Engineered T cell-derived extracellular vesicles for cancer immunotherapy
Rui Diao
1,2,#
,
Xiaozhou Zhang
1,2,#
,
Xudong Zhang
1,2,3,*

*Correspondence to:
Xudong Zhang, Shenzhen Key Laboratory for Systems Medicine in Inflammatory Diseases, School of Medicine, Shenzhen Campus of Sun Yat-Sen University, Sun Yat-Sen University, Shenzhen 518107, Guangdong, China; Department of Pharmacology, Molecular Cancer Research Center, School of Medicine, Shenzhen Campus of Sun Yat-Sen University, Sun Yat-sen University, Shenzhen 518107, Guangdong, China; Key Laboratory of Regenerative Medicine of Ministry of Education, Jinan University, Jinan 250022, Shandong, China.
E-mail: zhangxd56@mail.sysu.edu.cn
BME Horiz. 2026;4:202537. 10.70401/bmeh.2026.0017
Received: December 15, 2025Accepted: February 09, 2026Published: March 01, 2026
Abstract
T cell-derived extracellular vesicles (TcEVs) are nanoscale lipid bilayer-bound particles, including exosomes, microvesicles, apoptotic bodies, and T cell microvilli particles. TcEVs possess advantages such as high yield, favorable biocompatibility, low immunogenicity, and excellent solid tumor penetration, showing great potential in cancer immunotherapy. However, natural TcEVs exhibit weak targeting ability, limited immune activity, and low drug-loading capacity. To address these issues, several engineering strategies have been adopted to modify the vesicles through genetic engineering, surface modification, and drug-loading, thereby achieving effective treatment of both hematological and solid tumors. This review summarizes TcEVs’ classification, biological functions, engineering strategies, and applications in cancer immunotherapy, while discussing challenges and prospects to facilitate their clinical translation.
Keywords
Immunotherapy, T cells, extracellular vesicles, engineering modification
1. Introduction
Cancer immunotherapy has emerged as a revolutionary treatment modality, with chimeric antigen receptor T (CAR-T) cells and T cell receptor T (TCR-T) cells representing promising approaches. However, the clinical application of these cell therapies is hindered by issues such as off-target effects and severe side effects[1]. In recent years, extracellular vesicles (EVs) derived from T cells (TcEVs) have emerged as a promising “cell-free” alternative, integrating the immunological activity of parent T cells with the natural advantages of EVs. TcEVs overcome key limitations of current therapies: they lack proliferative capacity to avoid excessive immune activation, penetrate solid tumors more efficiently due to their nanoscale size, avoid programmed death-ligand 1 (PD-L1)-mediated inhibition, and enables standardized production and preservation[2]. These unique properties make TcEVs a versatile platform bridging cell therapy and targeted drug delivery, providing great potential to address unmet clinical needs in both hematological and solid tumors (Table 1).
Table 1. Advantages and disadvantages of current cancer immunotherapy technologies and strategies.
| Therapeutic Strategy | Main Advantages | Main Disadvantages |
| CAR-T Cell Therapy | Potent targeted killing, long-term memory effect | Off-target toxicity, CRS/ICANS, poor solid tumor penetration, personalized production |
| TCR-T Cell Therapy | Recognition of endogenous antigens, low off-target risk | Strict antigen restriction, sensitivity to TME suppression |
| Immune Checkpoint Inhibitors | Broad applicability, simple operation | Low response rate, immune-related adverse reactions |
| Oncolytic Viruses | Dual effects of oncolysis and immune activation | Pre-existing immunity interference, insufficient targeting |
| Cancer Vaccines | Active immunity, long-term protection | Weak immunogenicity, low response rate |
| TcEVs Therapy | No proliferative toxicity, strong solid tumor penetration, standardized production, low immunogenicity | Weak natural targeting, limited immune activity |
CAR-T: chimeric antigen receptor T; TCR-T: T cell receptor T; TcEVs: T cell-derived extracellular vesicles; TME: tumor microenvironment; CRS: cytokine release syndrome; ICANS: immune effector cell-associated neurotoxicity syndrome.
