induced by GI lesions
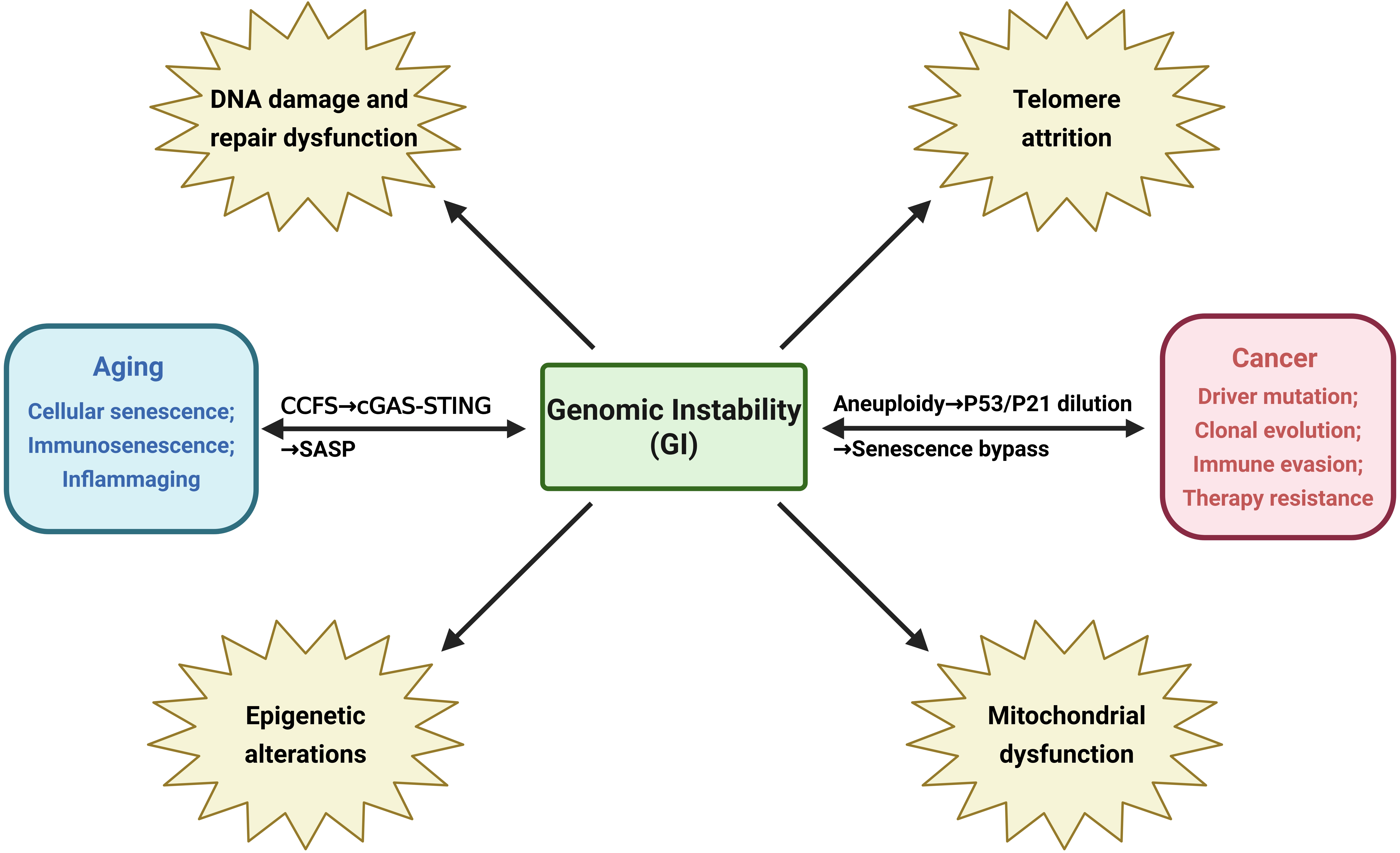
Figure 2. Key biological processes associated with GI. CCFS: cytoplasmic chromatin fragments; cGAS-STING: cyclic GMP-AMP synthase–stimulator of interferon genes;
GI plays a central role in aging, primarily driven by four interrelated mechanisms: accumulation of DNA damage and dysfunction of repair systems, telomere shortening, epigenetic dysregulation, and mitochondrial dysfunction. These processes collectively form a self-reinforcing network that leads to a gradual imbalance in cellular homeostasis.
The four axes of GI do not operate in isolation but converge at three critical decision nodes: (i) The
7. Genomic Instability as A Direct Oncogenic Catalyst: Beyond Senescence
GI is not merely a trigger for senescence, a transient barrier to tumorigenesis, but functions as an autonomous, progressive oncogenic engine that directly shapes malignant evolution independent of, or even in defiance of, senescence checkpoints. Three non-redundant mechanisms underpin this direct catalytic role:
7.1 Clonal expansion of driver mutations via GI-enabled mutational burst
Age-associated accumulation of DNA damage and repair defects (e.g., POLE exonuclease domain mutations, MMR deficiency) generates mutational signatures with high functional impact. In HCC, whole-genome sequencing reveals that tumors arising from senescence-bypassed, metabolically stressed hepatocytes exhibit C > T-dominant mutational spectra (SBS1/SBS5), mirroring human HCC and directly implicating oxidative base damage in CTNNB1, ARID1A, and TP53 driver acquisition[56,61]. This demonstrates that GI provides the substrate for Darwinian selection, not just cellular arrest.
7.2 Aneuploidy and polyploidy as senescence-bypass mechanisms
CIN induces whole-chromosome or segmental aneuploidy, which dilutes the stoichiometric threshold required for p53/p21 or p16/Rb checkpoint activation. Polyploid giant cancer cells, frequently observed after chemotherapy-induced senescence, evade growth arrest by buffering lethal gene dosage imbalances and subsequently undergoing depolyploidization to generate highly heterogeneous, therapy-resistant progeny[62]. Thus, GI does not always culminate in senescence; it can instead *license uncontrolled proliferation through karyotypic chaos.
