Macrophage-targeted mRNA therapeutics: Emerging strategies for cancer immunotherapy
Kedan Gu
1,2

,
Sitao Xie
1,2,3,*
*Correspondence to:
Sitao Xie, Zhejiang Cancer Hospital, The Key Laboratory of Zhejiang Province for Aptamers and Theranostics, Hangzhou Institute of Medicine (HIM), Chinese Academy of Sciences, Hangzhou 310022, Zhejiang, China; School of Molecular Medicine, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, Zhejiang, China; Zhejiang Key Lab of Vaccine, Infectious Disease Prevention and Control, Zhejiang Provincial Center for Disease Control and Prevention, Hangzhou 310015, Zhejiang, China.
E-mail: xiesitao@him.cas.cn
BME Horiz. 2026;4:202601. 10.70401/bmeh.2026.0023
Received: January 08, 2026Accepted: April 03, 2026Published: April 07, 2026
This article belongs to the Special lssue Engineering RNA Delivery Technologies for Vaccine and Therapeutic Development
Abstract
Tumor-associated macrophages (TAMs) are pivotal regulators of the immunosuppressive tumor microenvironment and major contributors to resistance against conventional and immunotherapeutic interventions. Rather than eliminating TAMs, emerging strategies aim to functionally reprogram them toward an antitumor phenotype, a therapeutic objective uniquely enabled by the precise, transient, and non-integrating nature of mRNA, which allows reversible modulation without genomic risk. Recent progress in nanocarrier design has improved selective delivery to TAMs through both passive uptake and active targeting, with administration routes tailored to tumor location. Precise immunomodulatory interventions in macrophages, accomplished by mRNA payloads designed to induce pro-inflammatory polarization, enhance phagocytosis, or block immunosuppressive signals, thereby remodel the tumor immune microenvironment and generate synergy with established treatments. Future efforts might concentrate on macrophage heterogeneity, carrier immunogenicity, and scalable formulation development to advance clinical translation, not only in cancer but also in other diseases shaped by dysregulated macrophage function.
Keywords
mRNA delivery, macrophage-targeted, cancer immunotherapy
1. Introduction
Tumor-associated macrophages (TAMs) are among the most abundant myeloid cells in the tumor microenvironment (TME) and play a critical role in cancer initiation, progression, metastasis, and therapeutic resistance[1,2]. Functioning as key bridges between innate and adaptive immunity, TAMs engage in extensive crosstalk with T cells, natural killer cells, and dendritic cells (DCs), modulating immune responses through cytokine secretion, expression of immune checkpoint ligands, and metabolic competition, hallmarks of the non-resolving inflammation that characterizes cancer. TAMs exhibit remarkable plasticity[3,4], polarizing into proinflammatory M1-like states that enhance antigen presentation and secrete TNF-α and IL-12 to support antitumor immunity, or more commonly into immunosuppressive M2-like states (induced by IL-4) that secrete IL-10 and TGF-β, recruit regulatory T cells via CCL22, and deplete essential amino acids via arginase 1 and indoleamine 2,3-dioxygenase (IDO). This enzymatic activity exhausts amino acid reserves, inhibits T cell expansion, and broadly suppresses immune effector functions[5-7]. While first-generation IDO1 inhibition failed in the ECHO-301/KEYNOTE-252 melanoma trial[8], preclinical and human data still validate the pathway’s immunoregulatory potential. Future strategies will require validated targets and biomarker stratification. Beyond immunosuppression, TAM-derived mediators, such as IL-1β, IL-6, and TNF, directly fuel tumorigenesis by activating oncogenic pathways, inducing DNA damage via reactive oxygen species, and promoting mutagenesis through the ectopic expression of activation-induced cytidine deaminase[9,10].
