Engineering mRNA therapeutics for protein supplementation: Challenges and future horizons
Yiyang Tao
1
,
Yue Zhao
1
,
Qia Su
1
,
Jianxin Pan
2
,
Jianguo Yin
3,4,*
,
Congcong Xu
1,5,6,7,*

*Correspondence to:
Jianguo Yin, Wisdom Lake Academy of Pharmacy, Xi’an Jiao Tong Liverpool University, Suzhou 215123, Jiangsu, China; Institute of Systems, Molecular and Integrative Biology, Faculty of Health & Life Sciences, University of Liverpool, Liverpool L69 3BX, UK.
E-mail: jianguoy@liverpool.ac.uk
Congcong Xu, International College of Pharmaceutical Innovation, Soochow University, Suzhou 215222, Jiangsu, China; Biomedical Polymers Laboratory, College of Chemistry Chemical Engineering and Materials Science and State Key Laboratory of Radiation Medicine and Protection, Soochow University, Suzhou 215123, Jiangsu, China; College of Pharmaceutical Sciences, Soochow University, Suzhou 215123, Jiangsu, China; Suzhou Institute of Pharmaceutical Innovation, Suzhou 215123, Jiangsu, China. E-mail: xucc@suda.edu.cn
Congcong Xu, International College of Pharmaceutical Innovation, Soochow University, Suzhou 215222, Jiangsu, China; Biomedical Polymers Laboratory, College of Chemistry Chemical Engineering and Materials Science and State Key Laboratory of Radiation Medicine and Protection, Soochow University, Suzhou 215123, Jiangsu, China; College of Pharmaceutical Sciences, Soochow University, Suzhou 215123, Jiangsu, China; Suzhou Institute of Pharmaceutical Innovation, Suzhou 215123, Jiangsu, China. E-mail: xucc@suda.edu.cn
BME Horiz. 2026;4:202604. 10.70401/bmeh.2026.0025
Received: January 13, 2026Accepted: April 14, 2026Published: April 16, 2026
This article belongs to the Special lssue Engineering RNA Delivery Technologies for Vaccine and Therapeutic Development
This manuscript is made available in its unedited form to allow early access to the
reported findings. Further editing will be completed before final publication. As
such,
the content may include errors, and standard legal disclaimers are applicable.
Abstract
Messenger RNA (mRNA) protein replacement therapy harnesses synthetic mRNA to direct endogenous protein synthesis, offering a versatile approach to restore or substitute proteins that are absent or dysfunctional in disease. Here, we review recent advances that have transformed this concept into a promising therapeutic platform, summarizing progress in mRNA design, delivery technologies, and preclinical and clinical applications across metabolic, oncological, and cardiovascular disorders. We also examine persistent challenges, including achieving precise tissue targeting, extending expression duration, and balancing immune tolerance with translation efficiency, that define the next frontier for clinical translation. By systematically analyzing these obstacles and evaluating emerging solutions, such as
Graphical Abstract
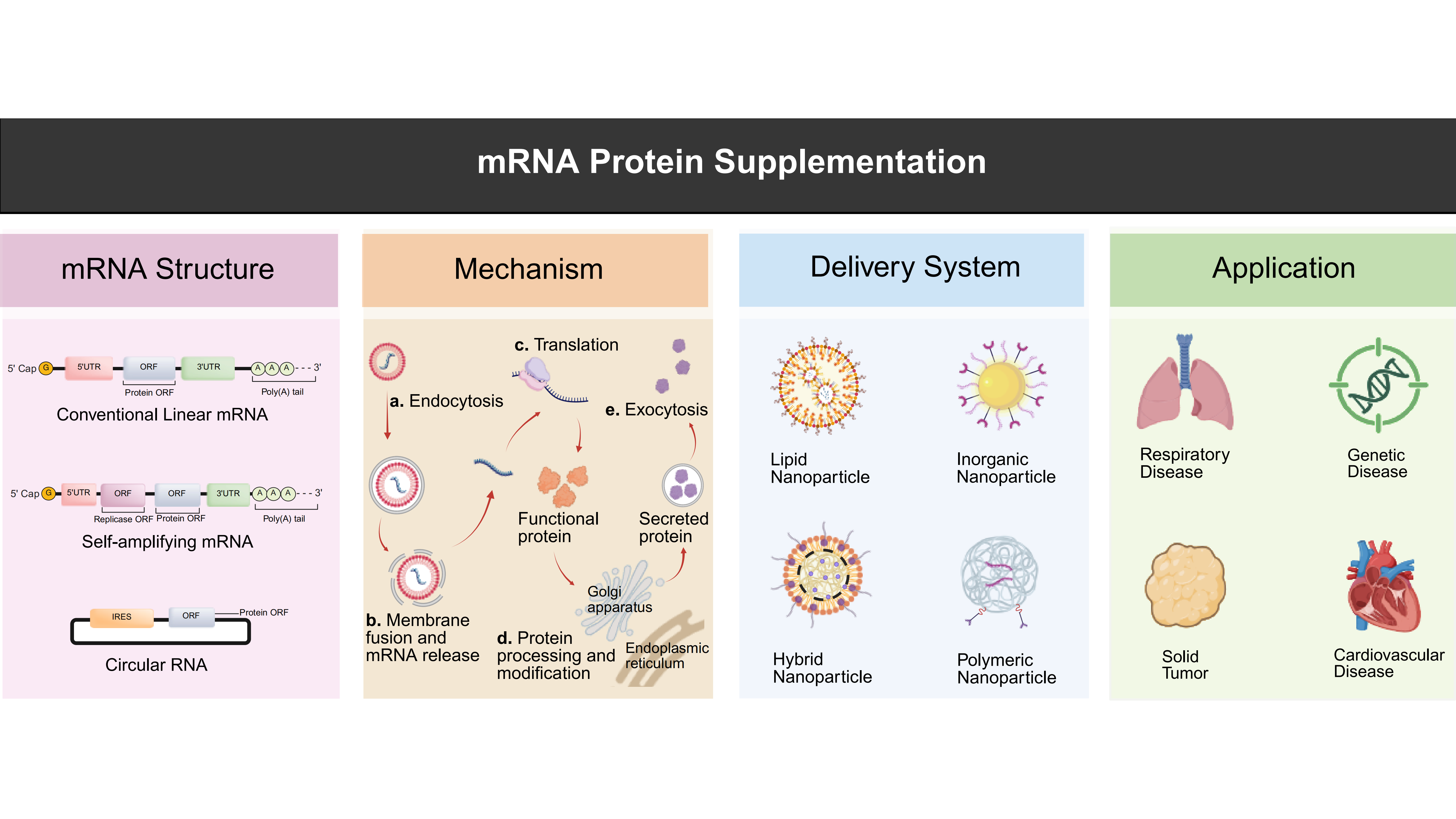
Keywords
mRNA therapy, protein replacement, gene therapy, lipid nanoparticles, in vivo expression, mRNA vaccines
1. Introduction
Traditional protein replacement and gene therapy have made significant progress in recent years in treating a variety of hereditary and enzyme deficiency diseases[1], however, it still faces several shared challenges such as high production costs of therapeutic proteins, short half-lives requiring frequent administration, and potential immune reactions that could decrease the treatment’s effectiveness[2]. To overcome these limitations, messenger RNA (mRNA) has gained widespread attention as an emerging therapeutic tool due to its remarkable ability to directly promote protein synthesis within the body.
mRNA protein replacement therapy leverages synthetic, sequence-optimized mRNA to drive the intracellular synthesis of functional, full-length proteins in the human body, thereby restoring the expression of missing or dysfunctional proteins caused by genetic defects or acquired diseases. Compared to traditional approaches, mRNA therapy not only provides more sustained therapeutic effects than direct protein administration, but it also avoids the genomic integration risks associated with DNA-based gene therapies. Furthermore, it significantly reduces production costs while offering enhanced safety and flexibility[3]. The ability to rapidly synthesize mRNA and tailor it to target specific diseases allows for an adaptable therapeutic strategy, which is particularly advantageous in situations where rapid response is critical, such as in infectious disease outbreaks[4], allowing for potentially more effective treatment options[5].
Current studies have shown that mRNA therapies exhibit great efficacy in treating genetic disorders, various forms of cancer, and infectious diseases such as COVID-19[3,6]. However, despite the promising results, research in this field still faces numerous challenges, including the stability of mRNA, delivery efficiency, immunogenicity, and the complexity of large-scale manufacturing processes[4,6]. Addressing these challenges is essential to unlock the full potential of mRNA therapies. As these obstacles are overcome, mRNA therapies could revolutionize the landscape of modern medicine, providing new hope for patients suffering from a wide range of genetic and acquired conditions. Here, we systematically examine the molecular design and delivery strategies of therapeutic mRNA, summarize recent preclinical and clinical advances across a spectrum of diseases including rare genetic disorders, cancers, and cardiovascular conditions, and critically analyze persistent challenges such as tissue-specific delivery, immunogenicity, and stability.
2. Fundamental Aspects of mRNA-Based Protein Supplementation
2.1 Structure and design of mRNA
mRNA is a single-stranded ribonucleic acid molecule responsible for transmitting genetic information and directing protein synthesis. Its molecular architecture typically includes a 5′ cap structure, an open reading frame (ORF), and a 3′ polyadenylate tail (poly(A) tail)[7,8]. The 5′ cap protects transcripts from exonucleolytic degradation and facilitates ribosomal recognition, thereby promoting both stability and translation efficiency[9,10]. Likewise, the poly(A) tail contributes to mRNA persistence in the cytoplasm, even though the precise correlation between tail length and transcript half-life is not yet fully elucidated[11].
Beyond serving as a mere carrier of genetic information, therapeutic mRNA also regulates translation efficiency and protein folding by virtue of its sequence and structural properties[12-14]. Thus, a deep understanding of its structural elements is fundamental to therapeutic applications, particularly in contexts such as protein replacement therapy. At present, three main mRNA formats are used or under investigation: traditional linear mRNA, self-amplifying mRNA (saRNA), and circular RNA (circRNA). Traditional linear mRNA is the form employed in currently approved vaccines, consisting of a 5′cap, coding region, and 3′poly(A)tail (Table 1, Figure 1a). It enables rapid protein production and is relatively simple to manufacture, though its transient expression requires stabilization strategies[15]. saRNA contains additional replicase elements that allow intracellular RNA replication after delivery, leading to much higher protein expression from minimal doses (Table 1, Figure 1b)[16]. In contrast, circRNA is a covalently closed RNA without free ends, making it highly resistant to degradation. It supports prolonged expression and lower immune activation, emerging as a promising next-generation platform for therapeutics (Table 1, Figure 1c)[17,18].