7.3 Premalignant field formation in stem/progenitor compartments with intrinsic senescence evasion
GI in tissue-resident stem cells (e.g., hepatic progenitors, intestinal crypt base columnar cells) creates clonally expanded, genomically scarred fields where senescence is actively suppressed but not induced. For instance, aged hematopoietic stem cells with clonal hematopoiesis harbor DNMT3A or TET2 mutations that impair epigenetic silencing of pro-proliferative genes while dampening DDR signaling, permitting survival and expansion of damaged clones[13,63]. Similarly, in pancreatic intraepithelial neoplasia, ARID1A loss attenuates KRAS-induced senescence, enabling precancerous lesions to progress without triggering p16-dependent arrest[15]. This underscores that GI’s oncogenic power is maximized when it occurs in compartments where senescence machinery is developmentally or epigenetically constrained.
8. Genomic Instability Drives Oncogenic Evolution: From Molecular Lesions to Tissue-Level Malignancy
p16/Rb activation and telomere shortening are tumor-suppressive mechanisms of cell cycle arrest, while the resulting SASP, immune evasion, and microenvironment remodeling are pro-cancerous. While cellular senescence plays a crucial tumor-suppressive role by inhibiting the proliferation of damaged cells, the long-term retention of SnCs can promote the formation of a carcinogenic microenvironment through four main mechanisms: (1) chronic inflammation and matrix remodeling driven by the SASP; (2) evasion of immune surveillance; (3) depletion of stem cells and abnormal tissue regeneration; (4) epigenetic plasticity and cellular dedifferentiation. These mechanisms reflect the dual role of aging in cancer development (Table 2).
Table 2. Dual role of aging in cancer development.
| Aspect | Pro-tumorigenic Effects | Anti-tumorigenic Effects |
| Inflammatory microenvironment | SASP secretes pro-inflammatory factors, remodels the immune microenvironment, promoting tumor growth and metastasis | Cell cycle arrest |
| Immune suppression | Accumulation of driver mutations leads to proto-oncogene activation / tumor suppressor gene inactivation | Immune activation |
| Regenerative impairment | Decline in immune surveillance function (immunosenescence) facilitates tumor immune escape | Homeostasis maintenance |
| Dedifferentiation | Clearing senescent cells may reduce tumor risk | Identity locking |
SASP: senescence-associated secretory phenotype.
9. SASP: A Pro-Tumorigenic Secretome
Although the SASP is generally recognized for its pro-carcinogenic properties, its components are highly dependent on the cell type and microenvironment. For instance, fibroblast-derived SASP is enriched in IL-6, which drives epithelial-mesenchymal transition (EMT), while endothelial cell-derived SASP primarily promotes angiogenesis through vascular endothelial growth factor[16,64]. SASP comprises a heterogeneous mixture of pro-inflammatory cytokines (e.g., IL-6, IL-8), chemokines, growth factors, and proteases. While some components may transiently support tissue repair, the net effect of persistent SASP secretion in aging is the creation of a chronically inflamed, immunosuppressive, and growth-promoting microenvironment conducive to tumor development[65]. The components of SASP are highly heterogeneous, making its tumor-promoting mechanisms complex and context-dependent. For example, IL-6 and IL-8 are frequently upregulated in aggressive breast cancers and correlate with poor prognosis by enhancing stemness and suppressing anti-tumor immunity[16,64,66-68]. Notably, treatment-induced senescence, such as that triggered by CDK4/6 inhibitors, can inadvertently promote tumor relapse through SASP-mediated immunosuppression and stromal remodeling[17,69-71]. illustrating the therapeutic dilemma posed by senescence induction. Crucially, the SASP is context- and cell type-dependent, suggesting that selective modulation, not broad suppression, may be required for effective intervention. The pro-tumorigenic impact of SASP is not intrinsic but spatially constrained: stromal IL-6 promotes EMT and immunosuppression, whereas
10. Evasion of Immune Surveillance
Aging drives immunosenescence and chronic inflammation (“inflammaging”), impairing T/NK-cell function and promoting immune evasion via expansion of myeloid-derived suppressor cells (MDSCs), which upregulate PD-L1, CXCR2, arginase-1, iNOS, and ROS, thereby facilitating T-cell exhaustion and pre-metastatic niche formation[72,73]. Senescent stromal and macrophage populations
11. Exhaustion of Stem Cells and Abnormal Tissue Regeneration
Aging impairs stem/progenitor cell self-renewal and differentiation through epigenetic reprogramming and accumulated DNA damage, leading to functional exhaustion (e.g., reduced neurogenesis, vascular repair, colonic epithelial renewal) and aberrant regeneration, which collectively foster precancerous field formation[76-81]. Critically, accelerated epigenetic aging in histologically normal colonic epithelium directly predicts future colorectal cancer risk, confirming that epigenetic erosion in stem cell niches is an early, field-defining event preceding neoplasia[14].