Given their multifaceted contributions to therapy resistance, spanning chemotherapy, radiotherapy, and immune checkpoint blockade, TAMs have emerged as pivotal targets for improving clinical outcomes in cancer[11,12]. Strategies aimed at depleting TAMs, blocking their recruitment (for example, via CSF-1R inhibitors), or disrupting “don’t-eat-me” signals (such as CD47-blocking antibodies) have already entered clinical trials, underscoring their translational relevance[13-16]. However, accumulating evidence indicates that the balance between SPP1+ and CXCL9+ macrophage states firmly stratifies the TME and predicts responsiveness to immunotherapy[17]. At the same time, SPP1+ macrophages exhibit an immunosuppressive signature linked to poor prognosis, whereas CXCL9+ TAMs represent an IFN-γ-polarized phenotype associated with anti-tumor immunity[17]. This highlights the plasticity of TAMs, cautioning against discrete categorization since macrophage function is highly context-dependent and suggests that a more nuanced approach, functionally reprogramming TAMs rather than simply removing them, may yield more durable antitumor responses. In this context, RNA-based therapeutics, particularly macrophage-targeted mRNA delivery platforms, offer a robust and programmable means to modulate TAM function precisely. By enabling the transient, tunable expression of immunomodulatory proteins or silencing key polarization drivers, mRNA technology represents a frontier strategy for remodeling the TME and overcoming current limitations in cancer immunotherapy.
2. Engineering Targeted Delivery to Macrophages
Macrophages’ intrinsic phagocytic activity provides a natural gateway for nanoparticle internalization, enabling the efficient passive delivery of nucleic acid cargos without the need for mandatory surface functionalization. Delivery vehicles cause massive endosomal rupture in macrophages via proton sponge or phase transition mechanisms, but cargo escape to the cytosol remains highly inefficient[18-20]. This principle underpins the success of multiple platforms, including lipid nanoparticles (LNPs) and polymeric carriers, which are readily engulfed by TAMs in vivo (Figure 1A).
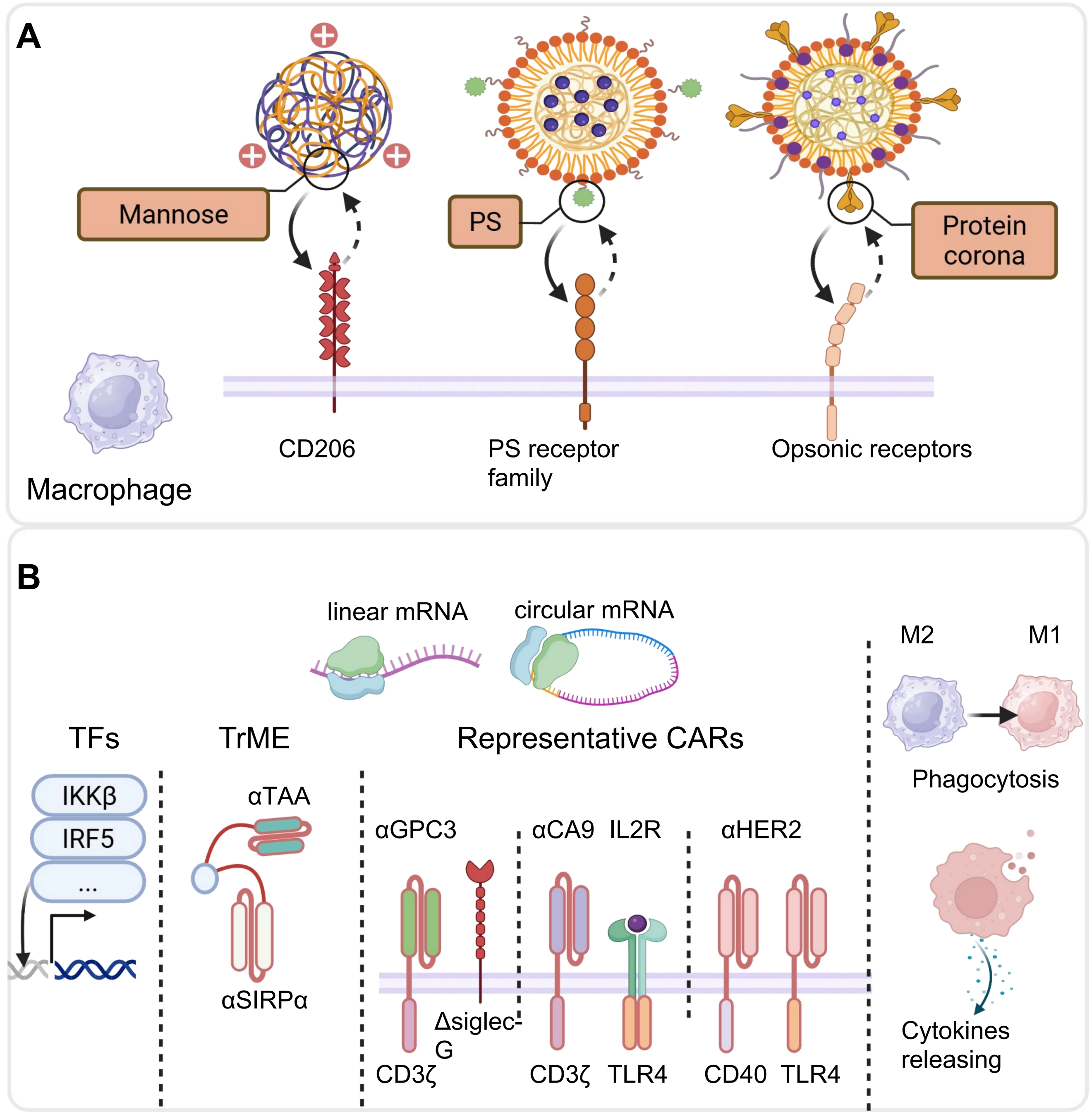
