Advances in catalytic asymmetric hydrogenation of third-row heteroatom-substituted alkenes
Jian Zhang
1,2,#
,
Ye Chen
1,#
,
Zhenfeng Zhang
2,*

,
Wanbin Zhang
2,3,*
*Correspondence to:
Wanbin Zhang, Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Pharmaceutical Sciences, Shanghai Jiao Tong University, Shanghai 200240, China.
E-mail: wanbin@sjtu.edu.cn
Zhenfeng Zhang, Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Pharmaceutical Sciences, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: zhenfeng@sjtu.edu.cn
Zhenfeng Zhang, Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Pharmaceutical Sciences, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: zhenfeng@sjtu.edu.cn
Chiral Chem. 2025;1:202507. 10.70401/cc.2025.0002
Received: October 09, 2025Accepted: November 25, 2025Published: November 27, 2025
Abstract
The asymmetric hydrogenation of vinyl silanes, vinyl sulfides, vinyl phosphines, and vinyl chlorides, those substituted with heteroatoms from the third-row of the periodic table, has emerged as a valuable and environmentally friendly method for the construction of the related optically active organosilanes, organosulfides, organophosphine, and organochlorides. These compounds have shown considerable potential for preparing functional molecules and synthesizing natural products. Over the past few decades, considerable research efforts have focused on the design and development of transition-metal catalysts featuring chiral ligands for the asymmetric hydrogenation of such substrates. In parallel, in-depth mechanistic studies have been conducted to elucidate the pathways of these enantioselective hydrogenation reactions, significantly advancing the understanding of their catalytic behavior and stereocontrol. This review focuses on the recent momentum and key advancements in the enantioselective hydrogenation of vinyl silanes, vinyl sulfides, and vinyl chlorides. In addition, given the widespread industrial interest in these compounds, the practical utility of this transformation in the synthesis of chiral silanes, chiral thioethers, chiral alkyl chlorides, as well as related derivatives, is also discussed.
Graphical Abstract

Keywords
Asymmetric hydrogenation, alkenes, chiral alkyl chlorides, chiral silanes, chiral thioethers
1. Introduction
Asymmetric hydrogenation (AH) is widely recognized as one of the most pivotal transformations in enantioselective catalysis, providing atom-efficient and high-yielding solutions for the synthesis of chiral molecules[1-9]. The field was revolutionized by the pioneering contributions of Knowles, who introduced Rh/DIPAMP catalysts[10], and Noyori, who developed Ru/BINAP systems for the asymmetric hydrogenation of C=C and C=O bonds[11], respectively. These landmark discoveries laid the foundation for the design of diverse transition-metal-based catalysts, predominantly incorporating rhodium, ruthenium, and iridium centers coordinated with chiral ligands. Owing to their tunable steric and electronic properties, these catalysts exhibit exceptional chemo-, regio-, and enantioselectivity, enabling the efficient hydrogenation of a broad range of unsaturated substrates[12-14].
The collective efforts of researchers have firmly established AH of C=C, C=O, and C=N bonds as a highly efficient strategy for constructing chiral compounds, particularly those featuring one or two consecutive stereogenic centers[15,16]. This methodology has been widely applied in the stereoselective synthesis of natural products, pharmaceuticals, agrochemicals, chiral ligands, and functional materials, demonstrating its broad utility in synthetic chemistry[17-40]. The precise control over stereochemistry in these transformations has rendered asymmetric hydrogenation indispensable in modern synthesis, especially within the pharmaceutical industry, where enantiopurity is often a stringent requirement[41-44].
Despite these advances, research efforts have predominantly focused on substituted alkenes, particularly investigating the impact of carbon-based substituents at various positions of the C=C bond[45]. While this focus has enabled remarkable progress in constructing complex chiral architectures, the asymmetric hydrogenation of heteroatom-substituted alkenes remains underexplored. This area presents substantial challenges due to the electronic and steric perturbations introduced by heteroatoms, which often hinder effective catalyst-substrate coordination (Figure 1). Specifically, the lone-pair electrons of heteroatoms (e.g., S or P) elevate the electron density of the C=C bond through p–π conjugation while simultaneously reducing coordination efficiency via competitive binding to the metal center. These dual effects collectively diminish the hydrogenation reactivity of the C=C bond[46]. Additionally, the steric bulk of heteroatoms with larger atomic radii (e.g., Si or Cl) further obstructs both metal-alkene coordination and the subsequent hydride transfer step, ultimately compromising reactivity and enantioselectivity in these transformations[47].
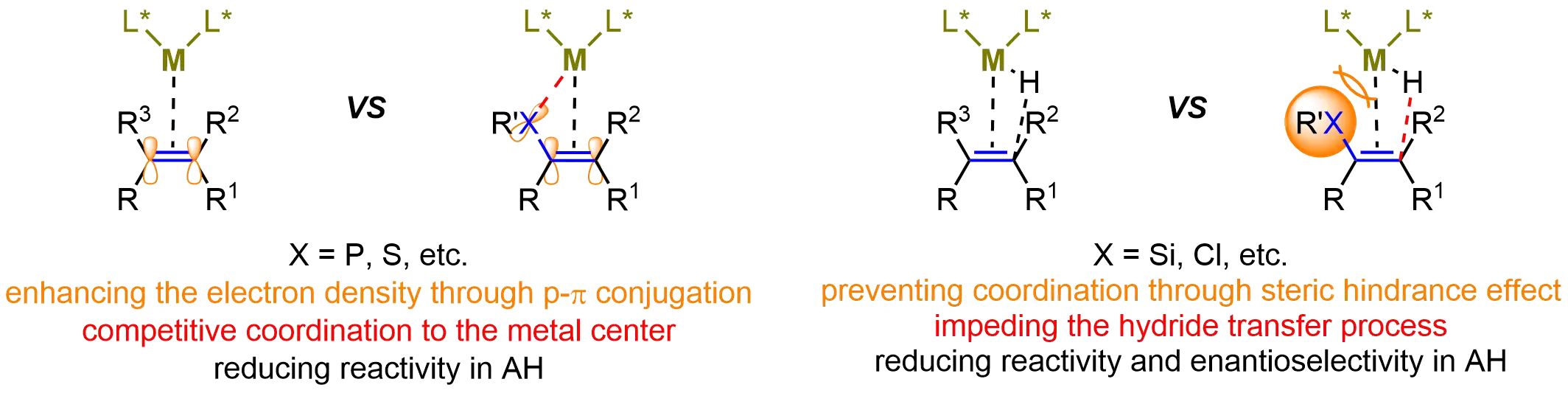
Figure 1. The difficulties in catalytic asymmetric hydrogenation of heteroatom-substituted alkenes. Created in ChemDraw. AH: asymmetric hydrogenation.
Given these challenges, developing novel catalytic systems and strategies for the asymmetric hydrogenation of heteroatom-substituted alkenes represents a critical research direction. Progress in this area would significantly expand the synthetic utility of asymmetric hydrogenation, enabling efficient access to a more diverse range of stereochemically complex molecules. A fundamental understanding of the unique challenges presented by these substrates, including their electronic and steric properties, is therefore essential for advancing the field and fully realizing the transformative potential of asymmetric hydrogenation in modern synthetic chemistry.
Over recent decades, significant advances have been achieved in the asymmetric hydrogenation of heteroatom-substituted alkenes. To overcome the characteristic low reactivity and modest stereoselectivity of these transformations, researchers have developed several effective strategies. These include: (1) the rational design of high-performance chiral catalysts to improve reaction efficiency, and (2) structural modifications of the alkene substrates through the introduction of electron-withdrawing groups or auxiliary coordinating moieties to enhance both reactivity and enantiocontrol. Remarkable progress has been documented for alkenes bearing various heteroatom substituents, including boron (B), nitrogen (N), oxygen (O), fluorine (F), silicon (Si), phosphorus (P), sulfur (S), and chlorine (Cl). While comprehensive reviews have covered the asymmetric hydrogenation of B-[48], N-[49], O-[9], F-[50], and P-[51]substituted alkenes, the corresponding transformations of Si-, S-, and Cl-substituted alkenes remain systematically unexplored in the review literature, to the best of our knowledge.
This review highlights significant advances in this rapidly developing field, demonstrating how asymmetric hydrogenation of Si-, S-, and Cl-substituted alkenes provides an efficient strategy for accessing enantioenriched Si-, S-, and Cl-functionalized compounds (Scheme 1). We systematically summarize key progress in the hydrogenation of C=C bonds containing these heteroatomic substituents. To offer a comprehensive survey of synthetic methodologies for constructing diverse Si-, S-, and Cl-containing scaffolds, the transformations are categorized based on the different heteroatom-substituted alkenes and the various transition-metal catalysts.
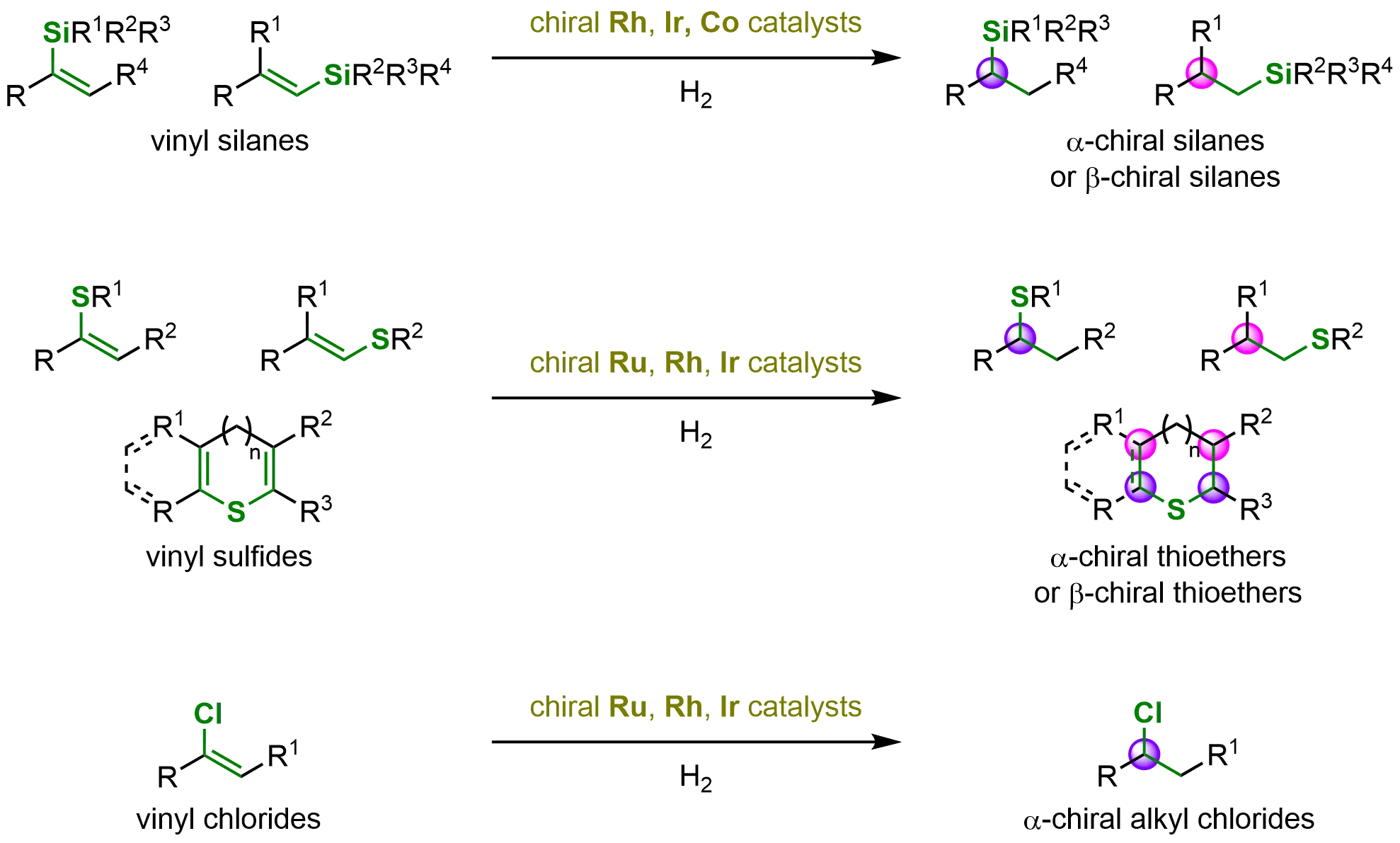
Scheme 1. Catalytic asymmetric hydrogenation of vinyl silanes, vinyl sulfides and vinyl chlorides. Created in ChemDraw.
2. Asymmetric Hydrogenation of Vinyl Silanes
Chiral organosilanes represent crucial synthetic intermediates for both chiral catalyst development and bioactive molecule modification through silicon substitution, particularly in medicinal chemistry applications[52]. As shown in Figure 2, several representative compounds highlight the broad utility of chiral organosilanes across chemical disciplines. For instance, the Sieburth silicon analog and sila-HNE inhibitor are often referred to as the “silicon switch” in bioactive molecules, where carbon-to-silicon substitution modulates biological activity[53]. These silicon-based analogs have gained increasing importance in medicinal chemistry, where such substitutions can enhance or precisely tune bioactivity. Furthermore, prolinol silyl ethers have emerged as particularly valuable reagents due to their wide application in aldehyde-involving asymmetric transformations[54]. Their synthetic versatility, high efficiency, excellent selectivity, and remarkable stability have established them as indispensable tools for constructing complex chiral architectures.
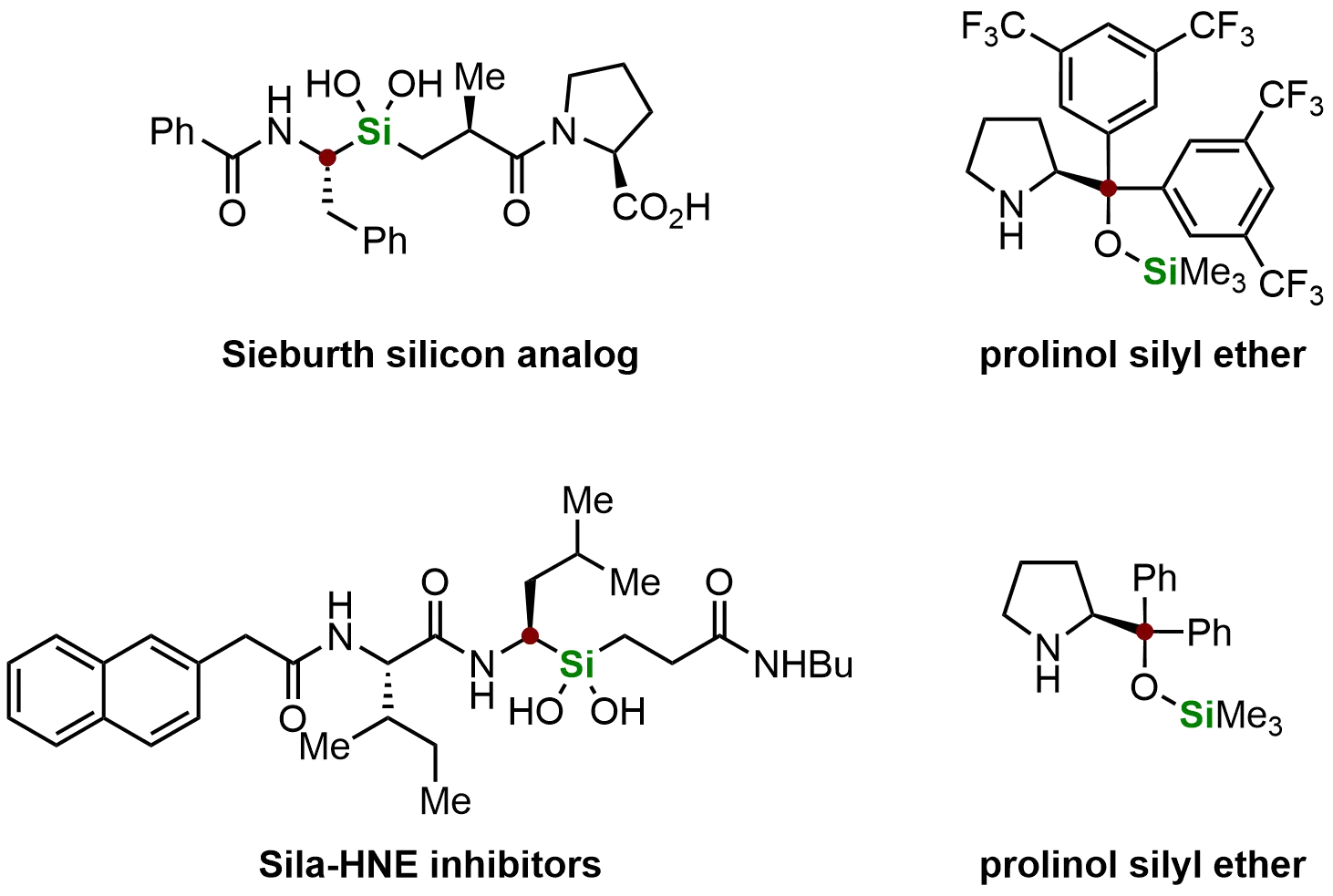
Figure 2. Typical examples of chiral organosilanes in medicinal chemistry and asymmetric catalysis. Created in ChemDraw.
While various methods for preparing chiral organosilanes have been developed, there remains a continuous demand for more efficient and innovative strategies to synthesize these compounds[55-58]. One particularly promising avenue is the transition-metal-catalyzed asymmetric hydrogenation of vinyl silanes, which has emerged as an effective and atom-economical approach. This method enables the direct, straightforward, and highly selective formation of chiral organosilanes, thereby offering significant advantages in asymmetric synthesis. The ability to achieve high enantioselectivity, coupled with the simplicity of the reaction, makes this strategy a powerful tool in both academic research and industrial applications. Specifically, although silyl groups are widely used as functional groups in synthetic chemistry, the coordination of silicon atoms in silyl-substituted alkenes with transition-metal catalysts is generally not observed. The large size of silicon atoms inhibits the chelation of transition metals with silyl-substituted alkenes, making their asymmetric hydrogenation particularly challenging (Figure 3).
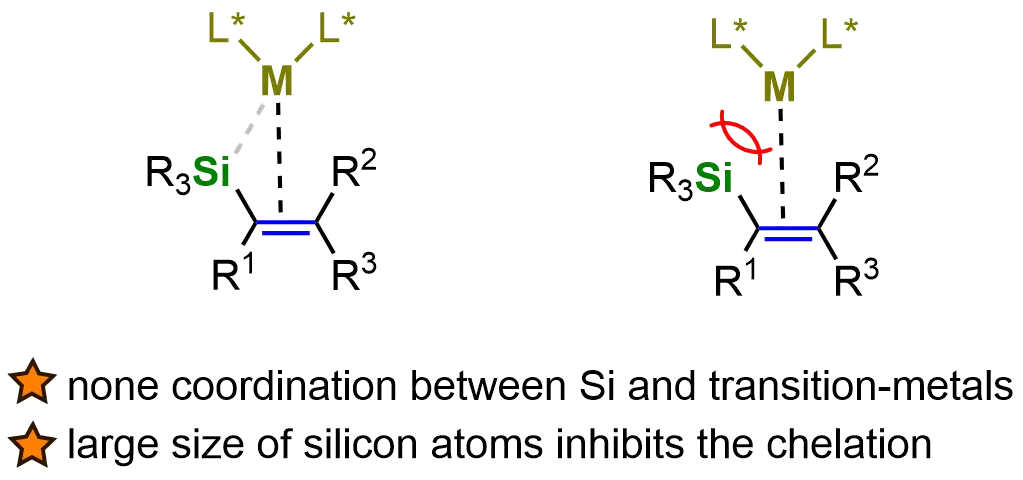
Figure 3. The low reactivity of vinyl silanes in AH. Created in ChemDraw. AH: asymmetric hydrogenation.
2.1 Rh-catalyzed asymmetric hydrogenation of vinyl silanes
In 2005, Feringa, Vries, Minnaard, and co-workers reported the enantioselective rhodium-catalyzed hydrogenation of enol carbamates using monodentate phosphoramidites (Scheme 2)[59]. Their study demonstrated that the sterically hindered trimethylsilyl (TMS) substituted enol N,N-diethylcarbamate (S1) could be hydrogenated to full conversion under mild conditions, albeit with modest enantioselectivity (43%). The reactivity of the asymmetric hydrogenation (Int2) was significantly enhanced by the strong coordination between the carbonyl functionality and the rhodium center. In contrast, the weak silicon-rhodium interaction failed to establish an analogous coordination model (Int1). This pioneering work not only underscored the potential of phosphoramidite-ligated rhodium catalysts for the hydrogenation of challenging substrates but also opened up new avenues for exploring the catalytic asymmetric hydrogenation of silicon-substituted alkenes. Since then, this area has attracted considerable attention in both synthetic and mechanistic studies.
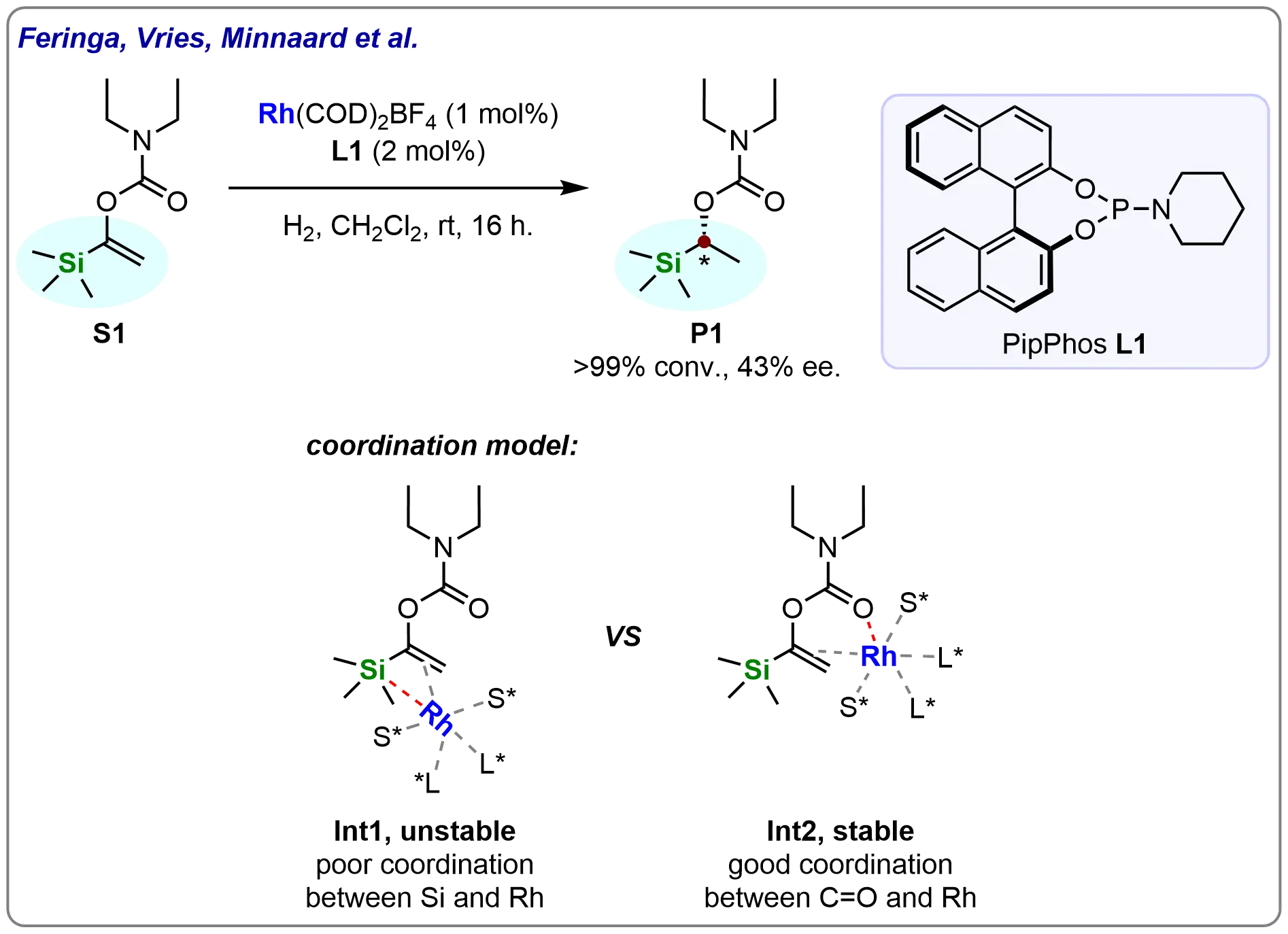
Scheme 2. Rh-catalyzed asymmetric hydrogenation of TMS-substituted enol N,N-diethylcarbamate. Created in ChemDraw.
In 2017, Zhang et al. successfully developed an asymmetric hydrogenation method for β-silyl-α,β-unsaturated esters, providing access to chiral 3-substituted-3-silylpropionic ester derivatives[60]. This transformation was catalyzed by a rhodium complex coordinated with a bisphosphine-thiourea ligand (ZhaoPhos) (Scheme 3a). Notably, the Si-protecting group in the substrates exhibited minimal influence on either reactivity or enantioselectivity. For instance, when the Si-protecting group was varied from triethylsilyl (S2a) to dimethylphenylsilyl (S2b), the corresponding hydrogenated products (P2a and P2b) were obtained in excellent yields (96%-97%) and enantioselectivities. Moreover, the methodology demonstrated broad compatibility with substrates bearing diverse electronic properties. Both electron-rich (S2c and S2d) and electron-deficient (S2e) phenyl-substituted substrates underwent efficient hydrogenation, affording products with near-perfect enantioselectivities (99% ee). Remarkably, even alkyl-substituted substrates (S2f) were well-tolerated, yielding the desired product in 95% yield and 89% ee. These results highlight the versatility of this catalytic system for the asymmetric hydrogenation of β-silyl-α,β-unsaturated esters, offering a robust strategy for synthesizing enantiomerically enriched 3-silylpropionic ester derivatives.