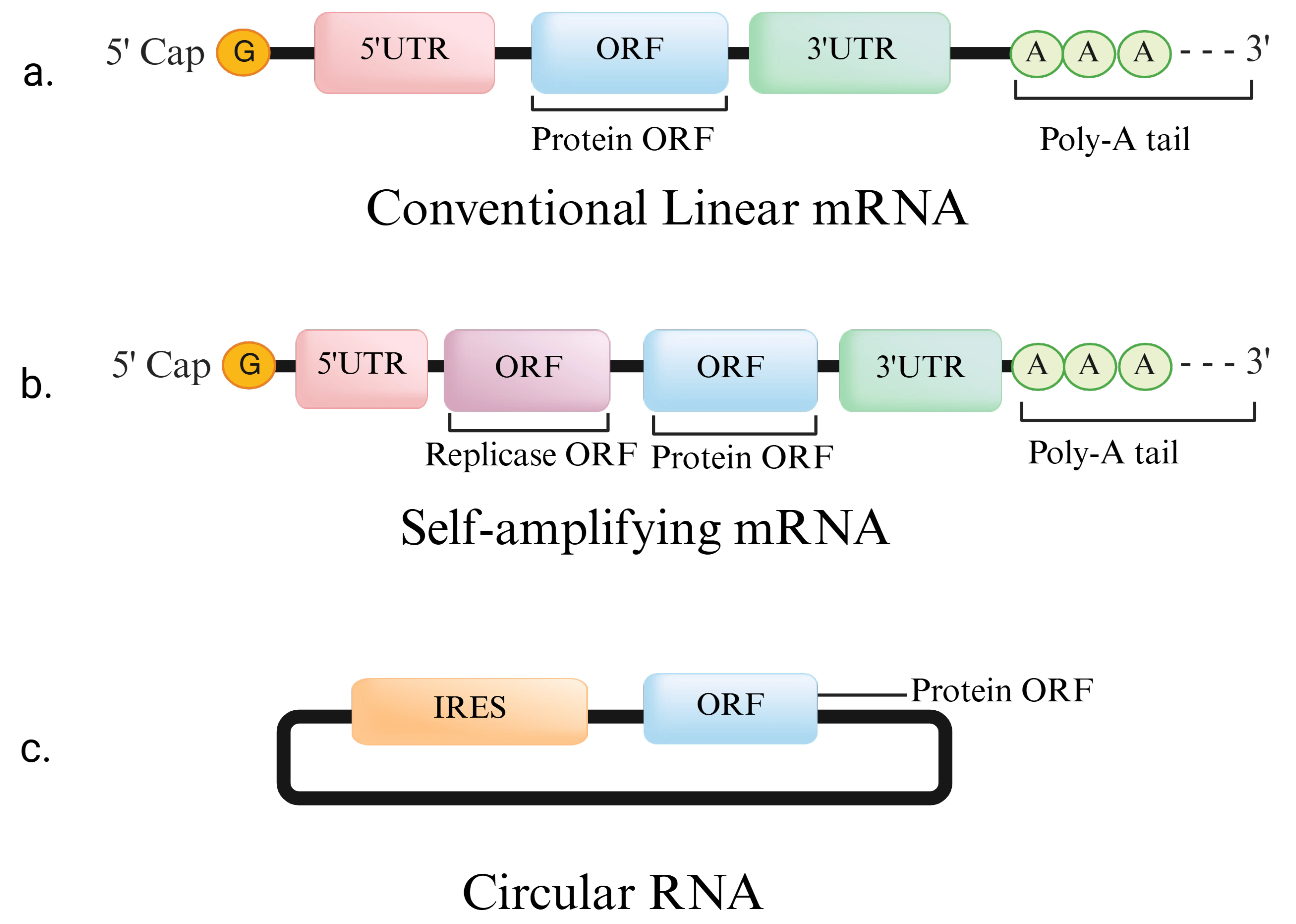
Figure 1. Structural comparison of mRNA modalities for protein replacement therapy. (a) Conventional linear mRNA contains a 5′ cap, 5′ UTR, ORF encoding the therapeutic protein, 3′ UTR and a poly(A) tail; (b) saRNA additionally harbours a replicase ORF that drives intracellular amplification of the antigen transcript, permitting dose-sparing;
Table 1. Comparison of different mRNA platforms.
| Type of mRNA | Structural features | Advantages | Disadvantages |
| Linear mRNA | 5' cap, 5'-UTR, coding sequence, 3'-UTR, and poly(A) tail | Simple manufacturing; shorter length; low immunogenicity after modification | Short half-life |
| Self-amplifying mRNA | Alphavirus virus-derived sequence | Longer half-life | Higher immunogenicity; toxicity concerns |
| Circular RNA | Without 5' cap and 3' UTR and poly(A) tail; internal ribosome entry site element | Higher stability | Complicate manufacturing process |
mRNA: messenger RNA; UTR: untranslated region.
To achieve optimal performance in vivo, dedicated sequence optimization strategies are applied[19]. Adjustments in the untranslated regions (UTRs) are central: the 5′ UTR affects ribosome binding and initiation efficiency, and structural elements including internal ribosome entry sites or unstructured domains can modulate both translation and immune suppression[20-22]. Recent advances in mRNA sequence engineering have refined 5′ UTR design, incorporating optimized Kozak sequences and minimizing inhibitory secondary structures to improve ribosomal scanning and translation initiation. Meanwhile, the 3′ UTR contains numerous regulatory elements that influence transcript stability and post-transcriptional control. Incorporating stabilizing motifs or extending the poly(A) tail can significantly prolong expression in cells[20]. In addition, newly developed branched, chemically modified poly(A)tails have been shown to markedly enhance translation capacity and mRNA longevity, providing an emerging strategy for next-generation mRNA therapeutics[23].
Codon optimization and GC content adjustment are widely adopted to maximize protein yield and reduce translational pausing[24-26]. Furthermore, nucleotide modifications such as pseudouridine and N1-methylpseudouridine enhance mRNA stability, suppress cellular innate immune responses, and thus improve translational efficiency and therapeutic effect[8,27,28]. The integration of such chemical strategies has now become clinical practice for therapeutic mRNA design[29]. Collectively, these combined optimizations ensure that therapeutic mRNA not only encodes the correct protein sequence but also possesses the structural robustness required for protein replacement applications.
Together, these structural, sequence, and chemical optimization strategies establish a foundation for efficient, stable, and
2.2 Delivery system for mRNA therapeutics
Efficient delivery remains the bottleneck for translating mRNA protein replacement therapy into the clinic. Exogenous mRNA is inherently unstable and negatively charged, hindering cellular uptake, and thus requires carrier systems to protect its integrity and ensure functional translation[30].
Viral vectors, such as retroviruses, adenoviruses, and adeno-associated viruses, provide high transduction efficiency and tropism. Nevertheless, concerns over genomic integration, long-term expression, and host immune responses limit their utility for transient protein replacement therapy[31,32].
By contrast, non-viral systems have advanced rapidly. Lipid nanoparticles (LNPs) have become the leading platform, especially for liver-targeted protein replacement therapies, with optimized formulations of four key components, each serving distinct functions and subject to specific optimization strategies (Table 2, Figure 2a). Ionizable lipids are designed to encapsulate mRNA at an acidic pH and facilitate endosomal escape in the cytosol; their optimization often involves tuning the pKa and modifying lipid tails to enhance biodegradability and transfection efficiency. Cholesterol provides structural stability and promotes membrane fusion, with structural analogs being explored to adjust nanoparticle rigidity and cellular uptake. Helper lipids (e.g., 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC)) support the formation of the lipid bilayer and stabilize the overall LNP architecture, with optimization focusing on varying lipid structures to influence biodistribution. Finally, polyethylene glycol (PEG)-lipids control particle size, prevent aggregation, and shield the nanoparticles from premature immune clearance (stealth effect). The optimization of PEG-lipids typically focuses on adjusting the PEG chain length and lipid anchor to balance systemic circulation half-life with efficient cellular uptake. Together, these meticulously engineered components enable protection from nuclease degradation and efficient endosomal escape after cellular uptake[33,34]. Their success in licensed COVID-19 vaccines demonstrates scalability and translational viability[35]. Importantly, for protein replacement applications, design objectives differ: whereas vaccines prioritize robust immune engagement, protein supplementation requires controlled, often tissue-specific delivery with sustained but reversible expression. LNPs naturally accumulate in the liver through apolipoprotein E (ApoE)-mediated targeting, which explains their nearly exclusive use for liver metabolic diseases[36]. This strong and specific hepatic tropism driven by ApoE makes LNPs uniquely well suited for liver-directed mRNA protein replacement therapy.
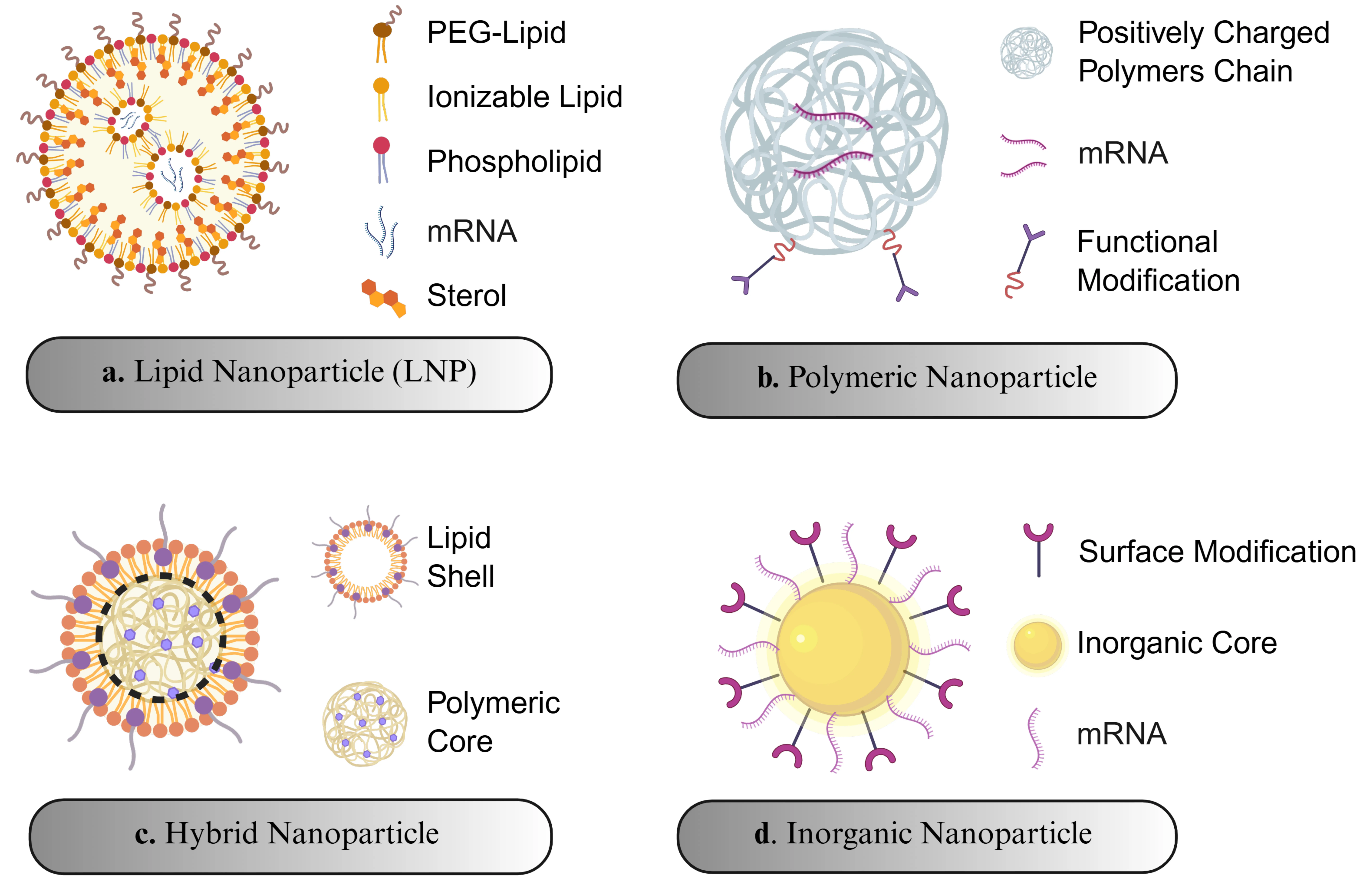
Figure 2. Representative nanocarriers for mRNA delivery. (a) LNPs composed of ionizable lipids, cholesterol, helper phospholipids, and PEG-lipids, providing efficient encapsulation and promoting endosomal escape; (b) Polymeric nanoparticles that are electrostatically complex with mRNA, conferring stability and allowing sustained or controlled release; (c) Hybrid nanoparticles combining lipid and polymer components to integrate stability, biocompatibility, and delivery efficiency; (d) Inorganic nanoparticles, such as gold or silica nanomaterials, surface-modified to adsorb or conjugate mRNA, offering high stability and multifunctionality. mRNA: messenger RNA; LNPs: lipid nanoparticles; PEG: polyethylene glycol.