12. Epigenetic Plasticity and Cellular Dedifferentiation
Aging-associated epigenetic drift, including global hypomethylation, promoter-specific hypermethylation (e.g., CDKN2A), histone modification imbalances (e.g., loss of H3K9me3, gain of H3K27ac), and chromatin disorganization, disrupts transcriptional fidelity and promotes cellular dedifferentiation: a reversion to stem-like states that enhances lineage plasticity, therapy resistance, metastasis, and immune evasion[38,82-88]. This epigenetic “permissive state” mirrors cancer epigenomes and is mechanistically reinforced by metabolic reprogramming (e.g., Warburg effect), which alters acetyl-CoA availability and reshapes histone acetylation
Critically, the pro-tumorigenic impact of aging-associated mechanisms is not uniform across malignancies. For instance, melanoma, characterized by high mutational burden, constitutive MAPK activation, and intrinsic resistance to senescence, often exhibits attenuated response to CDK4/6 inhibitor–induced senescence and may even exploit SASP-mediated immunosuppression to evade checkpoint blockade[17,71]. In contrast, low-grade serous ovarian carcinoma or indolent chronic lymphocytic leukemia frequently harbor age-acquired TP53 mutations or NOTCH1 alterations that impair senescence execution, rendering them resistant to senescence-inducing therapies but potentially vulnerable to senolytic clearance of pre-malignant stromal niches[13,14]. Furthermore, molecular subtypes defined by epigenetic regulators (e.g., EZH2-mutant follicular lymphoma) or DNA repair defects (e.g., POLE-mutant endometrial cancer) exhibit distinct dependencies on SASP components: EZH2-mutant cells show heightened sensitivity to JAK/STAT inhibition due to IL-6-driven survival signaling, whereas POLE-mutant tumors accumulate CCFs and respond preferentially to
13. Functional Imbalance of the p53/p16-Rb Pathway
Oncogenes may also trigger cellular defense responses. For instance, the aberrant activation of the proto-oncogene “ras” not only drives tumorigenesis but also induces a phenomenon known as “oncogene-induced senescence” (OIS), which leads to a
This phenomenon of “senescence reversal”, while potentially improving tissue function in the short term, allows cells with DNA damage to continue dividing, thus accumulating more mutations and significantly increasing the risk of HCC. In a mouse model of liver fibrosis induced by CCl4, activated hepatic stellate cells enter a state of senescence[100]. Compared to the control group of
14. NF-κB and Activation of the Inflammatory Signaling Network
The NF-κB signaling network plays a central role in the formation of the aging-associated inflammatory microenvironment. IL-17A can induce endothelial cell senescence through the NF-κB/p53/Rb signaling pathway[101]. In hepatocytes, GATA4 can directly regulate NF-κB activation to induce premature senescence, a mechanism independent of the classical p53-p21 pathway. Hexavalent chromium triggers hepatocytes[102]. Studies have also found that Astragaloside IV alleviates inflammation by inhibiting the
15. Dual Regulatory Role of the mTOR Autophagy Pathway
The mTOR autophagy pathway exhibits dual regulatory characteristics in aging and carcinogenesis. Research indicates that taurine can inhibit the Akt/mTOR signaling by enhancing PTEN activity, thereby promoting the dephosphorylation of ULK1 and ATG13 to activate autophagy, ultimately reducing inflammation damage caused by infection[106]. In glioblastoma, inhibition of the
16. Towards Precision Interventions
16.1 Combination of biomarkers for early risk prediction
GI-related aging characteristics, particularly DNA methylation signatures, provide a novel, quantifiable source of biomarkers for early cancer warning. As comprehensively summarized by Chen et al., aberrant methylation patterns (e.g., LINE-1 hypomethylation, ELOVL2 hypermethylation) exhibit robust associations with both biological age acceleration and tissue-specific cancer incidence, supporting their integration into multi-modal risk models[110]. Studies have shown that accumulated DNA damage signals (such as γH2AX) in SnCs and ROS produced by mitochondrial dysfunction can form specific molecular characteristics[15]. By integrating telomere length detection, CIN scoring, and epigenetic clock analysis, a multi-parameter risk assessment model can be established[7]. It is noteworthy that different tumor types exhibit age-specific genomic variation profiles, such as age-related copy number variation patterns in gliomas and endometrial cancer[8], providing a basis for developing tissue-specific prediction tools.
16.2 Development and clinical trials of SASP inhibitors