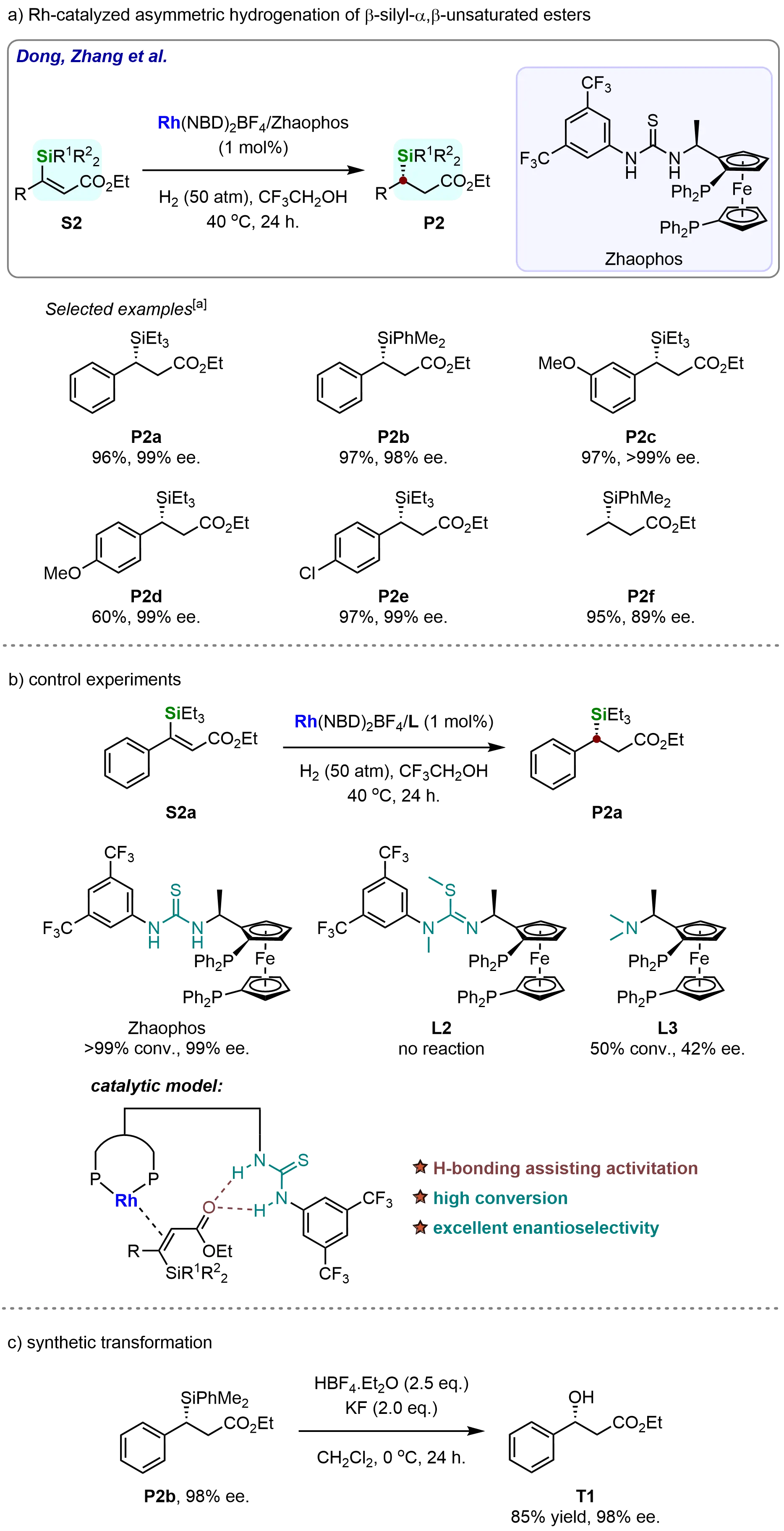
Scheme 3. Rhodium/ZhaoPhos-catalyzed enantioselective synthesis of chiral 3-substituted-3-silypropionic esters. Created in ChemDraw. [a]: Isolated yield was recorded.
To further investigate the crucial role of the thiourea motif in the ZhaoPhos ligand, control experiments were conducted (Scheme 3b). Ligands L2 and L3, which lack the thiourea functionality, were tested under the optimized reaction conditions for the asymmetric hydrogenation of β-triethylsilyl-α,β-unsaturated ethyl ester (S2a). As shown in Scheme 2b, no conversion was observed with ligand L2, confirming the essential role of the thiourea group. In contrast, ligand L3 afforded modest conversion and enantioselectivity (50% conversion, 42% ee), further demonstrating the necessity of the thiourea motif for achieving high reactivity and enantioselectivity. These results indicate that the thiourea group contributes to substrate activation, likely through hydrogen bonding with the substrate, thereby enabling the observed high enantioselectivity.
To further demonstrate the synthetic utility of the developed catalytic methodology, a subsequent transformation was performed to construct another synthetically valuable molecule (Scheme 3c). The hydrogenation product P2b was successfully converted into ethyl (R)-3-hydroxy-3-phenylpropanoate (T1) via Tamao oxidation, without any loss in enantiomeric excess (ee).
2.2 Ir-catalyzed asymmetric hydrogenation of vinyl silanes
In 2006, Andersson and colleagues made a significant contribution to catalytic asymmetric hydrogenation by developing an iridium-catalyzed asymmetric hydrogenation of vinyl silanes, providing an efficient method for synthesizing various optically active silanes (Scheme 4a)[61]. The researchers demonstrated that an iridium thiazole complex (C1) was highly effective for this transformation, offering a versatile catalytic system compatible with a broad range of vinyl silane substrates. This system achieved excellent conversions for the corresponding optically active silanes, though enantioselectivities varied substantially, from poor to excellent. Notably, the β-stereogenic silane (P3a) was obtained with full conversion and a remarkable enantiomeric excess (ee) of 98%. In contrast, α-stereogenic silanes (P3b-P3d) were produced with lower enantioselectivities, ranging from 28% to 58% ee.
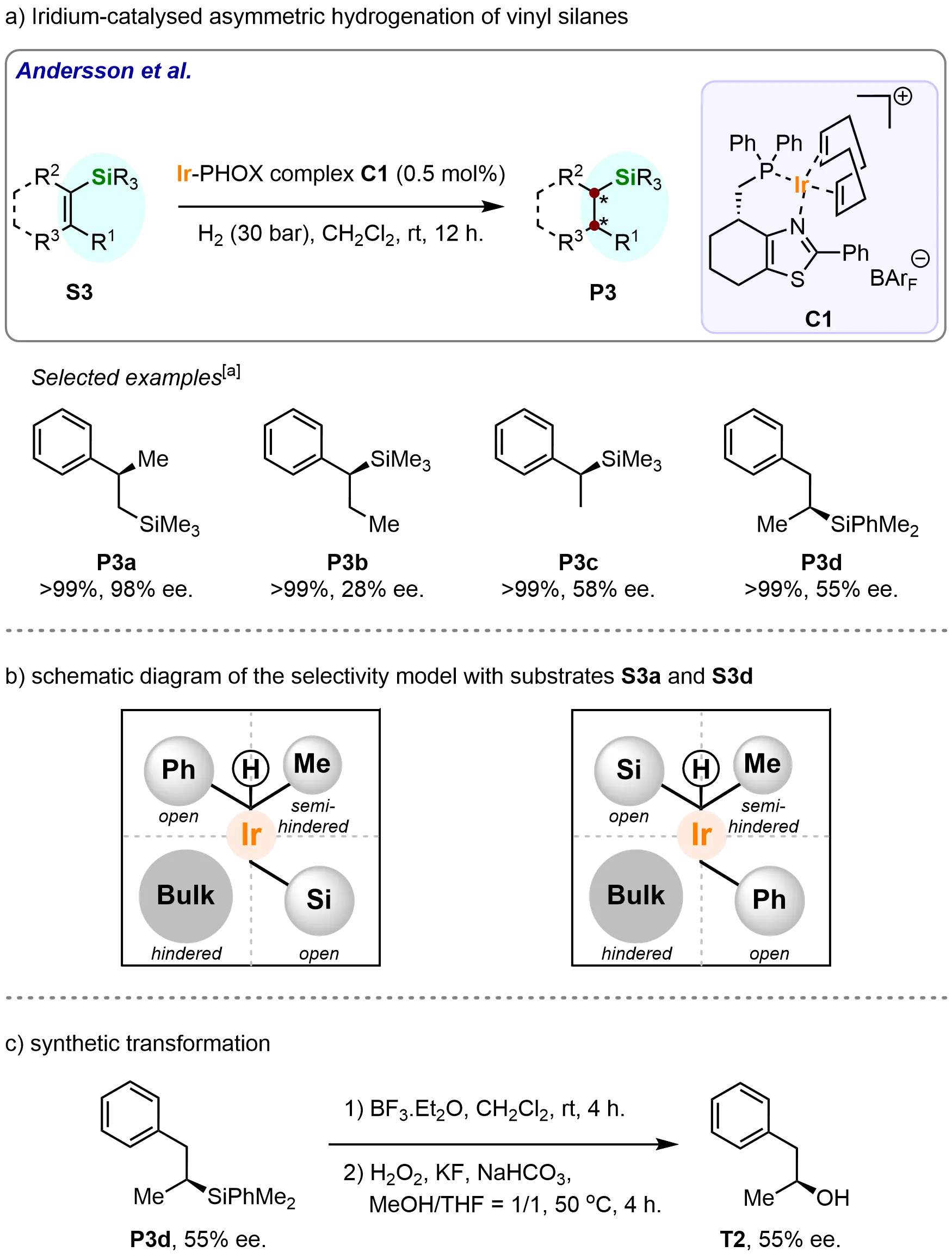
Scheme 4. Ir-catalyzed asymmetric hydrogenation of vinyl silanes. Created in ChemDraw. [a]: Conversions were recorded.
To further understand the observed selectivities, the researchers applied their previously proposed selectivity model (Scheme 4b), which rationalizes the stereochemical outcome of the reaction. According to this model, the substrate arrangement critically influences enantioselectivity. Specifically, when the vinylic proton is positioned in the hindered quadrant while the bulky silyl and phenyl groups occupy the more accessible open quadrants, the model correctly predicted the stereochemical outcome for all substrates with known absolute configurations. This rationalization not only provided mechanistic insight but also underscored the significance of substrate orientation in achieving high enantioselectivity.
Oxidative cleavage of the dimethylphenylsilyl group, carried out using a modified version of the original Fleming-Tamao oxidation, afforded compound T2 while maintaining its enantiomeric excess (ee) (Scheme 4c). This result further demonstrates the synthetic utility of the developed catalytic methodology.
In 2010, Mazuela et al. achieved a significant breakthrough in asymmetric catalysis by successfully employing a biaryl phosphite-thiazole ligand (L4) in the iridium-catalyzed asymmetric hydrogenation of β-branched vinyl silane (S4) (Scheme 5a)[62]. This catalytic system demonstrated exceptional performance, delivering the desired β-stereogenic silane ((R)-P4) with complete conversion and excellent enantioselectivity (98% ee). This outstanding performance was attributed to the distinctive properties of the phosphite-thiazole ligand, which provides optimal steric and electronic environments for the iridium catalyst.
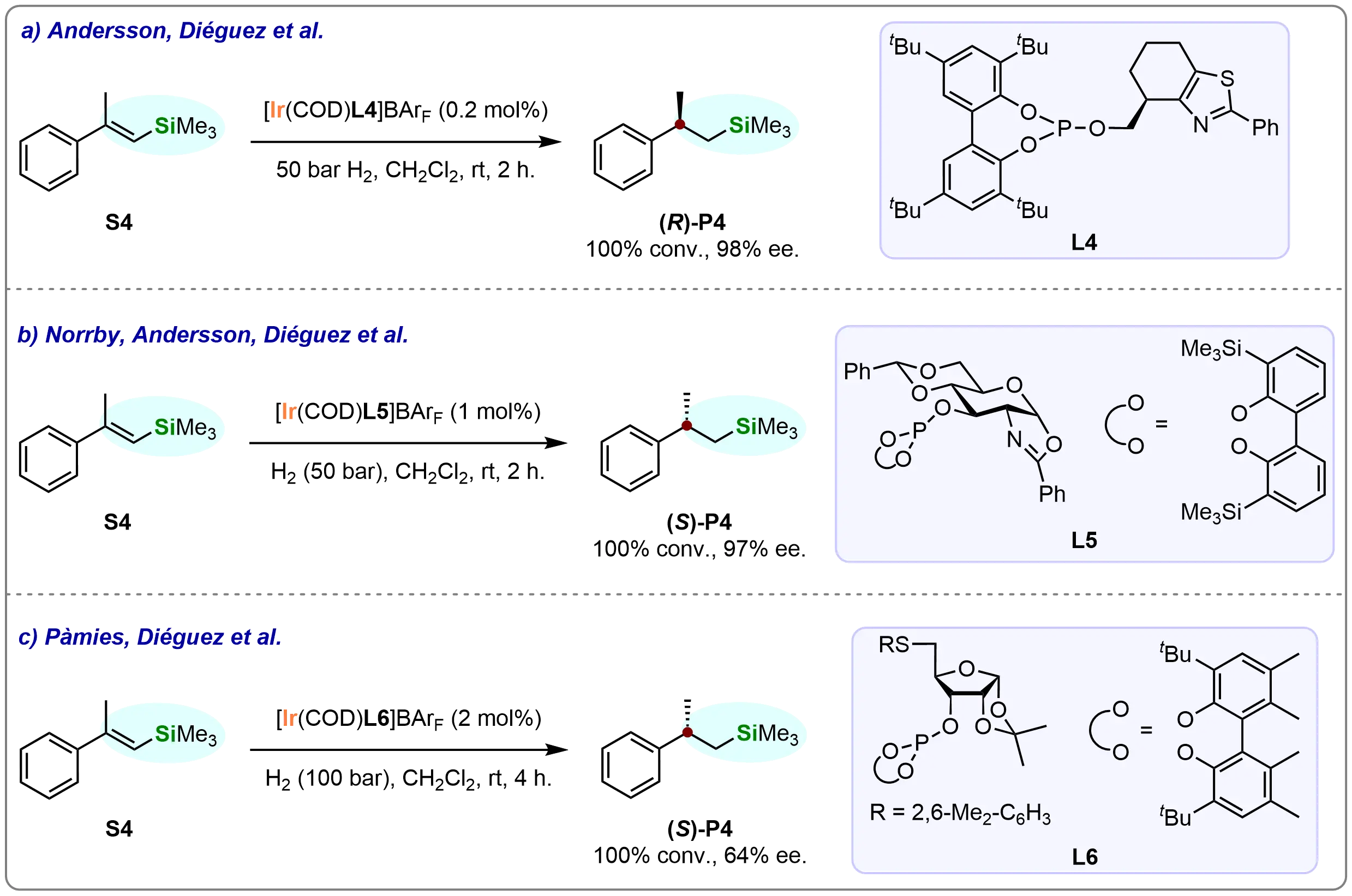
Scheme 5. Ir-catalyzed asymmetric hydrogenation of β-branched vinyl silane (S4) via virous ligands. Created in ChemDraw.
In 2011, Mazuela et al. advanced their pioneering work in asymmetric catalysis through the development of novel chiral ligands for the enantioselective iridium-catalyzed hydrogenation of minimally functionalized olefins (Scheme 5b)[63]. A key achievement was the successful design of the pyranoside phosphite-oxazoline ligand (L5), which elegantly combines the advantageous features of phosphite and sugar moieties. This ligand is not only readily accessible from inexpensive feedstocks but also exhibits high resistance to oxidation, along with a straightforward, modular construction that facilitates its widespread use in catalysis. These attributes establish L5 as an optimal ligand for asymmetric hydrogenation reactions requiring both robustness and high efficiency.
When the pyranoside phosphite-oxazoline ligand (L5) was employed in the iridium-catalyzed asymmetric hydrogenation of β-branched vinyl silane (S4), the desired β-stereogenic silane ((S)-P4) was obtained in full conversion with excellent enantioselectivity (97% ee) (Scheme 5b). This remarkable result demonstrates the high efficacy of L5 in enhancing both the rate and selectivity of the hydrogenation process, further advancing the utility of iridium-based catalysts in the synthesis of chiral organosilicon compounds.
Complementing experimental studies, the Mazuela et al.[63] conducted density functional theory (DFT) calculations to elucidate the mechanism of the iridium-catalyzed olefin hydrogenation. The computational results revealed an Ir(III/V) catalytic cycle with hydride migratory insertion as the stereodetermining step (Figure 4). This mechanistic understanding not only accounts for the observed enantioselectivity but also provides fundamental insights into how ligand structure governs asymmetric hydrogenation outcomes. These findings establish a framework for designing improved catalysts and ligands with enhanced selectivity and efficiency.
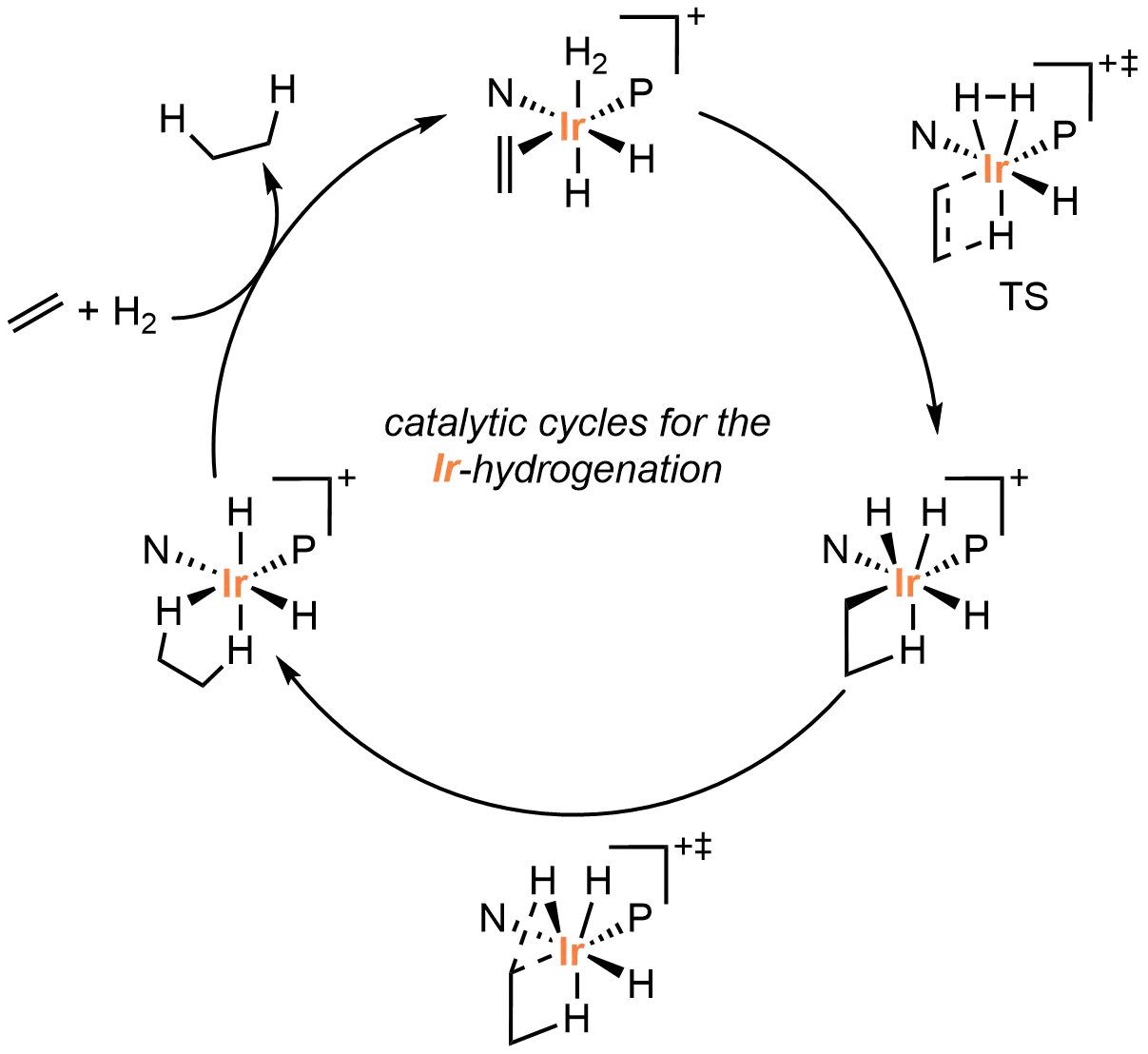
Figure 4. Computational study for the mechanism of Ir-catalyzed asymmetric hydrogenation by Norrby, Andersson, Diéguez, and co-workers. Created in ChemDraw.
In 2013, Coll et al. extended their research by developing a novel furanoside thioether-phosphite ligand (L6) for iridium-catalyzed asymmetric hydrogenation of minimally functionalized olefins[64]. The ligand was efficiently synthesized from commercially available D-(+)-xylose, offering practical advantages for scale-up due to its accessible precursor and cost efficiency. The furanoside thioether-phosphite architecture combines beneficial features of both structural motifs, creating an optimized framework for asymmetric hydrogenation catalysis.
The furanoside thioether-phosphite ligand (L6) achieved complete conversion in the iridium-catalyzed asymmetric hydrogenation of β-branched vinyl silane (S4), yielding the β-stereogenic silane product ((S)-P4). While the reaction proceeded quantitatively, the enantioselectivity remained moderate (Scheme 5c). Although lower than previously reported systems, these results establish L6 as a practical ligand for asymmetric hydrogenations where moderate enantioselectivity suffices or where further optimization may be pursued.
In 2016, Ma et al. reported a significant advance in asymmetric catalysis through N,P-ligated iridium-catalyzed asymmetric hydrogenation of diverse 1-silyl-1-substituted alkenes[65]. This catalytic system demonstrated broad substrate scope, producing different optically active silanes in good to excellent yields (74%-99%) and enantioselectivities (66%-99% ee) (Scheme 6). Notably, 1-TMS-1-phenyl ethylene (S5a) was hydrogenated on a 1.0 mmol scale to afford (P5a) in 86% yield with 99% ee. Similarly, the substrate 1-TMS-1-para-methylphenyl ethylene (S5b) was well-tolerated under the optimized reaction conditions, delivering the desired optically pure silane (P5b) in 83% yield with 99% ee. The naphthalene-substituted silane (P5c) was obtained in 99% yield and 97% ee, further highlighting the system’s capability to handle sterically demanding aromatic substrates effectively. However, the hydrogenation of alkyl-substituted substrates (S5d and S5e) revealed some limitations of the catalytic system. These substrates required longer reaction times to achieve full conversion, suggesting lower reactivity under standard conditions. For the triethylsilyl (TES) substituted substrate (S5e), the reaction necessitated the use of 5 mol% catalyst and higher temperatures, ultimately yielding the corresponding product with reduced enantioselectivity (66% ee). Similarly, the trimethoxylsilyl-substituted alkene (S5f) was also tolerated by the catalytic system but afforded the product (P5f) with moderate yield (74%) and enantioselectivity (78% ee).
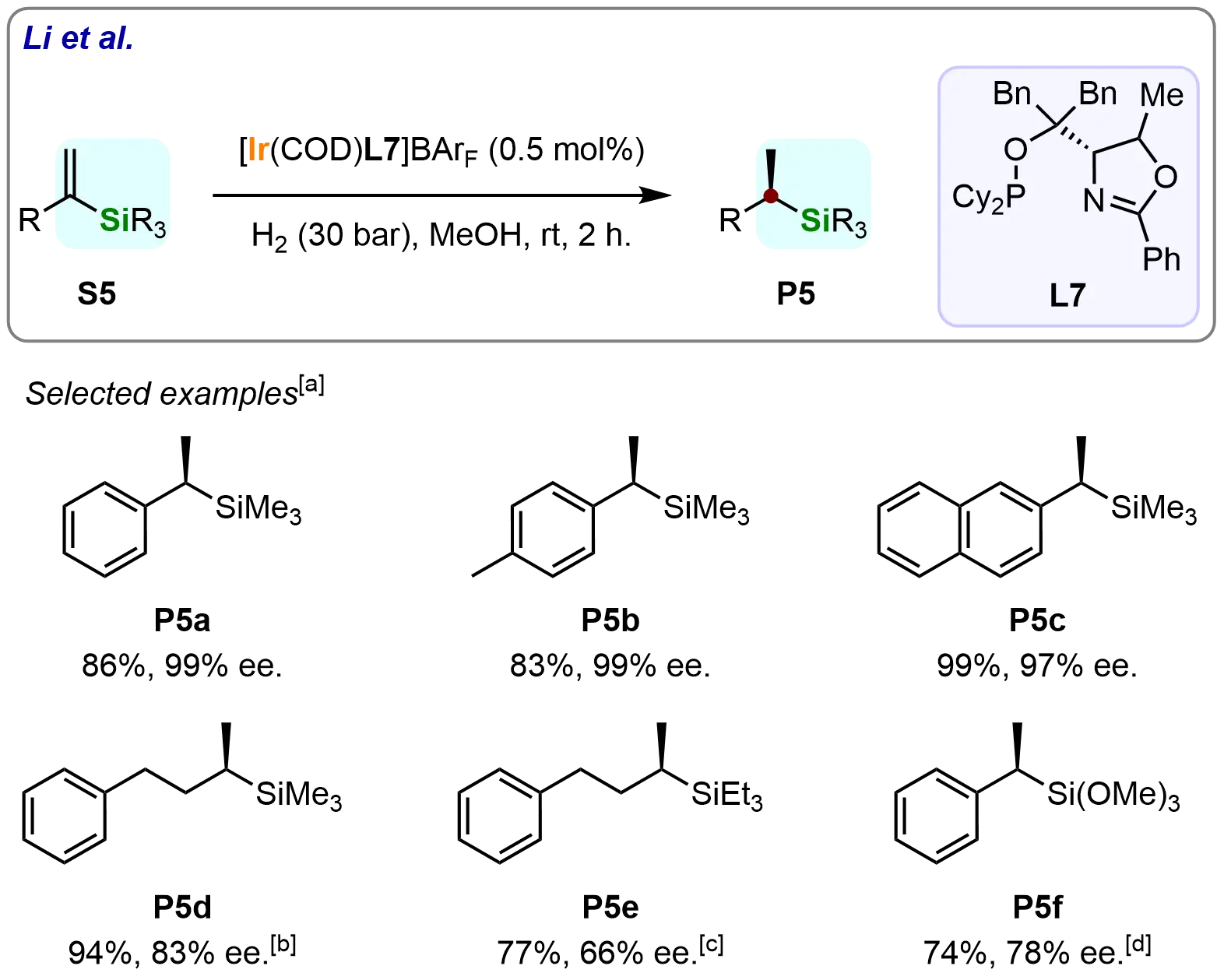
Scheme 6. Ir-catalyzed asymmetric hydrogenation of 1-silyl-1-substituted alkenes. Created in ChemDraw. [a]: Isolated yield was recorded; [b]: Reaction was performed for 20 h; [c]: Reaction was performed with [Ir(COD)L7]BArF (5.0 mol%), H2 (45 bar), 50 ℃, 24 h; [d]: Reaction was performed with [Ir(COD)L7]BArF (1.0 mol%).
To expand the substrate scope of asymmetric hydrogenation of vinyl silanes, Wang et al.[66] systematically evaluated a library of chiral N,P-ligated iridium catalysts. These complexes facilitated highly enantioselective hydrogenation of diverse vinyl silanes, including trisubstituted and terminal disubstituted alkenes with aryl, alkyl, ethoxycarbonyl, and hydroxymethyl substituents. This study represents a major advance in catalytic systems for selective vinyl silane hydrogenation, offering a general strategy to access chiral silanes with broad functional group tolerance.
The researchers first employed pyridine-based N,P-ligands (L8 and L9) in the asymmetric hydrogenation of β-branched vinyl silanes (Scheme 7)[66]. Notably, substrates containing non-aromatic substituents, such as (S6b) and (S6d), were also well-tolerated under the developed catalytic conditions, yielding the corresponding β-stereogenic silanes (P6b and P6d) in full conversion with excellent enantioselectivities (95-96% ee). This outcome demonstrates the robustness of the catalytic system and its ability to accommodate substrates with diverse electronic and steric properties, thereby broadening the substrate scope for asymmetric hydrogenation reactions involving vinyl silanes.