1.1 Overview of EVs
EVs are nanoscale particles enclosed by a lipid bilayer and released by cells, playing a pivotal role in intercellular communication[3,4]. Based on size, biological characteristics, and biogenesis mechanisms, EVs can be roughly categorized into three classes: exosomes (Exos), microvesicles (MVs), and apoptotic bodies (ABs)[5,6]. Exos, with a diameter of approximately 30-150 nm, originate from intraluminal vesicles released by the fusion of multivesicular bodies with the plasma membrane, and their biogenesis relies on the endosomal sorting complex required for transport (ESCRT), lipid, or tetraspanin pathways[7-9]. Exos are enriched in tetraspanin family members (e.g., CD63, CD81, CD9), ESCRT-related proteins (ALIX, TSG101), and heat shock proteins (HSP70, HSP90)[10,11]. MVs, with a diameter of approximately 100-1,000 nm, are directly produced by plasma membrane budding; their composition is similar to that of the parent cell membrane, carrying a large number of membrane proteins and phospholipids[12]. ABs range from 1-5 μm in diameter and are vesicular structures formed by the plasma membrane wrapping nuclear fragments, organelles, etc., during programmed cell death[13,14].
In recent years, a specialized type of T cell-derived EV, namely T cell microvilli particles (TMPs), has been reported. T cells are rich in microvilli on their surface; TMPs are membrane particles formed by the fragmentation and shedding of microvilli after T cells adhere to antigen-presenting cells (APCs), with a diameter of approximately 20-500 nm. They are enriched in T cell receptor complexes (TCRα/β, CD3ζ/ε/γ) and adhesion molecules (CD2, CD44, L-selectin). Notably, cholesterol sulfate can directly induce microvilli fragmentation to release TMPs[15-17] (Figure 1b, Table 2).
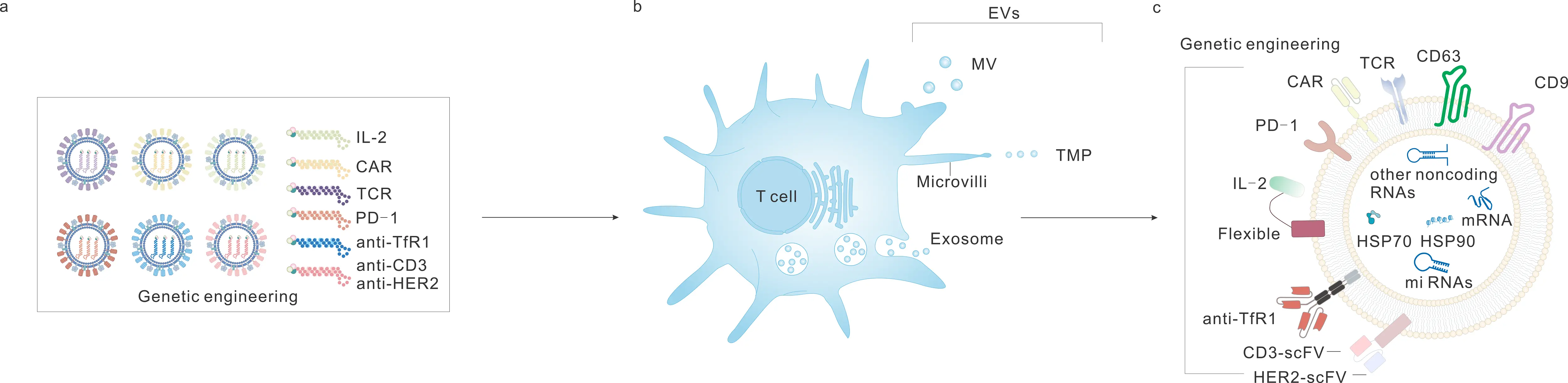
Figure 1. Genetic engineering and biogenesis of engineered TcEVs. This diagram illustrates the generation of engineered TcEVs from T cells. (a) Genetic engineering: Key functional molecules (IL-2, CAR, TCR, PD-1, anti-TfR1, anti-CD3, anti-HER2) are introduced into T cells, such as via lentiviral transduction, to modify their membrane components; (b) Biogenesis of TcEVs: T cells secrete diverse TcEV subtypes, including MV, exosomes, and TMP, all of which inherit the engineered surface molecules from parental T cells; (c) Molecular composition of engineered TcEVs: The phospholipid bilayer integrates genetically modified surface components (CAR, TCR, PD-1, IL-2, anti-TfR1, CD3-scFV, HER2-scFV) and typical exosome markers (CD63, CD9). The vesicle lumen also contains diverse intrinsic cargos (mRNA, miRNAs, other noncoding RNAs, HSP70, HSP90), forming the structural and functional basis for subsequent modification. TcEVs: T cell-derived extracellular vesicles; CAR: chimeric antigen receptor; TCR: T cell receptor; TfR1: transferrin receptor 1; MV: microvesicles; TMP: T cell microvilli particles; mRNA: messenger RNA; miRNAs: microRNAs; HSP: heat shock proteins; PD-1: programmed cell death protein 1.