Figure 1. Illustration of mRNA-based strategies for reprogramming macrophages against tumors. (A) Targeted mRNA delivery to macrophages via ligand–receptor interactions, including mannose–CD206 engagement[21]. Republished with permission from[22], PS binding to PS receptors[23], and protein-corona recognition by opsonic receptors. Republished with permission from[24]; (B) Translation of mRNA-encoded proteins, including CARs, TFs and TrME, reprograms macrophages, driving M2-to-M1 polarization, enhancing phagocytosis, and promoting cytokine release to support anti-tumor immunity[21,23]. Republished with permission from[24,25]. Created in BioRender.com. PS: phosphatidylserine; CARs: chimeric antigen receptors; TFs: transcription factors; TrME: trispecific macrophage engager.
To enhance specificity, particularly toward immunosuppressive M2-like TAMs that overexpress the mannose receptor CD206, active targeting strategies have been widely adopted. For instance, mannose-conjugated poly(β-amino ester) nanoparticles have been developed, achieving selective macrophage transfection in tumors with minimal induction of systemic inflammatory cytokines[21,22]. Remarkably, intraperitoneal (i.p.) delivery has emerged as a highly effective route for peritoneal malignancies: a recent study showed that i.p. administered mRNA-LNPs with phosphatidylserine modification efficiently reprogrammed TAMs into chimeric antigen receptor macrophages (CAR-M) directly within the peritoneal cavity, bypassing systemic exposure[23]. Notably, even systemically administered, non-targeted LNPs can achieve selective uptake by liver-resident macrophages through endogenous biological cues: after intravenous infusion, specific plasma proteins, including apolipoproteins, complement components, and vitronectin, adsorb onto the LNP surface to form a protein corona, which is recognized by endogenous receptors abundantly expressed on Kupffer cells and hepatic TAMs[24]. This biomimetic mechanism enables preferential mRNA delivery to liver macrophages without requiring exogenous targeting ligands, as demonstrated in recent work using LNP-mediated co-delivery of CAR and immunomodulatory mRNAs for the treatment of hepatocellular carcinoma (HCC). Beyond ligand-receptor interactions, physicochemical parameters such as size (< 200 nm), charge neutrality, and moderate stiffness further refine biodistribution and cellular uptake[26].
3. mRNA Cargo Design for Functional Reprogramming
The therapeutic power of macrophage-targeted delivery lies not only in where the cargo goes, but also in what it encodes (Figure 1B). Recent advances demonstrate that mRNA can be precisely engineered to reprogram TAMs from pro-tumorigenic cells into multifunctional antitumor effectors (Table 1).