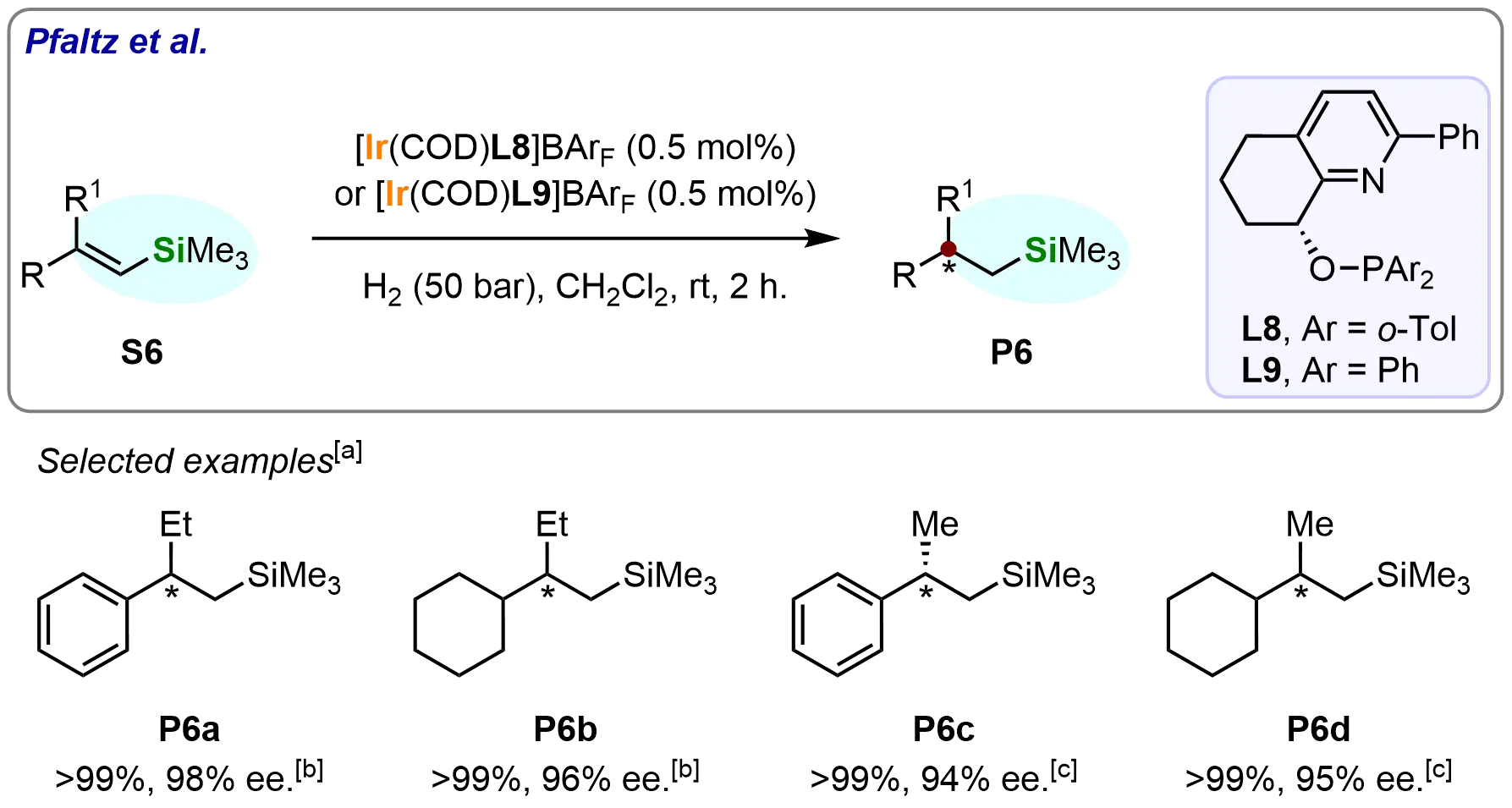
Scheme 7. Iridium-catalyzed asymmetric hydrogenation of vinyl silanes with trisubstituted C=C bonds. Created in ChemDraw. [a]: Conversions were recorded; [b]: Reaction was performed with L8; [c]: Reaction was performed with L9.
Building on these results, the team further investigated iridium-catalyzed asymmetric hydrogenation of terminal vinyl silanes using a pyridine-phosphinite ligand (L10) (Scheme 8)[66]. α-Trimethylsilylstyrene (S7a) converted to the α-stereogenic silane (P7a) with complete conversion and 88% ee. The system also accommodated cyclohexyl-substituted substrates (S7b and S7c), yielding chiral silanes with 90-91% ee. These results demonstrate the catalytic system’s versatility in hydrogenating diverse vinyl silanes across varying steric environments.
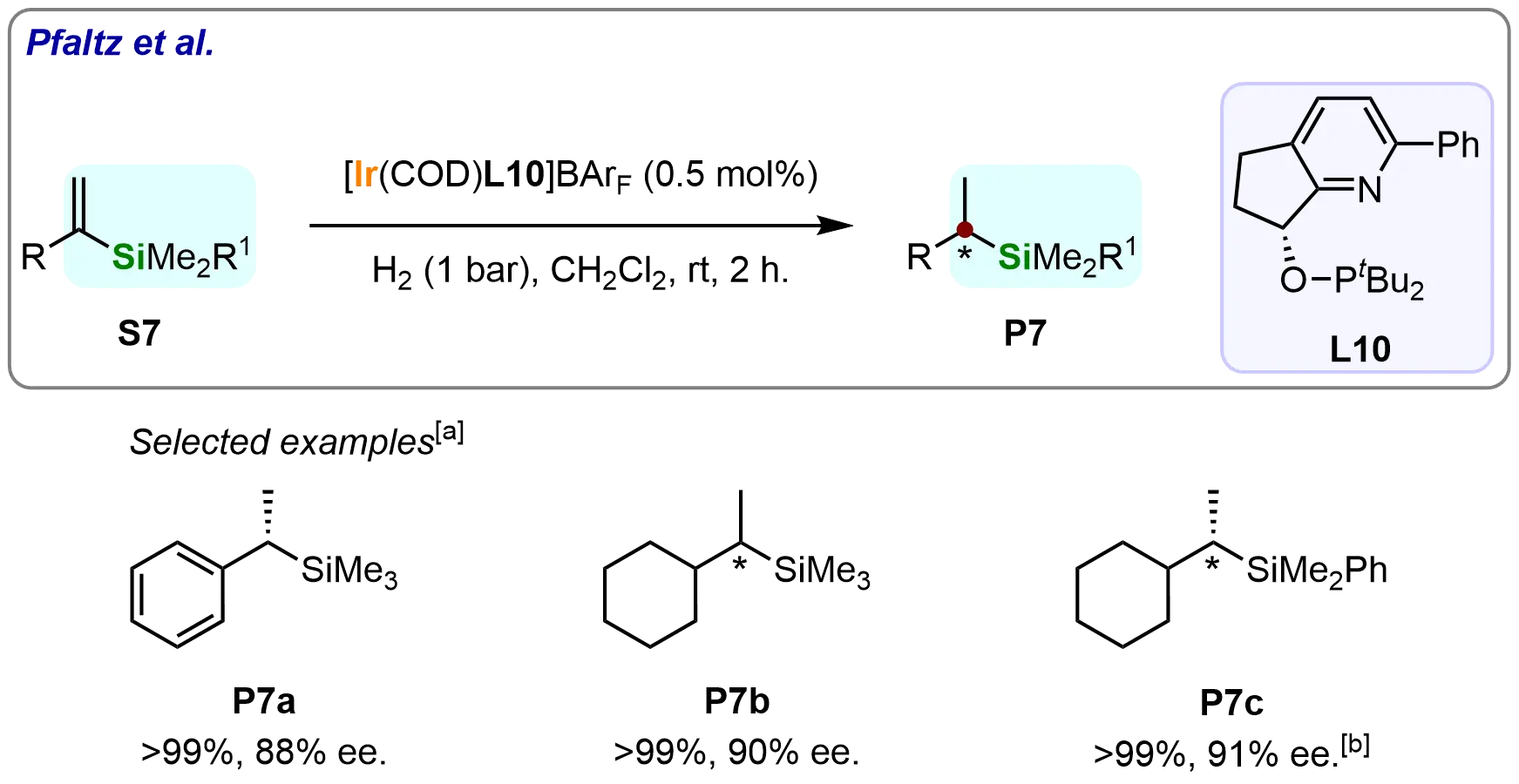
Scheme 8. Iridium-catalyzed asymmetric hydrogenation of terminal vinyl silanes. Created in ChemDraw. [a]: Conversions were recorded; [b]: Reaction was performed with H2 (5 bar).
Furthermore, Pfaltz et al. extended the methodology to α-alkyl substituted trialkoxy(vinyl)silanes using PHOX ligand (L11) and its simplified derivative (L12) in iridium-catalyzed asymmetric hydrogenation (Scheme 9)[66]. The system efficiently converted various α-alkyl substrates (S8) to α-stereogenic silanes (P8) with complete conversion and 70-88% ee. This successful application to complex alkyl-substituted trialkoxy(vinyl)silanes further establishes the broad utility of the iridium catalytic system for diverse chiral silane synthesis.
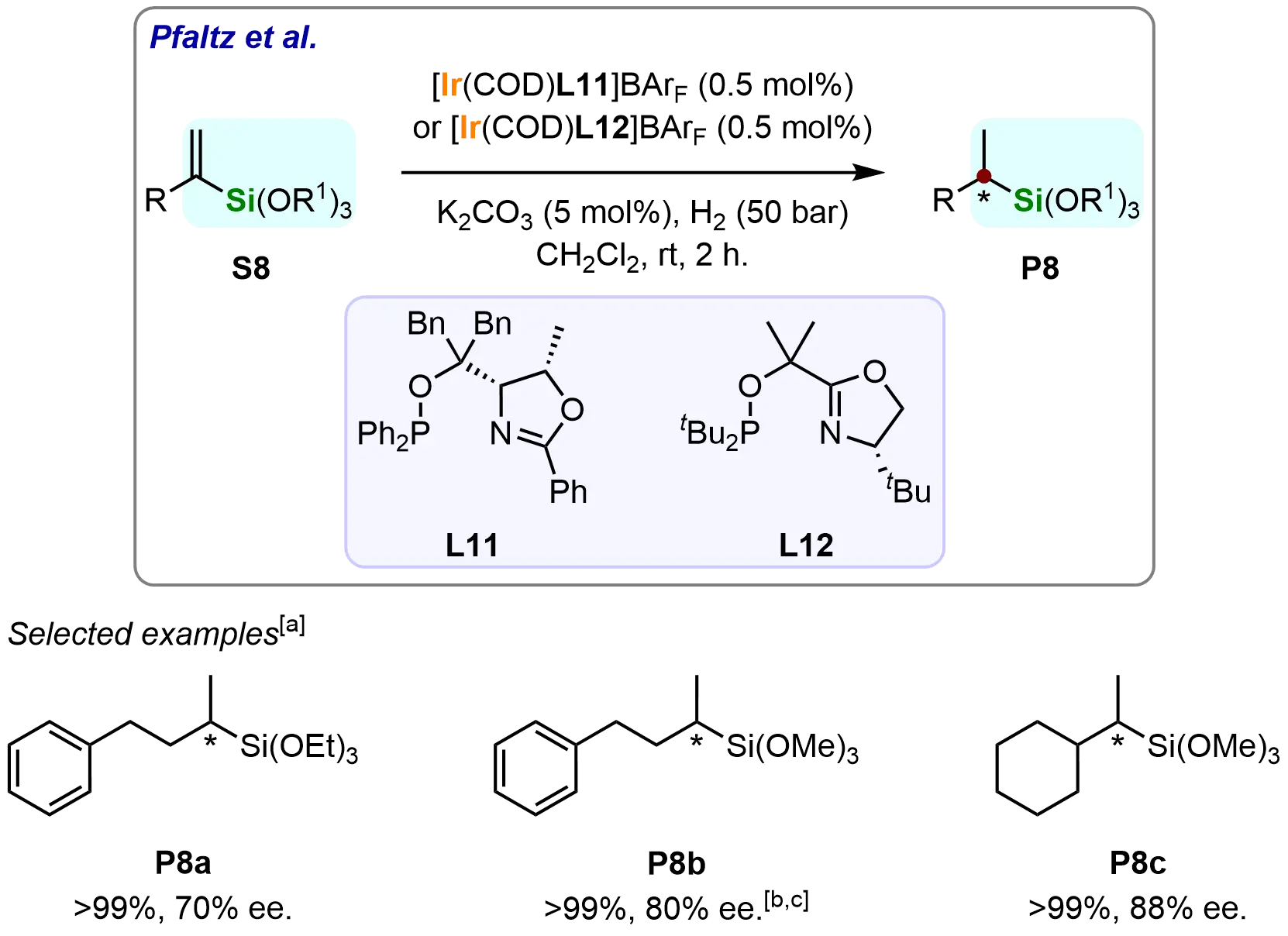
Scheme 9. Iridium-catalyzed asymmetric hydrogenation of trialkoxy(vinyl)silanes. Created in ChemDraw. [a]: Conversions were recorded; [b]: Reaction was performed with L12; [c]: Reaction was performed in the absence of K2CO3, for 16 h.
The researchers further evaluated challenging substrates under optimized conditions (Scheme 10)[66]. Vinyl siletane was hydrogenated using the Ir-PHOX complex (L13), affording product (P9) in 93% conversion and 83% ee. The system also accommodated functionalized vinyl silanes, including α,β-unsaturated esters (S10), which were reduced with 100% conversion and 89% ee using pyridine-phosphinite ligand (L14). Similarly, an allylic alcohol substrate (S11) achieved complete conversion with 86% ee when catalyzed by a Neo-PHOX-derived complex (L15). These results demonstrate the Ir-PHOX system’s versatility in processing diverse functionalized substrates.
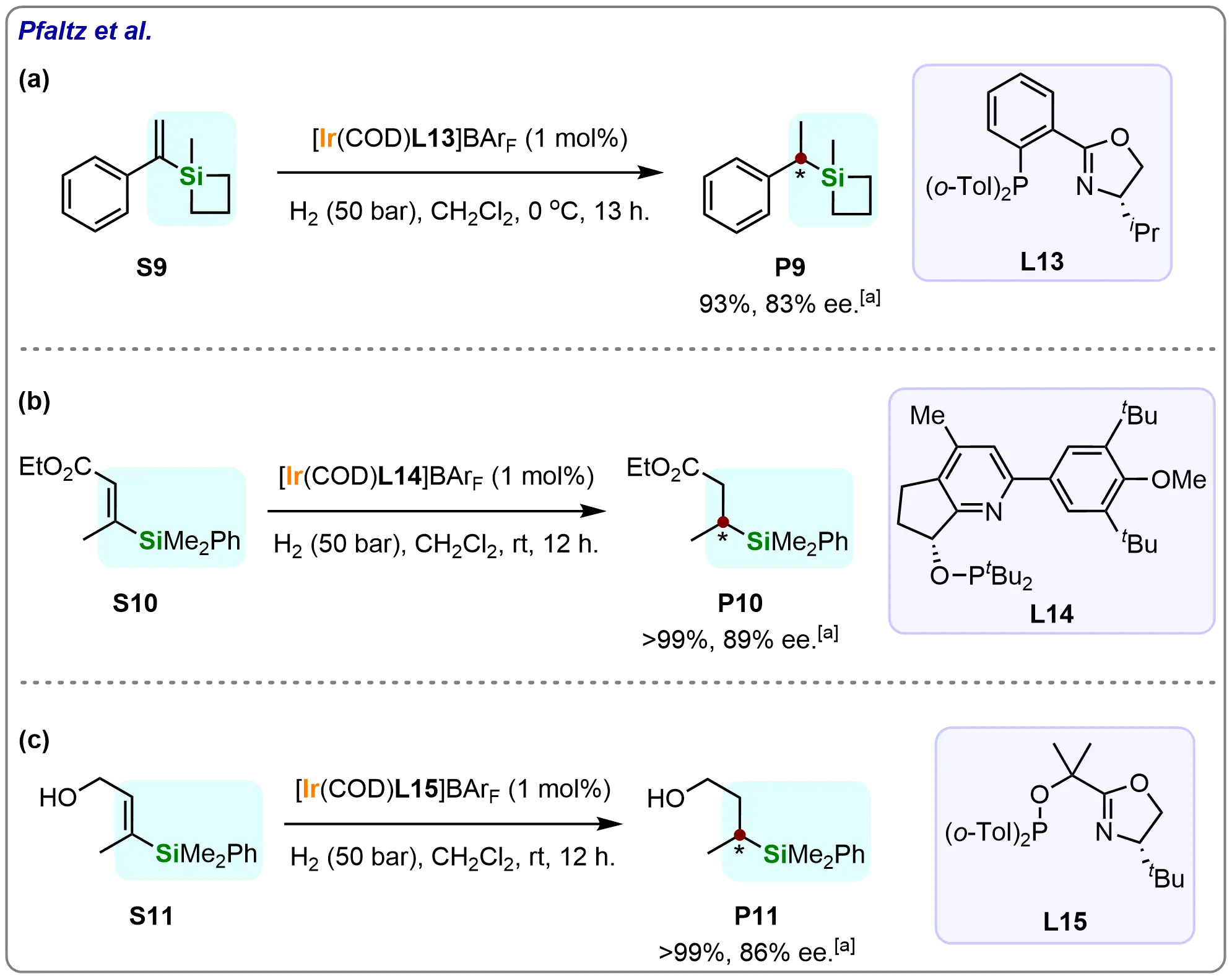
Scheme 10. Iridium-catalyzed asymmetric hydrogenation of several challenging substrates. Created in ChemDraw. [a]: Conversions were recorded.
In short, Wang et al.[66] work constitutes a significant advance in asymmetric catalysis, specifically for vinyl silane hydrogenation. The developed catalytic system combines broad substrate scope, including challenging functional groups, with high enantioselectivity, establishing its utility for chiral silane synthesis. This methodology’s capacity to deliver precise stereocontrol across diverse substrates renders it particularly valuable for constructing complex chiral silanes.
2.3 Co-catalyzed asymmetric hydrogenation of vinyl silanes
In 2017, Guo et al. developed a cobalt-catalyzed sequential hydrosilylation/hydrogenation of alkynes that proceeds with high regio- and enantioselectivity (Scheme 11)[67]. This methodology employs commercially available alkynes, silanes, and H2 to efficiently construct chiral silanes. The reaction exhibited broad substrate scope, delivering products in 74-95% yield with 85% to > 99% ee. The cobalt precatalyst (C2) proved crucial for achieving high enantioselectivity. Notably, the system accommodated 5-indolyl groups, yielding (P13f) with 85% yield and 91% ee. The protocol was further applicable to alkynes in bioactive scaffolds (P13i and P13j), demonstrating late-stage functionalization potential (Scheme 11a). Ketones, alkenes, and ethers remained intact under the reaction conditions, highlighting the method’s functional group tolerance.
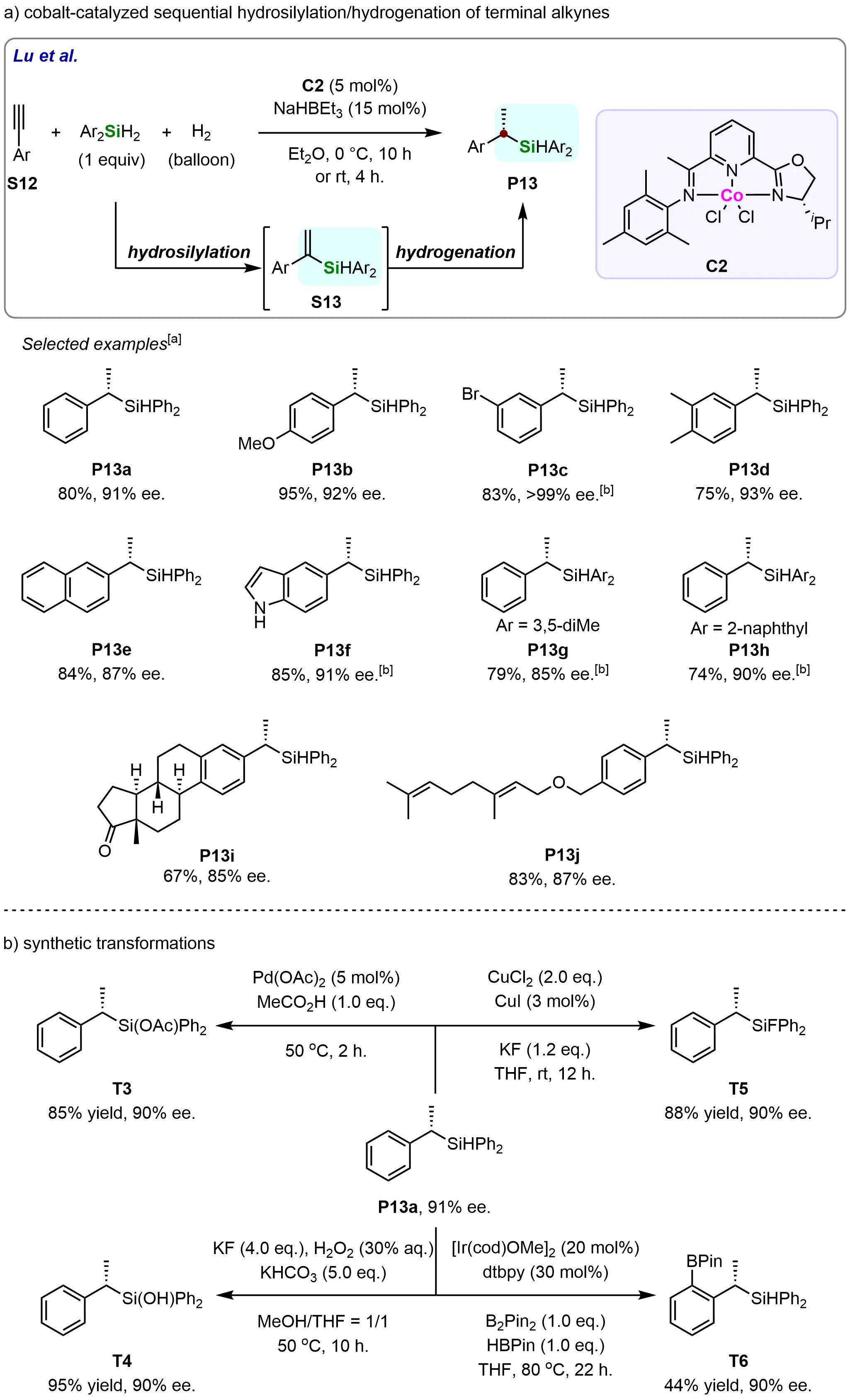
Scheme 11. Regio- and enantioselective cobalt-catalyzed sequential hydrosilylation/hydrogenation of terminal alkynes. Created in ChemDraw. [a]: Isolated yield was recorded; [b]: Reaction was performed with 1 mmol scale, Et2O (0.5 M), rt, 4 h.
To further demonstrate the versatility and synthetic utility of the developed methodology, several downstream transformations were performed by the researchers (Scheme 11b). The chiral silane P13a was successfully converted into a series of valuable derivatives, including silyl esters (T3), silanols (T4), silyl fluorides (T5), and C–H borylation products (T6), under appropriate conditions while largely preserving the enantiomeric excess (ee) values.
The proposed mechanism for the cobalt-catalyzed hydrogenation of vinyl silanes is illustrated in Figure 5. The reaction begins with the coordination of the vinyl silane (S13) to the cobalt species (A), followed by alkene insertion into the cobalt-hydrogen bond to form cobalt-carbon intermediates (C and D). Both intermediates are capable of reacting with H₂ to yield the desired product and regenerate the cobalt-hydride species. β-Hydrogen elimination from intermediate D may lead to the formation of species E, which can undergo rapid insertion of the C=Si double bond, generating the cobalt silyl species (F). This species (F) is then quenched by H₂ to release the final product (P13) and regenerate the cobalt-hydride species (A). Importantly, most of the reaction steps are reversible, providing a dynamic catalytic cycle. While this mechanistic framework rationalizes the reaction pathway, further studies are required to fully elucidate the enantioselective process. This cobalt-catalyzed methodology constitutes a significant advance in the asymmetric synthesis of chiral silanes, combining broad functional group tolerance with operational simplicity—attributes particularly valuable for complex molecule synthesis.
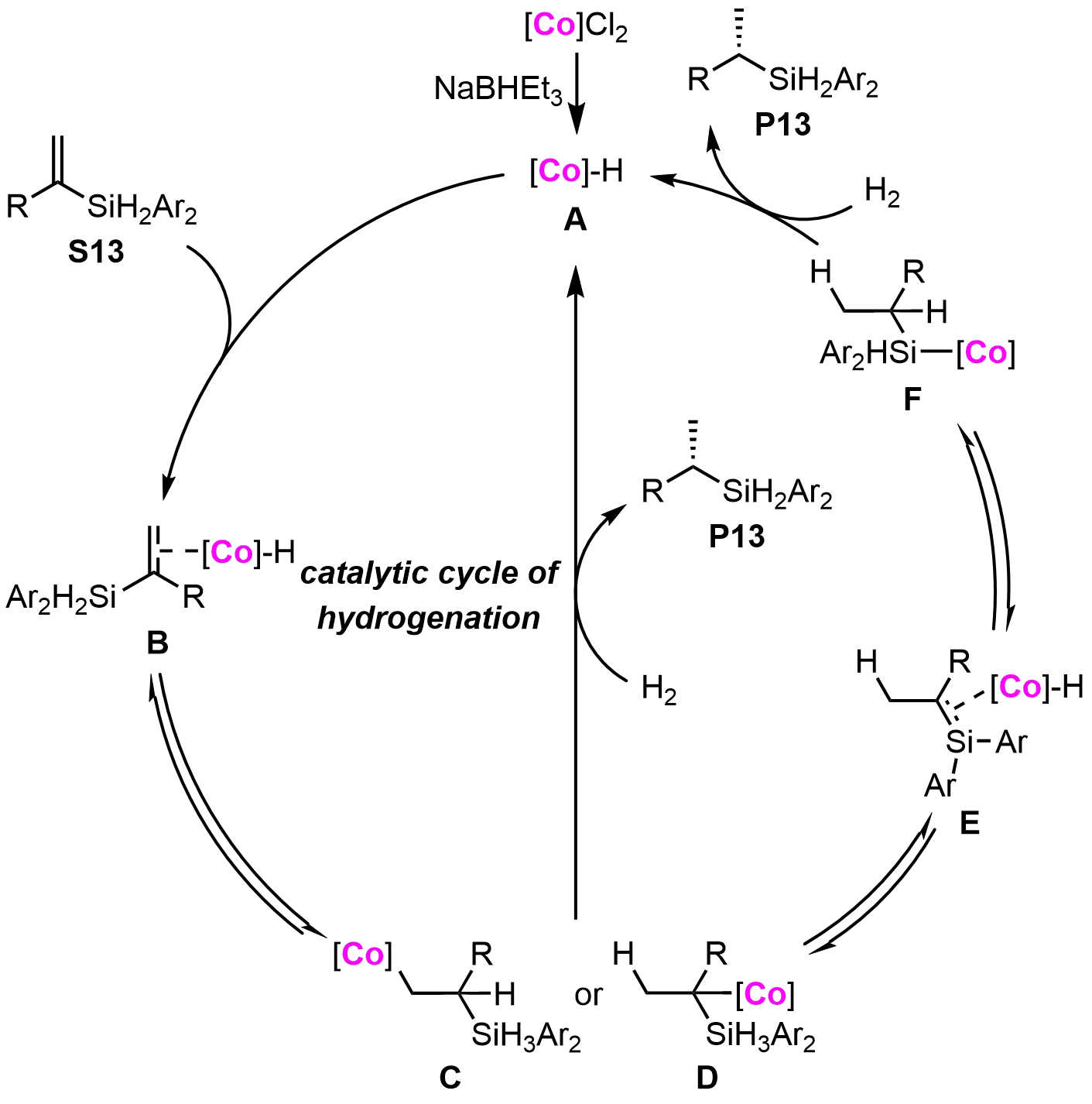
Figure 5. Proposed mechanism of co-catalyzed hydrogenation reported by Lu and co-workers. Created in ChemDraw.