Table 2. Summary of applications of different types of extracellular vesicles in cancer immunotherapy.
| EV Type | Diameter Range | Biogenesis Pathway | Core Functions | Potential Applications | Key References |
| Exos | 30-150 nm | Fusion of MVBs with the plasma membrane | Antigen presentation, immune signal transduction | Off-target toxicity, CRS/ICANS, poor solid tumor penetration, personalized production | [6-8] |
| MVs | 100-1,000 nm | Direct budding from the plasma membrane | Targeted killing, inflammation regulation | Strict antigen restriction, sensitivity to TME suppression | [11] |
| ABs | 1-5 μm | Membrane encapsulation during cellular apoptosis | Antigen cross-presentation, immune tolerance induction | Low response rate, immune-related adverse reactions | [12,13] |
| TMPs | 200-500 nm | Fragmentation and shedding of T cell microvilli | TCR enrichment, antigen recognition | Pre-existing immunity interference, insufficient targeting | [14-16] |
Exos: exosomes; MVs: microvesicles; ABs: apoptotic bodies; TMPs: T cell microvilli particles; MVBs: multivesicular bodies; TCR: T cell receptor; TME: tumor microenvironment; CRS: cytokine release syndrome; ICANS: immune effector cell-associated neurotoxicity syndrome.
1.2 Biological functions and therapeutic potential of TcEVs
As natural nanocarriers, EVs possess unique biological characteristics that endow them with significant advantages in cancer immunotherapy.
1.2.1 Targeted killing of cancer cells or pathogen-infected cells
Compared with EVs derived from other immune cells, TcEVs carry TCR/CD3ζ complexes on their surface, which can specifically recognize the “peptide-MHC-I complexes (pMHC-I)” on the surface of target cells[18]. Among TcEVs, those secreted by antigen-activated cytotoxic T lymphocytes (CTLs) serve as crucial carriers mediating cytotoxicity[19]. More precisely, CTL-derived TcEVs encapsulate highly active granzyme and perforin: upon binding to target cells, perforin forms pores in the cell membrane, allowing granzyme to enter the cytoplasm and activate apoptotic pathways, thereby inducing the death of cancer cells or virus-infected cells[20].
In contrast to traditional CAR-T cell therapy, EVs have no proliferative capacity and do not trigger excessive immune activation[21]. Furthermore, EVs lack expression of programmed cell death protein 1 (PD-1), and thus will not be bound by PD-L1 in the tumor microenvironment (TME), avoiding functional inhibition and reducing the risk of immune escape[22]. With a diameter ranging from 30-1,500 nm, EVs can penetrate the dense extracellular matrix and tumor-associated fibroblasts barrier of solid tumors, infiltrating deep into the tumor tissue with strong penetrability[23].
Moreover, unlike personalized CAR-T therapy, TcEVs can be mass-produced, strictly quality-controlled, and stored for a long time, making them more conducive to standardization and clinical application[24]. Studies have found that storing EVs in phosphate-buffered saline supplemented with human albumin and trehalose can support stable preservation of EVs at -80 °C for 2 years, which is expected to further promote clinical translation[25].
1.2.2 Maintenance of immune homeostasis
Derived from autologous cells, EVs retain the TCR, costimulatory molecules (e.g., CD28), and adhesion molecules (e.g., LFA-1) of the parent T cells[26]. This enables them to more effectively target and retain on the surface of APCs or cancer cells, simulating some functions of T cells[27]. They possess higher targeting ability, circulatory stability, and lower immunogenicity without the need for complex chemical modifications[28]. Additionally, their natural bilayer phospholipid membrane structure can effectively protect their luminal contents from degradation by enzymes in the extracellular environment and endow them with stronger biocompatibility. They can simultaneously load small-molecule drugs, small interfering RNA (siRNA), gene editing tools (CRISPR/Cas9), and others[29,30].
TcEVs carry microRNAs (miRNAs) (e.g., miR-155, miR-146a). After being phagocytosed by macrophages and dendritic cells (DCs), they can inhibit the secretion of pro-inflammatory cytokines (e.g., tumor necrosis factor-α, interleukin-6) and prevent excessive T cell proliferation[31]. Activated T cells (e.g., activated T helper 1 (Th1)/CTLs) can release EVs containing Fas ligand (FasL) and Apo2L, also known as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). These EVs induce apoptosis of overactivated T cells themselves or adjacent ones through the Fas-FasL or TRAIL-DR4/DR5 pathway, thereby terminating the immune response[32].