Table 2. Comparison of mRNA delivery systems.
| Delivery system | Composition | Core advantages | Limitations |
| LNP | Ionizable lipids Cholesterol Helper lipids PEG-lipids | Structural stability, controlled expression, low immunogenicity, scalable manufacturing | Strong liver tropism |
| Polymeric nanoparticle | Biodegradable polymers such as PLGA, PEG, chitosan, or PCL | Easy to functionalize, large specific surface area | Low transfection efficiency, toxicity concerns |
| Hybrid nanoparticle | Organic and inorganic, multiple inorganic or multiple organic components | High stability, controlled release, tunable structure | Lack of scalable manufacturing standards Structural complexity |
| Inorganic nanoparticle | Gold, magnetic materials, quantum dots | High stability, easy functionalization, magnetic guidance, optical properties, easy to functionalize,good biocompatibility. | Unclear long-term toxicity Complex in vivo metabolic pathways Insufficient reproducibility |
mRNA: messenger RNA; LNP: lipid nanoparticle; PEG: polyethylene glycol; PLGA: poly(lactic-co-glycolic acid); PCL: poly(ε-caprolactone).
Polymeric nanoparticles such as polyethylenimine (PEI), poly(amidoamine) (PAMAM) dendrimers, and natural polymers (e.g., chitosan) offer tunable electrostatic interactions with mRNA and functional modifiability, yet typically suffer from lower stability and variable biocompatibility (Table 2, Figure 2b)[37,38]. Hybrid lipid–polymer nanoparticles combine structural stability with efficient cellular uptake and present a promising platform for more complex therapeutic constructs (Table 2, Figure 2c)[39]. Inorganic nanoparticles, including gold and silica-based systems, enable surface modifications and therapeutic–diagnostic integration, though their limited degradability restricts clinical deployment (Table 2, Figure 2d). Notably, none of these systems possess the intrinsic ApoE-mediated liver-targeting capacity that makes LNPs ideal for hepatic disorders.
These properties make LNPs highly suitable for protein supplementation, where stable and sustained expression with minimized immune activation is preferred over the high immunogenicity often desired in vaccine applications.
Alternative vectors, including polymeric and inorganic nanoparticles as well as viral systems, have also been explored. Polymeric nanoparticles offer design flexibility but often show lower transfection efficiency or potential toxicity[40]. Inorganic nanoparticles, such as gold or silica-based carriers, provide stability but are limited by poor biodegradability[41]. Viral vectors enable high expression efficiency, yet their integration risk and immunogenicity make them less suitable for protein supplementation[42]. In contrast to vaccines or cancer immunotherapy, where immune activation can be beneficial, the delivery systems for protein replacement must focus on safety, tissue specificity, and controlled protein expression[43].
Overall, the delivery demands of protein replacement therapy, requiring precise biodistribution, low immunogenicity, and controllable expression, distinguish it from other therapeutic contexts such as vaccines. The dominant choice of LNPs for liver metabolic diseases directly stems from their ApoE-mediated hepatic targeting mechanism[36]. Continuous refinement of LNPs and development of innovative hybrid or next-generation carriers are key to realizing the clinical potential of mRNA protein supplementation.
The distinct properties of LNPs, polymeric, hybrid, and inorganic delivery platforms outlined above determine their biodistribution and fate in vivo across disease models including liver metabolic disorders, lung diseases, and cancer. These differences directly underpin the intracellular mechanisms by which mRNA exerts its therapeutic function, as discussed in the following section.
2.3 Mechanism of mRNA protein replacement therapy
Exogenous mRNA exerts its therapeutic action by being delivered into the cytoplasm, where it is translated by ribosomes into functional proteins that can substitute for missing or dysfunctional endogenous proteins[44]. This principle has particular relevance in hereditary enzyme deficiencies, where supplementation of the encoded protein may restore normal metabolic function.
Taking LNPs as an example, particle size, surface charge, and hydrophobicity critically determine biodistribution and therapeutic activity[45]. After endocytosis, LNPs facilitate endosomal escape, enabling the release of intact mRNA into the cytoplasm for translation (Figure 3)[45,46]. This release process is considered one of the key efficiency-limiting steps for lipid nanoparticle–mediated delivery. If endosomal escape is inefficient or delayed, LNPs remain trapped as early endosomes mature into late endosomes and subsequently fuse with lysosomes. Within the highly acidic and enzyme-rich lysosomal compartment, the encapsulated mRNA is rapidly degraded by resident nucleases and acid hydrolases, completely abrogating its therapeutic potential. Mechanistically, the ionizable lipids within LNPs are largely neutral at physiological pH, which minimizes systemic toxicity and nonspecific interactions, but become protonated in the acidic endosomal environment. The resulting cationic lipids interact electrostatically with anionic phospholipids on the endosomal membrane, disrupting its integrity through fusion and phase transition events[47]. The efficiency of endosomal escape is strongly influenced by multiple factors, including the apparentpKaof ionizable lipids, helper lipid composition (such as
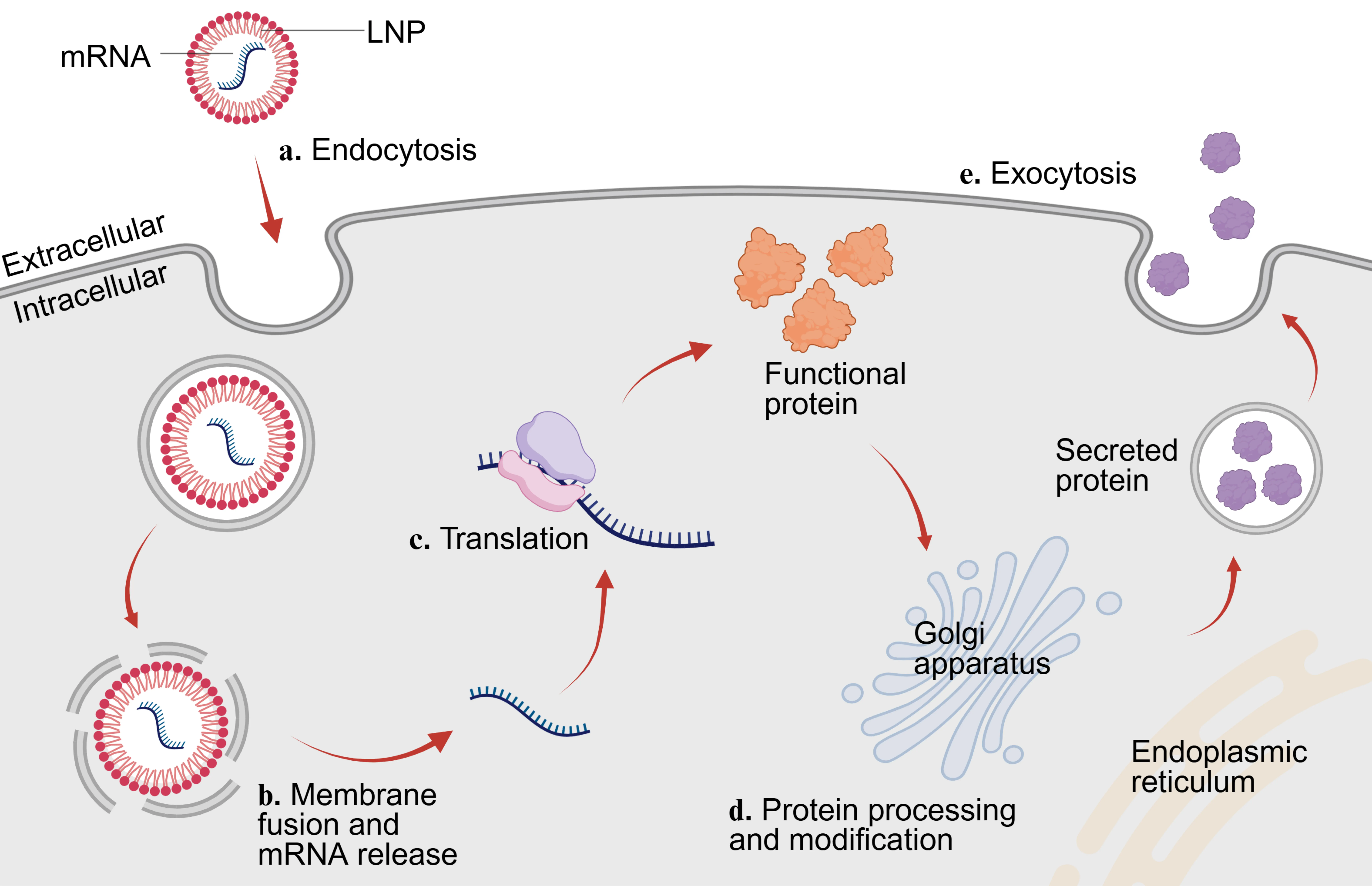
Figure 3. Intracellular pathway of mRNA delivery. (a) Cellular uptake of mRNA-loaded nanoparticles through endocytosis; (b) Endosomal escape via membrane fusion and release of mRNA into the cytoplasm; (c) Translation of mRNA by ribosomes to synthesize functional proteins; (d) Post-translational processing and modification of nascent proteins in the endoplasmic reticulum and Golgi apparatus; (e) Transport and secretion of mature proteins from the cell via exocytosis to exert their physiological functions. mRNA: messenger RNA; LNP: lipid nanoparticle.
Once successfully released inside the cell, mRNA is translated by ribosomes to synthesize target proteins. After entering the cytoplasm, mRNA binds to ribosomes and transfer RNA (tRNA) to initiate translation. With the assistance of the ribosome, tRNA delivers amino acids in the sequence specified by the mRNA codons, assembling a polypeptide chain. Upon encountering a stop codon, the newly synthesized polypeptide chain is released. It then enters the lumen of the endoplasmic reticulum for initial modification and processing, followed by further maturation in the Golgi apparatus.
The expressed proteins can restore their normal functions, thereby contributing to disease treatment[49]. For example, in patients with various genetic disorders, the missing or mutated proteins can be replenished through mRNA supplementation. In practice, these newly synthesized proteins exert their therapeutic effects through two major mechanisms: (1)intracellular functional restoration,for cytosolic or organelle-localized enzymes such as metabolic enzymes, the translated protein remains in the cell and directly restores normal biochemical pathways[15]; (2)supplementation of secreted proteins,for proteins normally secreted into the bloodstream, such as coagulation factors or hormones, the translated mature proteins undergo exocytosis and enter systemic circulation, where they perform their physiological functions[50]. Through these routes, mRNA therapeutics can effectively compensate for lost or dysfunctional proteins in diverse pathological settings. Moreover, these pathways not only ensure the synthesis of therapeutic proteins but also guarantee their proper processing and trafficking, which is critically advantageous for enzymes requiring post-translational modifications or specific subcellular localization. Research indicates that mRNA protein supplementation therapy shows great promise in treating various hereditary diseases, tumors, and cardiovascular diseases, offering new hope for patients[49,51-53]. To date, multiple preclinical studies and clinical trials have demonstrated the broad applicability and significant role of mRNA in disease treatment[54].
3. Current Progress
3.1 Preclinical research and disease models
With clear understanding of mRNA design principles, delivery vehicle characteristics, and intracellular mechanisms, extensive preclinical studies have been conducted in models of inherited metabolic diseases, cancer, cardiovascular disorders, and other conditions (Table 3). These investigations systematically validate the efficacy and feasibility of mRNA protein replacement therapy and provide essential support for subsequent clinical translation.