As a key nexus connecting aging and cancer, the SASP has become an important target for drug development. Preclinical studies have shown that targeting the NF-κB or IL-6/JAK/STAT pathways can effectively block the tumor-promoting effects of SASP[111]. Currently, several small molecule inhibitors have entered clinical trial phases, including p38 MAPK inhibitors (such as Losmapimod) and JAK inhibitors (such as Ruxolitinib) (Figure 3)[42]. However, it is important to note that complete inhibition of SASP may affect its physiological roles in tissue repair. Therefore, developing selective strategies to precisely modulate the secretion of specific cytokines has become a research focus[112]. Moreover, non-pharmacological interventions may effectively regulate the SASP and the aging process. Recent studies indicate that acute exercise can trigger a temporary stress response, while long-term regular exercise can induce adaptive changes, significantly reducing the accumulation of SnCs in tissues and suppressing chronic inflammation. This effect may be related to the enhanced metabolism of betaine[60]. Betaine can directly bind to and inhibit the activity of TBK1, a key node in inflammatory pathways such as cGAS-STING, which is involved in the activation of SASP.
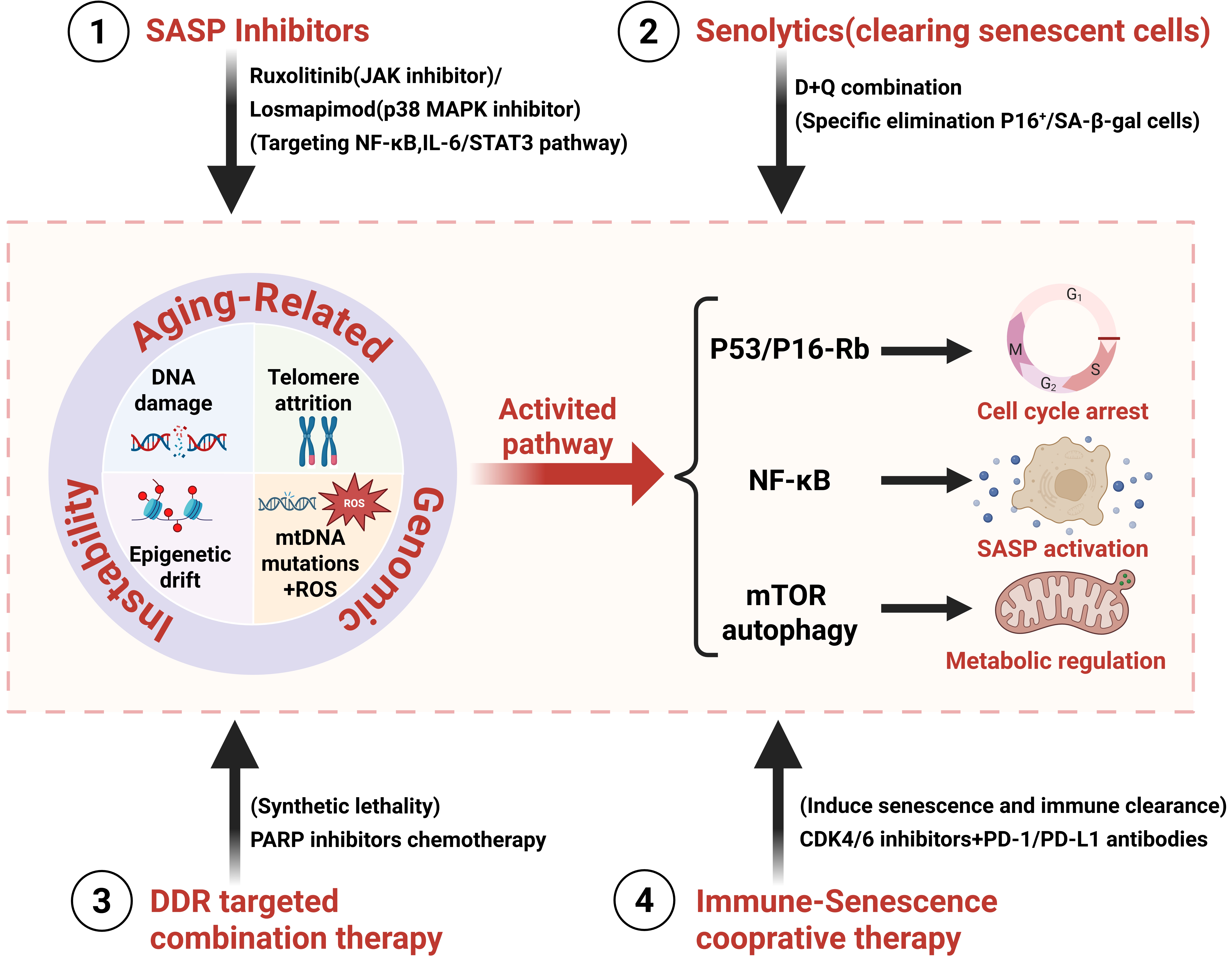
Figure 3. Integrated therapeutic strategies targeting the senescence-GI-cancer axis in aging. GI: genomic instability; SASP: senescence-associated secretory phenotype;
SASP inhibition: This strategy targets the paracrine and systemic tumor-promoting effects of SnCs, rather than eliminating the cells themselves. SnCs secrete a complex mixture of pro-inflammatory cytokines (e.g., IL-6, IL-8), chemokines, growth factors, and proteases, the SASP, which creates a chronically inflamed, immunosuppressive, and tissue-remodeling microenvironment. This microenvironment directly fuels cancer cell proliferation, EMT, therapy resistance, and immune evasion.
Senolytic Therapy (Senescent Cell Clearance): This strategy aims to eliminate SnCs from tissues, thereby removing the source of the harmful SASP and other senescence-associated dysfunctions.
Combined DDR Targeting: This strategy exploits the inherent genomic instability and DNA repair defects that are hallmarks of both aging and SnCs, using them as an “Achilles’ heel” for selective therapeutic intervention.
Synergistic Immunotherapy and Senescence Induction: This strategy represents a sophisticated “double-edged sword” approach that leverages the immune-modulatory duality of cellular senescence.
16.3 Combined therapy targeting DDR
Based on the unique DNA repair defects in SnCs, combined therapy strategies targeting the DDR show promising prospects. Clinical observations have found that chemotherapy-induced senescent tumor cells often exhibit increased GI, making them prone to treatment resistance[113]. By combining PARP inhibitors with conventional chemotherapeutic drugs, it is possible to selectively eliminate senescent cancer cells with DNA repair defects[62]. Recent studies have also discovered that the accumulation of CCFs in SnCs can serve as specific targets, enhancing treatment sensitivity through the activation of the p53 pathway[15,62].
16.4 Synergistic strategy of immunotherapy and senescence induction
The dynamic interplay between the senescent microenvironment and the immune system offers new avenues for combined therapy. On one hand, inducing senescence can enhance tumor antigen presentation and activate innate immune responses[111]. On the other hand, senescence-associated chronic inflammation can promote the formation of an immunosuppressive microenvironment[7]. Preclinical models have shown that combining senescence inducers (such as CDK4/6 inhibitors) with PD-1/PD-L1 inhibitors can significantly improve T-cell infiltration and extend survival[114]. However, SnCs induced by CDK4/6 inhibitors can secrete SASP (such as IL-6 and TGF-β), which not only inhibits T cell function but may also protect residual tumor cells by remodeling the stromal barrier, this ‘double-edged effect’ has been identified as a key safety endpoint in combination trials such as NCT045[69,114]. Current explorations are focusing on combination strategies that enhance immunotherapy efficacy by eliminating senescence-associated MDSCs[42].
While senolytic strategies hold promise, their clinical translation faces three non-trivial barriers: First, pharmacodynamic specificity: Dasatinib inhibits senescence-resistant human mesenchymal progenitor cells (SRC)-family kinases essential for NK-cell
17. Unresolved Questions
17.1 Threshold definition for senescence-cancer transformation
There is significant debate in the academic community regarding the critical threshold for the transformation of SnCs into cancer cells. Research indicates that SnCs, through GI and a chronic inflammatory microenvironment, can both inhibit tumorigenesis and promote the progression of certain malignant tumors[114]. The ‘senescence-to-cancer switch’ lacks quantitative thresholds: Is it defined by CCF load (> 5/cell?), telomere dysfunction burden (≥ 3 uncapped ends/cell?), or SASP cytokine stoichiometry
17.2 Mechanistic elucidation of tissue-specific differences
There is significant heterogeneity in the senescence-cancer association across different tissue types, and the molecular basis for this remains an unresolved issue. In skeletal muscle, senescent myogenic progenitor cells can reverse their senescent phenotype through NANOG overexpression[117], whereas pancreatic tissue shows that specific gene expression can attenuate senescence-induced tumorigenesis[15]. This difference may stem from: i) Specific regulation of DDR by the tissue stem cell microenvironment[14].