1.2.3 Participation in immune synapse signal transduction
After forming immune synapses, T cells directionally release EVs enriched with immune signal molecules such as TCR, CD40 ligand (CD40L), and costimulatory molecule ligands on their surface, providing a material basis for signal transduction. The released EVs can act as “signal carriers” and be taken up by APCs such as B cells and DCs. They activate APCs through the CD40L-CD40 pathway, enabling DCs to upregulate costimulatory molecules like intercellular adhesion molecule 1 and CD86. As such, the activation efficiency of DCs on other T cells is enhanced, and the coordination of humoral immunity and cellular immunity is promoted[27].
However, natural TcEVs have weak targeting ability, limited immune activity, and low drug-loading capacity due to MHC restriction. To maximize the biological functions of TcEVs, engineering modification is indispensable.
2. Engineering Strategies and Applications
2.1 Genetic engineering strategy
The core of the genetic engineering strategy is to edit the genome of EV donor cells (e.g., CAR-T cells, TCR-T cells, immune cells) to regulate the cargo composition, surface molecule expression, or secretion efficiency of EVs, endowing EVs with customized functions such as precise targeting, potent killing, or immune regulation. Among them, construction of personalized CAR-T cells and TCR-T cells is currently the most promising direction for clinical translation (Figure 1).
2.1.1 Genetic engineering modification of CAR-T cells and TCR-T cells
At present, researchers genetically engineer CAR-T cells to enhance their cellular therapeutic efficacy. For instance, introduction of coding gene of the granzyme B (GzmB) inhibitor SerpinB9 into CAR-T cells can significantly reduce CAR-T cell fratricide induced by tumor-derived EVs, and enhance the tumor infiltration ability and vitality of CAR-T cells; when used in combination with PD-1 antibodies, their inhibitory effect on solid tumors is notably enhanced[33]. Delivery of the coding gene of C-X-C motif chemokine ligand 13 into CAR-T cells can improve the mitochondrial function of CAR-T cells through the AKT-mammalian target of rapamycin (mTOR) signaling pathway, enhance their central memory phenotype, and reduce exhaustion. In in vivo experiments, their anti-tumor activity is enhanced, and they exert a synergistic effect with PD-1 inhibitors[34]. However, these studies have not explored the therapeutic efficacy of CAR-T cell-derived extracellular vesicles (CAR-TEVs).
After CAR-T cells are genetically edited to express CARs, the secreted EVs retain CAR molecules and the cytotoxic characteristics of the parent cells, while avoiding common side effects of CAR-T therapy, such as cytokine release syndrome and immune effector cell-associated neurotoxicity syndrome[22,35]. For example, EVs derived from mesothelin (MSLN)-targeted CAR-T cells stably express CAR and CD3 molecules on their surface. They can specifically inhibit the growth of MSLN-positive triple-negative breast cancer cells through the perforin/GzmB-mediated apoptotic pathway, with no obvious side effects observed in in vivo experiments[36]. The GzmB level of EVs secreted by human epidermal growth factor receptor 2 (HER-2)-targeted CAR-T cells is increased by ≥ 20 times compared with the unstimulated group. They can specifically penetrate HER-2-positive target cells and induce target cell apoptosis within 60-90 hours, demonstrating a killing efficiency comparable to that of CAR-T cells while producing no acute toxicity[35].
The genetic engineering of TCR-T cells focuses on optimizing the sorting and secretion of TCRs in EVs. Studies have found that after TCR binds to pMHC of APCs, MVs rich in TCR are polarized and released at the immune synapse. Regulating the expression of TSG101 (responsible for TCR sorting) and vacuolar protein sorting 4 (mediating MV separation) through genetic modification can enhance the release efficiency of TCR-positive EVs, thereby strengthening antigen-dependent transcellular signal transduction and activating downstream immune responses[37].
2.1.2 Genetic engineering of immune checkpoints
In terms of ligand modification, studies have engineered T cells to overexpress PD-1, enabling the secreted EVs to stably display PD-1 molecules on their surface[2]. These PD-1-expressing EVs can bind to cancer cells through PD-1/PD-L1 specific interaction, blocking the PD-1/PD-L1 pathway to restore the proliferative activity and cytotoxicity of CD8+ T cells. They can also directly inducing cancer cell apoptosis through carried FasL and GzmB, significantly inhibiting the subcutaneous growth and lung metastasis of melanoma[2].