Table 3. Preclinical studies involving mRNA treatment.
| Condition | mRNA | Route of administration | Delivery system | Encoded protein | Ref |
| PKU | Linear | IV | LNP | PAH/PAL | [55-58] |
| PA | Linear | IV | LNP | hPCCA and hPCCB | [57,59,60] |
| MMA | Linear | IV | LNP | hMUT | [57,61] |
| CF | Linear | IV/Intratracheal instillation | Biodegradable chitosan-coated PLGA (NP)/LNP | CFTR | [62,63] |
| Fabry Disease | Linear | IV | LNP | human α-galactosidase enzyme | [64,65] |
| HemA | Linear | IV | LNP | Factor VIII | [66] |
| AATD | Linear | IV | LNP | AAT | [67,68] |
| Diabetic-wound healing/Angiogenesis | Linear | Topical | LNP | VEGF | [69] |
| OTC deficiency | Linear | IV | HMT (Hydroxyethyl starch-based Micelle-like Targeted carrier) | hOTC | [70] |
| Hemophilia B (Factor IX deficiency) | Linear | IV | LNP | hFIX | [71] |
| OA | Circular ivcRNA/linear | IA | LNP | Musashi2/Zdhhc11 | [72,73] |
| Arginase deficiency | Linear | IV | LNP | ARG1 | [74] |
| ASA | Linear | IV/IP | Liver-targeted LNP | ASL | [75,76] |
| Glioblastoma | Linear | Intracranial (i.c.) | PBAE nanoparticles | Tau-specific antibody | [77] |
| Arginase-1 deficiency | Linear | IV | Liver-targeted LNP | hPCCA and hPCCB | [74] |
| Solid tumors | Linear | Intratumoral | LNP | IL-12 | [75] |
| MAFL) and MAFLD-related HCC | Linear | IV | Def-LNP (VEPC-based) | TCPTP | [76] |
| Cardiac Fibrosis/Hypertensive injury | Linear | intravenous | CD5-targeted LNP | CAR (anti-FAP) | [77] |
| MI | modRNA | intracardiac | naked | CCN5 | [78] |
| Pressure overload (TAC) | modRNA | intracardiac | naked | Pip4k2c | [79] |
| Complete AV block | Linear mRNA | intramyocardial | naked | TBX18 | [80] |
| Cardiovascular disease | modRNA | intravenous | LNP | Relaxin-2 fusion protein (Rel2-vlk) | [81] |
mRNA: messenger RNA; PKU: phenylketonuria; LNP: lipid nanoparticle; PA: propionic acidemia; MMA: methylmalonic acidemia; CF: cystic fibrous; NP: nanoparticles;
Extensive preclinical investigations have provided compelling evidence supporting the therapeutic promise of mRNA-based protein replacement. In phenylketonuria (PKU) models, repeated intravenous administration of phenylalanine hydroxylase (PAH)-encoding mRNA encapsulated in LNPs successfully restored hepatic enzyme activity, reduced circulating phenylalanine levels, and improved metabolic balance. Additional studies using Arcturus’ LUNAR delivery platform demonstrated that both wild-type and mutant forms of PAH mRNA could induce dose-dependent therapeutic responses in PKU mice, further validating this approach. Comparable progress has been achieved in propionic acidemia (PA): intravenous injection of mRNA-3927, encoding the propionyl-CoA carboxylase alpha subunit (PCCA) and propionyl-CoA carboxylase beta subunit (PCCB), as subunits, reconstituted propionyl-CoA carboxylase activity, normalized key biomarkers, and improved survival in murine models, with benefits persisting for several weeks. Similar success has been observed in methylmalonic acidemia (MMA), where methylmalonyl-CoA mutase (MMUT)-encoding mRNA enhanced enzyme activity and reduced toxic metabolite accumulation.
Beyond metabolic disorders, mRNA therapy has also been investigated in other genetic disease models. In ornithine transcarbamylase (OTC), delivery of OTC-encoding mRNA restored urea cycle function in knockout mice. In cystic fibrosis, administration of cystic fibrosis transmembrane conductance regulator (CFTR) mRNA reinstated chloride channel activity within 72 hours of treatment, while in hemophilia models, infusion of FVIII-encoding mRNA rapidly increased circulating FVIII levels, correcting coagulation defects and normalizing bleeding phenotypes. From a clinical perspective, these disease contexts underscore the potential advantages of mRNA-based protein replacement over conventional protein therapeutics. For example, in hemophiliaA, the short plasma half-life of FVIII (approximately12hours) necessitates frequent intravenous infusions, often every few days, to maintain hemostasis[82]. In contrast, mRNA delivery enables sustained endogenous FVIII expression in situ, potentially maintaining therapeutic levels for weeks or even months after a single administration[83]. In OTC deficiency, direct enzyme replacement is challenged by limited cellular uptake and poor mitochondrial targeting of recombinant protein; mRNA therapy circumvents these hurdles by producing the functional enzyme directly inside hepatocytes, enabling proper localization to the mitochondria and effective restoration of metabolic activity[84].
Beyond cataloging preclinical successes, these data reveal a fundamental decision-making framework for mRNA protein replacement therapies. Rather than a one-size-fits-all approach, design principles must be dictated by the spatial, functional, and pharmacokinetic requirements of the target protein, stratified into three therapeutic paradigms:
(1) The “Systemic Bioreactor” model for secreted proteins: For conditions like hemophilia, the exact cellular target is flexible. Leveraging its massive synthetic capacity and the natural tropism of standard LNPs, the liver serves as an ideal endogenous factory. The priority here is maximizing translational yield and secretion rather than strict cellular targeting.
(2) The “In Situ Restoration” model for intracellular and organelle-specific enzymes: In metabolic disorders (e.g., OTC deficiency, PA, MMA), missing enzymes must function within specific subcellular compartments like mitochondria. Because recombinant proteins cannot cross membranes, mRNA must be delivered precisely into affected cells, relying on encoded signal peptides and intrinsic sorting machinery for correct localization.
(3) The “Extrahepatic and Barrier-Restricted” model for membrane proteins: For diseases like cystic fibrosis, the target protein (CFTR) functions at specific epithelial membranes, rendering systemic liver-targeted LNPs ineffective. This mandates localized delivery systems (e.g., inhaled nanoparticles) capable of penetrating mucosal barriers to transfect refractory extrahepatic cells.
By applying this framework, researchers can align mRNA engineering and delivery selection with the specific biological demands of the disease, moving from empirical screening toward rational therapeutic design[85].
3.2 Ongoing clinical trials
Building on encouraging preclinical results, several mRNA-based protein replacement therapies have already progressed into clinical trials, spanning metabolic, cardiovascular, oncological, and even infectious and neurological indications (Table 4, Figure 4). In the field of inherited metabolic disorders, mRNA-3927 for PA is undergoing multiple phase I/II studies in genetically confirmed patients, with dose-escalation protocols designed to optimize pharmacodynamics and safety[86]. A parallel study is further assessing the performance of this candidate in patients who had previously completed earlier treatment regimens, thereby providing insight into long-term outcomes.
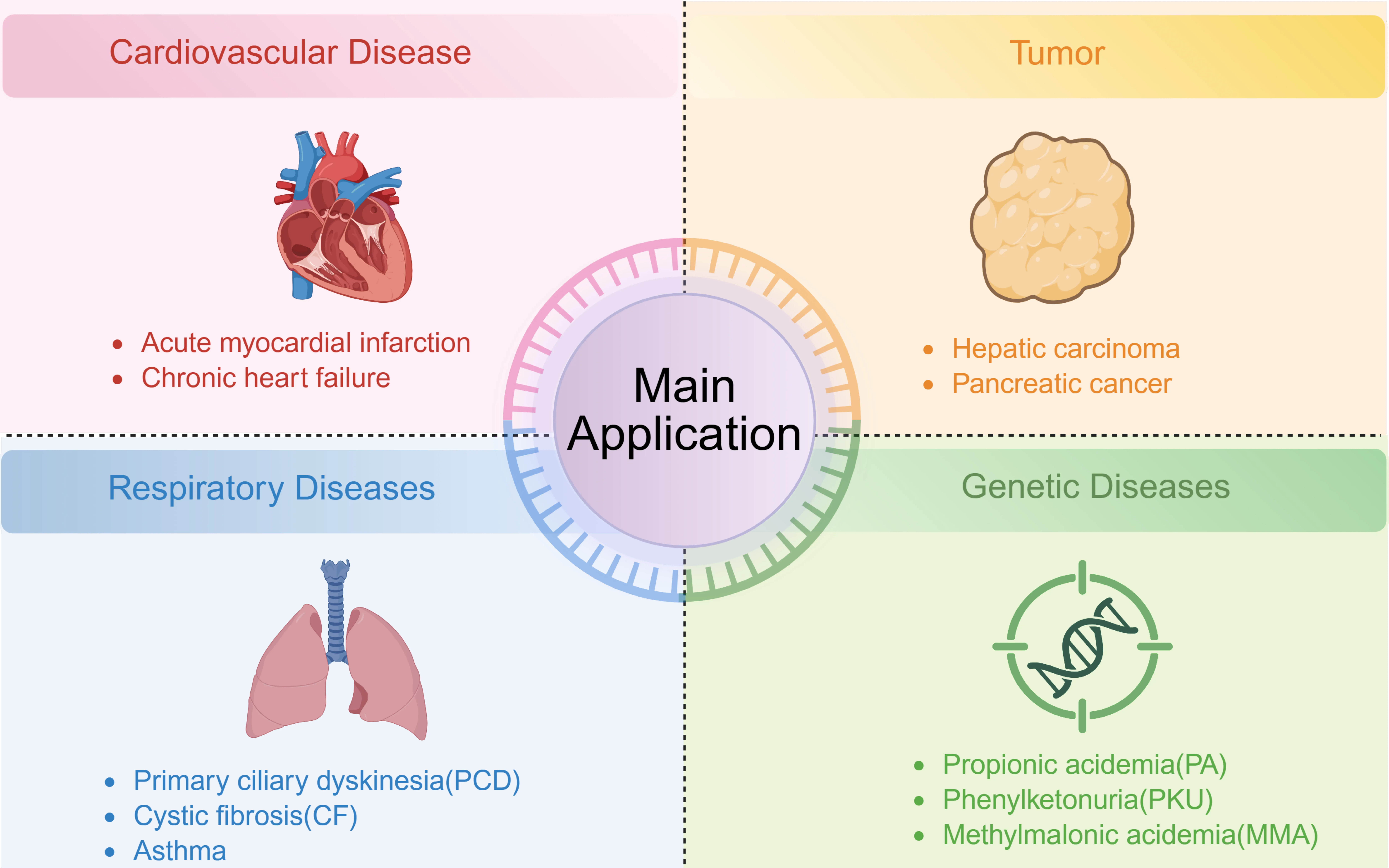
Figure 4. Therapeutic indications of mRNA protein replacement therapy. mRNA: messenger RNA.