17.3 Potential risks of therapeutically induced senescence
Anti-cancer strategies that aim to induce senescence face significant safety concerns. The main risks include: i) Persisting SnCs may promote the recurrence of residual tumor cells through SASP[116]. ii) Senescence induced by DNA-damaging agents may exacerbate GI and lead to secondary carcinogenesis[66]. Adjuvant chemotherapy in breast cancer patients induces rapid acceleration of epigenetic age in peripheral blood correlating with persistent inflammation and frailty, highlighting therapy-induced ‘biological aging’ as a tangible contributor to long-term morbidity and potential relapse risk[119]. Subsequent work further revealed that even localized radiotherapy triggers acute, transient spikes in epigenetic age acceleration in circulating leukocytes, suggesting that DNA damage burden, irrespective of systemic exposure, can rapidly reprogram the epigenome toward a pro-inflammatory, senescence-prone state[120]. Research indicates that the balance between p53-mediated senescence and apoptosis is crucial for therapeutic outcomes[75], but there is no clear method for precisely regulating this balance. Furthermore, the heterogeneity of SnCs results in individual differences in response to anti-senescence drugs[121], adding additional challenges to clinical translation. The interaction between mitochondrial dysfunction and inflammatory pathways is also considered a key node in therapy-related risks[9,122]. The safety of CDK4/6 inhibitors as senescence inducers is further complicated by inter-patient heterogeneity in DDR proficiency. Patients with germline ATM mutations exhibit profound resistance to palbociclib-induced senescence[21], rendering such therapy ineffective or even selecting for ATM-null clones with heightened GI. Hence, biomarker-stratified trials (e.g., ATM protein expression by IHC) are mandatory before broad application.
17.4 The quantitative paradox of senescence: When does SASP switch from tumor-suppressive to oncogenic?
Current evidence reveals no universal SASP ‘dose threshold’ for tumor promotion. In murine models, IL-6 > 100 pg/mL in serum correlates with accelerated lung metastasis[16], yet in human melanoma, high IL-6 associates with improved CD8+ T-cell infiltration[17]. This context-dependency likely stems from spatial compartmentalization: stromal SASP may suppress immunity, while
18. Future Research Directions
18.1 Development and application of spatiotemporal dynamic analysis techniques
With advancements in single-cell sequencing and live-cell imaging technologies, developing new analytical tools capable of capturing the spatiotemporal dynamics of genome instability and the aging process will become a key research direction. Recent studies have shown that the accumulation of aging-related genome instability exhibits significant tissue specificity and temporal heterogeneity[7,123]. In the future, it will be necessary to develop spatiotemporal analysis platforms that integrate
18.2 Cross-scale mechanism integration research
Current research has revealed multi-layered aging mechanisms from molecular damage to tissue microenvironment remodeling, but the causal relationships between these scales remain unclear. In the future, it will be necessary to establish an integrated cross-scale research framework that encompasses DNA repair defects (molecular scale), cellular senescence (cellular scale), and immune microenvironment changes (systemic scale)[7,66]. Key focal points include: i) Developing organoid co-culture systems capable of simultaneously monitoring genome instability and microenvironment changes[126]. ii) Constructing multi-dimensional data analysis models that include epigenetic clocks, metabolomic profiles, and inflammatory factors[39,83]. iii) Using computational biology methods to analyze the quantitative relationships between DNA damage signals and the activation of pathways such as NF-κB[9]. This
18.3 Precision design of personalized intervention strategies
Based on the heterogeneous characteristics of aging-carcinogenesis association mechanisms, developing personalized intervention strategies tailored to different genetic backgrounds and age stages will become a focus in clinical translation. Research indicates that imbalances in the p53/p16-Rb pathway and telomere dysfunction exhibit significant differences among individuals[75,128], suggesting the need to: i) Establish predictive models integrating biomarkers of genome instability (such as SCNAs, mutation profiles) and epigenetic age[39,42]. ii) Develop targeted intervention strategies for specific DNA repair defect subgroups (such as POLE mutation carriers)[59,129]. iii) Explore the temporal combination strategies of SASP modulators with immunotherapy to balance the anti-cancer effects induced by aging with tumor-promoting risks[66,125]. Moreover, metformin exhibits notable neuroprotective effects, significantly slowing aging markers, particularly reversing brain aging, maintaining brain structure, and enhancing cognitive
19. Conclusions
Cellular senescence is not a one-way dead end, but rather a stalled journey requiring continuous “maintenance”; once this maintenance fails, cells may re-enter the cell cycle in a more aggressive “post-senescence” state with profound biological and clinical consequences[130]. Through DNA damage accumulation, telomere attrition, epigenetic dysregulation, and mitochondrial dysfunction, aging creates a fertile ground for oncogenic transformation. SnCs, while initially protective, become drivers of malignancy via the SASP, immune evasion, and regenerative failure. Central signaling nodes, including the p53/p16-Rb, NF-κB, and mTOR-autophagy pathways, orchestrate this transition, offering promising targets for intervention.
GI fulfills the criteria of a unifying driver: it is causally upstream of both aging hallmarks (senescence, mitochondrial decline) and cancer hallmarks (sustained proliferation, invasion, immune evasion). It is a therapeutic nexus because: (i) Its lesions are quantifiable biomarkers for risk stratification (e.g., epigenetic clocks + SCNA scoring); (ii) Its effectors (ATM, cGAS, STING) are druggable nodes whose inhibition redirects cell fate from pro-tumorigenic inflammation to immunogenic clearance; and (iii) Its dynamics (not just presence), such as rate of telomere shortening or CCF turnover, predict therapeutic windows for senolytics vs. DDR inhibitors. Thus, targeting GI, not merely its downstream phenotypes, is the most direct path to precision prevention of age-associated cancer.
Despite progress, critical questions remain about the thresholds governing the switch from tumor suppression to promotion, the basis of tissue-specific vulnerability, and the safety of senescence-inducing therapies. Addressing these will require integrative approaches combining single-cell resolution, organoid modeling, and longitudinal biomarker tracking. Ultimately, targeting the shared mechanisms of aging and cancer holds transformative potential for extending healthspan and preventing age-related malignancies.
20. From Biomarker to Bedside: The Clinical Translation of GI
GI has transcended theoretical concepts and entered clinical practice: Diagnostic aspect: The burden of SCNAs in liquid biopsies, abnormalities in cfDNA fragmentomes, and dynamic monitoring of telomere length have been incorporated into NCCN guidelines for cancer risk stratification in elderly patients[8,42];
Therapeutic aspect: The success of PARP inhibitors in BRCA-mutant (essentially HR-GI) tumors has validated the paradigm of “targeting GI defects”; the proposed classification of GI subtypes (e.g., CCF-high vs. telomere-short tumors) is driving the design of novel clinical trials (e.g., NCT045, targeting cGAS inhibitors combined with PD-1 antibodies);
Preventive aspect: GI-based “aging clocks” (e.g., GrimAge, DNAmTL) are better predictors of cancer occurrence than traditional phenotypic age, providing a basis for precision chemoprevention[39,42].