2.1.3 Other genetic engineering modifications
Beyond modifying CAR-T cells and TCR-T cells to improve the targeting of TcEVs, the functions of EVs can be optimized by genetically engineering cytokines or ligands on parent cells. For instance, by fusing IL-2 with the transmembrane domain of the platelet-derived growth factor receptor via a lentiviral vector to construct Jurkat T cells with membrane-bound IL-2, the secreted IL2-small EVs (sEVs) upregulate molecules such as miR-181a-3p through autocrine, enhance CD8+ T cell activity, and downregulate melanoma PD-L1 expression. Moreover, as IL-2 is anchored to the membrane, the systemic toxicity of free IL-2 is avoided[38].
Genetic engineering enables antibody modification of vesicles by engineering the parental cells. Antibody modification achieves precise localization of EVs to cancer cells by conjugating tumor antigen-specific antibodies or receptor antibodies to the EV surface. For example, T cell-derived sEVs can be modified on their surface with anti-transferrin receptor 1 (TfR1) antibodies to construct TcEVs with significantly improved delivery efficiency to 6 types of cancer cells, including breast cancer, lung cancer, and skin cancer. The engineered TcEVs can downregulate the expressions of PD-L1 and Rab27a in cancer cells, reduce the cells’ EV secretion, and enhance their sensitivity to CD8+ T cell cytotoxicity[39]. Bispecific antibody modification has more advantages. Relevant studies have engineered EVs to simultaneously display anti-CD3 antibodies and anti-HER2 antibodies on their surface, constructing “synthetic multivalent antibody-retargeted exosomes”, which redirect cytotoxic T cells to HER2-positive cancer cells, achieving specific killing both in vitro and in vivo[40].
2.2 Biological and chemical modifications of the membrane surface
One of the most widely used strategies in EV engineering involves introducing exogenous targeting ligands, cytokines, or immunomodulators into the EV membrane to improve their targeted delivery, immune regulation, or tumor recognition specificity, while preserving the natural structure of EVs (Figure 2).
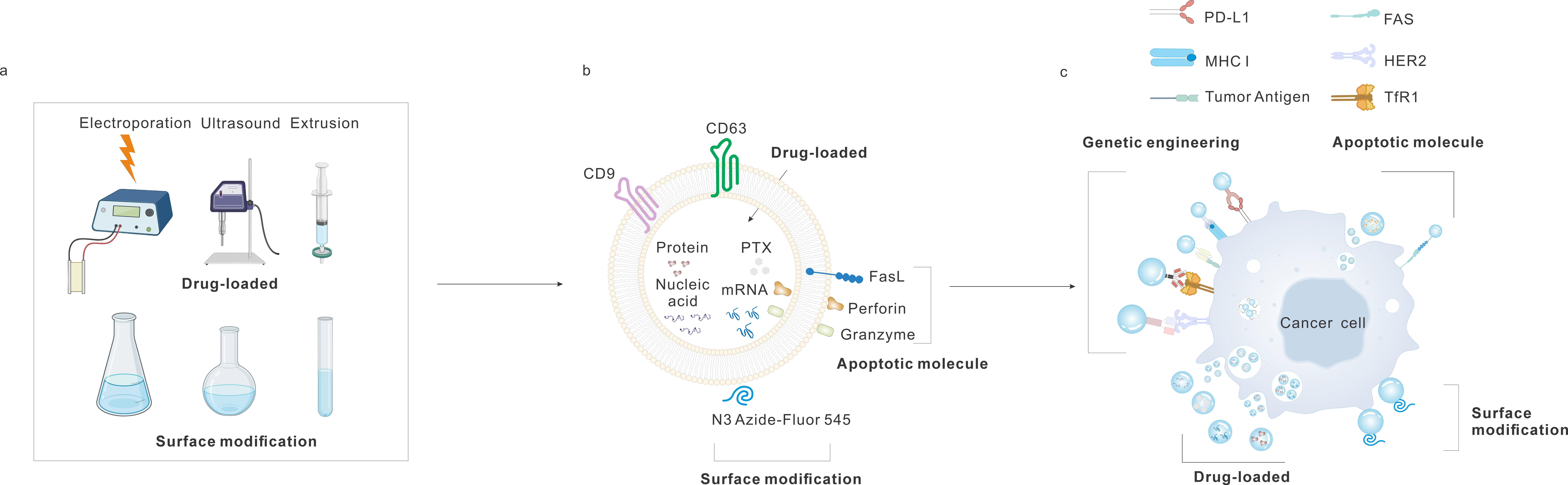
Figure 2. Functional modification and anti-tumor mechanism of engineered TcEVs. This diagram depicts the post-secretion modification and anti-tumor action of TcEVs. (a) Cargo loading and surface modification: Cargos are encapsulated into TcEVs via electroporation, ultrasound, or extrusion, while targeted molecules (e.g., N3 Azide-Fluor 545) are conjugated onto the vesicle surface to enhance targeting capability; (b) Molecular composition of modified TcEVs: The vesicle lumen contains loaded cargos (proteins, nucleic acids, mRNA, PTX) and intrinsic apoptotic effectors (FasL, perforin, granzyme), with surface-modified molecules integrated into the bilayer; (c) Anti-tumor interaction: Engineered TcEVs bind to ligands (MHC I, PD-L1, HER2, TfR1) on cancer cells via their surface components, deliver encapsulated cargos into cancer cells, and induce apoptosis through apoptotic effectors, thereby mediating targeted anti-tumor immune signaling. TcEVs: T cell-derived extracellular vesicles; mRNA: messenger RNA; PTX: paclitaxel; FasL: Fas ligand; PD-L1: programmed death-ligand 1; TfR1: transferrin receptor 1.