Table 1. Representative macrophage-targeted mRNA studies for cancer immunnotherapy.
| Cargo | Target | ICD | Platform | Macrophage transfection effiency | In vivo model | Injection | Dosage | Ref. |
| TFs (mRNA) | IRF5/IKKβ | / | Mannose-modified PBAE polymer | 46% | C57BL/6, ID8 ovarian tumor | i.p. for ovarian cancer, i.v. for melanoma lung metastases or glioblastoma | two 50 μg mRNA doses/week for 4 weeks | [21] |
| CAR (mRNA) | CD47 HER2 | CD3z, Dectin-1, CD40, TLR4 | PSβ-LNP | 99% | C57BL/6, PAN02-luc pancreatic tumor; Balb/c, CT26-luc colon carcinoma | i.p. | 5 ug mRNA dose/day for 7 days | [23] |
| TrME (mRNA) | B7H3, LRP1, SIRPα | / | LNP | / | C57BL/6, GL261 GBM model; MB49 bladder tumor | i.v. | 0.5 or 2.5 mg/kg | [25] |
| CAR/CSR (circle RNA) | IL13Rα2, Sialic acid | CD40, TLR4 | MLNP@Gel System | 47% | C57BL/6, CT2A and GL261 murine GBM model | intracranial | 0.5 mg/kg for CAR/MLNP and 0.5 mg/kg for CSR/MLNP | [27] |
ICD: intracellular domain; TFs: transcription factors; IRF5: interferon regulatory factor 5; IKKβ: inhibitor of nuclear factor kappa-B kinase subunit beta; PBAE: poly(β-amino ester); CAR: chimeric antigen receptor; LNP: lipid nanoparticle; TrME: trispecific macrophage engager; CSR: synthetic chimeric IL-2 receptor; i.p.: intraperitoneal; i.v.: intravenous.
Notably, the MAGE platform, developed by Ping et al., employs macrophage-targeted delivery of programmable RNA-editing components to selectively disrupt the immunosuppressive phenotype, encoding two key proteins: interferon regulatory factor 5 (IRF5) and inhibitor of nuclear factor kappa-B kinase subunit beta[22]. This co-expression reprograms immunosuppressive, pro-tumor M2-like TAMs into pro-inflammatory, anti-tumor M1-like macrophages by activating NF-κB signaling and enhancing IRF5-driven transcriptional programs. As a result, the reprogrammed macrophages secrete inflammatory cytokines (e.g., TNF-α, IL-12), promote T cell activation, and remodel the immunosuppressive TME, thereby significantly inhibiting tumor growth in multiple mouse models. Another recent study presents a novel cancer immunotherapy strategy combining a trispecific macrophage engager (TrME) with mRNA-LNP. The TrME molecule acts as a logic gate, simultaneously activating pro-phagocytic signals and blocking anti-phagocytic signals on macrophages to enhance tumor cell phagocytosis[25].
CAR-M therapy has emerged as a highly promising immunotherapeutic strategy for the treatment of solid malignancies[28,29]. Recent advances have leveraged mRNA-LNP platforms to achieve in situ delivery and expression of CARs, thereby enabling targeted reprogramming of endogenous macrophages. For example, even more sophisticated approaches have emerged: in HCC, researchers co-delivered two mRNAs via LNPs, one encoding a CAR specific for the HCC-associated antigen glypican-3 (GPC3), and another encoding a Siglec-G lacking immunoreceptor tyrosine-based inhibition motifs, to simultaneously confer tumor-targeting specificity and disable the CD24–Siglec-G “don’t eat me” checkpoint[24]. This dual-mRNA strategy directly converts endogenous TAMs within the TME into GPC3-targeted CAR-M with enhanced phagocytic activity. In multiple orthotopic and metastatic HCC mouse models, this approach markedly suppresses tumor growth and metastasis. Another innovative in situ macrophage engineering strategy for renal cell carcinoma (RCC) therapy, combining circular RNA (circRNA)-loaded LNPs with a thermosensitive hydrogel to locally reprogram TAMs into potent CAR-Ms. The platform co-delivers two circRNA constructs, one encoding a tumor-targeted CAR and the other a synthetic chimeric IL-2 receptor (CSR) alongside IL-2 via intratumoral hydrogel injection. This approach enables sustained, localized expression of both CAR and CSR, driving TAMs toward a proinflammatory, tumoricidal phenotype while avoiding systemic toxicity. In orthotopic RCC models, the treatment significantly suppresses primary tumor growth and lung metastasis, reshaping the immunosuppressive TME[30]. Furthermore, the i.p. administration of mRNA-loaded LNPs encoding a CD47/HER2-targeted CAR with a CD3ζ/TLR4/CD40 signaling domain successfully generated functional CAR-M directly within phagocytosis and pro-cytokine release, resulting in enhanced tumor eradication, remodeling of the TME, and synergistic effects with anti-PD-1 therapy[23]. Together, these studies demonstrate how the rational design of mRNA payloads enables the precise, multimodal reprogramming of macrophages, transforming them into dynamic, tumor-directed immune engines for the treatment of solid cancers.