Table 4. Clinical studies involving mRNA treatment.
| Indication | NCT number/phase | Company | Encoded Protein | Delivery System | Subjects | Status |
| PA | NCT04159103/Phase1/2 | Moderna | PCC, PCCB (Propionyl-CoA carboxylase α/β) | LNPs | 36 patients ≥ 1 year with PA | Recruiting |
| PA | NCT05130437/Phase1/2 | Moderna | PCC, PCCB | LNPs | PA patients who finished trial NCT04159103 | Recruiting |
| Male subjects with T2DM | NCT02935712/Phase1 | AstraZeneca | VEGF-A | Naked mRNA | 60 male T2DM patients (18-65 yrs) | Completed |
| Heart failure | NCT03370887/Phase1 | AstraZeneca | VEGF-A | Naked mRNA | 24 CABG surgery patients with moderate dysfunction | Completed |
| MMA | NCT03810690/Phase1/2 | Moderna | MUT | LNPs | MMA patients aged 1-18 yrs | Withdrawn |
| OTCD | NCT03767270/Phase1/2 | Translate Bio | OTC | LNPs | Patients with OTCD | Withdrawn |
| CF | NCT03375047/Phase1/2 | Sanofi | CFTR | LNPs | About 40 adult subjects with CF | Unknown |
| Prevention of Chikungunya Virus Infection | NCT03829384/Phase1 | Moderna | Chikungunya virus neutralizing antibody | LNPs | 39 healthy adults | Completed |
| PCD | NCT05737485/Phase1 | ReCode Therapeutics | DNAH5/CFAP300-related genes | Inhaled RCT1100 administered via nebulizer. | 9 healthy subjects & PCD patients | Completed |
| RIX | NCT06714253/Phase1/2 | RiboX Therapeutics | Aquaporin-1 | LNPs | About 42 patients with radiation-induced salivary gland injury | Recruiting |
| Ischemic Heart Disease | NCT06621576/Phase1 | Circode | VEGF-A | Circular RNA/LNP-like delivery system | 3 patients with Ischemic Heart Failure | Active, not recruiting |
mRNA: messenger RNA; PA: propionic acidaemia; LNPs: lipid nanoparticles; MMA: methylmalonic acidemia; CF: cysticfibrosis; PCD: primary ciliary dyskinesia; OTCD: ornithine transcarbamylase deficiency; OTC: ornithine transcarbamylase; T2DM: type II diabetes; MUT: methylmalonyl-CoA mutase; RIX: radiation-induced Xerostomia and hyposalivation; PCC: propionyl-CoA carboxylase alpha subunit; PCCB: propionyl-CoA carboxylase beta subunit; VEGF: vascular endothelial growth factor; CABG: coronary artery bypass grafting; CFTR: cystic fibrosis transmembrane conductance regulator.
The clinical pipeline also includes oncology applications, such as mRNA-4157, which is being tested alone or in combination with checkpoint inhibitors in patients with metastatic or unresectable solid tumors, including melanoma and gastrointestinal cancers. In cardiovascular medicine, AZD8601, an mRNA encoding VEGF-A, has been evaluated in type 2 diabetes patients and in individuals undergoing surgery for heart failure. Completed trials have shown acceptable tolerability and signals of improved cardiac function. For other monogenic disorders, such as MMA and ornithine transcarbamylase deficiency (OTCD), early clinical studies were initiated but later discontinued due to dose-limiting toxicities and narrow therapeutic windows. A critical pharmacological analysis of these clinical failures, particularly Translate Bio’s phase I/II trial for OTCD (MRT5201), reveals that the high and repeated mRNA doses required to achieve therapeutic intracellular enzyme levels triggered systemic inflammatory responses and significant elevations in liver enzymes (ALT/AST). These adverse events are primarily driven by the inherent immunogenicity of the high-dose mRNA payloads and the cellular stress caused by the accumulation of LNP lipid components in hepatocytes[87]. This underlines the need for
Importantly, the reach of mRNA therapeutics is expanding beyond rare genetic diseases. A prophylactic candidate, mRNA-1944, designed for Chikungunya virus prevention, has successfully completed a phase I trial[88], while exploratory work in Alzheimer’s disease is underway[89].
mRNA-based protein replacement strategies are being developed for a wide spectrum of diseases, including cardiovascular, respiratory, neoplastic, and genetic disorders. These categories highlight the versatility of mRNA modalities in addressing both acquired and inherited conditions, underscoring their potential to transform therapeutic approaches across diverse clinical fields.
4. Challenges
4.1 Delivery efficiency and targeting issues
Despite the substantial promise demonstrated in preclinical and clinical studies, further translation of mRNA protein replacement therapy faces several critical challenges. Among these, insufficient delivery efficiency and targeting specificity represent the primary bottlenecks restricting broader clinical application. The distribution and uptake efficiency of mRNA in the body are influenced by several factors, including the nature of the delivery system, the characteristics of cell membranes, and the existence of physiological barriers. Different tissues and cells react significantly differently to mRNA delivery systems, making the selection and design of the delivery system particularly important in the treatment of specific diseases. This variability can lead to inconsistent therapeutic outcomes, emphasizing the need for a comprehensive understanding of how different mRNA formulations interact with diverse biological environments.
To achieve specific cell recognition within these complex environments, a primary challenge lies in the rational selection and optimization of targeting ligands. Effective targeting ligands must possess high specificity and affinity to ensure accurate recognition and binding to target cells[90]. Many potential targeting ligands perform well in in vitro studies, but their targeting effects may decline a lot in vivo due to the complex physiological environment[90]. Additionally, the synthesis and optimization processes of targeting ligands may be hindered by technical limitations, obstructing their promotion in clinical applications[90].
Beyond ligand optimization, ensuring effective delivery requires overcoming formidable physiological barriers. In vivo, mRNA molecules and their delivery systems need to navigate cell membranes, vascular endothelium, and interstitial tissues. The presence of these physiological barriers not only affects the distribution of the delivery system but may also lead to insufficient drug concentration in the target tissues. Particularly in tumor therapy, the unique properties of the tumor microenvironment, such as hypoxia, acidity, and high interstitial fluid pressure, can all impact the effectiveness of the delivery system and cellular uptake[91].
Another factor is the different response of various cell types to mRNA delivery systems. The physiological characteristics of different tissues and cell types vary, which means that the same delivery system may perform differently in different cells[92,93]. For instance, variations in endocytic pathways, cell-surface receptor expressions, and intracellular trafficking mechanisms mean that highly phagocytic cells (like macrophages) may rapidly internalize nanoparticles, whereas immune cells (like T cells) typically exhibit poor uptake and require specialized formulation strategies for effective transfection. This differential response makes it extremely challenging to develop a universal and effective targeted delivery system, especially in the treatment of complex diseases. Ensuring that mRNA can be effectively delivered to all target cells remains a challenge[92,93].
4.2 Immune response and safety issues
The immunogenicity of mRNA is a critical safety concern that must be addressed during therapy. Unmodified mRNA is easily recognized by intracellular immune receptors, such as Toll-like receptors (TLRs), triggering immune responses that lead to inflammation and mRNA degradation. Studies have shown that receptors such as TLR3, TLR7, and TLR8 can recognize specific structures within mRNA molecules, thereby eliciting a strong immune response[94]. Furthermore, certain structures in the mRNA sequence, such as cytosine-phosphate-guanine (CpG) dinucleotides, can significantly enhance the intensity of the immune
To reduce immunogenicity, chemical modifications have emerged as an effective strategy. By using modified nucleotides such as pseudouridine and N1-methylpseudouridine, the risk of mRNA being recognized by TLRs can be significantly reduced[96], thereby lowering the incidence of immune responses. Additionally, optimizing the mRNA sequence to decrease the content of CpG motifs is another effective means to further reduce immunogenic activity[95]. However, these modifications also present certain limitations. While they can effectively lower immunogenicity, they may impact mRNA translation efficiency and stability[96]. Therefore, when designing modification strategies, it is essential to find a balance between reducing immunogenicity and maintaining translational capacity.
In terms of safety assessment, long-term safety studies of mRNA protein supplementation therapy are still insufficient. Unlike prophylactic vaccines that require only one or two doses, protein replacement therapy typically necessitates lifelong, frequent administration. This repeated dosing paradigm introduces unique safety challenges, most notably the long-term accumulation toxicity of LNP components (particularly in the liver, leading to chronic hepatotoxicity) and the Accelerated Blood Clearance (ABC) phenomenon driven by the generation of anti-PEG antibodies upon repeated exposure. These factors make it difficult to comprehensively assess potential risks based solely on short-term studies. In particular, off-target effects and gene toxicity issues that may arise from mRNA therapies remain key points of research. mRNA may express in non-target cells, leading to unnecessary biological responses and even causing damage to healthy cells[97]. Moreover, immune responses may result in allergic reactions and autoimmune responses, especially in patients sensitive to mRNA or components of its delivery system, potentially increasing the risk of severe adverse reactions[98]. Consequently, designing therapeutic mRNA requires careful optimization to maximize protein expression while minimizing recognition by innate immune sensors, thereby ensuring both high therapeutic efficacy and reduced adverse immune responses, which is fundamentally different from the aims of different from the aims of vaccine design, where robust immunogenicity is deliberately sought to elicit strong protective immune responses.
4.3 Stability and expression efficiency of mRNA
The stability and expression efficiency of mRNA are two core indicators for measuring therapeutic efficacy. The degradation mechanism of mRNA in the body is complex and primarily affected by ribonucleases (RNases)[99]. Specifically, mRNA turnover is driven by endonucleases that cleave internal phosphodiester bonds, as well as exonucleases such as XRN1 (which mediates 5'-to-3' decay) and the RNA exosome complex (which mediates 3'-to-5' decay). These enzymes can rapidly degrade unprotected mRNA, leading to a significant reduction in the availability of functional mRNA for translation. To counteract this, the intrinsic structural elements of mRNA, namely the 5' cap and the 3' poly(A) tail, play critical protective roles. The 5' cap prevents 5'-to-3' exonuclease attack and recruits translation initiation factors, while the poly(A) tail binds poly(A)-binding proteins (PABPs) to shield the 3' end from rapid deadenylation. This rapid degradation poses a challenge for achieving sustained therapeutic effects, as the half-life of mRNA can be significantly shortened in the presence of these enzymes. Additionally, the varying levels of RNase activity in different tissues further complicate the ability to predict mRNA behavior in vivo, highlighting the need for a deeper understanding of mRNA stability dynamics in therapeutic contexts.
Improving protein expression efficiency is another major challenge faced by mRNA therapies. Compared to traditional gene therapies, mRNA therapies often require higher dosages to achieve the same protein expression levels. Regulatory factors during translation initiation and elongation, such as the structure of the 5' UTR and the choice of codons, can all influence translation efficiency. Optimizing mRNA design, including structure, sequence, and modifications, will be a key focus of future research.
Within the cell, the enhancement of translation efficiency is also affected by the cellular environment. For instance, intracellular translation factors, energy supply, and other environmental factors can all impact the translation process[100]. For example, under conditions of cellular stress, such as hypoxia, lipid nanoparticle-induced toxicity, or amino acid starvation, the phosphorylation of eukaryotic initiation factor 2 alpha (eIF2α) is often triggered. This stress response globally represses translation initiation, thereby significantly reducing the yield of the therapeutic protein[101]. Conversely, highly metabolically active cells with abundant ATP, tRNA pools, and ribosomes, such as hepatocytes, generally exhibit much higher translation efficiencies compared to quiescent or
4.4 Challenges in the treatment of complex diseases
The treatment of complex diseases faces challenges from polygenic regulation and intricate pathological mechanisms. Polygenic diseases often involve interactions among multiple genes, leading to diverse phenotypic expressions. For instance, diabetes, cardiovascular diseases, and certain types of cancer are all polygenic diseases, making it difficult for a single mRNA protein supplementation therapy to effectively target all relevant genes[103]. In such cases, mRNA supplementation may not only fail to fully correct deficiencies but could also lead to imbalanced protein expression, potentially triggering other biological issues. To address these challenges, the exploration and research of combination treatment strategies have become important directions. Researchers are actively investigating the possibilities of combining mRNA protein supplementation therapy with traditional drugs or other treatment modalities, such as integrating mRNA therapy with immunotherapy to harness the power of the immune system for a more comprehensive treatment effect[104]. Simultaneously, the co-delivery of multiple mRNAs is also an emerging strategy that can deliver several mRNAs encoding different therapeutic proteins simultaneously, achieving intervention at multiple targets and enhancing overall treatment efficacy. Recent progress has extended this concept toward precision medicine. A recent study reported an in vivo translational regulation system that allows conditional protein expression in response to extracellular signals[105]. By co-delivering and sequence-engineering three mRNA species carrying specific receptor-responsive elements, the system achieved multiplexed sensing and selective translation control within living cells. Through ligand-dependent activation, the translational output of each mRNA could be dynamically tuned, enabling cells to modulate protein synthesis in real time according to their microenvironment.