2.2.1 Cytokine/ligand modification
Conjugating cytokines or immunomodulatory ligands to the EV surface can endow EVs with the ability to activate immune responses or reverse immune suppression. Currently, there are few studies on direct cytokine or ligand modification of T cell-derived vesicles, but mature applications have been achieved in other immune cells such as DCs. Studies have conjugated IL-12 and anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) antibodies to the surface of dendritic cell-derived EVs (DEVs). IL-12 can directly enhance T cell activation, while anti-CTLA-4 blocks immune checkpoint signals. The two synergistically improve the induction effect of DEVs on Th1 immune response and significantly reverse the exhaustion of CD8+ T cells in tumor-bearing mice[41].
2.2.2 Chemical conjugation
Chemical conjugation technology has become an important supplement for EV surface modification due to its mild reaction conditions and high specificity. For example, using copper-catalyzed azide-alkyne cycloaddition reaction, alkynyl groups are first cross-linked to the EV surface through carbodiimide chemistry, and then conjugated with azide-labeled fluorescein or targeting ligands, yielding N3 Azide-Fluor 545. This process does not change the size, structure, or adhesion/internalization ability of EVs with recipient cells, and each 150 kDa of EV protein can stably conjugate approximately 1.5 alkynyl-modified molecules, providing an efficient tool for multi-target modification and tracking of EVs[42].
2.3 Cargo loading into vesicles
As natural nanocarriers, EVs can load cargo such as chemotherapeutic drugs and biological macromolecules (messenger RNA, proteins, nucleic acids) through physical or chemical methods, reducing drug toxicity and improving targeting. They have unique advantages, especially in overcoming cancer multidrug resistance and cross-barrier delivery (Figure 2).
The loading of cargo into EVs is mostly achieved through methods such as electroporation or endogenous gene regulation[43]. Nucleic acid drug is one of the most extensively studied categories in EV drug delivery systems, achieving tumor-specific therapy by precisely targeting oncogenes or tumor suppressor pathways. For instance, CD47-modified EVs loaded with siRNA/small hairpin RNA targeting KRASG12D can effectively evade immune clearance, prolong in vivo circulation time, and enhance delivery efficiency[44]. Another study used plant-derived vesicles to load cancer-suppressive miRNAs such as miR-146a and miR-138-5p, which regulate the metabolism of cancer cells[45].
The loading of chemotherapeutic drugs into EVs mainly realizes membrane recombination through ultrasonic treatment, freeze-thaw cycles, or extrusion, improving drug encapsulation efficiency[46,47]. After being loaded into EVs, small-molecule chemotherapeutic drugs, such as doxorubicin and nedaplatin, can enhance tumor targeting and reduce systemic toxicity[48,49]. In T cell-derived vesicles, researchers have currently loaded paclitaxel (PTX) into a hybrid carrier of MSLN-targeting CAR-TEVs and lung-targeting liposomes. This carrier can achieve sequential targeted delivery of PTX through the lung targeting of liposomes and the tumor specificity of CAR-TEVs, while reducing systemic toxicity[50] (Table 3).