In 2019, Zuo et al. developed a novel C1-symmetric phosphine-pyridine-oxazoline cobalt complex, which was successfully applied in the asymmetric hydrogenation of vinyl silanes (Scheme 12)[68]. This new catalyst system demonstrated significant efficiency in the hydrogenation of a wide variety of vinyl silanes (S14), providing the corresponding tertiary silanes (P14) in high yields and with enantioselectivities ranging from moderate to excellent. Notably, substrates bearing both electron-donating and electron-withdrawing substituents on the vinyl group were effectively hydrogenated, affording the desired chiral products. The versatility of this catalytic system highlights its potential for fine-tuning the stereochemical outcomes of hydrogenation reactions involving vinyl silanes, making it a promising method for the synthesis of chiral silanes in synthetic and pharmaceutical chemistry (Scheme 12a). The C1-symmetric nature of the phosphine-pyridine-oxazoline ligand imparts excellent stereochemical control to the reaction, facilitating high enantioselectivities even with a broad range of functionalized vinyl silanes. This development marks an important step in the design of more efficient and tunable catalytic systems for asymmetric hydrogenation, as it offers a valuable alternative to existing methods that may be limited in substrate scope or enantioselectivity.
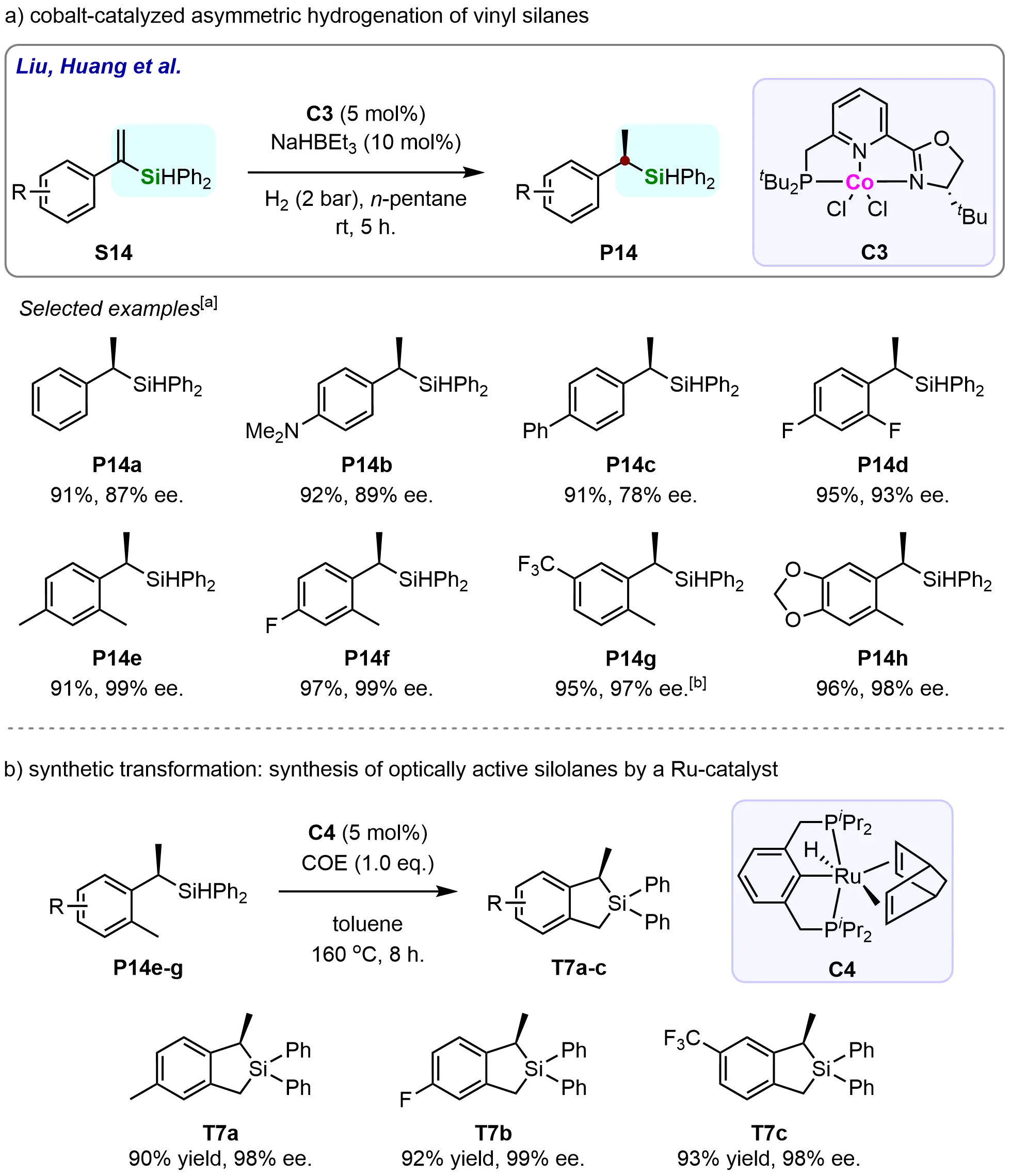
Scheme 12. Cobalt-catalyzed asymmetric hydrogenation of vinyl silanes with a phosphine-pyridine-oxazoline ligand. Created in ChemDraw. [a]: Isolated yield was recorded; [b]: Reaction was performed with H2 (4 bar).
To further demonstrate the synthetic utility of the developed methodology, the researchers achieved an intramolecular dehydrosilylation of o-methyl-substituted chiral benzylsilanes catalyzed by a pincer-type ruthenium complex (C4). This transformation efficiently afforded several five-membered silolanes (T7a-c) with excellent enantiopurity (98-99% ee) (Scheme 12b). Considering the scarcity of methods for constructing optically active silicon-containing heterocycles, the combination of Co-catalyzed asymmetric hydrogenation of α-vinylsilanes with the Ru-catalyzed dehydrosilylation represents a highly promising strategy for the enantioselective synthesis of silacyclic compounds.
2.4 Short summary of the asymmetric hydrogenation of vinyl silanes
Remarkable progress has been achieved in the asymmetric hydrogenation of vinyl silanes owing to the extensive efforts of numerous researchers. These advancements include the successful application of di- and trisubstituted vinyl silanes in metal-catalyzed asymmetric hydrogenation, leading to the efficient synthesis of various α- and β-chiral silanes. Moreover, a range of subsequent synthetic transformations has been developed, enabling access to structurally diverse and synthetically valuable compounds (Scheme 13a). Despite these significant achievements, several challenges remain in this field. In particular, the asymmetric hydrogenation of tetrasubstituted and cyclic vinyl silanes to construct complex molecules bearing chiral silicon centers is still yet to be fully realized (Scheme 13b). Furthermore, expanding the scope of catalysis beyond the currently dominant metal systems represents an exciting future direction. The exploration of other transition metals, such as ruthenium (Ru), palladium (Pd), nickel (Ni), manganese (Mn), and copper (Cu), may further enrich the catalytic toolbox for the enantioselective synthesis of chiral silanes.
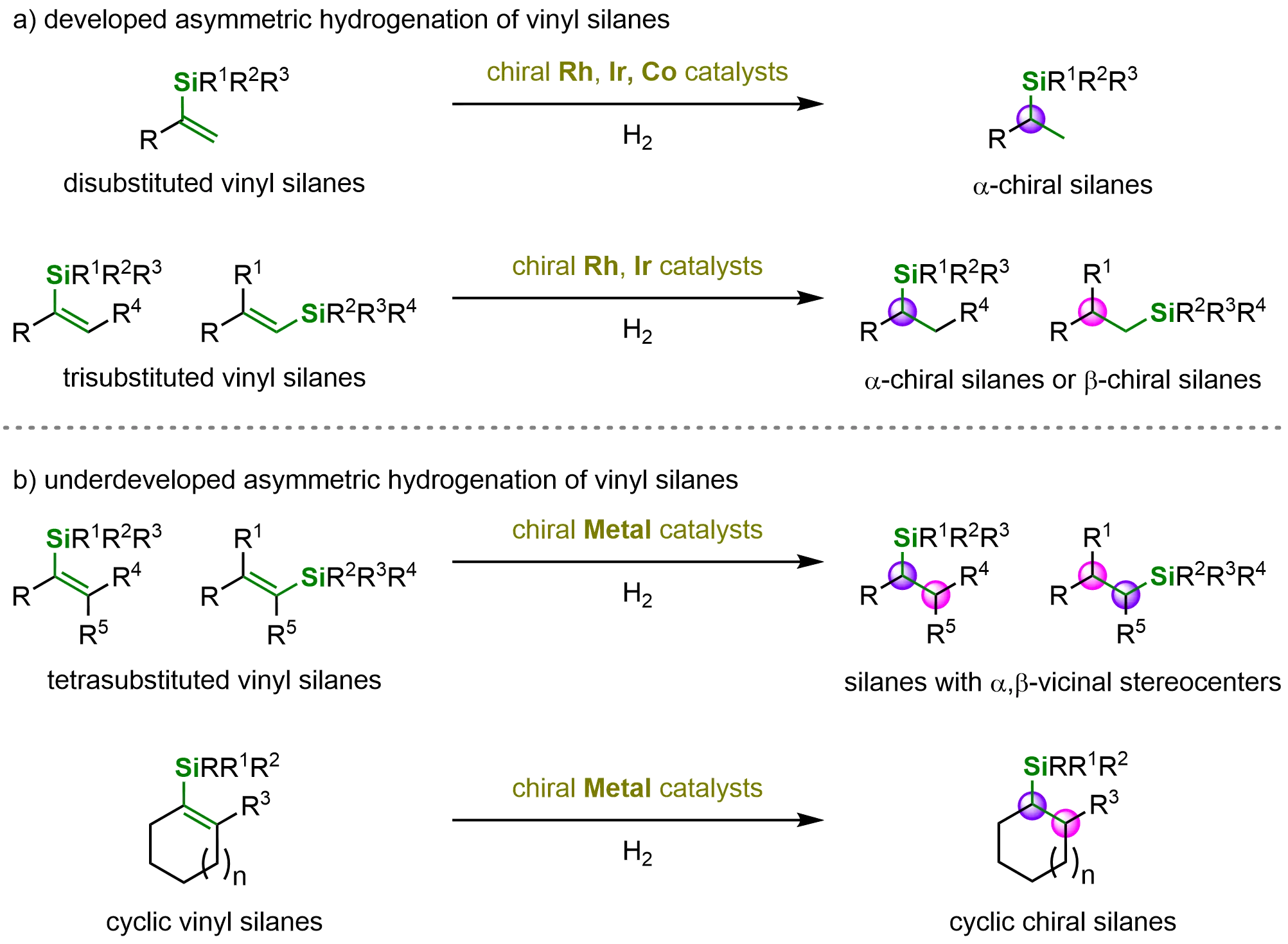
Scheme 13. Developed and underdeveloped metal-catalyzed asymmetric hydrogenation of vinyl silanes. Created in ChemDraw.
3. Asymmetric Hydrogenation of Vinyl Phosphines
Chiral phosphines have attracted considerable attention over the past several decades owing to their broad applications in biological, pharmaceutical, and chemical fields[51,69]. Consequently, a variety of efficient synthetic methodologies have been developed to access these valuable scaffolds. Among them, metal-catalyzed asymmetric hydrogenation (AH) of vinyl phosphine oxides has emerged as a highly efficient and stereoselective strategy. Numerous di- and trisubstituted vinyl phosphine oxides have been successfully employed in asymmetric hydrogenation reactions, utilizing various transition-metal catalysts, including ruthenium (Ru), rhodium (Rh), iridium (Ir), and nickel (Ni) (Scheme 14a)[26,37].
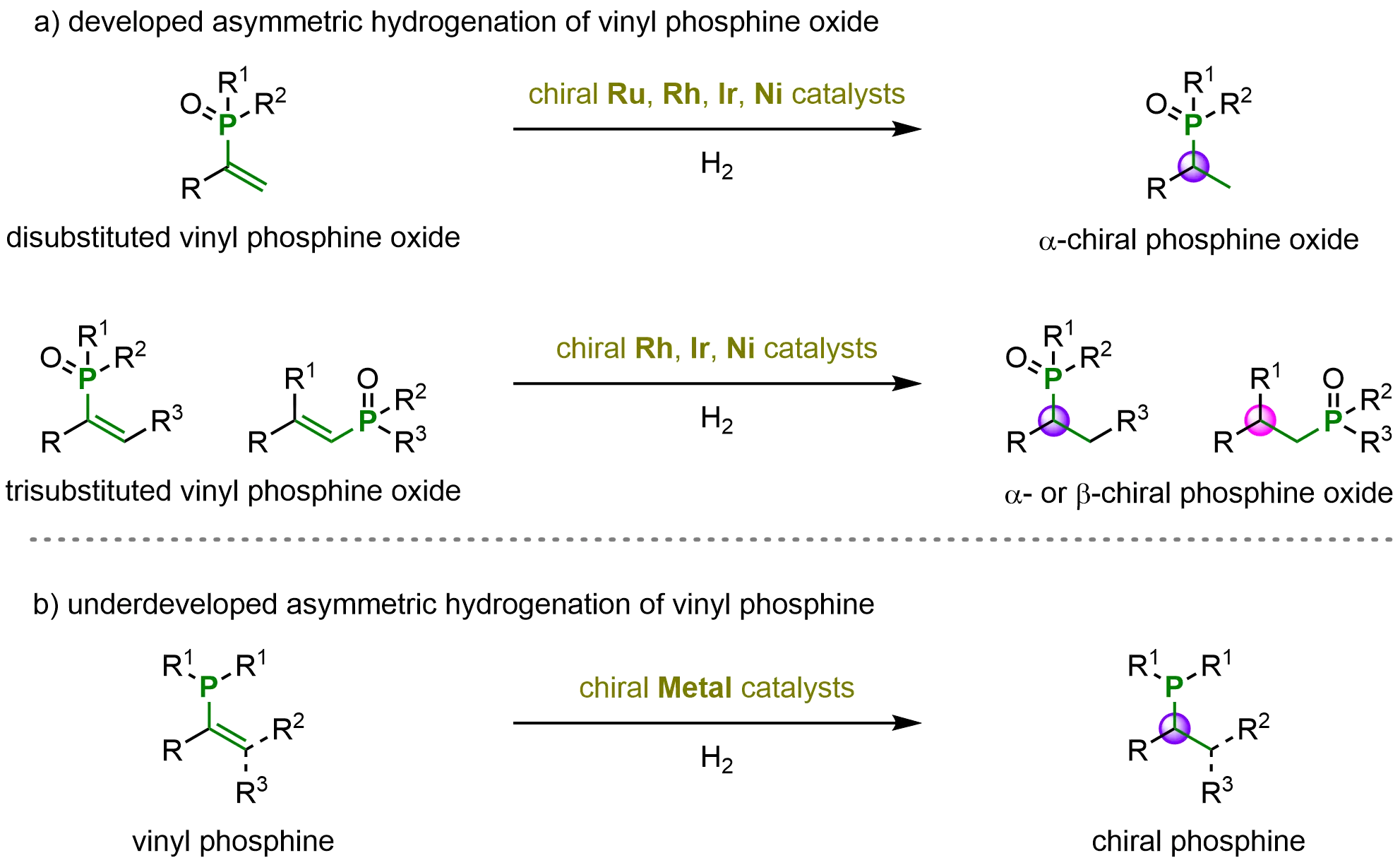
Scheme 14. Current research status of the asymmetric hydrogenation of vinyl phosphine oxides and vinyl phosphines. Created in ChemDraw.
To the best of our knowledge, in contrast to the well-developed asymmetric hydrogenation of vinyl phosphine oxides, the corresponding asymmetric hydrogenation of vinyl phosphines remains significantly underexplored (Scheme 14b). The strong p-π conjugation in vinyl phosphines, together with their potential phosphine-metal coordination, can interfere with the normal binding mode of the vinyl group to the catalyst. Such coordination effects greatly complicate both the activation of the vinyl moiety and the subsequent hydrogenation process. By contrast, in vinyl phosphine oxides, the oxygen atom can effectively coordinate with the metal center, facilitating catalysis. Moreover, the phosphoryl group, acting as a strong electron-withdrawing substituent, reduces the electron density of the alkene, thereby enhancing its reactivity toward hydrogenation relative to vinyl phosphines (Figure 6).
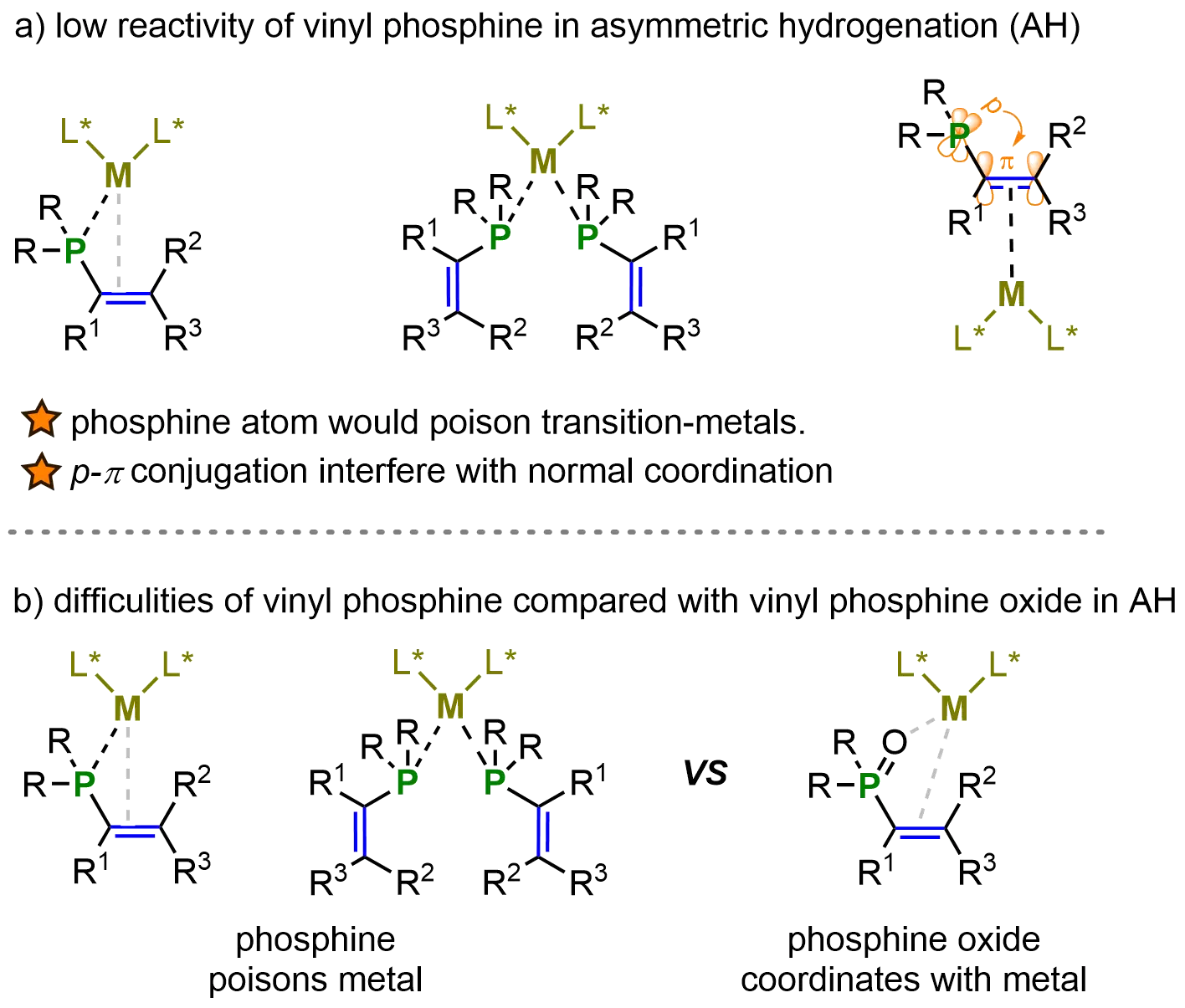
Figure 6. Challenges in the AH of vinyl phosphines. Created in ChemDraw. AH: asymmetric hydrogenation.
Given the higher intrinsic reactivity, lower synthetic complexity, and the extensive studies already reported on the metal-catalyzed asymmetric hydrogenation of vinyl phosphine oxides, this topic will not be discussed in detail in the present review. Instead, it is anticipated that future advances in the asymmetric hydrogenation of vinyl phosphines will enable the direct and efficient construction of chiral phosphine frameworks, thereby expanding the synthetic toolbox for enantioenriched phosphorus-containing molecules.
4. Asymmetric Hydrogenation of Vinyl Sulfides
Optically active chiral organosulfides represent a diverse class of compounds widely distributed in natural products. These molecules play pivotal roles in various scientific disciplines, including organic chemistry, pharmacology, and biochemistry. Owing to their distinct structural properties, chiral organosulfides are highly valued as chiral building blocks, auxiliaries, ligands, organocatalysts, and bioactive molecules, rendering them indispensable in both research and applied sciences[70].
As illustrated in Figure 7, several notable compounds exemplify the utility of chiral organosulfides. Montelukast, a potent cysteinyl leukotriene (cysLT) receptor antagonist, is widely used for asthma treatment. Biotin (vitamin B7 or H), an essential component of the B-vitamin complex, participates in multiple metabolic processes, including fat, carbohydrate, and amino acid metabolism. Dihydrobenzoxanthiin, a novel α-selective estrogen receptor modulator, exhibits significant antagonist activity, while thiazesim demonstrates antidepressant properties, collectively underscoring the therapeutic value of chiral sulfides[71].
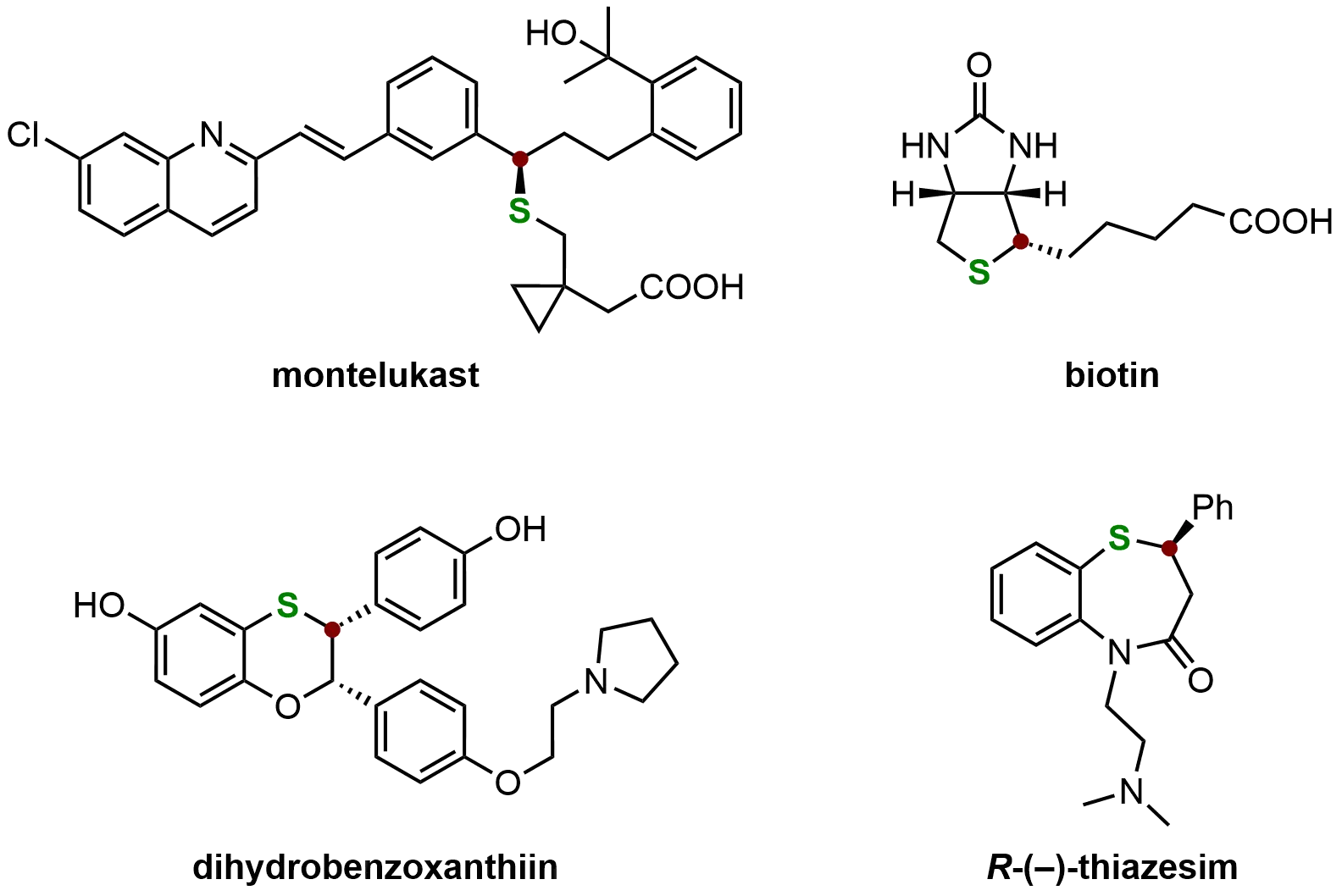
Figure 7. Typical examples of bioactive compounds containing chiral organosulfides. Created in ChemDraw.
Given the biological and synthetic significance of chiral organosulfides, extensive research has focused on developing efficient asymmetric catalytic methods for their synthesis. Transition-metal-catalyzed asymmetric hydrogenation of thioesters has proven particularly effective, offering a direct, atom-economical route to chiral organosulfides, thus highlighting its importance in asymmetric synthesis. However, vinyl sulfide hydrogenation presents unique challenges. The strong p–π conjugation in vinyl sulfides, coupled with potential sulfur-metal coordination, can disrupt normal vinyl-catalyst binding. These interactions may induce unwanted metal insertion into the C–S bond, leading to desulfurized byproducts. Such side reactions significantly complicate both vinyl group activation and the subsequent hydrogenation (Figure 8a).