Acknowledgements
The authors declare that ChatGPT was used solely for language polishing during the manuscript preparation process. All research content, including study design, data analysis, interpretations, figures, and tables, is original and was not generated using AI tools.
Authors contribution
Gu L: Conceptualization, writing-original draft, data curation.
Zhu Y: Conceptualization.
Xiong D: Writing-original draft, data curation.
Cheng L: Visualization, writing review & editing.
Conflicts of interest
Li Gu is a Youth Editorial Board Member of Ageing and Cancer Research & Treatment. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This study was supported by the National Nature Science Foundation of China (Grant Nos 82172990 and 82372838 to Yahui Zhu, Grant Nos. 32470830 and 92478135 to Li Gu), Sichuan Science and Technology Program (Grant No. 2025NSFJQ0031), and Sichuan University Interdisciplinary Innovation Fund.
Copyright
© The Author(s) 2026.
References
-
2. Laffon B, Bonassi S, Costa S, Valdiglesias V. Genomic instability as a main driving factor of unsuccessful ageing: Potential for translating the use of micronuclei into clinical practice. Mutat Res Mutat Res. 2021;787:108359.[DOI]
-
3. Siametis A, Niotis G, Garinis GA. DNA damage and the aging epigenome. J Investig Dermatol. 2021;141(4):961-967.[DOI]
-
4. Kallai A, Ungvari Z, Fekete M, Maier AB, Mikala G, Andrikovics H, et al. Genomic instability and genetic heterogeneity in aging: Insights from clonal hematopoiesis (CHIP), monoclonal gammopathy (MGUS), and monoclonal B-cell lymphocytosis (MBL). Geroscience. 2025;47(1):703-720.
-
11. Panier S, Wang S, Schumacher B. Genome instability and DNA repair in somatic and reproductive aging. Annu Rev Pathol Mech Dis. 2024;19:261-290.[DOI]
-
12. Bühler M, Stolz A. Estrogens: Origin of centrosome defects in human cancer? Cells. 2022;11(3):432.[DOI]
-
13. Al Zouabi L, Bardin AJ. Stem cell DNA damage and genome mutation in the context of aging and cancer initiation. Cold Spring Harb Perspect Biol. 2020;12(10):a036210.[DOI]
-
14. Huang Y, Che X, Wang PW, Qu X. p53/MDM2 signaling pathway in aging, senescence and tumorigenesis. Semin Cancer Biol. 2024;101:44-57.[DOI]
-
15. Liu S, Cao W, Niu Y, Luo J, Zhao Y, Hu Z, et al. Single-PanIN-seq unveils that ARID1A deficiency promotes pancreatic tumorigenesis by attenuating KRAS-induced senescence. eLife. 2021;10:e64204.[DOI]
-
17. Homann L, Rentschler M, Brenner E, Böhm K, Röcken M, Wieder T. IFN-γ and TNF induce senescence and a distinct senescence-associated secretory phenotype in melanoma. Cells. 2022;11(9):1514.[DOI]
-
18. De Majo F, Martens L, Hegenbarth JC, Rühle F, Hamczyk MR, Nevado RM, et al. Genomic instability in the naturally and prematurely aged myocardium. Proc Natl Acad Sci U S A. 2021;118(36):e2022974118.[DOI]
-
22. Yamauchi S, Sugiura Y, Yamaguchi J, Zhou X, Takenaka S, Odawara T, et al. Mitochondrial fatty acid oxidation drives senescence. Sci Adv. 2024;10(43):eado5887.[DOI]
-
23. Goutas A, Outskouni Z, Papathanasiou I, Georgakopoulou A, Karpetas GE, Gonos ES, et al. The establishment of mitotic errors-driven senescence depends on autophagy. Redox Biol. 2023;62:102701.[DOI]
-
24. Miller KN, Dasgupta N, Liu T, Adams PD, Vizioli MG. Cytoplasmic chromatin fragments: From mechanisms to therapeutic potential. eLife. 2021;10:e63728.[DOI]
-
26. Han JJ. The ticking of aging clocks. Trends Endocrinol Metab. 2024;35(1):11-22.[DOI]
-
27. Dalmasso G, Cougnoux A, Faïs T, Bonnin V, Mottet-Auselo B, Nguyen HT, et al. Colibactin-producing Escherichia coli enhance resistance to chemotherapeutic drugs by promoting epithelial to mesenchymal transition and cancer stem cell emergence. Gut Microbes. 2024;16(1):2310215.