Table 3. Summary of engineering strategies and applications of TcEVs.
| Engineering Strategy Type | Specific Methods | Core Functional Improvements | Application Scenarios | Key References |
| Genetic Engineering | CAR/TCR modification of T cells | Enhanced targeted recognition and killing | Antigen-positive tumor therapy | [21,34] |
| PD-1 overexpression | Blockade of the PD-1/PD-L1 pathway | PD-L1-positive tumors | [37] | |
| Membrane-bound IL-2 modification | Activation of CD8+ T cells, reduced systemic toxicity | TME with impaired immune function | [38] | |
| Antibody conjugation | Improved tumor targeting | Multiple types of solid tumors | [40] | |
| Surface Chemical Modification | Cytokine/ligand conjugation | Reversal of immune suppression | Immune-desert tumors | [41] |
| CuAAC-mediated conjugation | Efficient multi-target modification and in vivo tracking | Targeted delivery and mechanism research | [42] | |
| Cargo Loading | Electroporation/ultrasound-mediated loading of siRNA/miRNA | Regulation of tumor gene expression | Gene-mutated tumors | [44] |
| Extrusion/freeze-thaw-mediated loading of chemotherapeutic drugs | immunotherapy synergistic treatment | Chemotherapy-sensitive tumors | [50] |
TcEVs: T cell-derived extracellular vesicles; CAR: chimeric antigen receptor; TCR: T cell receptor; CuAAC: copper-catalyzed azide-alkyne cycloaddition; siRNA: small interfering RNA; miRNA: microRNA; PD-1: programmed cell death protein 1; PD-L1: programmed death-ligand 1; TME: tumor microenvironment.
3. Discussion and Perspective
Engineered TcEVs integrating the immunological activity of parent T cells and the natural delivery advantages of EVs, have emerged as a transformative candidate in tumor immunotherapy. Beyond cancer immunotherapy, engineered TcEVs hold potential in other immune-related diseases: (1) Autoimmune diseases: Treg-derived EVs engineered to overexpress IL-10 or TGF-β can suppress pathogenic T cell responses, offering a cell-free alternative for treating rheumatoid arthritis or multiple sclerosis[51]; (2) Chronic inflammatory diseases: TcEVs loaded with anti-inflammatory miRNAs (e.g., miR-146a) can modulate macrophage polarization in inflammatory bowel disease or psoriasis[52].
Despite promising preclinical data, the clinical translation of engineered TcEVs faces critical hurdles, such as heterogeneity, suboptimal pharmacokinetics, unclear mechanisms, and lack of standardization validation. Future translational efforts should focus on addressing these challenges through cross-disciplinary innovation: (1) developing precision separation technologies to reduce TcEV heterogeneity; (2) optimizing engineering strategies to improve in vivo stability and tumor tropism; (3) integrating multi-omics and live imaging to clarify cargo delivery and immune regulation mechanisms; (4) establishing unified production standards and quantitative bioassays for potency. In addition, combining TcEVs with immune checkpoint inhibitors or CAR-T cells may create synergistic therapeutic effects, and exploring their applications in hematological tumors and solid tumors will further expand their clinical value. Overall, engineered TcEVs represent a “cell-free” immunotherapy platform with both targeting and safety, and overcoming current technical bottlenecks will accelerate their transformation.
Authors contribution
Diao R, Zhang XZ: Investigation, data curation, formal analysis, writing-original draft, writing-review & editing.
Zhang XD: Writing-review & editing.
Conflicts of interest
Xudong Zhang is an Editorial Board Member of BME Horizon. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 32371425 and No. 32201084); Science, Technology & Innovation Commission of Shenzhen Municipality, Shenzhen Science and Technology Program (Grant Nos. JCYJ20240813151128037, RCYX20200714114643121, JCYJ20200109142610136, JCYJ20180507181654186, and ZDSYS20220606100803007); the Natural Science Foundation of Guangdong Province (Grant No. 2022A1515012289); Doctoral Personnel Scientific Research Start-up Fund project of Guangdong Medical University (Grant No. GDMUB2022037); Key Field Special Programs of Guangdong Provincial Ordinary Colleges and Universities (Grant No. 2024ZDZX2069); Research Grant of Key Laboratory of Regenerative Medicine of Ministry of Education, Jinan University (Grant No. ZSYXM202502); and Special Project for Clinical and Basic Sci & Tech Innovation of Guangdong Medical University (Grant No. GDMULCJC2024114).
Copyright
© The Author(s) 2026.