4. mRNA Modifications and Purification for Efficient Macrophage Transfection
The advent of single-cell RNA-sequencing and ATAC-sequencing technologies has unveiled the profound complexity of the macrophage compartment, particularly at the transcriptional and epigenetic levels[31,32]. RNA molecules can undergo over 150 distinct post-transcriptional modifications[33], catalyzed by specialized and often highly conserved enzymatic machinery. Recently, RNA modifications have also been found to significantly influence macrophage phenotypes by regulating RNA stability and translation efficiency in the context of different diseases[34].
For macrophage transfection, it is crucial to highlight the critical need for 5′-CAP and mRNA modifications (5-methyl-cytidine and pseudouridine or N1-methyl-pseudouridine)[23,35,36]. Intrinsic activation triggered by the carrier systems was negligible[36]. Except for the unmodified mRNA group in macrophages, the reagents alone showed no cytotoxicity or induction of CD80/TNF-α expression[36]. Unmodified in vitro transcription (IVT)-mRNA triggered a potent antiviral response, elevating IFN-β secretion by up to two orders of magnitude compared to modified counterparts. Conversely, replacing unmodified nucleosides with 5-methylcytidine and pseudouridine significantly attenuated the activation of DCs[37]. However, optimizing transfection protocols requires balancing competing demands, such as efficiency, viability, and the minimization of pleiotropic effects, which do not always align.
In addition, a significant challenge associated with this process is the generation of double-stranded RNA (dsRNA) byproducts. These dsRNA impurities are a critical quality attribute for mRNA therapeutics, as they can trigger innate immune activation, leading to the secretion of type I interferon and inflammatory cytokines, which in turn reduce translation activity[38]. Although various efforts have been made to optimize IVT synthesis to minimize dsRNA formation, these impurities cannot be fully resolved through process optimization alone. Consequently, the removal of dsRNA is essential to ensure the safety and efficacy of mRNA-based medicines and often requires complex purification processes, such as reversed-phase high-performance liquid chromatography or a rationally engineered mutant of T7 RNAP, to yield safe and effective products[38-40].
5. Clinical Translation of Macrophage-Targeted mRNA Therapeutics
Currently, two distinct technological approaches have advanced to clinical trials in the CAR-M space. Carisma Therapeutics’ CT-0508 represents the first-generation ex-vivo manufactured CAR-M platform, utilizing autologous monocyte-derived macrophages transduced with an Ad5.F35 adenoviral vector encoding an anti-HER2 CAR. In the completed Phase 1 trial (NCT04660929), CT-0508 demonstrated a favorable safety profile with no dose-limiting toxicities, grade ≥ 3 cytokine release syndrome, or neurotoxicity in 14 heavily pretreated patients with HER2-overexpressing solid tumors. However, the therapeutic efficacy was modest, with 44% of HER2 3+ patients achieving stable disease as the best response, but no objective responses per RECIST v1.1. The primary limitation was poor persistence, with CAR-M detectable in only 27% of tumor biopsies at week 4, correlating with transient ctDNA reduction followed by rebound[41]. This has prompted the development of CT-0525, a CAR-monocyte product with reportedly extended half-life, currently in Phase 1 (NCT06254807), though no clinical results have been disclosed to date.