-
29. Heba AC, Toupance S, Arnone D, Peyrin-Biroulet L, Benetos A, Ndiaye NC. Telomeres: New players in immune-mediated inflammatory diseases? J Autoimmun. 2021;123:102699.[DOI]
-
30. Lu X, Liu L. Genome stability from the perspective of telomere length. Trends Genet. 2024;40(2):175-186.[DOI]
-
31. Vessoni AT, Zhang T, Quinet A, Jeong HC, Munroe M, Wood M, et al. Telomere erosion in human pluripotent stem cells leads to ATR-mediated mitotic catastrophe. J Cell Biol. 2021;220(6):e202011014.[DOI]
-
33. Couteau F, Gagné LM, Boulay K, Rousseau P, Carbonneau M, McQuaid M, et al. R-2-hydroxyglutarate-mediated inhibition of KDM4A compromises telomere integrity. Nucleic Acids Res. 2025;53(11):gkaf512.[DOI]
-
36. Kapetanou M, Athanasopoulou S, Goutas A, Makatsori D, Trachana V, Gonos E. α-terpineol induces shelterin components TRF1 and TRF2 to mitigate senescence and telomere integrity loss via a telomerase-independent pathway. Antioxidants. 2024;13(10):1258.[DOI]
-
37. Rossiello F, Jurk D, Passos JF, d’Adda di Fagagna F. Telomere dysfunction in ageing and age-related diseases. Nat Cell Biol. 2022;24(2):135-147.[DOI]
-
46. Soto-Palma C, Niedernhofer LJ, Faulk CD, Dong X. Epigenetics, DNA damage, and aging. J Clin Investig. 2022;132(16):e158446.[DOI]
-
47. Ye T, Gao H, Zhang ZR, Ge Y, Liu YK, Yan JY, et al. Epigenetic regulation of cellular senescence in gastrointestinal cancer. Mol Cancer Ther. 2025;24(8):1145-1155.[DOI]
-
50. Miwa S, Kashyap S, Chini E, von Zglinicki T. Mitochondrial dysfunction in cell senescence and aging. J Clin Investig. 2022;132(13):e158447.[DOI]
-
51. Chang X, Liu J, Wang Y, Guan X, Liu R. Mitochondrial disorder and treatment of ischemic cardiomyopathy: Potential and advantages of Chinese herbal medicine. Biomed Pharmacother. 2023;159:114171.[DOI]
-
54. Yu T, Slone J, Liu W, Barnes R, Opresko PL, Wark L, et al. Premature aging is associated with higher levels of 8-oxoguanine and increased DNA damage in the Polg mutator mouse. Aging Cell. 2022;21(9):e13669.[DOI]
-
55. Rahman MA, Rahman MH, Biswas P, Hossain MS, Islam R, Hannan MA, et al. Potential therapeutic role of phytochemicals to mitigate mitochondrial dysfunctions in Alzheimer’s disease. Antioxidants. 2021;10(1):23.[DOI]
-
57. Terao R, Ahmed T, Suzumura A, Terasaki H. Oxidative stress-induced cellular senescence in aging retina and age-related macular degeneration. Antioxidants. 2022;11(11):2189.[DOI]
-
58. Lee YH, Kuk MU, So MK, Song ES, Lee H, Ahn SK, et al. Targeting mitochondrial oxidative stress as a strategy to treat aging and age-related diseases. Antioxidants. 2023;12(4):934.[DOI]
-
60. Geng L, Ping J, Wu R, Yan H, Zhang H, Zhuang Y, et al. Systematic profiling reveals betaine as an exercise mimetic for geroprotection. Cell. 2025;188(19):5403-5425.[DOI]
-
62. Patra S, Naik PP, Mahapatra KK, Alotaibi MR, Patil S, Patro BS, et al. Recent advancement of autophagy in polyploid giant cancer cells and its interconnection with senescence and stemness for therapeutic opportunities. Cancer Lett. 2024;590:216843.[DOI]
-
63. Laconi E, Cheri S, Fanti M, Marongiu F. Aging and cancer: The waning of community bonds. Cells. 2021;10(9):2269.[DOI]
-
66. Ngoi NY, Liew AQ, Chong SJF, Davids MS, Clement MV, Pervaiz S. The redox-senescence axis and its therapeutic targeting. Redox Biol. 2021;45:102032.[DOI]
-
67. Guo Z, Zhang Y, Gong Y, Li G, Pan J, Dou D, et al. Antibody functionalized curcuma-derived extracellular vesicles loaded with doxorubicin overcome therapy-induced senescence and enhance chemotherapy. J Control Release. 2025;379:377-389.[DOI]
-
68. Wang X, Shi W, Wang X, Lu JJ, He P, Zhang H, et al. Nifuroxazide boosts the anticancer efficacy of palbociclib-induced senescence by dual inhibition of STAT3 and CDK2 in triple-negative breast cancer. Cell Death Discov. 2023;9:355.[DOI]
-
72. Zhang J, Guan X, Zhong X. Immunosenescence in digestive system cancers: Mechanisms, research advances, and therapeutic strategies. Semin Cancer Biol. 2024;106:234-250.[DOI]
-
74. Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016;7:11762.[DOI]
-
77. Xiong J, Dong L, Lv Q, Yin Y, Zhao J, Ke Y, et al. Targeting senescence-associated secretory phenotypes to remodel the tumour microenvironment and modulate tumour outcomes. Clin Transl Med. 2024;14(9):e1772.[DOI]
-
78. Wang W, Wang Y, Duan C, Tian W, Gao L. LncRNA NEAT1-206 regulates autophagy of human umbilical cord mesenchymal stem cells through the WNT5A/Ca2+ signaling pathway under senescence stress. Non Coding RNA Res. 2025;11:234-248.[DOI]
-
80. Shimizu S, Iba T, Naito H, Rahmawati FN, Konishi H, Jia W, et al. Aging impairs the ability of vascular endothelial stem cells to generate endothelial cells in mice. Angiogenesis. 2023;26(4):567-580.[DOI]
-
83. Kordowitzki P, Grzeczka A. Unveiling the relation between cellular ageing, epigenetics and cancer. Aging Dis. 2025.[DOI]
-
84. Allegra A, Caserta S, Mirabile G, Gangemi S. Aging and age-related epigenetic drift in the pathogenesis of leukemia and lymphomas: New therapeutic targets. Cells. 2023;12(19):2392.[DOI]
-
85. Goel S, Bhatia V, Biswas T, Ateeq B. Epigenetic reprogramming during prostate cancer progression: A perspective from development. Semin Cancer Biol. 2022;83:136-151.[DOI]
-
88. Cheng J, Cao J, Yang Y, Wang Y, Hu X, Liu Z, et al. Multi-omics analysis unraveling stemness features associated with oncogenic dedifferentiation in 12 cancers. Cancer Lett. 2025;625:217816.[DOI]
-
90. Lazure F, Gomes AP. Cancer progression through the lens of age-induced metabolic reprogramming. Nat Rev Cancer. 2025;25(10):801-817.[DOI]
-
98. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479(7374):547-551.[DOI]
-
99. Saini N, Gordenin DA. Hypermutation in single-stranded DNA. DNA Repair. 2020;91:102868.[DOI]
-
102. Ma Y, Li S, Ye S, Hu D, Wei L, Xiao F. Hexavalent chromium triggers hepatocytes premature senescence via the GATA4/NF-κB signaling pathway mediated by the DNA damage response. Ecotoxicol Environ Saf. 2022;239:113645.[DOI]
-
103. Ying Y, Sun CB, Zhang SQ, Chen BJ, Yu JZ, Liu FY, et al. Induction of autophagy via the TLR4/NF-κB signaling pathway by astragaloside IV contributes to the amelioration of inflammation in RAW264.7 cells. Biomed Pharmacother. 2021;137:111271.[DOI]
-
106. Wang Z, Lan R, Xu Y, Zuo J, Han X, Phouthapane V, et al. Taurine alleviates streptococcus uberis-induced inflammation by activating autophagy in mammary epithelial cells. Front Immunol. 2021;12:631113.[DOI]
-
107. Tang Q, Cao H, Tong N, Liu Y, Wang W, Zou Y, et al. Tubeimoside-I sensitizes temozolomide-resistant glioblastoma cells to chemotherapy by reducing MGMT expression and suppressing EGFR induced PI3K/Akt/mTOR/NF-κB-mediated signaling pathway. Phytomedicine. 2022;99:154016.[DOI]
-
108. Li J, Gu X, Wan G, Wang Y, Chen K, Chen Q, et al. Rocuronium bromide suppresses esophageal cancer via blocking the secretion of C–X–C motif chemokine ligand 12 from cancer associated fibroblasts. J Transl Med. 2023;21(1):248.[DOI]
-
110. Chen L, Ganz PA, Sehl ME. DNA methylation, aging, and cancer risk: A mini-review. Front Bioinform. 2022;2:847629.[DOI]
-
111. Saleh T, Carpenter VJ, Bloukh S, Gewirtz DA. Targeting tumor cell senescence and polyploidy as potential therapeutic strategies. Semin Cancer Biol. 2022;81:37-47.[DOI]
-
113. Soto-Gamez A, Wang Y, Zhou X, Seras L, Quax W, Demaria M. Enhanced extrinsic apoptosis of therapy-induced senescent cancer cells using a death receptor 5 (DR5) selective agonist. Cancer Lett. 2022;525:67-75.[DOI]
-
114. Wang L, Lankhorst L, Bernards R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer. 2022;22(6):340-355.[DOI]
-
115. Fang Y, Medina D, Stockwell R, McFadden S, Quinn K, Peck MR, et al. Sexual dimorphic metabolic and cognitive responses of C57BL/6 mice to Fisetin or Dasatinib and quercetin cocktail oral treatment. GeroScience. 2023;45(5):2835-2850.[DOI]
-
117. Tobón-Cornejo S, Vargas-Castillo A, Juarez M, Acevedo-Carabantes JA, Noriega LG, Granados-Portillo O, et al. Metabolic reprogramming and synergistic cytotoxicity of genistein and chemotherapy in human breast cancer cells. Life Sci. 2025;370:123562.[DOI]
-
119. Sanoff HK, Deal AM, Krishnamurthy J, Torrice C, Dillon P, Sorrentino J, et al. Effect of cytotoxic chemotherapy on markers of molecular age in patients with breast cancer. J Natl Cancer Inst. 2014;106(4):dju057.[DOI]
-
120. Sehl ME, Carroll JE, Horvath S, Bower JE. The acute effects of adjuvant radiation and chemotherapy on peripheral blood epigenetic age in early stage breast cancer patients. npj Breast Cancer. 2020;6:23.[DOI]
-
121. Li C, Yuan Y, Jia Y, Zhou Q, Wang Q, Jiang X. Cellular senescence: From homeostasis to pathological implications and therapeutic strategies. Front Immunol. 2025;16:1534263.[DOI]
-
122. Urbancokova A, Hornofova T, Novak J, Salajkova SA, Hubackova SS, Uvizl A, et al. Topological stress triggers persistent DNA lesions in ribosomal DNA with ensuing formation of PML-nucleolar compartment. eLife. 2024;12:RP91304.[DOI]
-
123. Tsubosaka A, Komura D, Kakiuchi M, Katoh H, Onoyama T, Yamamoto A, et al. Stomach encyclopedia: Combined single-cell and spatial transcriptomics reveal cell diversity and homeostatic regulation of human stomach. Cell Rep. 2023;42(10):113236.[DOI]
-
127. Montégut L, López-Otín C, Kroemer G. Aging and cancer. Mol Cancer. 2024;23:106.[DOI]
-
129. Zhang P, Wu Z, Ju Q, Xu N, Chen X, Chen H, et al. POLE deficiency exacerbates diesel engine exhaust-induced genomic instability and malignant transformation of bronchial epithelial cells. Adv Sci. 2025;12(36):e15943.[DOI]
-
130. Reimann M, Lee S, Schmitt CA. Cellular senescence: Neither irreversible nor reversible. J Exp Med. 2024;221(4):e20232136.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Xiong D, Cheng L, Zhu Y, Gu L. From aging to cancer: Genomic instability as a unifying driver and therapeutic nexus. Ageing Cancer Res Treat. 2026;3:202524. https://doi.org/10.70401/acrt.2026.0019
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Core Mechanisms Linking GI-Driven Senescence
- 3. Accumulation of DNA Damage and Dysfunction of Repair Systems
- 4. Telomere Attrition
- 5. Epigenetic Dysregulation
- 6. Mitochondrial Dysfunction and Oxidative Stress
- 7. Genomic Instability as A Direct Oncogenic Catalyst: Beyond Senescence
- 8. Genomic Instability Drives Oncogenic Evolution: From Molecular Lesions to Tissue-Level Malignancy
- 9. SASP: A Pro-Tumorigenic Secretome
- 10. Evasion of Immune Surveillance
- 11. Exhaustion of Stem Cells and Abnormal Tissue Regeneration
- 12. Epigenetic Plasticity and Cellular Dedifferentiation
- 13. Functional Imbalance of the p53/p16-Rb Pathway
- 14. NF-κB and Activation of the Inflammatory Signaling Network
- 15. Dual Regulatory Role of the mTOR Autophagy Pathway
- 16. Towards Precision Interventions
- 17. Unresolved Questions
- 18. Future Research Directions
- 19. Conclusions
- 20. From Biomarker to Bedside: The Clinical Translation of GI
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Xiong D, Cheng L, Zhu Y, Gu L. From aging to cancer: Genomic instability as a unifying driver and therapeutic nexus. Ageing Cancer Res Treat. 2026;3:202524. https://doi.org/10.70401/acrt.2026.0019
copy
Share Link
copy