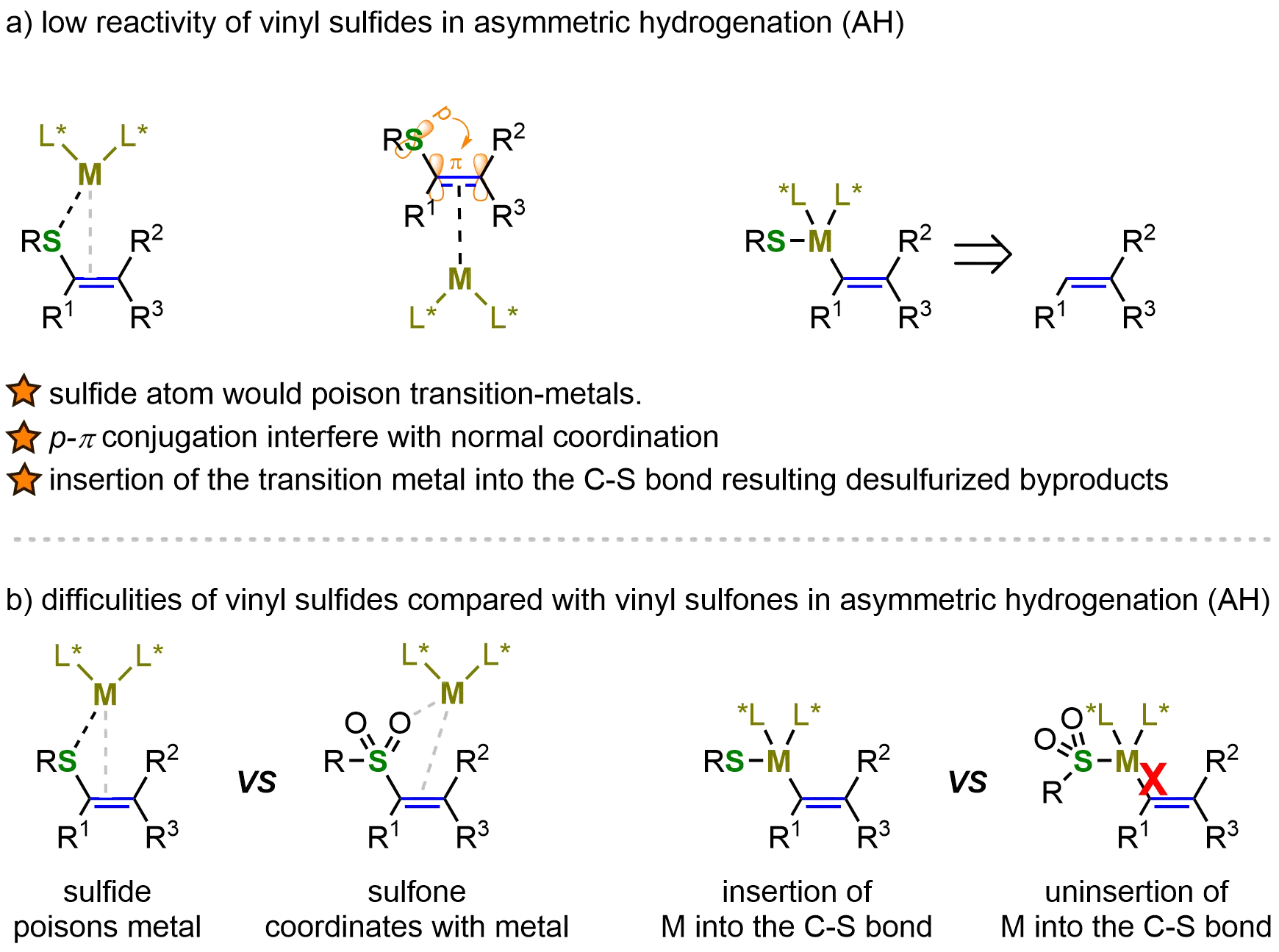
Figure 8. Challenges in the AH of vinyl sulfides: low intrinsic reactivity and greater difficulty compared with vinyl sulfones. Created in ChemDraw. AH: asymmetric hydrogenation.
In contrast, the asymmetric hydrogenation (AH) of vinyl sulfones, another class of sulfur-substituted alkenes, has been extensively developed[72-76]. In metal-catalyzed asymmetric hydrogenation of vinyl sulfones, the sulfone group can coordinate with the metal center, thereby stabilizing the catalytic intermediate. Additionally, as a strong electron-withdrawing group, the sulfone moiety decreases the electron density of the alkene, which enhances its reactivity toward hydrogenation compared with vinyl sulfides. Furthermore, transition-metal insertion into the C–S bond does not occur in vinyl sulfones, preventing the formation of undesired desulfurized byproducts (Figure 8b). Owing to their higher intrinsic reactivity, lower synthetic difficulty, and the wealth of existing studies on metal-catalyzed asymmetric hydrogenation of vinyl sulfones, this topic will not be discussed in detail in the present review. Instead, our focus will be directed toward the asymmetric hydrogenation of vinyl sulfides, which remains considerably more challenging and less explored.
4.1 Ru-catalyzed asymmetric hydrogenation of vinyl sulfides
In 2000, Iseki and co-workers successfully employed a Ru-(R)-biphemp catalyst for the asymmetric hydrogenation of a vinyl sulfide (S15) bearing a phenylthiol moiety at the olefinic carbon. This reaction afforded the desired hydrogenated product (P15) in 83% yield with 89% ee (Scheme 15)[77]. This pioneering work marked a significant advance in asymmetric hydrogenation by demonstrating the feasibility of vinyl sulfide hydrogenation. The phenylthiol substituent provided novel insights into achievable stereocontrol, highlighting ruthenium-catalyzed transformations’ potential for selective vinyl sulfide functionalization. Iseki’s study established foundational principles for vinyl sulfide asymmetric hydrogenation while opening avenues for more efficient syntheses of sulfur-containing chiral compounds.

Scheme 15. Ruthenium-catalyzed asymmetric hydrogenation of vinyl sulfides. Created in ChemDraw.
Glorius and co-workers developed an efficient, highly enantioselective hydrogenation of substituted thiophenes and benzothiophenes using a ruthenium-N-heterocyclic carbene (NHC) catalyst, establishing a novel strategy for synthesizing enantiomerically pure tetrahydrothiophenes and 2,3-dihydrobenzothiophenes[78]. A key achievement of this work is the reaction’s exceptional selectivity, with no observed side reactions (e.g., C–S bond metal insertion, hydrogenolysis, or hydrodesulfurization), demonstrating the catalytic system’s precision and robustness in achieving clean transformations.
To explore the scope of this methodology, the authors initially investigated the hydrogenation of various 2-alkyl substituted benzothiophenes under optimized reaction conditions. The corresponding 2,3-dihydrobenzothiophenes were obtained with exceptional enantioselectivities, as exemplified in Scheme 16. In particular, several 2-alkyl substituted benzothiophenes (S16a-S16d) were successfully hydrogenated, yielding the corresponding products (P16a-P16d) in good to high yields and with outstanding enantioselectivities (ranging from 96% to 98%). While 2-isobutyl- (S16e) and 2-benzylbenzothiophene (S16f) gave modest yields (45-62%), both maintained > 95% ee, confirming the catalyst’s robust stereocontrol across structurally diverse substrates.
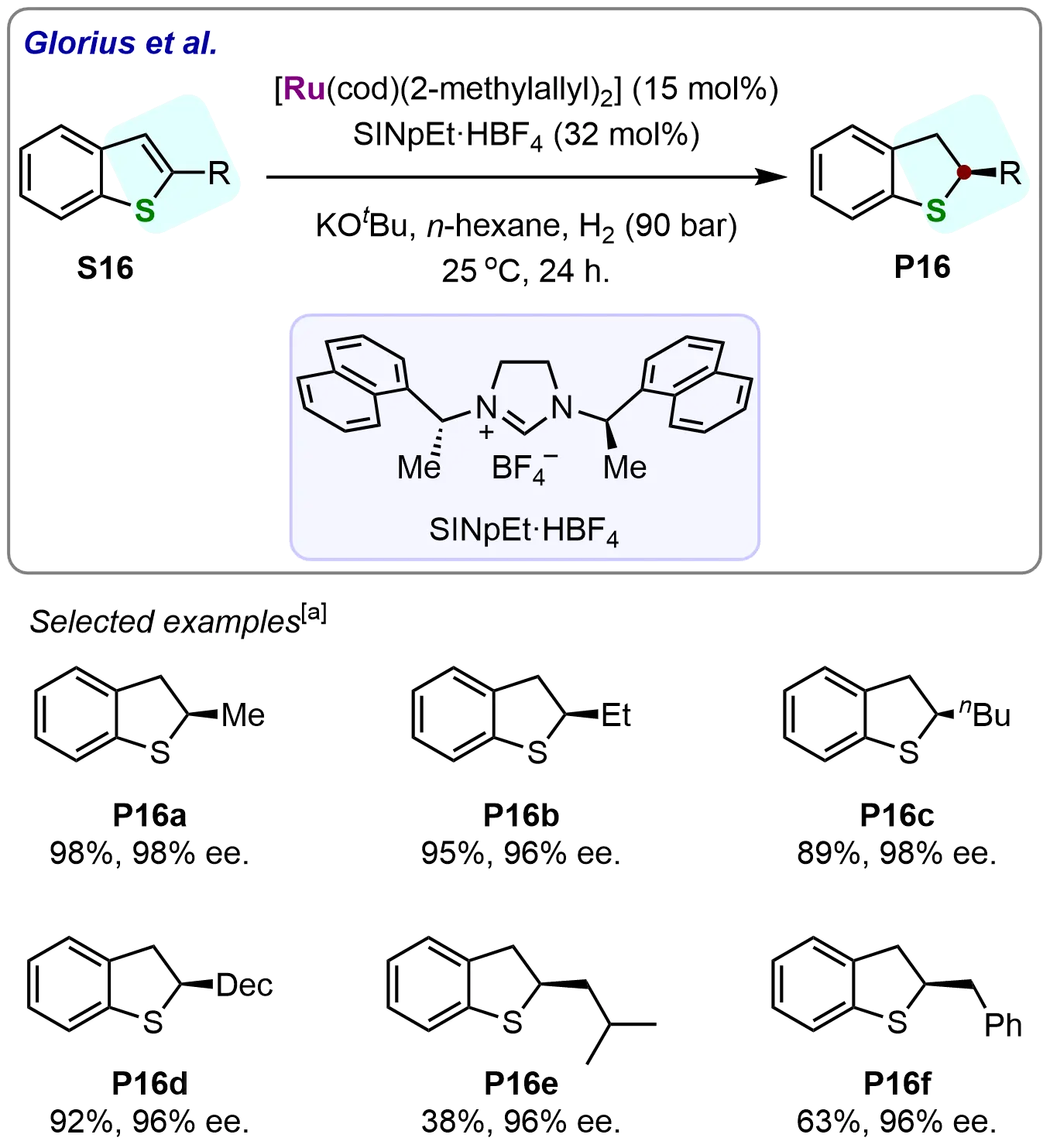
Scheme 16. Ruthenium-NHC-catalyzed asymmetric hydrogenation of benzothiophenes. Created in ChemDraw. [a]: Isolated yield was recorded.
The methodology was further extended to disubstituted thiophenes under identical optimized conditions, efficiently yielding tetrahydrothiophene derivatives (Scheme 17). This work represents the first successful homogeneous hydrogenation of disubstituted thiophenes, confirming the Ru–NHC catalyst’s versatility. While phenyl-substituted thiophene (S17a) gave modest yields, electron-deficient substrates like 2-fluorothiophene (S17b) achieved excellent conversions, with both cases exhibiting > 95:5 er and complete cis-diastereoselectivity. The protocol also accommodated regioisomers (e.g., 3,4-disubstituted S17c), albeit with lower selectivity (63:37 er). This study establishes Ru–NHC systems as highly efficient and selective catalysts for asymmetric hydrogenation of sulfur heterocycles across diverse substitution patterns.
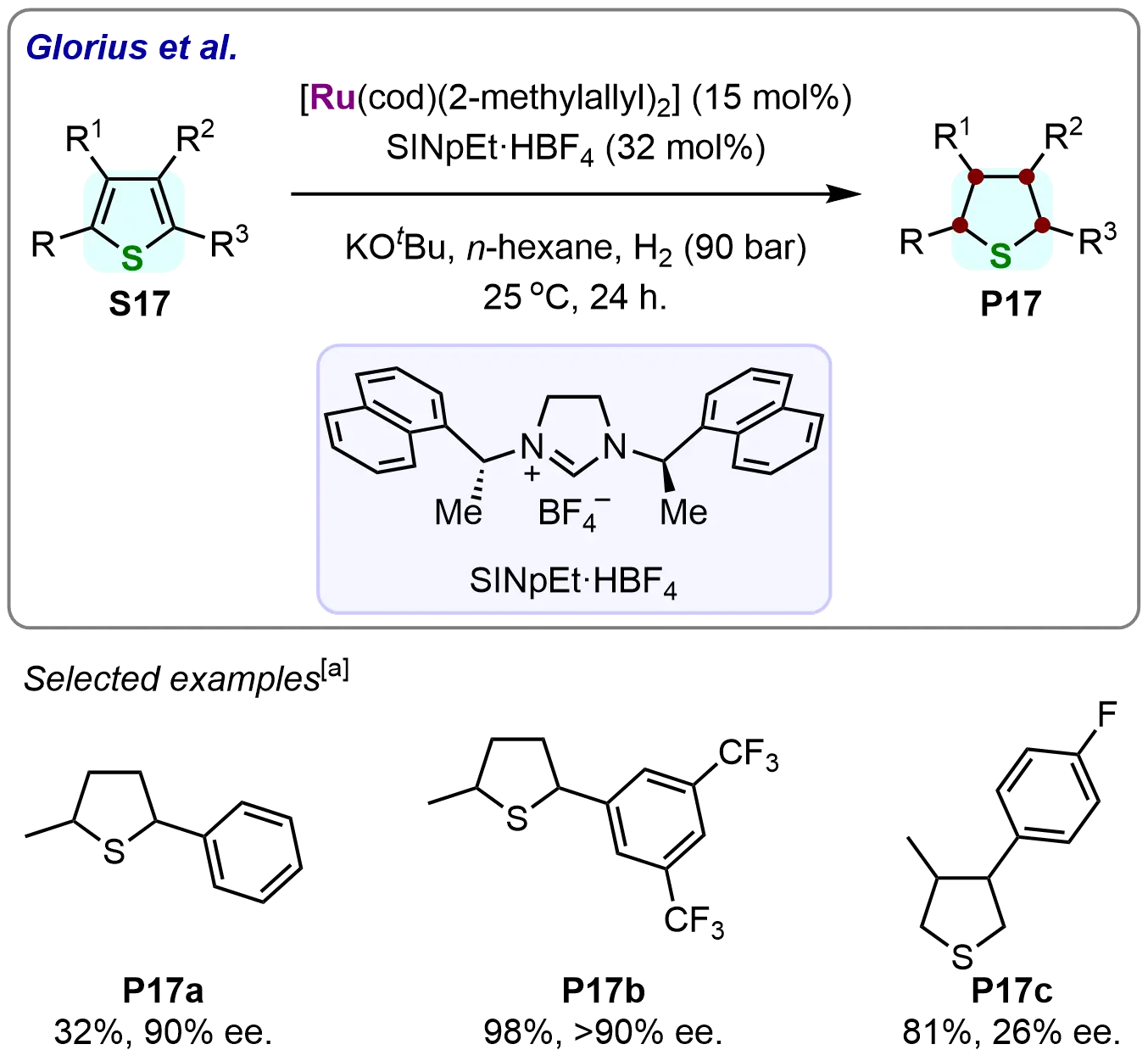
Scheme 17. Ruthenium-NHC-catalyzed asymmetric hydrogenation of disubstituted thiophenes. Created in ChemDraw. [a]: Isolated yield was recorded.
Optically active 1,5-benzothiazepine, featuring a seven-membered ring with a sulfur-substituted stereocenter, constitutes a privileged pharmacophore in pharmaceutical research. This scaffold plays a pivotal role in bioactive molecule development, particularly in drug design, owing to its broad pharmacological profile. Despite increasing demand for efficient syntheses of this core structure, asymmetric catalytic methods remain scarce. Addressing this gap, Glorius and co-workers developed an innovative asymmetric hydrogenation of vinyl thioethers using a Ru-NHC (N-heterocyclic carbene) catalyst (Scheme 18)[79].
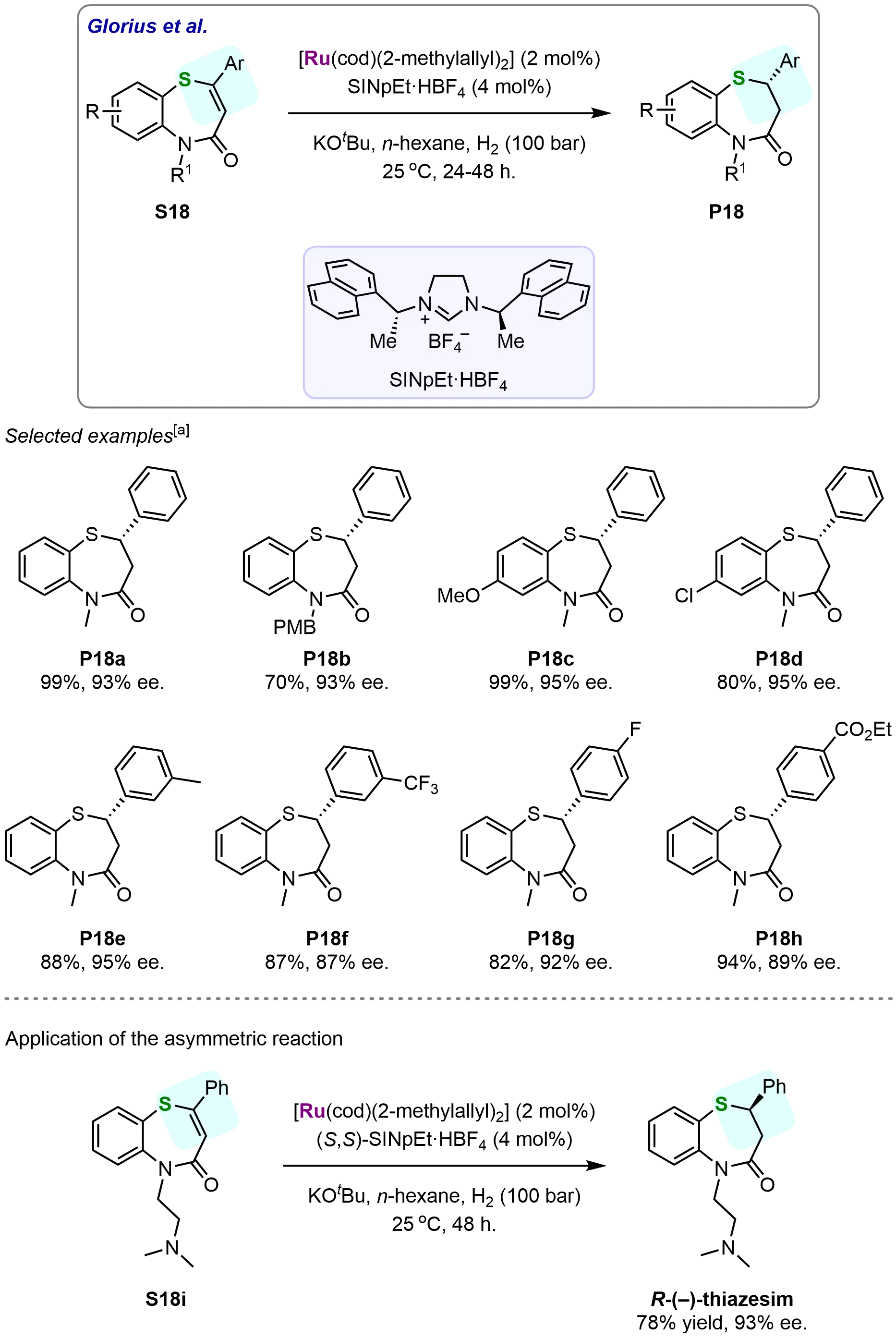
Scheme 18. Ruthenium-NHC-catalyzed asymmetric hydrogenation of vinylthioethers for efficient synthesis of optically active 1,5-benzothiazepine derivatives. Created in ChemDraw. [a]: Isolated yield was recorded.
This strategy enabled the synthesis of various optically active 1,5-benzothiazepines, each featuring stereocenters at C–S bonds, with high yields (up to 99%) and excellent enantioselectivities (up to 95% ee). For instance, when the N-methyl-protected substrate S18a was subjected to this catalytic system, the corresponding 2,3-dihydro-1,5-benzothiazepinone P18a was isolated in an impressive yield of 99%, with an enantiomeric excess (ee) of 93%. Similarly, hydrogenation of the N-4-methoxybenzyl-protected substrate S18b led to the desired product, albeit with a reduced yield of 70%, while maintaining a high ee of 93%. Further investigation into the electronic effects of substituents on the aminothiophenol ring revealed that electron-rich substrates afforded the corresponding 2,3-dihydro-1,5-benzothiazepinones with excellent yields (99%) and enantioselectivities (95% ee), as exemplified by substrate S18c, while electron-poor substrates, such as S18d, gave lower yields (80%), although enantioselectivity was largely preserved. Moreover, the study explored the impact of various functional groups, including electron-donating and electron-withdrawing substituents, at the meta- and para-positions of the aromatic ring. Substrates bearing substituents such as CF3, F, and CO2Me exhibited excellent compatibility with the catalytic system, yielding the corresponding 2,3-dihydro-1,5-benzothiazepinones in good to excellent yields (82%-94%) and enantioselectivities (P18e-P18h). These results demonstrate the broad scope and robustness of the reaction, making it a valuable tool for the synthesis of structurally diverse 1,5-benzothiazepine derivatives. To further highlight the synthetic utility of this methodology, the unsaturated 1,5-benzothiazepinone S18i, which contains a tertiary amine moiety, was successfully hydrogenated under the developed conditions. This reaction afforded the seven-membered heterocyclic antidepressant drug (R)-(–)-thiazesim in 78% yield with 93% ee, showcasing the practical applicability of the method in the context of drug development. These findings underscore not only the efficiency and selectivity of the ruthenium NHC-catalyzed asymmetric hydrogenation but also suggest its potential routes for the preparation of complex bioactive compounds with high enantiomeric purity.
Paul et al. have developed a highly effective and versatile chiral Ru-NHC catalyst for the enantioselective hydrogenation of heteroaromatic compounds bearing sulfur atoms. In all cases, the NHC ligand SINpEt (N,N'-bis(naphthylethyl)imidazolidinium-2-ylidene) demonstrated superior reaction performance, compatible with various substrate classes. Despite the catalyst’s unique reactivity profile and overall versatility, there has been limited structural and mechanistic information available regarding the Ru-SINpEt catalyst system[80]. In this study, Glorius and co-workers present the synthesis, characterization, and structural identification of precatalyst 19, featuring an unusual doubly cyclometallated NHC ligand (Scheme 19a). The hydrogenation of 2-methylbenzofuran under 10 bar H₂ was halted after 2.5 hours, and an aliquot was analyzed. A second batch of substrate was subsequently added to resume the reaction, with the catalyst retaining activity, achieving full conversion of the second batch of benzofuran with consistent enantioselectivity (Scheme 19b). Electrospray ionization mass spectrometry (ESI-MS) analysis identified complex 21 as the sole detectable species after both the first and second reactions. The researchers demonstrated that complex 21, which could be generated from precatalyst 19, serves as the active catalyst in the asymmetric hydrogenation process (Scheme 19c).
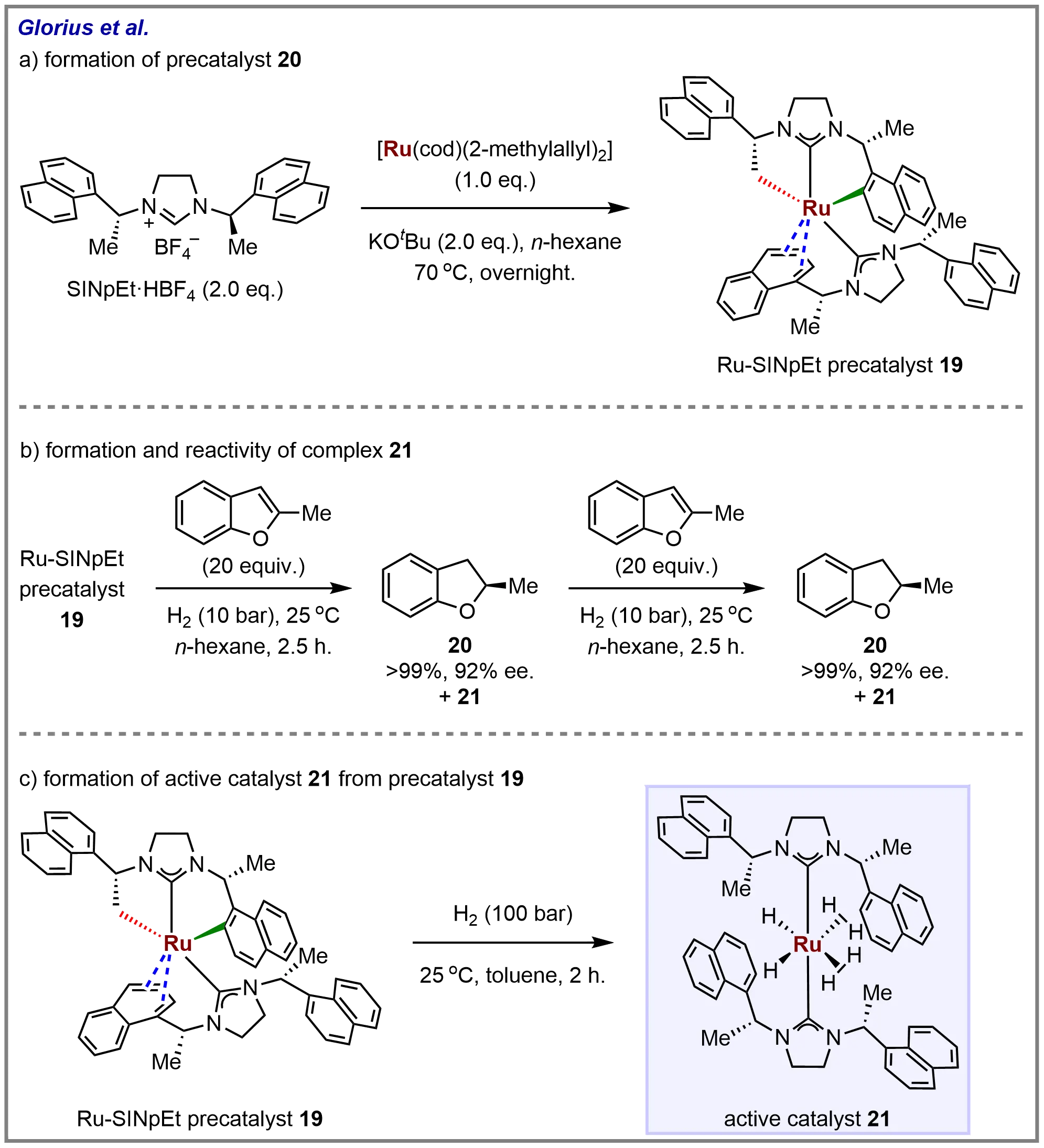
Scheme 19. Ruthenium-NHC precatalyst for the asymmetric hydrogenation of heteroarene and its activation pathway described by Glorius and co-workers. Created in ChemDraw.
4.2 Rh-catalyzed asymmetric hydrogenation of vinyl sulfides
In 2007, Meindertsma et al. employed a rhodium/Josiphos catalyst system for the asymmetric hydrogenation of α-branched vinyl sulfides, efficiently affording α-stereogenic thioethers (Scheme 20a)[81]. After thorough optimization of reaction conditions, the desired hydrogenated product (P22) was obtained in full conversion with 60% enantiomeric excess (ee). This work represents a significant advancement in the asymmetric hydrogenation of sulfur-containing compounds. Notably, the formation of desulfurized byproducts (P23 and P23’) was observed, which prompted further investigation into the reaction mechanism. Control experiments elucidated the byproduct origin and provided mechanistic insights into the catalytic process. These results supported a proposed mechanistic pathway (Scheme 20b). The mechanism involves the insertion of the rhodium species into the carbon-sulfur bond of the substrate, which may lead to a desulfurization process, or alternatively, a syn-elimination of a rhodium sulfide intermediate following the addition of the rhodium hydride to the olefin. These findings advance the understanding of rhodium catalysis in sulfur-containing systems and may guide the design of improved vinyl thioether hydrogenation catalysts.