Concurrently, Myeloid Therapeutics has pioneered an alternative in situ reprogramming strategy through its MT series (MT-302, MT-303), employing systemically administered lipid nanoparticle-formulated mRNA encoding CAR constructs (Table 2). This approach circumvents the manufacturing complexity and scalability constraints inherent to ex vivo cellular products. MT-302 (anti-TROP2) and MT-303 (anti-GPC3) are under evaluation in Phase 1 trials for epithelial tumors and HCC, respectively (NCT05969041, NCT06478693). To date, no specific clinical efficacy or safety data from these mRNA-LNP programs have been publicly disclosed, and their comparative therapeutic potential relative to ex-vivo CAR-M approaches remains to be established through ongoing clinical investigation.
Table 2. Representative ongoing clinical studies.
| CAR-M | Clinical trial number | Type of administration | Target | Platform | Dosage | Study Phase | Objective | Sponsor |
| MT-302 | NCT05969041 | intravenous infusion | TROP2 | LNP | Every 14 days for 3 doses, followed by administration once every 28 days for three doses. | Phase 1 | To assess the safety, tolerability, and define the RP2D of MT-302 in participants with advanced epithelial cancer. | Myeloid Therapeutics |
| MT-303 | NCT06478693 | intravenous infusion | GPC3 | LNP | / | Phase 1 | To assess the safety, tolerability and define the RP2D of MT-303 alone (Module 1) and in combination with Atezolizumab/Bevacizumab (Module 2) in participants with advanced hepatocellular carcinoma expressing GPC3. | Myeloid Therapeutics |
CAR-M: chimeric antigen receptor macrophages; LNP: lipid nanoparticle; GPC3: glypican-3.
In situ macrophage reprogramming strategies tailor delivery to anatomical niches to balance efficacy with safety. For HCC, systemic LNPs utilize a protein corona to target liver macrophages with dual mRNA, achieving tumor clearance without systemic cytokine release syndrome[24]. In glioblastoma, biomimetic hydrogels implanted in the surgical cavity enable localized retention of nanoparticles carrying CAR-encoding plasmids or circRNA, reprogramming local myeloid cells while sparing healthy brain tissue[27,42]. For peritoneal metastasis, i.p. mRNA-LNP injection leverages compartmentalized delivery to induce potent local immunity[23].
Collectively, despite employing distinct CAR-M preparation strategies, two preclinical studies reveal that CAR-M therapy acts as a pivotal immune modulator within the TME[23,43]. They demonstrate that CAR-Ms not only exert direct phagocytosis but also function as potent immune activators by reprogramming immunosuppressive M2 macrophages into pro-inflammatory M1 phenotypes and facilitating antigen cross-presentation to induce robust T-cell responses and long-term immunological memory. Furthermore, they establish a compelling synergistic mechanism between CAR-M and PD-1/PD-L1 blockade, where CAR-M therapy converts non-responsive tumors into pro-inflammatory states, thereby sensitizing resistant solid tumors to immune checkpoint inhibitors. These findings collectively validate the superior efficacy of tailored CAR designs and in situ programming strategies, positioning the combination of CAR-M with checkpoint immunotherapy as a transformative approach for treating refractory solid tumors.
6. Conclusions and Future Outlook
Looking ahead, macrophage-targeted mRNA therapeutics are poised to evolve beyond proof-of-concept studies toward clinically viable strategies, yet key challenges remain. The profound heterogeneity of TAM subsets, specifically the functional dichotomy between immunosuppressive SPP1+ macrophages and immunostimulatory CXCL9+ TAMs, necessitates more precise stratification strategies that move beyond broad markers like CD206. This complexity calls for patient stratification guided by advanced single-cell profiling to identify those most likely to benefit from specific reprogramming interventions. While current platforms, ranging from mannose-conjugated nanoparticles to systemic LNPs with protein coronas, show promise, the field must address the translational hurdles highlighted by early clinical data. The limited in vivo persistence observed in first-generation ex vivo products like CT-0508 underscores the need for next-generation designs, such as the in situ LNP-mediated delivery strategies exemplified by Myeloid Therapeutics’ MT series. In this landscape, repeated dosing emerges as a pivotal strategy, not only does it help mitigate anti-carrier immunity through tolerance induction, but it also sustains the in vivo persistence of CAR-M cells via repeated infusions.