This ligand-responsive and programmable framework represents a shift from static mRNA expression to context-adaptive gene regulation. It offers a means to enhance therapeutic precision and safety by restricting protein production to disease-relevant conditions. Integrating such controllable systems with lipid nanoparticle delivery may facilitate personalized interventions that respond to complex pathological cues, marking a significant advance in the evolution of mRNA therapeutics.
However, this combination treatment model also faces numerous challenges, including the optimization of treatment protocols, efficacy evaluation, and management of potential side effects[3]. Clinical research requires substantial experimental data support to validate the safety and efficacy of different treatment combinations. Moreover, individual patient differences complicate the formulation of universal treatment regimens. Therefore, mRNA protein supplementation therapy targeting complex diseases needs constant adjustment and optimization in clinical trials to achieve optimal therapeutic effects[3].
5. Future Directions and Prospects
5.1 Next-generation mRNA technologies and delivery systems
Next-generation mRNA technology is driving a revolution in the field of biomedicine, especially in the applications of vaccines and gene therapy. To achieve efficient and low-immunogenicity mRNA design and synthesis, researchers are exploring various strategies. In the optimization of mRNA sequences, scientists utilize computational biology tools to predict the impact of different sequences on translation efficiency and stability. Recently, advanced deep generative learning models have been introduced to design mRNA sequences with enhanced translational capacity and stability, by integrating information on codon usage, secondary structure, and untranslated region elements[106].
Furthermore, immunogenicity can be significantly reduced by modifying nucleotide structures, such as using modified nucleotides (e.g., pseudo-uridine and 5-methylcytidine), thereby enhancing safety[8,28]. Beyond nucleotide chemistry, researchers have proposed ACEmRNA (AdditionalChimericElement incorporatedIVTmRNA), in which short RNA/DNA chimeric elements are introduced into the invitro-transcribed molecule to modulate innate immune activation[107]. This structural refinement helps balance immune sensing with translational efficiency, further improving overall mRNA performance. In addition, new progress has been made in optimizing the 5′region of mRNA. A 2025study demonstrated that specific structural modifications at the 5′end can enhance cap-independent translation of linearmRNAs, significantly increase protein yield while maintaining low immunogenicity[108].
In the development of delivery systems, future efforts will focus on intelligent and targeted delivery mechanisms that directly address the unique challenges of chronic dosing and tissue specificity outlined previously. Advances in nanotechnology enable researchers to design more efficient carrier nanoparticles and liposomes. For instance, to mitigate the long-term accumulation toxicity associated with lifelong administration, the development of rapidly biodegradable lipids is a critical future direction. These lipids are designed to be swiftly metabolized and cleared from the liver after releasing their payload. Furthermore, to overcome the limitations of inherent hepatic tropism and address targeting challenges, extrahepatic delivery technologies, such as Selective Organ Targeting (SORT) LNPs, are being actively engineered to precisely route mRNA expression to specific extrahepatic tissues like the lungs or spleen. Additionally, research on targeted delivery using biomarkers is rapidly evolving; by surface modification, delivery systems can be coupled with specific receptors, enhancing therapeutic efficacy. For instance, targeted delivery based on tumor cell surface characteristics can greatly improve the selectivity and safety of mRNA therapies[109].
Furthermore, the potential of nanoparticle-mediated delivery of antigen-coding mRNAs into tumors has emerged as a promising strategy to modulate the immunosuppressive tumor microenvironment and elicit robust anti-tumor immune responses[110].
5.2 Integration with personalized and precision medicine
The integration of personalized treatment and precision medicine will play an increasingly important role in mRNA therapeutic strategies. With the widespread adoption of genomic sequencing technologies, researchers can obtain more comprehensive genomic information from patients, providing a foundation for personalized mRNA therapy development. For example, mRNA vaccines tailored to specific genetic mutations can significantly increase treatment specificity, particularly in cancer and hereditary diseases[109,111].
In the future, bioinformatics will play a critical role: by analyzing patients’ whole-genome, transcriptome, and metabolome data, researchers can identify disease-related features, thereby formulating more precise treatment plans. This personalized approach not only enhances efficacy but also reduces side effects. Moreover, clinical trial designs will increasingly account for individual differences, employing adaptive trial designs to adjust treatment strategies in real-time, ensuring that each patient receives optimal therapeutic outcomes.
5.3 Ethical, regulatory, and social considerations
As mRNA protein replacement therapies progress, ethical concerns and societal acceptance have become critical issues. Currently, the U.S.Food and Drug Administration (FDA) has issued guidance mainly focused on mRNA vaccines for infectious disease prevention. Dedicated regulatory guidelines for non-vaccine mRNA therapeutics, such as protein replacement and cancer treatment, have not yet been published; these products are presently evaluated under the existing framework for cell and gene therapy. Key challenges include balancing technological innovation with patient safety, managing the risks associated with gene editing, and safeguarding patient privacy regarding genetic data. Establishing a transparent regulatory framework is essential for ensuring the safety and efficacy of these therapies, incorporating rigorous oversight of clinical trials and evaluations of market access, intellectual property rights, and patient protections. Enhanced international cooperation among regulatory agencies is vital to address the complexities of cross-border drug development and facilitate the global sharing of mRNA technologies. By creating comprehensive ethical guidelines and support mechanisms, public trust in mRNA therapies can be strengthened, thereby promoting their clinical adoption and expanding treatment options for patients.
6. Discussion
In recent years, mRNA protein supplementation therapy has shown great potential in treating rare genetic disorders, cancers, and cardiovascular diseases, offering clear advantages over traditional protein supplementation approaches. By introducing synthetic mRNA, cells can continuously produce deficient or dysfunctional proteins, offering longer-lasting and more controllable therapy compared with conventional protein injections. This approach is flexible, scalable, and cost-effective, representing a valuable complement to gene and protein therapies.
Despite sharing a common molecular foundation with mRNA vaccines, mRNA protein supplementation therapy differs in several critical aspects that define its development and clinical application. First, safety requirements are far stricter. Protein supplementation usually requires higher mRNA doses to maintain therapeutic protein levels, meaning that the delivery carriers, especially LNPs, must undergo rapid metabolism and clearance to avoid long-term accumulation and potential toxicity. Achieving high-dose safety therefore depends on optimizing lipid composition, biodegradability, and systemic tolerance.
In addition, the desired sites of protein expression differ markedly. Vaccines rely on intramuscular delivery to induce systemic immune responses, while protein supplementation targets specific organs such as the liver, lung, or heart, where the restored proteins perform physiological functions. Achieving this requires carriers with precise biodistribution and tissue specificity, so that the therapeutic proteins are expressed in the right cells at sufficient levels. The success of this approach thus depends not only on the efficiency of cellular transfection but also on the accuracy of organ targeting.
Immune regulation provides another important distinction. While mRNA vaccines intentionally activate innate immune responses through pattern-recognition receptors to enhance immunogenicity and serve as self-adjuvants, protein supplementation therapy must avoid such activation altogether. Excessive stimulation of receptors such as TLR3, TLR7, or TLR8 can trigger cytokine release, inflammation, and degradation of therapeutic mRNA, leading to decreased protein output and adverse reactions. As a result, the mRNA used in protein supplementation often incorporates nucleotide modifications such as N1-methylpseudouridine or
Finally, the duration of translation represents a core difference in therapeutic design. Vaccines are optimized for short, high-level antigen production to stimulate adaptive immunity, but protein supplementation requires persistent and stable protein synthesis to maintain normal physiological function. Enhancing the structural stability of mRNA, through refined untranslated regions, codon usage, and extended poly(A) tails, promotes sustained expression without eliciting chronic inflammation. Prolonged translation and durable protein production are thus key requirements for achieving lasting therapeutic benefits.
Overall, these distinctions highlight that mRNA protein supplementation therapy is not merely an extension of vaccine technology but a distinct therapeutic paradigm. It requires a fundamentally different pharmacological perspective and decision-making framework. While vaccines operate on a “hit-and-run” principle, protein replacement demands precise control over pharmacokinetic and pharmacodynamic (PK/PD) profiles to ensure stable, long-term therapeutic protein levels without cumulative toxicity. Its success depends on the balance between safety, targeting accuracy, immune tolerance, and translation durability. Thanks to ongoing advances in biodegradable lipids, next-generation nanoparticles, and optimized mRNA designs, these challenges are gradually being addressed. As such innovations continue, mRNA protein supplementation is expected to play a pivotal role in precision medicine, providing long-term and precisely controlled protein restoration for a wide spectrum of human diseases.
Acknowledgements
The authors declare that AI tools were used solely for language polishing during the manuscript preparation process. All research content, including study design, data analysis, interpretations, figures, and tables, is original and was not generated using AI tools.
Authors contribution
Tao Y, Su Q: Conceptualization, visualization, writing-original draft.
Zhao Y, Pan J, Yin J: Writing-review & editing.
Xu C: Supervision, project administration, writing-review & editing.
Conflicts of interest
Congcong Xu is an Editorial Board Member of BME Horizon. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by Jiangsu Provincial Department of Science and Technology (Grant No. BG2025060), National Natural Science Foundation of China (Grant No. U25C2006), Science and Technology Program of Suzhou City (Grant No. SYW2025195), Jiangsu Province Youth Science and Technology Talent Support Program (Grant No. JSTJ-2025-128), Jiangsu Province Natural Science Foundation Program (Grant No. SBK20250405084).
Copyright
© The Author(s) 2026.