References
-
2. Li B, Fang T, Li Y, Xue T, Zhang Z, Li L, et al. Engineered T cell extracellular vesicles displaying PD-1 boost anti-tumor immunity. Nano Today. 2022;46:101606.[DOI]
-
5. Greening DW, Simpson RJ. Understanding extracellular vesicle diversity–current status. Expert Rev Proteom. 2018;15(11):887-910.[DOI]
-
7. Alonso R, Mazzeo C, Rodriguez MC, Marsh M, Fraile-Ramos A, Calvo V, et al. Diacylglycerol kinase α regulates the formation and polarisation of mature multivesicular bodies involved in the secretion of Fas ligand-containing exosomes in T lymphocytes. Cell Death Differ. 2011;18(7):1161-1173.
-
11. Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J Extracell Vesicle. 2014;3:26913.[DOI]
-
12. Clancy JW, Schmidtmann M, D’Souza-Schorey C. The ins and outs of microvesicles. FASEB Bioadv. 2021;3(6):399-406.[DOI]
-
13. Santavanond JP, Rutter SF, Atkin-Smith GK, Poon IKH. Apoptotic bodies: Mechanism of formation, isolation and functional relevance. In: Mathivanan S, Fonseka P, Nedeva C, Atukorala I, editors. New frontiers: Extracellular vesicles. Cham: Springer; 2021. p. 61-88.[DOI]
-
14. Zhou M, Li YJ, Tang YC, Hao XY, Xu WJ, Xiang DX, et al. Apoptotic bodies for advanced drug delivery and therapy. J Control Release. 2022;351:394-406.[DOI]
-
24. van de Wakker SI, van Oudheusden J, Mol EA, Roefs MT, Zheng W, Görgens A, et al. Influence of short term storage conditions, concentration methods and excipients on extracellular vesicle recovery and function. Eur J Pharm Biopharm. 2022;170:59-69.[DOI]
-
29. Kim SM, Yang Y, Oh SJ, Hong Y, Seo M, Jang M. Cancer-derived exosomes as a delivery platform of CRISPR/Cas9 confer cancer cell tropism-dependent targeting. J Control Release. 2017;266:8-16.[DOI]
-
30. van der Meel R, Fens MHAM, Vader P, van Solinge WW, Eniola-Adefeso O, Schiffelers RM. Extracellular vesicles as drug delivery systems: Lessons from the liposome field. J Control Release. 2014;195:72-85.[DOI]
-
31. Mittelbrunn M, Gutiérrez-Vázquez C, Villarroya-Beltri C, González S, Sánchez-Cabo F, González MÁ, et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun. 2011;2:282.[DOI]
-
32. Monleón I, Martínez-Lorenzo MJ, Monteagudo L, Lasierra P, Taulés M, Iturralde M, et al. Differential secretion of Fas ligand- or APO2 ligand/TNF-related apoptosis-inducing ligand-carrying microvesicles during activation-induced death of human T cells. J Immunol. 2001;167(12):6736-6744.
-
36. Yang P, Cao X, Cai H, Feng P, Chen X, Zhu Y, et al. The exosomes derived from CAR-T cell efficiently target mesothelin and reduce triple-negative breast cancer growth. Cell Immunol. 2021;360:104262.[DOI]
-
40. Shi X, Cheng Q, Hou T, Han M, Smbatyan G, Lang JE, et al. Genetically engineered cell-derived nanoparticles for targeted breast cancer immunotherapy. Mol Ther. 2020;28(2):536-547.[DOI]
-
46. Greening DW, Xu R, Rai A, Suwakulsiri W, Chen M, Simpson RJ. Clinical relevance of extracellular vesicles in cancer: Therapeutic and diagnostic potential. Nat Rev Clin Oncol. 2025;22(12):924-952.[DOI]
-
51. Ganeeva IA, Gilyazova EM, Khannanov AA, Nektorova ME, Rogov AM, Khaibullin TI, et al. Allogeneic Treg-derived artificial vesicles: A promising therapeutic modality for multiple sclerosis. BioImpacts. 2025;15:30880.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Diao R, Zhang XZ, Zhang XD. Engineered T cell-derived extracellular vesicles for cancer immunotherapy. BME Horiz. 2026;4:202537. https://doi.org/10.70401/bmeh.2026.0017
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Diao R, Zhang XZ, Zhang XD. Engineered T cell-derived extracellular vesicles for cancer immunotherapy. BME Horiz. 2026;4:202537. https://doi.org/10.70401/bmeh.2026.0017
copy
Share Link
copy