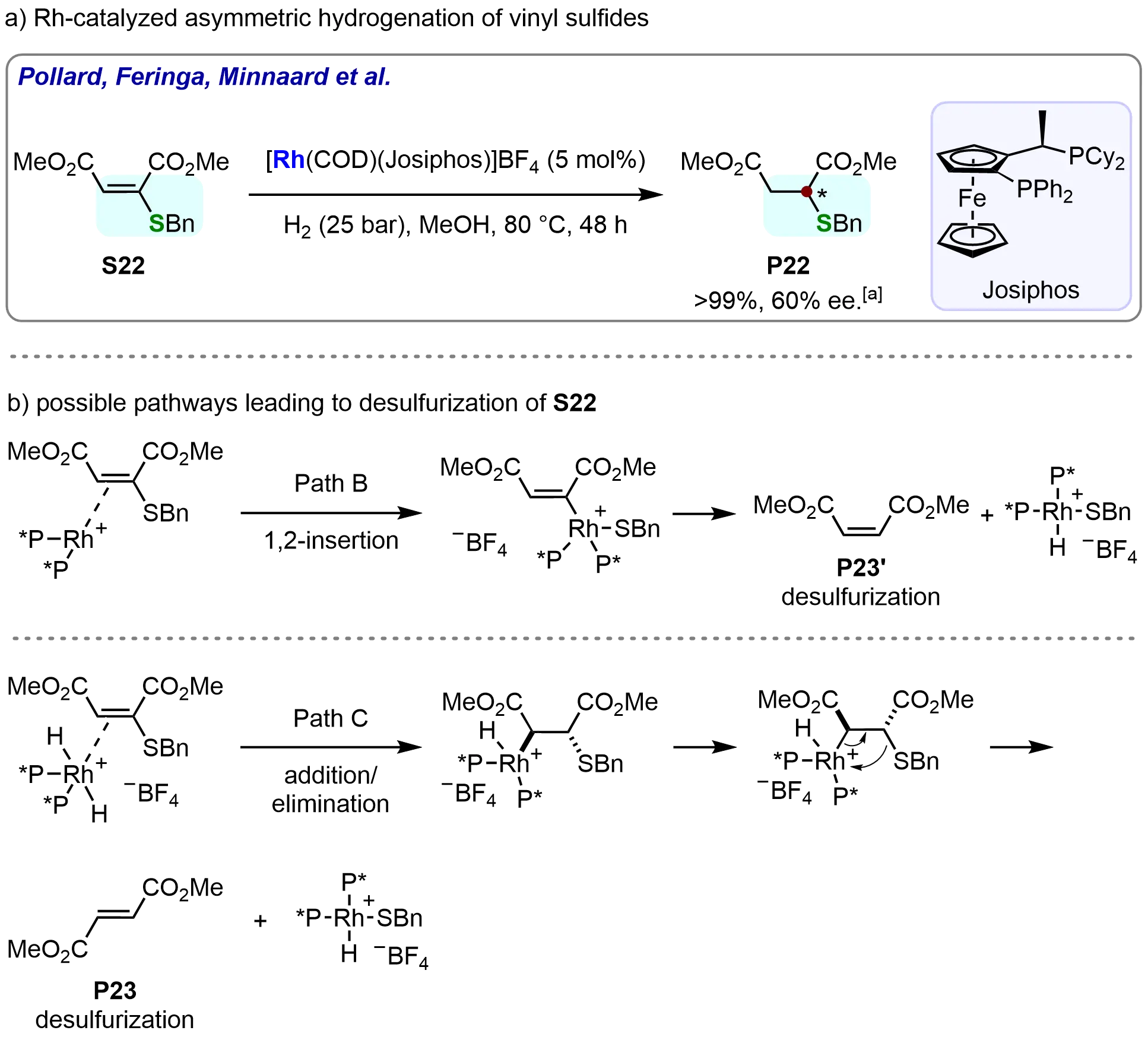
Scheme 20. Rhodium-catalyzed asymmetric hydrogenation of α-branched vinyl sulfides and related mechanism of desulfurization process. Created in ChemDraw. [a]: Conversions were recorded.
In 2017, Gao et al. developed an efficient Rh/DuanPhos-catalyzed asymmetric hydrogenation of β-acetylamino vinyl sulfides, affording β-stereogenic β-acetylamino sulfides with high yields and exceptional enantioselectivities (Scheme 21)[82]. This methodology represents a significant advancement in asymmetric catalysis, as it allows for the selective hydrogenation of β-acetylamino vinyl sulfides to produce optically active β-acetylamino sulfides, which are valuable intermediates in pharmaceutical synthesis. The developed reaction conditions exhibited broad adaptability, as a variety of β-acetylamino vinyl sulfides with diverse functional groups were efficiently hydrogenated to give the desired products in excellent yields (up to 99%) and enantioselectivities (up to 99%). Notably, the electronic nature of the substituents on the phenyl ring did not significantly affect the reactivity or enantioselectivity of the reaction. Whether electron-withdrawing or electron-donating groups were introduced at the ortho-, meta-, or para-positions of the phenyl group, the corresponding β-acetylamino sulfides (P24a–P24e) were obtained in high yields (94%-98%) and with excellent enantioselectivities (94%-98%). However, a slight decrease in yield was observed when a 2-naphthyl-substituted substrate was used. While the corresponding β-acetylamino sulfide (P24f) was obtained with good enantioselectivity (92%), the yield was only moderate (76%). Importantly, the reaction demonstrated excellent compatibility with heteroaryl-substituted β-acetylamino vinyl sulfides. These substrates were hydrogenated smoothly under the developed reaction conditions, leading to the desired products (P24g and P24h) in near-quantitative yields (99%) and with outstanding enantioselectivities (97%-99%) (Scheme 21a).
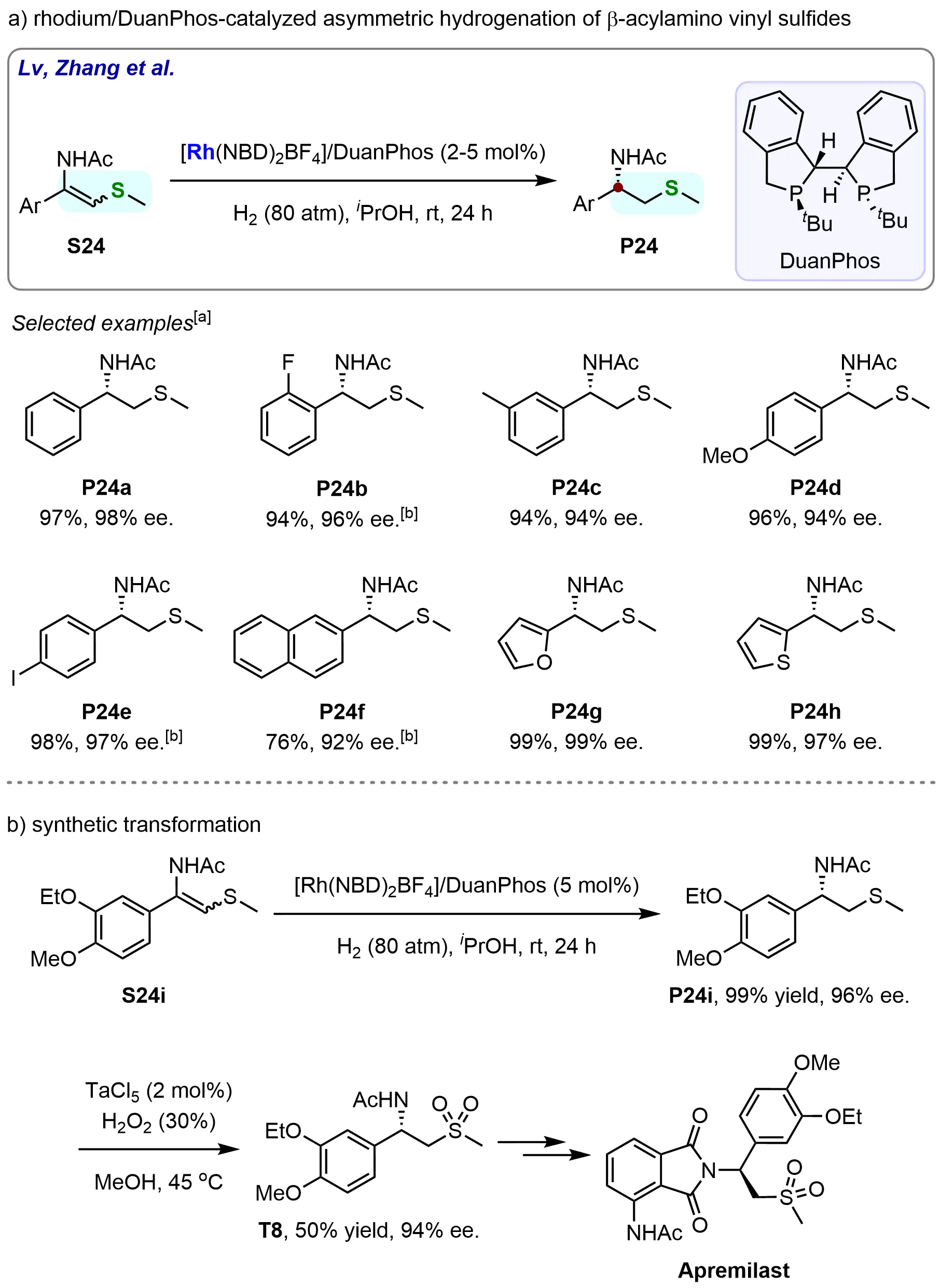
Scheme 21. Rhodium-catalyzed asymmetric hydrogenation of β-acylamino vinyl sulfides. Created in ChemDraw. [a]: Isolated yield was recorded; [b]: Reaction was performed with [Rh(NBD)2BF4]/DuanPhos (5 mol%), H2 (100 atm), at 30 ℃, for 30 h.
To further explore the synthetic utility of this methodology, a series of downstream derivatizations and applications were performed. As shown in Scheme 21b, the Rh-catalyzed asymmetric hydrogenation of S24i proceeded smoothly to afford the corresponding product P24i in 99% yield with 96% enantiomeric excess (ee). Subsequent oxidation of P24i with H2O2 in the presence of TaCl₅ furnished the β-acetylamino sulfone (T8) in 50% yield, without any erosion of enantiomeric purity. Notably, starting from T8, Apremilast, a drug approved by the U.S. Food and Drug Administration (FDA) in 2014 for the treatment of adult patients with active psoriatic arthritis, can be readily synthesized following reported procedures[83].
In 2018, Liu et al. developed a rhodium/ZhaoPhos-catalyzed asymmetric hydrogenation of (Z)- and (E)-β-substituted-β-thio-α,β-unsaturated esters, enabling the efficient and stereoselective synthesis of both enantiomers of ethyl β-substituted-β-thio-propanoates with excellent yields (up to 99%) and enantioselectivities (> 99% ee). This versatile method demonstrated broad substrate scope, successfully hydrogenating diverse β-thio-α,β-unsaturated esters while maintaining exceptional selectivity[84].
Initial evaluation of ethyl (Z)-3-substituted-3-thio-acrylates under optimized conditions (Scheme 22) yielded remarkable results. A series of chiral ethyl β-substituted-β-thio-propanoates were obtained in excellent yields (up to 99%) with high enantioselectivities (94% to > 99% ee). Notably, the phenyl ring substitution pattern on the arylthiol group showed minimal influence on the hydrogenation efficiency. Substrates bearing electron-neutral (S25a), electron-donating (S25b-c), or electron-withdrawing (S25d-e) groups all underwent smooth conversion, furnishing products P25a-P25e in 96-99% yields with 94% to > 99% ee. The method also accommodated the 2-naphthyl-substituted substrate S25f, achieving 99% yield and 96% ee. Particularly challenging substrates, including the alkyl-substituted ethyl (Z)-3-cyclohexyl-3-(phenylthio)acrylate (S25g) and the heteroaromatic ethyl (Z)-3-(phenylthio)-3-(pyridine-3-yl)acrylate (S25h), were successfully hydrogenated under the same conditions. The alkyl-substituted substrate (S25g) afforded the desired product (P25g) in 77% yield with 97% ee, while the heteroaromatic substrate (S25h) yielded the corresponding product (P25h) in 98% yield with > 99% ee. These results highlight the versatility of the catalytic system, demonstrating its broad applicability across various types of substituents, including both electron-rich and electron-deficient groups, as well as alkyl and heteroaromatic functionalities.
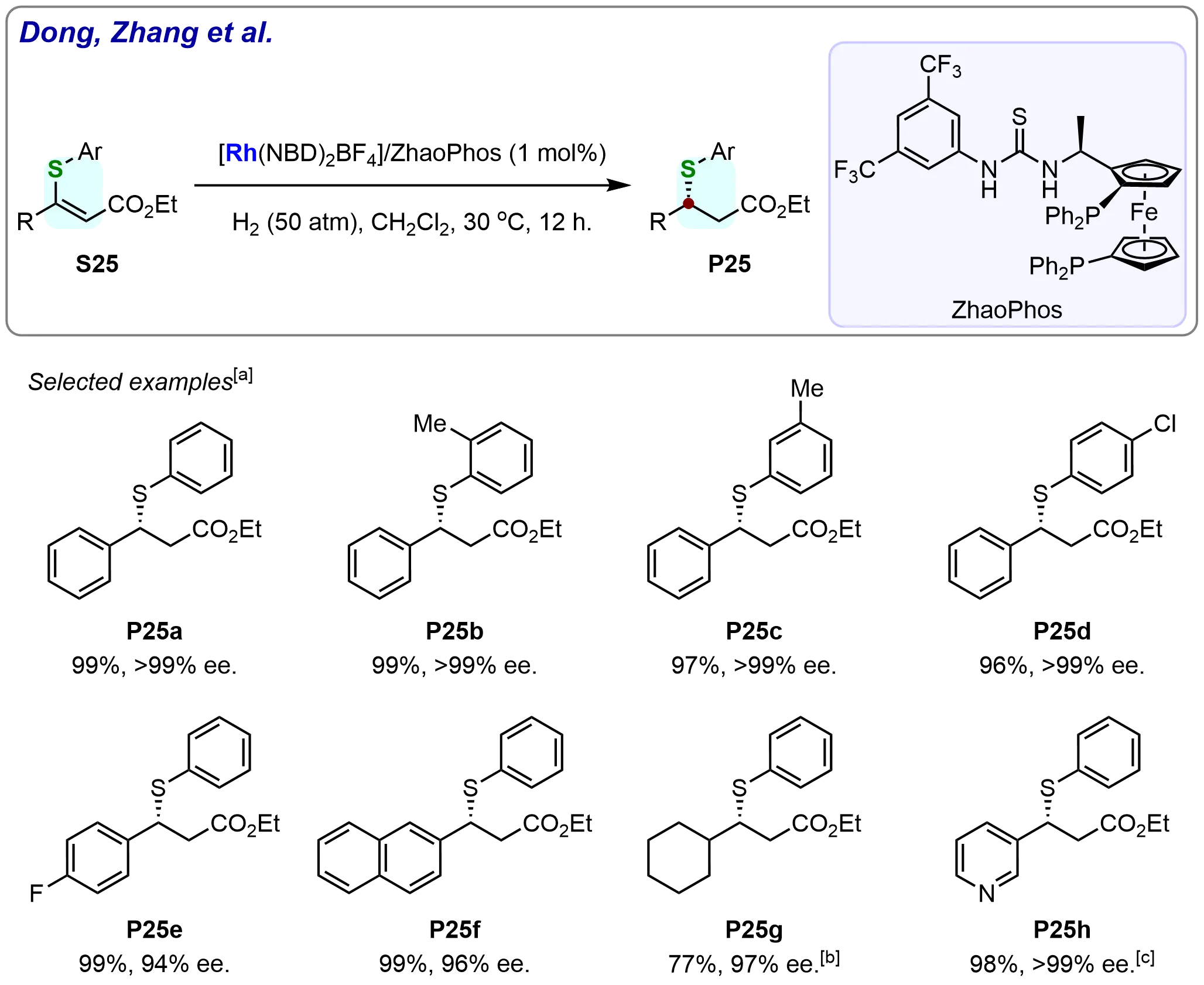
Scheme 22. Rhodium-catalyzed asymmetric hydrogenation of ethyl (Z)-3-substituted-3-thio-acrylates. Created in ChemDraw. [a]: Isolated yield was recorded; [b]: Reaction was carried out at 40 ℃ for 18 h; [c]: Reaction was carried out at 60 ℃ for 48 h.
Subsequently, the reactivity and enantioselectivity of various ethyl (E)-3-substituted-3-thio-acrylates were also evaluated under the optimized reaction conditions (Scheme 23). It was found that the substituents on both the phenyl ring of the arylthiol group and the 3-aryl ring had minimal effect on the outcome of the asymmetric hydrogenation. The hydrogenation of substrates (S26a-S26g) proceeded smoothly, with full conversions, yields ranging from 97% to 99%, and excellent enantioselectivities (94% to 98% ee). Moreover, the 2-naphthyl substituted substrate (S26h) was also successfully hydrogenated, providing the desired product (P26h) in 99% yield with 97% ee. These results confirm the robustness of the method, which can accommodate a wide variety of substituent patterns, thus offering a highly versatile and efficient strategy for the synthesis of chiral organic sulfides.
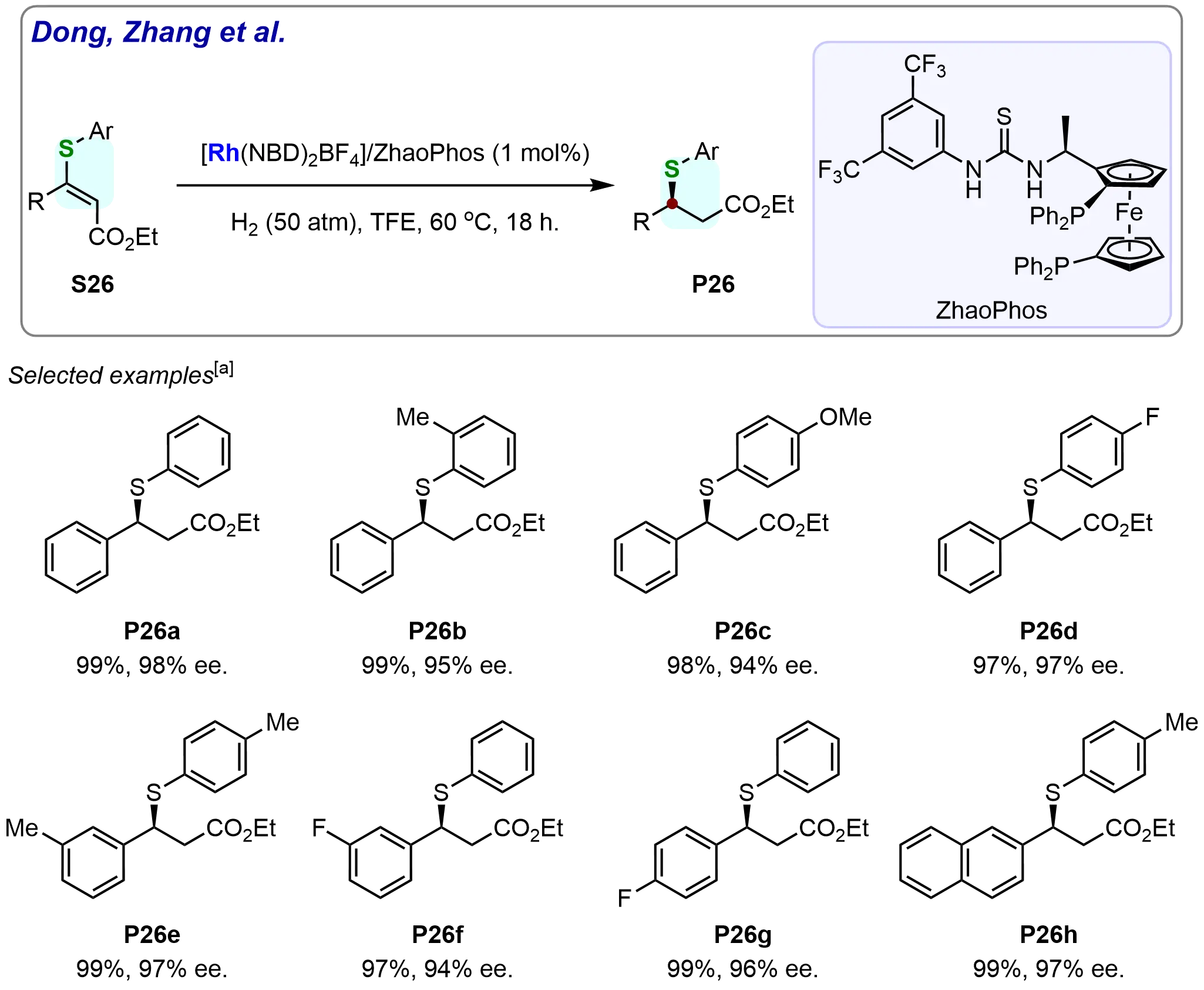
Scheme 23. Rhodium-catalyzed asymmetric hydrogenation of ethyl (E)-3-substituted-3-thio-acrylates. Created in ChemDraw. [a]: Isolated yield was recorded.
In 2020, Yin et al. introduced a rhodium-catalyzed asymmetric hydrogenation of unsaturated medium-ring NH lactams, achieving the highly enantioselective synthesis of N-unprotected 2,3-dihydro-1,5-benzothiazepinones (Scheme 24)[85]. The model substrate S27a was first examined under the optimized reaction conditions, employing ZhaoPhos as the best ligand. Under these conditions, the desired product, P27a, was obtained in an excellent yield of 95% with > 99% enantiomeric excess (ee). Further studies focused on the hydrogenation of substrates with various protective groups. For instance, the N-methyl-protected substrate S27b and the N-benzyl-protected substrate S27c both underwent hydrogenation smoothly, yielding the corresponding products (P27b and P27c) with exceptional enantioselectivities, ranging from 98% to > 99% ee. This outcome highlighted the robustness of the catalytic system and its tolerance toward different N-protecting groups. The effect of substituents on the fused benzene ring was also thoroughly investigated. Substrates bearing either electron-withdrawing (S27d) or electron-donating (S27e) substituents on the phenyl ring performed efficiently under the optimized reaction conditions, delivering the desired products with yields between 93% and 96% and enantioselectivities exceeding 99%. The introduction of an electron-withdrawing trifluoromethyl (CF3) group on the phenyl ring (S27f) resulted in a slight reduction in both reactivity and enantioselectivity. The hydrogenation product, P27f, was obtained in 81% yield with 95% ee. On the other hand, the introduction of an electron-donating methoxy (OMe) group on the phenyl ring (S27g) enhanced both reactivity and enantioselectivity. The corresponding hydrogenation product (P27g) was obtained in 90% yield with 98% ee. Additionally, the catalytic system was found to be compatible with alkyl-substituted substrates. The alkyl-substituted substrate S27h underwent hydrogenation smoothly, providing the desired product P27h in 95% yield with 95% ee. These findings collectively highlight the versatility and robustness of the rhodium-catalyzed asymmetric hydrogenation of unsaturated medium-ring NH lactams (Scheme 24a). The gram-scale asymmetric hydrogenation of S27a was carried out smoothly at an S/C ratio of 100, affording the desired product P27a in 99% yield with 99% ee. The hydrogenation product P27a was subsequently transformed into the antidepressant drug thiazesim in 85% yield while maintaining an enantiomeric purity of > 99% ee (Scheme 24b). The ability to synthesize N-unprotected 2,3-dihydro-1,5-benzothiazepinones with high yields and excellent enantioselectivities opens new avenues for the synthesis of chiral compounds, which are of significant interest in pharmaceutical and material science applications.
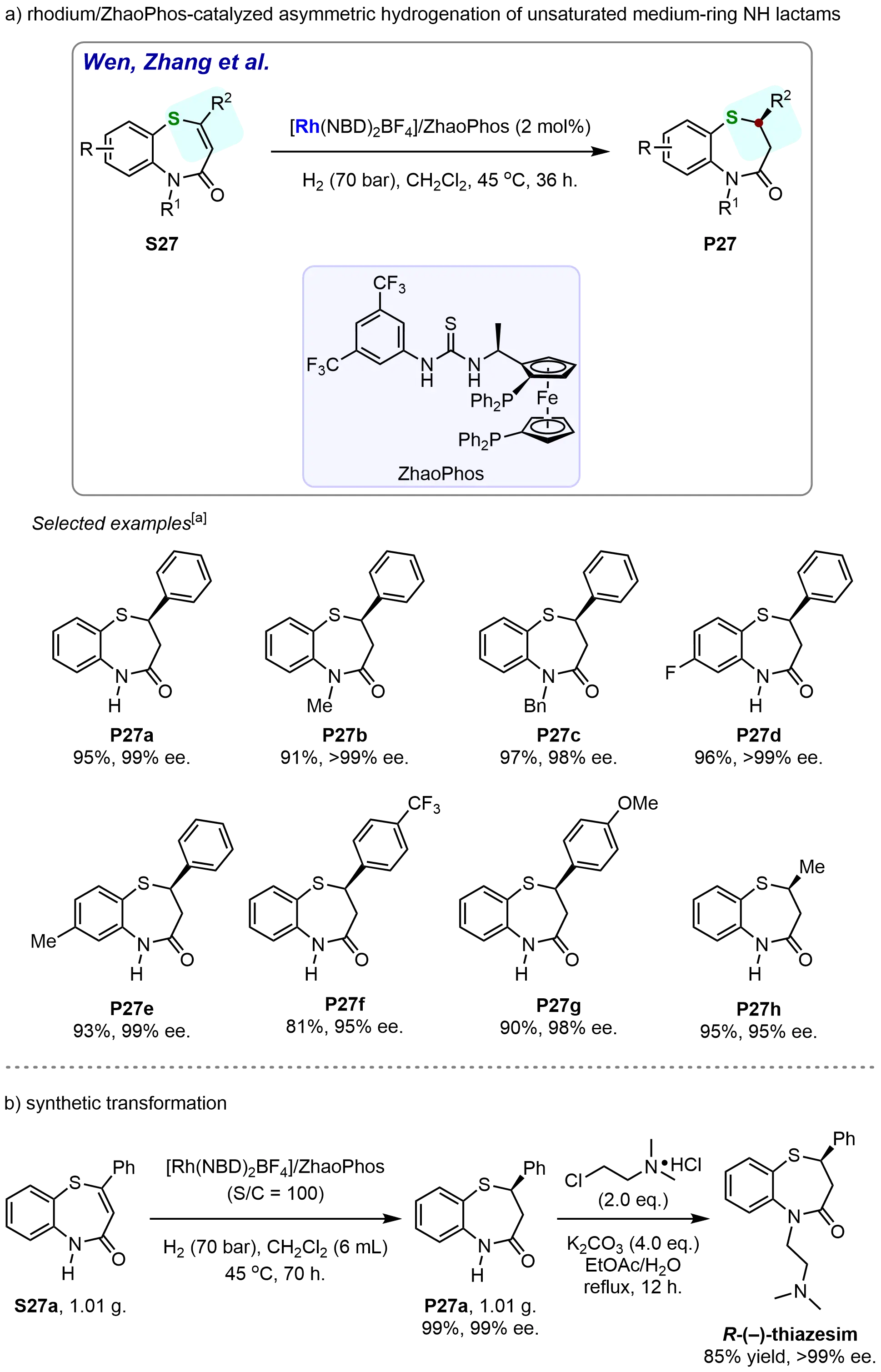
Scheme 24. Rhodium-catalyzed asymmetric hydrogenation of unsaturated medium-ring NH lactams. Created in ChemDraw. [a]: Isolated yield was recorded.