Critically, integrating mRNA-driven TAM reprogramming with checkpoint inhibitors offers a synergistic path forward, as preclinical data robustly demonstrate that CAR-M therapy can convert tumors into pro-inflammatory states, thereby sensitizing resistant solid tumors to PD-1/PD-L1 blockade. This positions macrophage-focused mRNA delivery not merely as a standalone treatment, but as a transformative pillar of next-generation immuno-oncology. Beyond oncology, this versatile platform holds promise for treating fibrosis, atherosclerosis, and neuroinflammatory diseases by reprogramming tissue-resident macrophages, potentially revolutionizing the treatment of chronic inflammatory conditions.
Acknowledgements
ChatGPT was used for language polishing of the manuscript. The authors take full responsibility for the accuracy and scientific content of the article.
Authors contribution
Gu K: Visualization, writing-original draft, writing review & editing.
Xie S: Conceptualization, supervision, writing review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Key Research and Development Program of China (Grant No. 2022YFC3401402).
Copyright
© The Author(s) 2026.
References
-
5. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19(6):369-382.[DOI]
-
8. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019;20(8):1083-1097.[DOI]
-
12. Jin R, Neufeld L, McGaha TL. Linking macrophage metabolism to function in the tumor microenvironment. Nat Cancer. 2025;6(2):239-252.[DOI]
-
14. Jarr KU, Nakamoto R, Doan BH, Kojima Y, Weissman IL, Advani RH, et al. Effect of CD47 blockade on vascular inflammation. N Engl J Med. 2021;384(4):382-383.[DOI]
-
20. Chatterjee S, Kon E, Sharma P, Peer D. Endosomal escape: A bottleneck for LNP-mediated therapeutics. Proc Natl Acad Sci U S A. 2024;121(11):e2307800120.[DOI]
-
28. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38(8):947-953.[DOI]
-
29. Lei A, Yu H, Lu S, Lu H, Ding X, Tan T, et al. A second-generation M1-polarized CAR macrophage with antitumor efficacy. Nat Immunol. 2024;25(1):102-116.[DOI]
-
30. Jing W, Han M, Wang G, Kong Z, Zhao X, Fu Z, et al. An in situ engineered chimeric IL-2 receptor potentiates the tumoricidal activity of proinflammatory CAR macrophages in renal cell carcinoma. Nat Cancer. 2025;6(5):838-853.[DOI]
-
31. Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. The role of M6A modification in the biological functions and diseases. Sig Transduct Target Ther. 2021;6:74.[DOI]
-
32. Wei J, He C. RNA modifications in gene regulation: Functions and pathways. Cell. 2026;189(6):1591-1619.[DOI]
-
33. Boccaletto P, Stefaniak F, Ray A, Cappannini A, Mukherjee S, Purta E, et al. MODOMICS: A database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022;50:D231-D235.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Gu K, Xie S. Macrophage-targeted mRNA therapeutics: Emerging strategies for cancer immunotherapy. BME Horiz. 2026;4:202601. https://doi.org/10.70401/bmeh.2026.0023
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Engineering Targeted Delivery to Macrophages
- 3. mRNA Cargo Design for Functional Reprogramming
- 4. mRNA Modifications and Purification for Efficient Macrophage Transfection
- 5. Clinical Translation of Macrophage-Targeted mRNA Therapeutics
- 6. Conclusions and Future Outlook
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Gu K, Xie S. Macrophage-targeted mRNA therapeutics: Emerging strategies for cancer immunotherapy. BME Horiz. 2026;4:202601. https://doi.org/10.70401/bmeh.2026.0023
copy
Share Link
copy