References
-
1. Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, et al. T lymphocyte-directed gene therapy for ADA-SCID: Initial trial results after 4 years. Science. 1995;270(5235):475-480.[DOI]
-
2. Genuth NR, Barna M. Heterogeneity and specialized functions of translation machinery: From genes to organisms. Nat Rev Genet. 2018;19(7):431-452.[DOI]
-
3. Vavilis T, Stamoula E, Ainatzoglou A, Sachinidis A, Lamprinou M, Dardalas I, et al. mRNA in the context of protein replacement therapy. Pharmaceutics. 2023;15(1):166.[DOI]
-
4. Youssef M, Hitti C, Puppin Chaves Fulber J, Kamen AA. Enabling mRNA therapeutics: Current landscape and challenges in manufacturing. Biomolecules. 2023;13(10):1497.[DOI]
-
5. Magadum A, Kaur K, Zangi L. mRNA-based protein replacement therapy for the heart. Mol Ther. 2019;27(4):785-793.[DOI]
-
6. Xu Z, Fisher DE. mRNA melanoma vaccine revolution spurred by the COVID-19 pandemic. Front Immunol. 2023;14:1155728.[DOI]
-
7. Gote V, Bolla PK, Kommineni N, Butreddy A, Nukala PK, Palakurthi SS, et al. A comprehensive review of mRNA vaccines. Int J Mol Sci. 2023;24(3):2700.[DOI]
-
8. Lu T, Chen A, Li C, Li K, Wang S, Zhang Y, et al. N1-methylpseudouridine mRNA modification enhances efficiency and specificity of gene overexpression by preventing Prkra-mediated global translation repression. Nucleic Acids Res. 2025;53(19):gkaf963.[DOI]
-
9. Cowling VH. Regulation of mRNA cap methylation. Biochem J. 2010;425(2):295-302.[DOI]
-
10. Mitchell SF, Walker SE, Algire MA, Park EH, Hinnebusch AG, Lorsch JR. The 5’-7-methylguanosine cap on eukaryotic mRNAs serves both to stimulate canonical translation initiation and to block an alternative pathway. Mol Cell. 2010;39(6):950-962.[DOI]
-
11. Chang H, Lim J, Ha M, Kim VN. TAIL-seq: Genome-wide determination of poly(A) tail length and 3′ end modifications. Mol Cell. 2014;53(6):1044-1052.[DOI]
-
12. Stupina VA, Yuan X, Meskauskas A, Dinman JD, Simon AE. Ribosome binding to a 5′ translational enhancer is altered in the presence of the 3′ untranslated region in cap-independent translation of turnip crinkle virus. J Virol. 2011;85(10):4638-4653.[DOI]
-
13. Polikanov YS, Steitz TA, Innis CA. A proton wire to couple aminoacyl-tRNA accommodation and peptide-bond formation on the ribosome. Nat Struct Mol Biol. 2014;21(9):787-793.[DOI]
-
14. Faure G, Ogurtsov AY, Shabalina SA, Koonin EV. Adaptation of mRNA structure to control protein folding. RNA Biol. 2017;14(12):1649-1654.[DOI]
-
15. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines: A new era in vaccinology. Nat Rev Drug Discov. 2018;17(4):261-279.[DOI]
-
16. Brito LA, Kommareddy S, Maione D, Uematsu Y, Giovani C, Scorza FB, et al. Self-amplifying mRNA vaccines. Adv Genet. 2015;89:179-233.[DOI]
-
17. Wesselhoeft RA, Kowalski PS, Anderson DG. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat Commun. 2018;9:2629.[DOI]
-
18. Qu L, Yi Z, Shen Y, Lin L, Chen F, Xu Y, et al. Circular RNA vaccines against SARS-CoV-2 and emerging variants. Cell. 2022;185(10):1728-1744.e16.[DOI]
-
19. Jin L, Zhou Y, Zhang S, Chen SJ. mRNA vaccine sequence and structure design and optimization: Advances and challenges. J Biol Chem. 2025;301(1):108015.[DOI]
-
20. Menendez-Gil P, Toledo-Arana A. Bacterial 3′UTRs: A useful resource in post-transcriptional regulation. Front Mol Biosci. 2021;7:617633.[DOI]
-
21. Hwang D, Park SA, Kim JH, Lee SY, Lee J, Kim HS, et al. Gold nanoparticle–mRNA conjugates encapsulated in lipid nanoparticles for coordinated codelivery of multiple mRNAs. ACS Omega. 2025;10(30):32998-33007.[DOI]
-
22. Leppek K, Das R, Barna M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol. 2018;19(3):158-174.[DOI]
-
23. Chen H, Liu D, Guo J, Aditham A, Zhou Y, Tian J, et al. Branched chemically modified poly(A) tails enhance the translation capacity of mRNA. Nat Biotechnol. 2025;43(2):194-203.[DOI]
-
24. Hanson G, Coller J. Codon optimality, bias and usage in translation and mRNA decay. Nat Rev Mol Cell Biol. 2018;19(1):20-30.[DOI]
-
25. Paremskaia AI, Kogan AA, Murashkina A, Naumova DA, Satish A, Abramov IS, et al. Codon-optimization in gene therapy: Promises, prospects and challenges. Front Bioeng Biotechnol. 2024;12:1371596.[DOI]
-
26. Hia F, Takeuchi O. The effects of codon bias and optimality on mRNA and protein regulation. Cell Mol Life Sci. 2021;78(5):1909-1928.[DOI]
-
27. Nance KD, Meier JL. Modifications in an emergency: The role of N1-methylpseudouridine in COVID-19 vaccines. ACS Cent Sci. 2021;7(5):748-756.[DOI]
-
28. Ho LLY, Schiess GHA, Miranda P, Weber G, Astakhova K. Pseudouridine and N1-methylpseudouridine as potent nucleotide analogues for RNA therapy and vaccine development. RSC Chem Biol. 2024;5(5):418-425.[DOI]
-
29. Andries O, Mc Cafferty S, De Smedt SC, Weiss R, Sanders NN, Kitada T. N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J Control Release. 2015;217:337-344.[DOI]
-
30. Nayerossadat N, Maedeh T, Ali P. Viral and nonviral delivery systems for gene delivery. Adv Biomed Res. 2012;1(1):27.[DOI]
-
31. Ginn SL, Amaya AK, Alexander IE, Edelstein M, Abedi MR. Gene therapy clinical trials worldwide to 2017: An update. J Gene Med. 2018;20(5):e3015.[DOI]
-
32. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415-419.[DOI]
-
33. Hou X, Zaks T, Langer R, Dong Y. Lipid nanoparticles for mRNA delivery. Nat Rev Mater. 2021;6(12):1078-1094.[DOI]
-
34. Samaridou E, Heyes J, Lutwyche P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv Drug Deliv Rev. 2020;154:37-63.[DOI]
-
35. Sahin U, Muik A, Derhovanessian E, Vogler I, Kranz LM, Vormehr M, et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature. 2020;586(7830):594-599.[DOI]
-
36. Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN, et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther. 2010;18(7):1357-1364.[DOI]
-
37. Ramamoorth M. Non viral vectors in gene therapy- an overview. J Clin Diagn Res. 2015;9(1).[DOI]
-
38. Kumar R, Santa Chalarca CF, Bockman MR, Van Bruggen C, Grimme CJ, Dalal RJ, et al. Polymeric delivery of therapeutic nucleic acids. Chem Rev. 2021;121(18):11527-11652.[DOI]
-
39. Zhang W, Jiang Y, He Y, Boucetta H, Wu J, Chen Z, et al. Lipid carriers for mRNA delivery. Acta Pharm Sin B. 2023;13(10):4105-4126.[DOI]
-
40. Li X, Qi J, Wang J, Hu W, Zhou W, Wang Y, et al. Nanoparticle technology for mRNA: Delivery strategy, clinical application and developmental landscape. Theranostics. 2024;14(2):738-760.[DOI]
-
41. Cooke JP, Youker KA. Future impact of mRNA therapy on cardiovascular diseases. Methodist DeBakey Cardiovasc J. 2022;18(5):64-73.[DOI]
-
42. Eralp Y. Application of mRNA technology in cancer therapeutics. Vaccines. 2022;10(8):1262.[DOI]
-
43. Lorenz C, Fotin-Mleczek M, Roth G, Becker C, Dam TC, Verdurmen WPR, et al. Protein expression from exogenous mRNA: Uptake by receptor-mediated endocytosis and trafficking via the lysosomal pathway. RNA Biol. 2011;8(4):627-636.[DOI]
-
44. Tenchov R, Bird R, Curtze AE, Zhou Q. Lipid Nanoparticles─From liposomes to mRNA vaccine delivery, a landscape of research diversity and advancement. ACS Nano. 2021;15(11):16982-17015.[DOI]
-
45. Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145(3):182-195.[DOI]
-
46. Dykman LA, Khlebtsov NG. Gold nanoparticles in biology and medicine: Recent advances and prospects. Acta Naturae. 2011;3(2):34-55.[DOI]
-
47. Sahay G, Querbes W, Alabi C, Eltoukhy A, Sarkar S, Zurenko C, et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol. 2013;31(7):653-658.[DOI]
-
48. Patel S, Ashwanikumar N, Robinson E, DuRoss A, Sun C, Murphy-Benenato KE, et al. Boosting intracellular delivery of lipid nanoparticle-encapsulated mRNA. Nano Lett. 2017;17(9):5711-5718.[DOI]
-
50. Sahin U, Karikó K, Türeci Ö. mRNA-based therapeutics: Developing a new class of drugs. Nat Rev Drug Discov. 2014;13(10):759-780.[DOI]
-
51. Suberi A, Grun MK, Mao T, Israelow B, Reschke M, Grundler J, et al. Polymer nanoparticles deliver mRNA to the lung for mucosal vaccination. Sci Transl Med. 2023;15(709):eabq0603.[DOI]
-
52. Kishore R, Magadum A. Cell-specific mRNA therapeutics for cardiovascular diseases and regeneration. J Cardiovasc Dev Dis. 2024;11(2):38.[DOI]
-
53. Monfrini E, Baso G, Ronchi D, Meneri M, Gagliardi D, Quetti L, et al. Unleashing the potential of mRNA therapeutics for inherited neurological diseases. Brain. 2024;147(9):2934-2945.[DOI]
-
54. Schürmann PJL, van Breda Vriesman SPE, Castro-Alpízar JA, Kooijmans SAA, Nieuwenhuis EES, Schiffelers RM, et al. Therapeutic application of mRNA for genetic diseases. WIREs Nanomed Nanobiotechnol. 2025;17(3):e70019.[DOI]
-
55. George LA, Sullivan SK, Giermasz A, Rasko JEJ, Samelson-Jones BJ, Ducore J, et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N Engl J Med. 2017;377(23):2215-2227.[DOI]
-
56. Fontanellas A, Berraondo P, Urigo F, Jericó D, Martini PGV, Pastor F, et al. RNA-based therapies in liver metabolic diseases. Gut. 2025;74(9):1514-1527.[DOI]
-
57. Friedrich MJ, Pham J, Tian J, Chen H, Huang J, Kehl N, et al. Transient hepatic reconstitution of trophic factors enhances aged immunity. Nature. 2026;650(8101):481-489.[DOI]
-
58. Yang T, Ge J, Huang L, Zhu X, Zhang D, Tang S, et al. Preclinical evaluation of AGT mRNA replacement therapy for primary hyperoxaluria type I disease. Sci Adv. 2025;11(15):eadt9694.[DOI]
-
59. Cacicedo ML, Weinl-Tenbruck C, Frank D, Limeres MJ, Wirsching S, Hilbert K, et al. Phenylalanine hydroxylase mRNA rescues the phenylketonuria phenotype in mice. Front Bioeng Biotechnol. 2022;10:993298.[DOI]
-
60. Diaz-Trelles R, Lee S, Kuakini K, Park J, Dukanovic A, Gonzalez JA, et al. Lipid nanoparticle delivers phenylalanine ammonia lyase mRNA to the liver leading to catabolism and clearance of phenylalanine in a phenylketonuria mouse model. Mol Genet Metab Rep. 2022;32:100882.[DOI]
-
61. Baek R, Coughlan K, Jiang L, Liang M, Ci L, Singh H, et al. Characterizing the mechanism of action for mRNA therapeutics for the treatment of propionic acidemia, methylmalonic acidemia, and phenylketonuria. Nat Commun. 2024;15:3804.[DOI]
-
62. Perez-Garcia CG, Diaz-Trelles R, Vega JB, Bao Y, Sablad M, Limphong P, et al. Development of an mRNA replacement therapy for phenylketonuria. Mol Ther Nucleic Acids. 2022;28:87-98.[DOI]