In 2024, Wu et al. developed a highly efficient rhodium-catalyzed asymmetric hydrogenation of 2-substituted 4H-thiochromenes, enabling the synthesis of chiral thiochromanes with high yields and excellent enantioselectivities (Scheme 25)[86]. After systematically screening various chiral ligands, the (R,R)-f-spiroPhos ligand was found to be the most effective, resulting in optimal catalytic performance. Under this optimized reaction conditions, chiral thiochromane P28a was synthesized in excellent yield (98%) and with excellent enantioselectivities (99%). The study also revealed that the reactivity and enantioselectivity of the reaction were largely unaffected by the position and electronic properties of the substituents on the aryl ring of the R1 group. Specifically, substrates bearing electron-donating (S28b) or electron-withdrawing (S28c-S28d) substituents on the 2-aryl group underwent smooth hydrogenation to afford the desired chiral products (P28b-P28d) in excellent yields (96%-98%) and with outstanding enantioselectivities (97%-99%). Of particular significance is the ability of the catalytic system to tolerate substrates containing heteroaromatic groups. Substrates with thiophenyl (S28e) or furanyl (S28f) substituents were successfully hydrogenated, yielding the corresponding chiral thiochromanes (P28e-P28f) in high yields (94%-98%) and excellent enantioselectivities (98%-99%). The reaction system also demonstrated tolerance for 2-alkenyl-substituted 4H-thiochromenes; substrate S28g, which contains a 2-alkenyl group, was hydrogenated efficiently, yielding the chiral 2-alkyl thiochromane (P28g) in good yield (93%) with good enantioselectivity (88%). Additionally, substrates bearing halogen substituents, such as the bromine-substituted compound S28h, exhibited comparable reactivity and enantioselectivity (Scheme 25a). Furthermore, the obtained hydrogenation products were selectively transformed into valuable chiral α-substituted sulfoxide and sulfone derivatives (Scheme 25b). The chiral 2-phenylthiochromane (P28a) was successfully oxidized with H2O2, affording the corresponding 2-phenylthiochromane 1-oxide (T9) in 84% yield with a diastereomeric ratio (d.r.) of 3.4:1 and 99% ee. In addition, the hydrogenation product P28e was readily converted into the chiral sulfone (T10) in 89% yield with 98% ee via mCPBA oxidation. These findings collectively highlight the excellent performance of the rhodium-catalyzed asymmetric hydrogenation of 2-substituted 4H-thiochromenes, offering a reliable and versatile method for the synthesis of chiral thiochromanes.
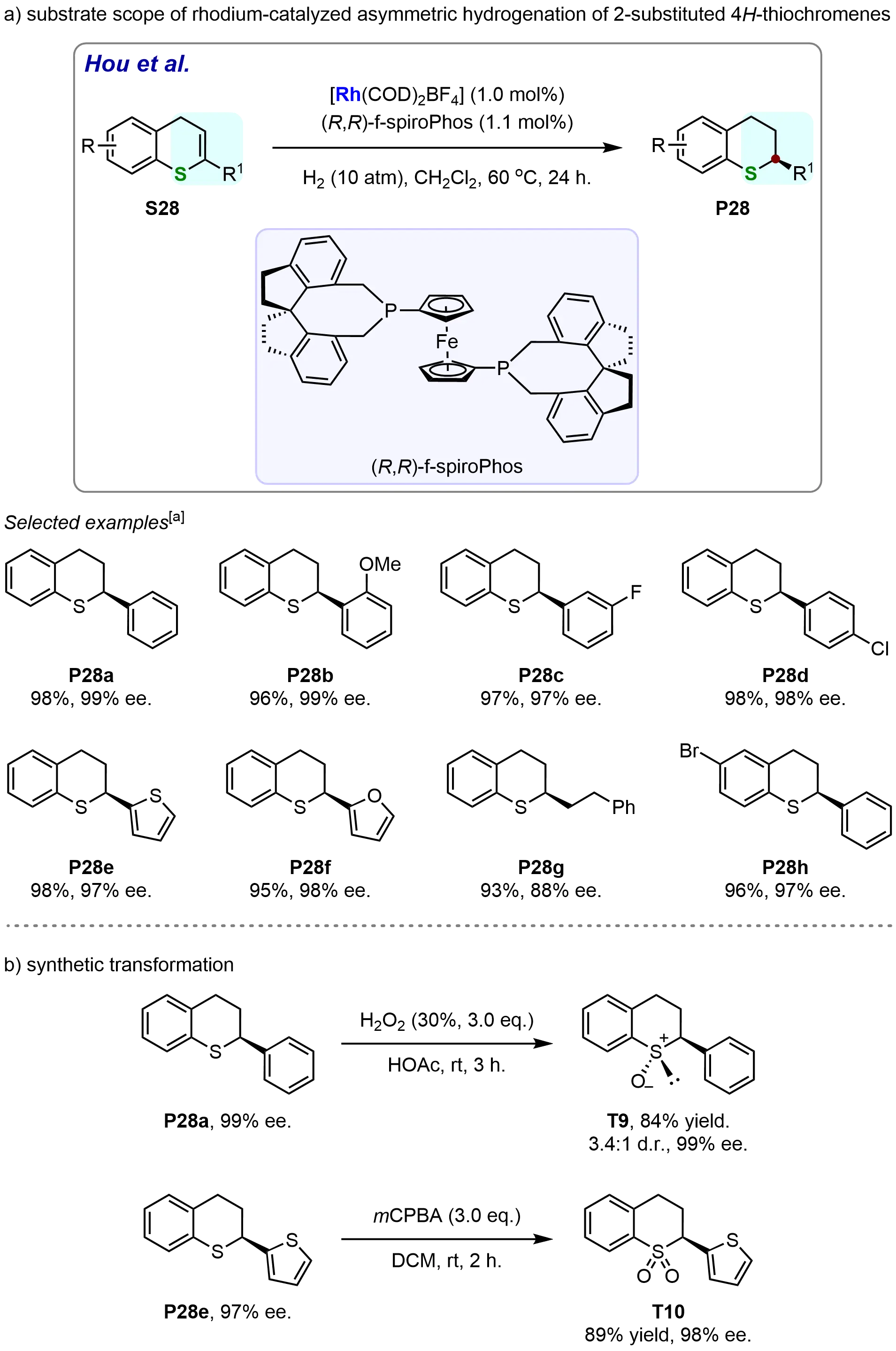
Scheme 25. Rhodium-catalyzed asymmetric hydrogenation of 2-substituted 4H-thiochromenes for synthesis of chiral thiochromanes. Created in ChemDraw. [a]: Isolated yield was recorded.
4.3 Ir-catalyzed asymmetric hydrogenation of vinyl sulfides
In 2008, Valla et al. successfully employed iridium complexes of chiral oxazoline-based P,N-ligands for the enantioselective hydrogenation of 2-phenyl-4H-thiochromene (S29) (Scheme 26)[87]. Under optimized reaction conditions, the desired α-stereogenic cyclothioether (P29) was obtained in 73% conversion with 91% ee. This work marked a significant advancement in the asymmetric hydrogenation of thiochromene derivatives, demonstrating the potential of iridium-catalyzed hydrogenation for the selective functionalization of sulfur-containing heterocycles.
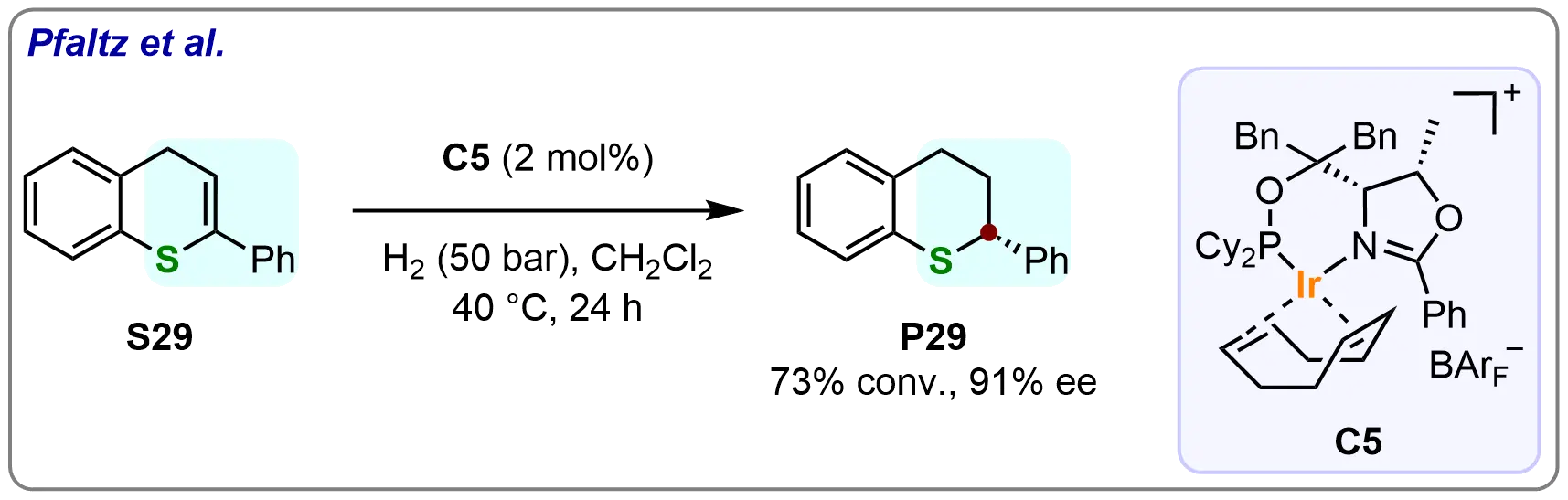
Scheme 26. Iridium-catalyzed asymmetric hydrogenation of thiochromene derivatives. Created in ChemDraw.
4.4 Short summary of the asymmetric hydrogenation of vinyl sulfides
Remarkable progress has been achieved in the asymmetric hydrogenation of vinyl sulfides, owing to the extensive efforts of numerous researchers. These advancements include the successful application of trisubstituted and cyclic vinyl sulfides in metal-catalyzed asymmetric hydrogenation, enabling the efficient synthesis of a variety of α- and β-chiral thioethers. Furthermore, a series of subsequent synthetic transformations has been developed, providing access to structurally diverse and synthetically valuable sulfur-containing compounds (Scheme 27a).
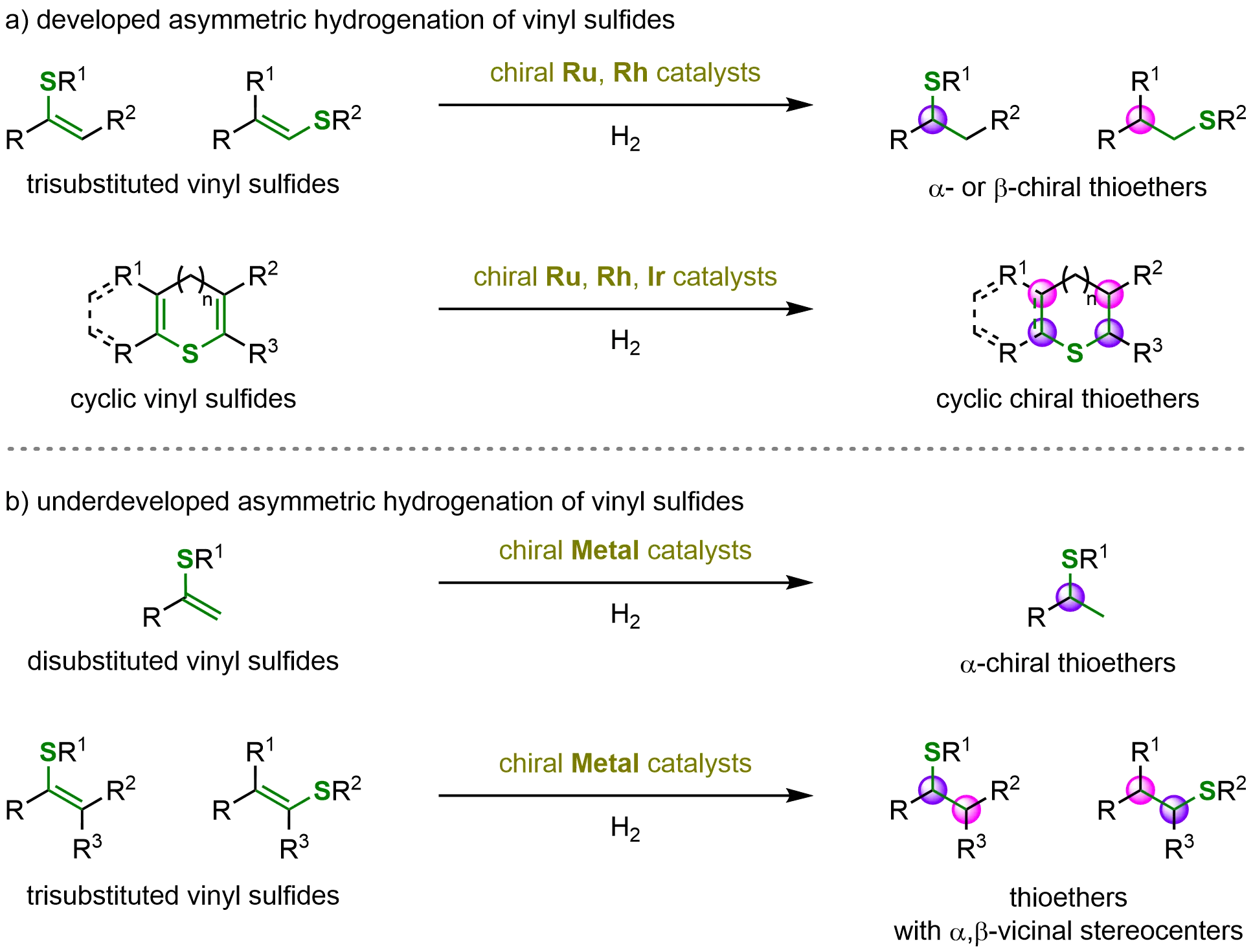
Scheme 27. Developed and underdeveloped metal-catalyzed asymmetric hydrogenation of vinyl sulfides. Created in ChemDraw.
Despite these significant accomplishments, several challenges persist in this field. In particular, the asymmetric hydrogenation of simple disubstituted and more demanding tetrasubstituted vinyl sulfides to construct molecules bearing chiral sulfur centers remains to be fully realized (Scheme 27b). Additionally, broadening the range of catalytic systems beyond the currently dominant transition metals presents an exciting future opportunity. The exploration of alternative catalysts based on palladium (Pd), nickel (Ni), cobalt (Co), manganese (Mn), and copper (Cu) may further expand the catalytic repertoire available for the enantioselective synthesis of chiral thioethers.
5. Asymmetric Hydrogenation of Vinyl Chlorides
With over 4,700 halogenated natural products reported to date, approximately half of which contain C(sp3)-chloride bonds (Figure 9), the role of halogens in drug discovery has become increasingly significant. Halogenated compounds often exhibit unique biological activities, making them valuable in the development of pharmaceuticals and agrochemicals. Furthermore, the potential of halogen-bearing stereogenic centers in asymmetric synthesis has spurred considerable interest, as the selective introduction of halogens can impart desirable structural and functional properties to target molecules. Consequently, the development of methods for the stereocontrolled introduction of carbon-halogen bonds has emerged as a dynamic and rapidly evolving area of research[88].
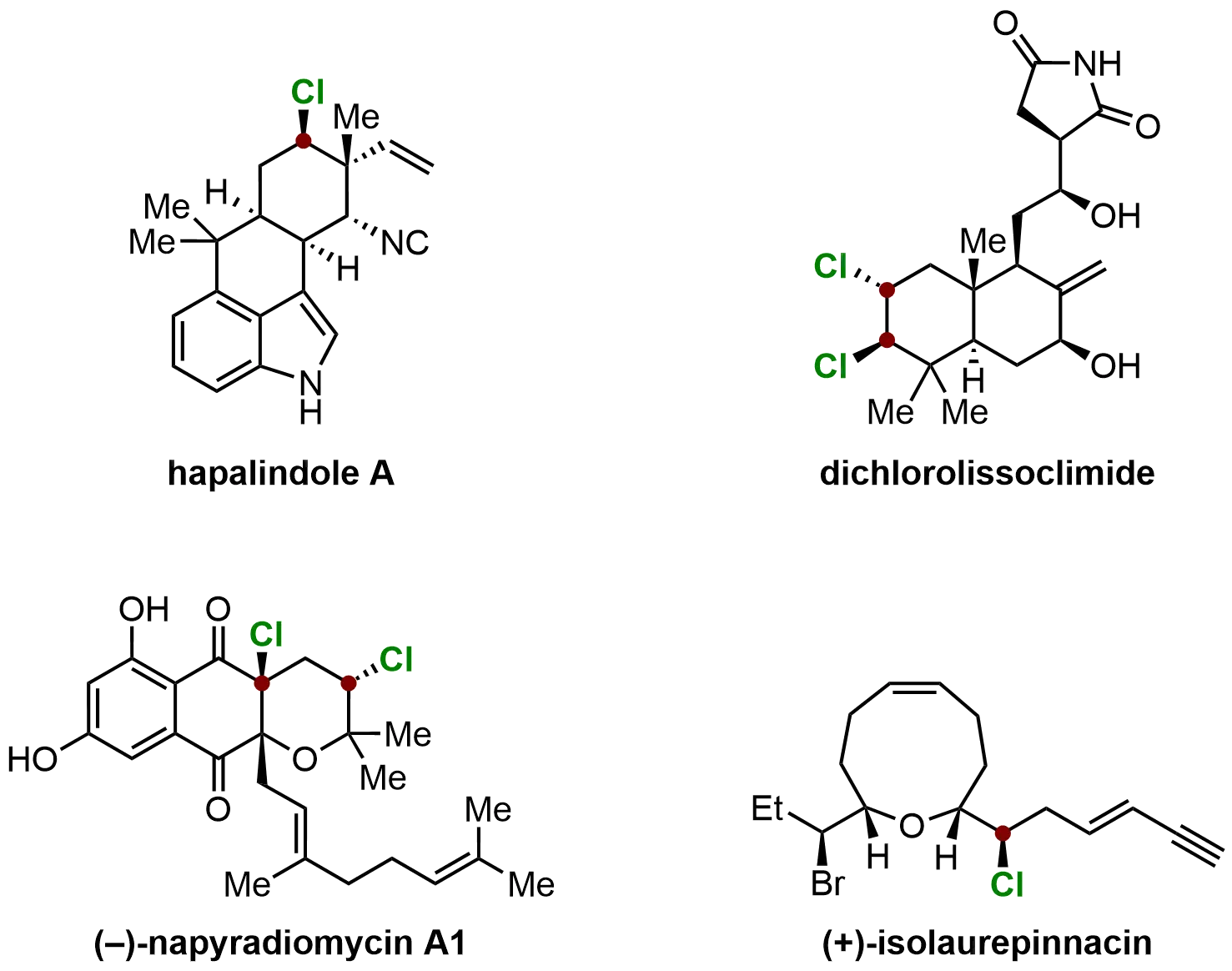
Figure 9. Typical examples of natural products containing chiral C(sp3)-chloride bonds. Created in ChemDraw.
A particularly promising approach in this field is the asymmetric hydrogenation of vinyl chloride, which holds the potential to create chloride-bearing stereogenic centers in a highly selective manner. This process has garnered significant attention due to its environmentally friendly and atom-economical nature, aligning with the growing demand for sustainable and efficient synthetic methodologies. Asymmetric hydrogenation, when applied to substrates such as vinyl chloride, offers a direct route to chiral chloride-bearing products without the need for elaborate protective group strategies or multi-step transformations. The simplicity and efficiency of this approach make it an attractive option for the synthesis of complex halogenated compounds, which are often difficult to achieve through traditional methods. However, the asymmetric hydrogenation of vinyl chlorides presents unique challenges. The large size of the chloride atom makes it difficult to form stable chelating coordination between the transition metal and the alkene. Furthermore, vinyl chlorides are prone to undergo dechlorination side reactions, which further hinder the desired catalytic process (Figure 10).
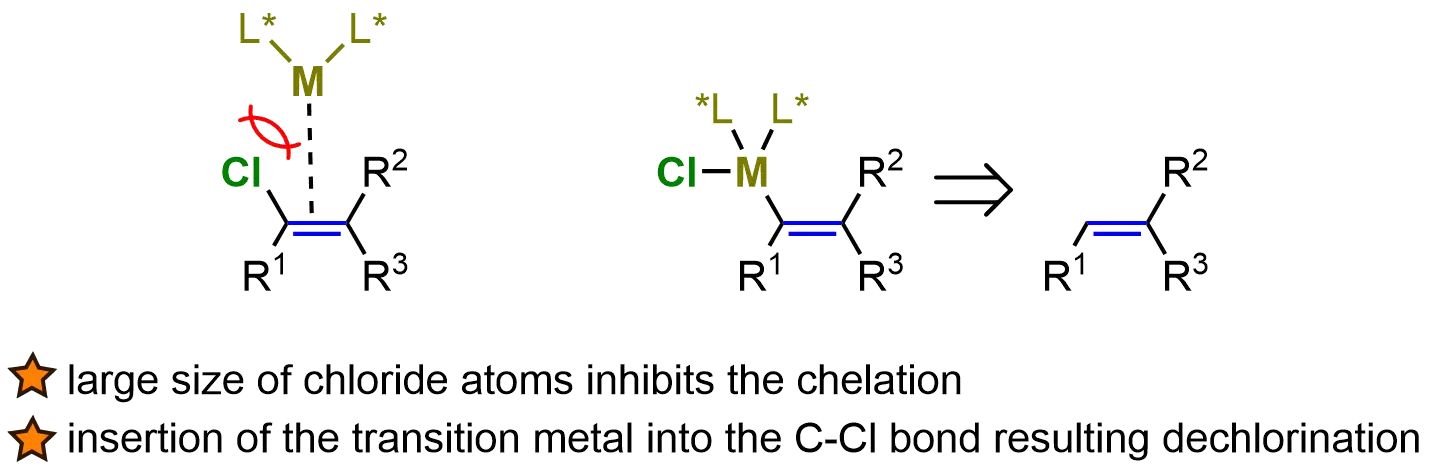
Figure 10. The low reactivity of vinyl chlorides in AH. Created in ChemDraw. AH: asymmetric hydrogenation.
5.1 Ru-catalyzed asymmetric hydrogenation of vinyl chlorides
In 2005, Wang et al. made a significant breakthrough in the field of asymmetric hydrogenation by developing a Ru-(S)-C3-tunephos catalyst system for the efficient hydrogenation of 2-chloroallylphthalimide (S30). This catalytic system enabled the high-yield synthesis of β-chloride-substituted β-stereogenic chiral amines, with the desired hydrogenated product (P30) being obtained in 97% yield and with an excellent enantiomeric excess (ee) of 93% (Scheme 28)[89]. This work represented the first successful example of asymmetric hydrogenation applied to chloride-substituted alkenes; a class of compounds that had previously been considered challenging. The impressive results not only demonstrated the effectiveness of the Ru-(S)-C3-tunephos catalyst system but also set a precedent for the development of new methodologies in the asymmetric hydrogenation of chloride-functionalized alkenes, opening up new avenues for the synthesis of chiral amines with chloride functional groups.
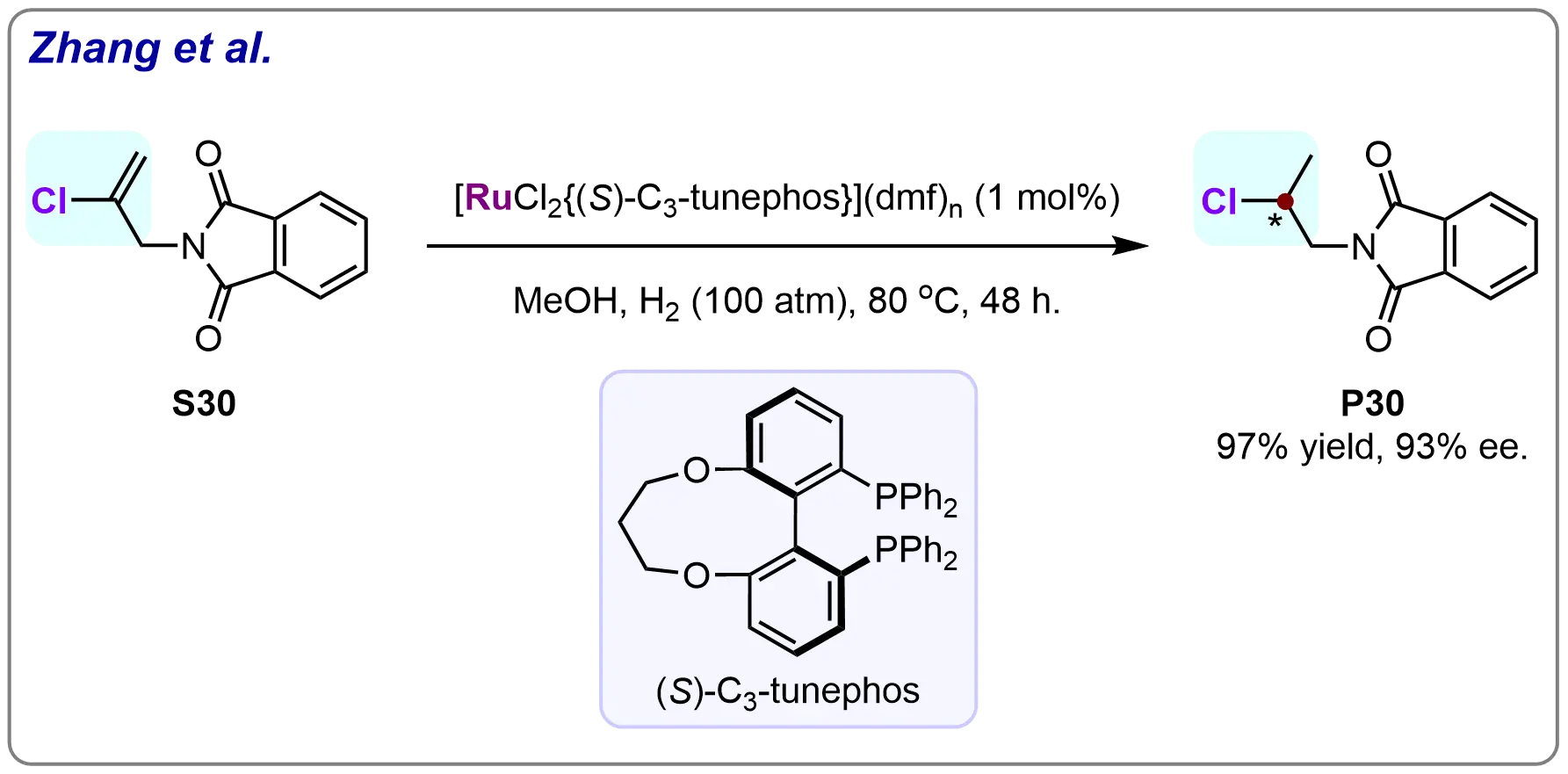
Scheme 28. Ruthenium-catalyzed asymmetric hydrogenation of 2-chloroallylphthalimide. Created in ChemDraw.
In 2008, Sun et al. made significant advancements in asymmetric hydrogenation by introducing a novel C3-tunephos-type chiral diphosphine ligand (L16) for use in ruthenium-catalyzed hydrogenation reactions (Scheme 29)[90]. Building on their previous work, they continuously applied this new ligand to the hydrogenation of 2-chloroallylphthalimide (S30). The results were remarkable, as the desired β-chloride-substituted β-stereogenic chiral amine (P30) was obtained with full conversion and a high enantiomeric excess of 94%.