-
63. Jiang L, Park JS, Yin L, Laureano R, Jacquinet E, Yang J, et al. Dual mRNA therapy restores metabolic function in long-term studies in mice with propionic acidemia. Nat Commun. 2020;11:5339.[DOI]
-
64. Attarwala H, Lumley M, Liang M, Ivaturi V, Senn J. Translational pharmacokinetic/pharmacodynamic model for mRNA-3927, an investigational therapeutic for the treatment of propionic acidemia. Nucleic Acid Ther. 2023;33(2):141-147.[DOI]
-
65. An D, Schneller JL, Frassetto A, Liang S, Zhu X, Park JS, et al. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep. 2017;21(12):3548-3558.[DOI]
-
66. Haque AKMA, Dewerth A, Antony JS, Riethmüller J, Schweizer GR, Weinmann P, et al. Chemically modified hCFTR mRNAs recuperate lung function in a mouse model of cystic fibrosis. Sci Rep. 2018;8:16776.[DOI]
-
67. Robinson E, MacDonald KD, Slaughter K, McKinney M, Patel S, Sun C, et al. Lipid nanoparticle-delivered chemically modified mRNA restores chloride secretion in cystic fibrosis. Mol Ther. 2018;26(8):2034-2046.[DOI]
-
68. DeRosa F, Smith L, Shen Y, Huang Y, Pan J, Xie H, et al. Improved efficacy in a fabry disease model using a systemic mRNA liver depot system as compared to enzyme replacement therapy. Mol Ther. 2019;27(4):878-889.[DOI]
-
69. Zhu X, Yin L, Theisen M, Zhuo J, Siddiqui S, Levy B, et al. Systemic mRNA therapy for the treatment of fabry disease: Preclinical studies in wild-type mice, fabry mouse model, and wild-type non-human Primates. Am J Hum Genet. 2019;104(4):625-637.[DOI]
-
70. Chen CY, Tran DM, Cavedon A, Cai X, Rajendran R, Lyle MJ, et al. Treatment of hemophilia a using factor VIII messenger RNA lipid nanoparticles. Mol Ther Nucleic Acids. 2020;20:534-544.[DOI]
-
71. Connolly B, Isaacs C, Cheng L, Asrani KH, Subramanian RR. SERPINA1 mRNA as a treatment for alpha-1 antitrypsin deficiency. J Nucleic Acids. 2018;2018:8247935.[DOI]
-
72. Karadagi A, Cavedon AG, Zemack H, Nowak G, Eybye ME, Zhu X, et al. Systemic modified messenger RNA for replacement therapy in alpha 1-antitrypsin deficiency. Sci Rep. 2020;10:7052.[DOI]
-
73. Liu J, Zhang Y, Liu C, Jiang Y, Wang Z, Guo Z, et al. A single dose of VEGF-A circular RNA sustains in situ long-term expression of protein to accelerate diabetic wound healing. J Control Release. 2024;373:319-335.[DOI]
-
74. Prieve MG, Harvie P, Monahan SD, Roy D, Li AG, Blevins TL, et al. Targeted mRNA therapy for ornithine transcarbamylase deficiency. Mol Ther. 2018;26(3):801-813.[DOI]
-
75. Ramaswamy S, Tonnu N, Tachikawa K, Limphong P, Vega JB, Karmali PP, et al. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc Natl Acad Sci U S A. 2017;114(10):E1941-E1950.[DOI]
-
76. Suo J, Li L, Tan W, Yin X, Wang J, Shao R, et al. Circular RNA-based protein replacement therapy mitigates osteoarthritis in male mice. Nat Commun. 2025;16:8480.[DOI]
-
77. Wang K, He W, Gong Z, Gao J, Gao T, Pan N, et al. ZDHHC11-mediated palmitoylation alleviates chondrocyte senescence and serves as a therapeutic target for osteoarthritis. Nat Aging. 2025;5(11):2228-2246.[DOI]
-
78. Truong B, Allegri G, Liu XB, Burke KE, Zhu X, Cederbaum SD, et al. Lipid nanoparticle-targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency. Proc Natl Acad Sci U S A. 2019;116(42):21150-21159.[DOI]
-
80. Yu X, Qi S, Cao W, Cheng M, Zhang W, Wang Y, et al. Metabolism-programming mRNA-lipid nanoparticles remodel the immune microenvironment to improve immunotherapy against MAFLD. Sci Transl Med. 2025;17(827):eadv2293.[DOI]
-
81. Rurik JG, Tombácz I, Yadegari A, Méndez Fernández PO, Shewale SV, Li L, et al. CAR T cells produced in vivo to treat cardiac injury. Science. 2022;375(6576):91-96.[DOI]
-
82. Song MH, Yoo J, Kwon DA, Chepurko E, Cho S, Fargnoli A, et al. Modified mRNA-mediated CCN5 gene transfer ameliorates cardiac dysfunction and fibrosis without adverse structural remodeling. Int J Mol Sci. 2024;25(11):6262.[DOI]
-
83. Magadum A, Singh N, Kurian AA, Sharkar MTK, Sultana N, Chepurko E, et al. Therapeutic delivery of Pip4k2c-modified mRNA attenuates cardiac hypertrophy and fibrosis in the failing heart. Adv Sci. 2021;8(10):2004661.[DOI]
-
84. Wolfson DW, Kim NK, Lee KH, Beyersdorf JP, Langberg JJ, Fernandez N, et al. Transient pacing in pigs with complete heart block via myocardial injection of mRNA coding for the T-box transcription factor 18. Nat Biomed Eng. 2024;8(9):1124-1141.[DOI]
-
85. Kaushal N, Attarwala H, Iqbal MJ, Saini R, Van L, Liang M. Translational pharmacokinetic/pharmacodynamic model for mRNA-0184, an investigational therapeutic for the treatment of heart failure. Clinical Translational Sci. 2024;17(8):e13894.[DOI]
-
86. Koeberl D, Schulze A, Sondheimer N, Lipshutz GS, Geberhiwot T, Li L, et al. Interim analyses of a first-in-human phase 1/2 mRNA trial for propionic acidaemia. Nature. 2024;628(8009):872-877.[DOI]
-
87. Sabnis S, Kumarasinghe ES, Salerno T, Mihai C, Ketova T, Senn JJ, et al. A novel amino lipid series for mRNA delivery: Improved endosomal escape and sustained pharmacology and safety in non-human Primates. Mol Ther. 2018;26(6):1509-1519.[DOI]
-
88. August A, Attarwala HZ, Himansu S, Kalidindi S, Lu S, Pajon R, et al. A phase 1 trial of lipid-encapsulated mRNA encoding a monoclonal antibody with neutralizing activity against Chikungunya virus. Nat Med. 2021;27(12):2224-2233.[DOI]
-
89. Shao L, Zhang Y, Yang Z, Shi C, Yue X, Li C, et al. Synthetic efferocytic receptor microglia enhances anti-inflammatory clearance of amyloid-β for AD treatment in mice. Sci Adv. 2025;11(28):eads6613.[DOI]
-
90. Song JY, Aravand P, Nikonov S, Leo L, Lyubarsky A, Bennicelli JL, et al. Amelioration of neurosensory structure and function in animal and cellular models of a congenital blindness. Mol Ther. 2018;26(6):1581-1593.[DOI]
-
91. Thomas OS, Weber W. Overcoming physiological barriers to nanoparticle delivery: Are we there yet? Front Bioeng Biotechnol. 2019;7:415.[DOI]
-
92. Hatit MZC, Lokugamage MP, Dobrowolski CN, Paunovska K, Ni H, Zhao K, et al. Species-dependent in vivo mRNA delivery and cellular responses to nanoparticles. Nat Nanotechnol. 2022;17(3):310-318.[DOI]
-
93. Lv Z, Dai Y. mRNA vaccines and SiRNAs targeting cancer immunotherapy: Challenges and opportunities. Discov Onc. 2025;16:1265.[DOI]
-
94. Bagheri-Hosseinabadi Z, Rezazadeh Zarandi E, Mirabzadeh M, Amiri A, Abbasifard M. mRNA expression of toll-like receptors 3, 7, 8, and 9 in the nasopharyngeal epithelial cells of coronavirus disease 2019 patients. BMC Infect Dis. 2022;22:448.[DOI]
-
95. Krieg AM, Wu T, Weeratna R, Efler SM, Love-Homan L, Yang L, et al. Sequence motifs in adenoviral DNA block immune activation by stimulatory CpG motifs. Proc Natl Acad Sci U S A. 1998;95(21):12631-12636.[DOI]
-
96. Karikó K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther. 2008;16(11):1833-1840.[DOI]
-
97. Acevedo-Whitehouse K, Bruno R. Potential health risks of mRNA-based vaccine therapy: A hypothesis. Med Hypotheses. 2023;171:111015.[DOI]
-
98. Bhujel R, Enkmann V, Burgstaller H, Maharjan R. Artificial intelligence-driven strategies for targeted delivery and enhanced stability of RNA-based lipid nanoparticle cancer vaccines. Pharmaceutics. 2025;17(8):992.[DOI]
-
99. Hui MP, Foley PL, Belasco JG. Messenger RNA degradation in bacterial cells. Annu Rev Genet. 2014;48:537-559.[DOI]
-
100. Lima BFC, Ramos DC, Barbiero JK, Pulido L, Redgrave P, Robinson DL, et al. Partial lesion of dopamine neurons of rat substantia nigra impairs conditioned place aversion but spares conditioned place preference. Neuroscience. 2017;349:264-277.[DOI]
-
101. Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17(10):1374-1395.[DOI]
-
102. Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell. 2009;136(4):731-745.[DOI]
-
103. Bredemeyer AL, Helmink BA, Innes CL, Calderon B, McGinnis LM, Mahowald GK, et al. DNA double-strand breaks activate a multi-functional genetic program in developing lymphocytes. Nature. 2008;456(7223):819-823.[DOI]
-
104. Pérez-Núñez I, Rozalén C, Palomeque JÁ, Sangrador I, Dalmau M, Comerma L, et al. LCOR mediates interferon-independent tumor immunogenicity and responsiveness to immune-checkpoint blockade in triple-negative breast cancer. Nat Cancer. 2022;3(3):355-370.[DOI]
-
105. Nakanishi H, Itaka K. Extracellular ligand-responsive translational regulation of synthetic mRNAs using engineered receptors. NPG Asia Mater. 2025;17:25.[DOI]
-
106. Zhang H, Liu H, Xu Y, Huang H, Liu Y, Wang J, et al. Deep generative models design mRNA sequences with enhanced translational capacity and stability. Science. 2025;390(6773):eadr8470.[DOI]
-
107. Son S, Park M, Kim J, Lee K. ACE mRNA (Additional Chimeric Element incorporated IVT mRNA) for Enhancing Protein Expression by Modulating Immunogenicity. Adv Sci. 2024;11(18):2307541.[DOI]
-
108. Golojuch S, Largey B, El-Sagheer AH, Brown T. Enhancing cap-independent translation of linear mRNA. Nat Commun. 2025;16:9205.[DOI]
-
109. Leong KY, Tham SK, Poh CL. Revolutionizing immunization: A comprehensive review of mRNA vaccine technology and applications. Virol J. 2025;22:71.[DOI]
-
110. Huang X, He T, Liang X, Xiang Z, Liu C, Zhou S, et al. Advances and applications of nanoparticles in cancer therapy. MedComm Oncol. 2024;3:e67.[DOI]
-
111. Montin D, Santilli V, Beni A, Costagliola G, Martire B, Mastrototaro MF, et al. Towards personalized vaccines. Front Immunol. 2024;15:1436108.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Tao Y, Zhao Y, Su Q, Pan J, Yin J, Xu C. Engineering mRNA therapeutics for protein supplementation: Challenges and future horizons. BME Horiz. 2026;4:202604. https://doi.org/10.70401/bmeh.2026.0025
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Fundamental Aspects of mRNA-Based Protein Supplementation
- 3. Current Progress
- 4. Challenges
- 5. Future Directions and Prospects
- 6. Discussion
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Tao Y, Zhao Y, Su Q, Pan J, Yin J, Xu C. Engineering mRNA therapeutics for protein supplementation: Challenges and future horizons. BME Horiz. 2026;4:202604. https://doi.org/10.70401/bmeh.2026.0025
copy
Share Link
copy