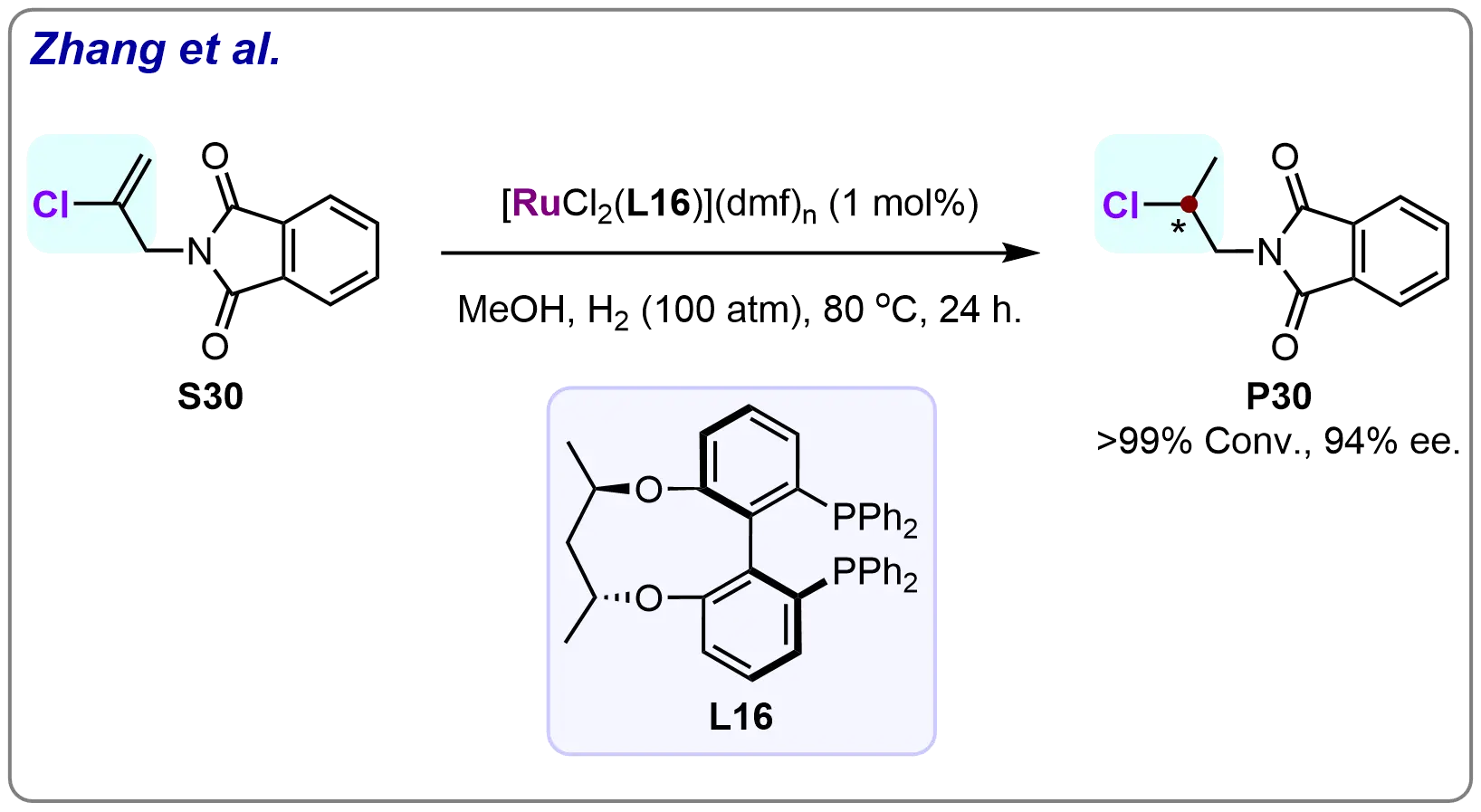
Scheme 29. Application of tunephos-type chiral diphosphine ligand in highly enantioselective ruthenium-catalyzed hydrogenation of 2-chloroallylphthalimide. Created in ChemDraw.
5.2 Rh-catalyzed asymmetric hydrogenation of vinyl chlorides
During their total synthesis of (–)-Acutumine and (–)-Dechloroacutumine, Herzon and co-workers successfully carried out the homogeneous hydrogenation of substrate S31, which contains a vinyl chloride functional group. The hydrogenation was catalyzed by the complex [Rh(nbd)(dppb)]BF4 under 300 psi H₂, yielding (–)-Acutumine (P31) in 17% yield. In contrast, when a heterogeneous hydrogenation approach was employed (using H₂ and Pd/C), (–)-Dechloroacutumine (P31’) was obtained in a significantly higher yield of 60% (Scheme 30)[91]. This comparison underscores the inherent challenges associated with the asymmetric hydrogenation of vinyl chloride, a reaction that requires careful optimization of both catalyst and reaction conditions to achieve selective transformations. The low yield in the homogeneous hydrogenation suggests the difficulties in selectively hydrogenating the C=C bond without affecting the chlorinated functional group, highlighting the complexity of such transformations in synthetic chemistry.
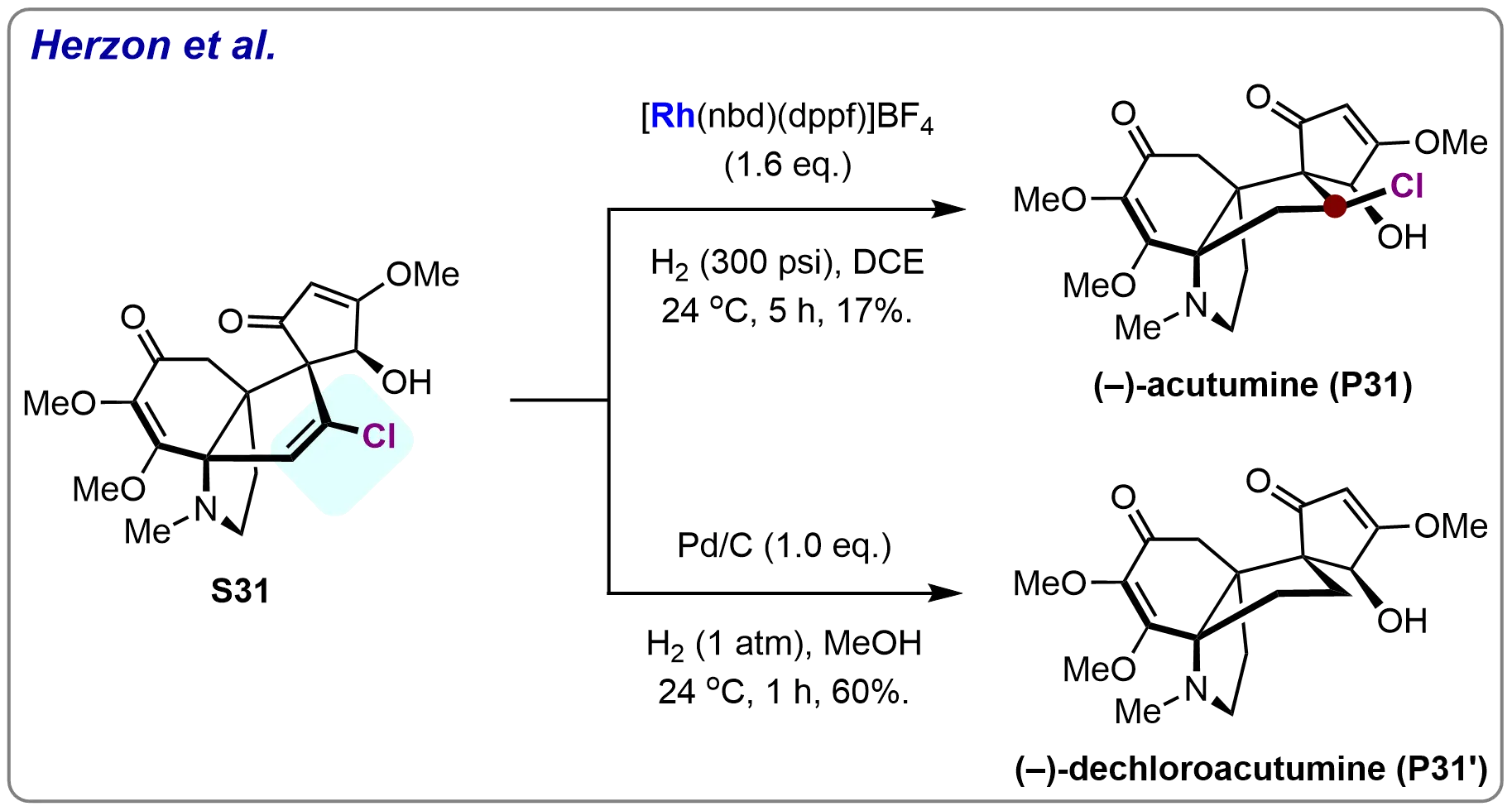
Scheme 30. Hydrogenation of vinyl chloride to realize total synthesis of (–)-Acutumine and (–)-Dechloroacutumine. Created in ChemDraw.
5.3 Ir-catalyzed asymmetric hydrogenation of vinyl chlorides
In 2012, Gazić Smilović et al. made significant strides in the development of iridium-based catalysts by incorporating a P,N-ferrocenyl-oxazolino-phosphinite ligand, which enabled the chemoselective and enantioselective hydrogenation of (1-chloro-1-alkenyl) boronic esters[92]. This novel catalytic system was highly effective, providing a range of α-stereogenic α-chloro boronic esters in moderate to good yields (70%-91%) and enantioselectivities (83%-93%). Furthermore, it demonstrated excellent chemoselectivity, with only 3%-14% of dechlorinated byproducts being isolated, highlighting the catalyst’s ability to selectively target the hydrogenation of the C=C bond while minimizing side reactions. A broad range of substrates, including aliphatic derivatives with varying steric hindrances and aromatic derivatives with differing electronic properties, was tested under optimized reaction conditions. Among these, sterically hindered substrates, particularly aromatic derivatives (S32e-S32h) and, in particular, substrate S32b, required higher catalyst loadings to achieve optimal results. However, by carefully adjusting reaction parameters, the researchers were able to reduce all substrates (S32a-S32h) to their corresponding products (P32a-P32h), with full conversion and high enantioselectivity achieved in all cases, except for S32c. Notably, the presence of bulky alkyl or aryl groups in the substrates was found to enhance enantiodifferentiation, suggesting that steric effects play a significant role in the enantioselective hydrogenation process. In contrast, the dechlorination levels appeared to be influenced by both electronic and steric effects, although no clear trend could be established. These findings are encapsulated in Scheme 31, which illustrates the scope and selectivity of the catalytic system.
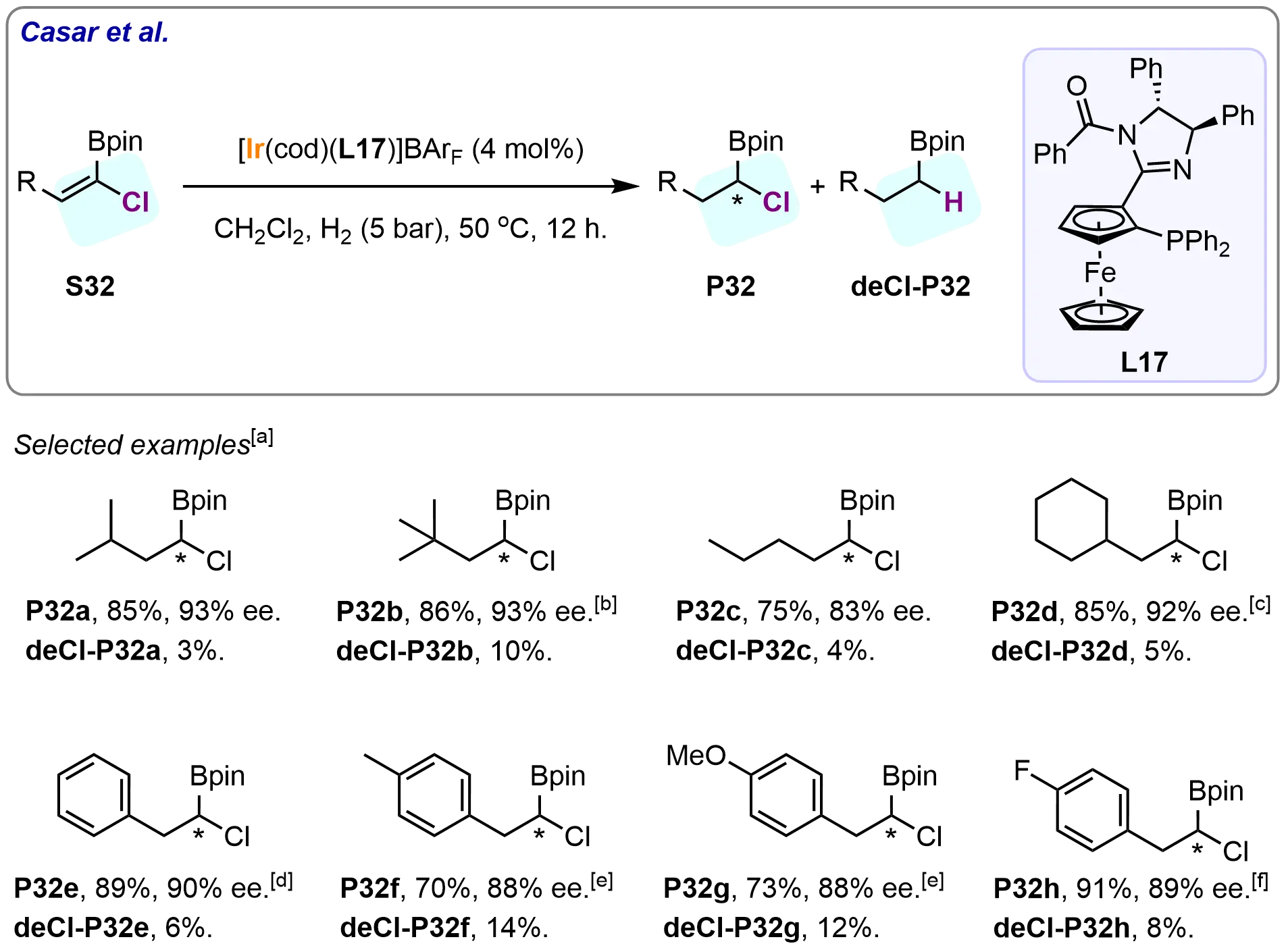
Scheme 31. Iridium-catalyzed chemo- and enantioselective hydrogenation of (1-chloro-1-alkenyl) boronic esters. Created in ChemDraw. [a]Isolated yield was recorded; [b]: Reaction was carried out with 14 mol% catalyst and 10 bar H2; [c]: Reaction was carried out with 30 bar H2 for 18 h; [d]: Reaction was carried out with 10 mol% catalyst and 10 bar H2; [e]: Reaction was carried out with 10 mol% catalyst and 30 bar H2 for 18 h. [f]Reaction was carried out with 10 mol% catalyst.
5.4 Short summary of the asymmetric hydrogenation of vinyl chlorides
Remarkable progress has been achieved in the asymmetric hydrogenation of vinyl chlorides, owing to the dedicated efforts of numerous researchers. These advancements include the successful application of di- and trisubstituted vinyl chlorides in metal-catalyzed asymmetric hydrogenation, enabling the efficient synthesis of a variety of α- and β-chiral alkyl chlorides. However, only a limited range of substrate types has been explored to date, and further expansion of substrate scope and synthetic applications is highly desirable in this research area (Scheme 32a).
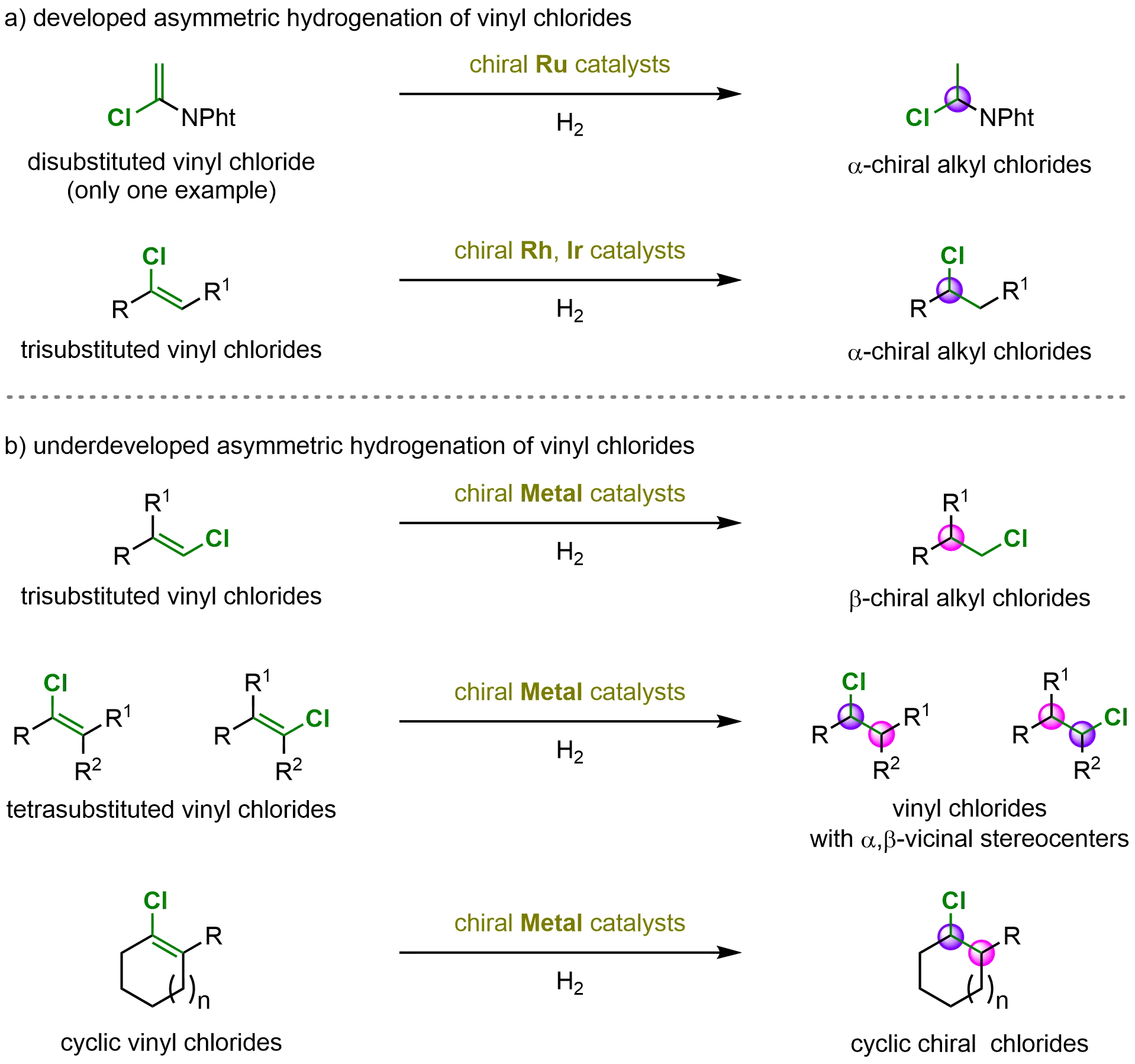
Scheme 32. Developed and underdeveloped metal-catalyzed asymmetric hydrogenation of vinyl chlorides. Created in ChemDraw.
Despite these notable accomplishments, several challenges remain. In particular, the asymmetric hydrogenation of tetrasubstituted and cyclic vinyl chlorides to construct complex molecules bearing chiral alkyl chloride centers has yet to be realized (Scheme 32b). Moreover, broadening the range of catalytic systems beyond the currently dominant transition-metal platforms represents an exciting direction for future exploration. The development of catalytic systems based on palladium (Pd), nickel (Ni), cobalt (Co), manganese (Mn), and copper (Cu) could further enrich the toolbox for enantioselective synthesis of chiral alkyl chlorides and enhance the versatility of asymmetric hydrogenation chemistry.
6. Summary and Outlook
As demonstrated in this review, the asymmetric hydrogenation of vinyl silanes, vinyl sulfides, and vinyl chlorides, traditionally regarded as challenging substrates, has garnered increasing attention in recent years. The notable progress in this field can largely be attributed to the discovery of novel catalysts and catalytic systems that exhibit exceptional activity, regioselectivity, and stereoselectivity. These advances have not only enhanced synthetic methodology but also have played a pivotal role in the asymmetric total synthesis of bioactive molecules.
For instance, the Co-pincer catalyst has been explored for its high reactivity, regioselectivity, and enantioselectivity in the sequential hydrosilylation/hydrogenation of alkynes, enabling the efficient synthesis of chiral silanes. Similarly, the chiral Ru-NHC catalyst has demonstrated remarkable diastereoselectivity and enantioselectivity in the hydrogenation of disubstituted thiophenes, 2-alkyl-substituted benzothiophenes, and N-unprotected 2,3-dihydro-1,5-benzothiazepinones, generating various chiral thioethers. These advancements have significantly contributed to the synthesis of valuable synthetic building blocks and bioactive compounds. Moreover, the Iridium-P,N-ferrocenyl-oxazolino-phosphinite catalyst has proven highly efficient in the chemoselective and enantioselective hydrogenation of vinyl chlorides, yielding chiral organochlorides with high efficiency.
Despite these substantial advances, numerous challenges remain that limit the broader application of asymmetric hydrogenation, particularly in the context of unfunctionalized vinyl silanes, vinyl sulfides, and vinyl chlorides. One of the primary challenges is the need for the development of new chiral catalysts or catalytic systems with enhanced activity and enantioselectivity. Special emphasis should be placed on exploring catalysts derived from earth-abundant metals, which offer the potential for more sustainable and cost-effective processes. Additionally, improving catalyst loadings in reaction systems is crucial to enhancing their practical utility, especially in large-scale applications.
In parallel, given the inherently low reactivity of vinyl silanes, vinyl sulfides, and vinyl chlorides in asymmetric hydrogenation, the development of effective substrate activation strategies is essential to overcome this challenge. Activation approaches utilizing Brønsted acids, Lewis acids, or organic activators could significantly improve the reactivity of these substrates and thus facilitate more efficient hydrogenation processes. Furthermore, expanding the substrate scope of these reactions remains a critical area of future research, as it is essential to meet the growing demand for a diverse range of optically pure organosilanes, organosulfides, and organochlorides.
In conclusion, while the asymmetric hydrogenation of vinyl silanes, vinyl sulfides, and vinyl chlorides has seen substantial progress, it is clear that the continued development of more effective catalysts, innovative substrate activation strategies, and expanded reaction scopes is crucial for advancing the field. Addressing these challenges will not only improve the synthetic efficiency and selectivity of these transformations but will also play a pivotal role in enabling their broader application in the synthesis of complex molecules for pharmaceutical and industrial purposes.
Authors contribution
Zhang J, Chen Y: Investigation, writing-original draft, writing-review & editing.
Zhang Z, Zhang W: Conceptualization, writing-review & editing.
Conflict of interests
Jian Zhang is an employee of Changzhou Pharmaceutical Factory Co., Ltd. Wanbin Zhang is an Editorial Board Member of Chiral Chemistry. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Key R & D Program of China (2021YFA1500200), the National Natural Science Foundation of China (22371182), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, the China Postdoctoral Science Foundation (2024M760299) and the Changzhou Municipal Leading Enterprise Talent Program (CQ20250029).
Copyright
© The Author(s) 2025.
References
-
1. Wang H, Wen J, Zhang X. Chiral tridentate ligands in transition metal-catalyzed asymmetric hydrogenation. Chem Rev. 2021;121(13):7530-7567.[DOI]
-
2. Wen J, Wang F, Zhang X. Asymmetric hydrogenation catalyzed by first-row transition metal complexes. Chem Soc Rev. 2021;50(5):3211-3237.[DOI]
-
3. Wang Y, Wang M, Li Y, Liu Q. Homogeneous manganese-catalyzed hydrogenation and dehydrogenation reactions. Chem. 2021;7(5):1180-1223.[DOI]
-
4. Cabré A, Verdaguer X, Riera A. Recent advances in the enantioselective synthesis of chiral amines via transition metal-catalyzed asymmetric hydrogenation. Chem Rev. 2022;122(1):269-339.[DOI]
-
5. Yang F, Xie J-H, Zhou Q-L. Highly efficient asymmetric hydrogenation catalyzed by iridium complexes with tridentate chiral spiro aminophosphine ligands. Acc Chem Res. 2023;56(3):332-349.[DOI]
-
6. Lückemeier L, Pierau M, Glorius F. Asymmetric arene hydrogenation: Towards sustainability and application. Chem Soc Rev. 2023;52(15):4996-5012.[DOI]
-
7. He YM, Cheng YZ, Duan Y, Zhang YD, Fan QH, You SL, et al. Recent progress of asymmetric catalysis from a Chinese perspective. CCS Chem. 2023;5(12):2685-2716.[DOI]
-
8. Chen Q, Han Z, Ding K. Transition metal catalyzed carbocycle-selective asymmetric hydrogenation of aromatic rings. Chin J Org Chem. 2024;44:2063-2076.[DOI]
-
9. Zhang J, Song Y, Zhang Z, Zhang W. Advances in asymmetric hydrogenation of unfunctionalized enol ethers. Asian J Org Chem. 2025;14(2):e202400584.[DOI]
-
10. Knowles WS. Asymmetric Hydrogenations (Nobel Lecture). Angew Chem Int Ed. 2002;41(12):1998-2007.[DOI]
-
11. Noyori R. Facts are the enemy of truth—reflections on serendipitous discovery and unforeseen developments in asymmetric catalysis. Angew Chem Int Ed. 2013;52(1):79-92.[DOI]
-
12. Ma Y, Liu K, He L, Lv H. Divergent synthesis of chiral amines via Ni-catalyzed chemo- and enantioselective hydrogenation of alkynone imines. Sci China Chem. 2023;66(11):3186-3192.[DOI]
-
13. Liu G, Yang X, Gu P, Wang M, Zhang X, Dong X-Q. Challenging task of ni-catalyzed highly regio-/enantioselective semihydrogenation of racemic tetrasubstituted allenes via a kinetic resolution process. J Am Chem Soc. 2024;146(11):7419-7430.[DOI]
-
14. Zhang Z, Zhu B, Yi Z, Fang T, Jin Z, He L, et al. Catalytic asymmetric synthesis and applications of stereogenic β’-methyl enones and β,β’-dimethyl ketones. Angew Chem Int Ed. 2025;64(5):e202414449.[DOI]
-
15. Ratovelomanana-Vidal V, Phansavath P. Aymmetric Hydrogenation and Transfer Hydrogenation. New York: Wiley-VCH; 2021.
-
16. Imamoto T. P-stereogenic phosphorus ligands in asymmetric catalysis. Chem Rev. 2024;124(14):8657-8739.[DOI]
-
17. Hu Y, Chen J, Li B, Zhang Z, Gridnev ID, Zhang W. Nickel-catalyzed asymmetric hydrogenation of 2‐amidoacrylates. Angew Chem Int Ed. 2020;59(13):5371-5375.[DOI]
-
18. Fan D, Zhang J, Hu Y, Zhang Z, Gridnev ID, Zhang W. Asymmetric hydrogenation of α-boryl enamides enabled by nonbonding interactions. ACS Catal. 2020;10(5):3232-3240.[DOI]
-
19. iu D, Li B, Chen J, Gridnev ID, Yan D, Zhang W. Ni-catalyzed asymmetric hydrogenation of N-aryl imino esters for the efficient synthesis of chiral α-aryl glycines. Nat Commun. 2020;11(1):5935.[DOI]
-
20. Hu Y, Zhang Z, Liu Y, Zhang W. Cobalt‐catalyzed chemo‐ and enantioselective hydrogenation of conjugated enynes. Angew Chem Int Ed. 2021;60(31):16989-16993.[DOI]
-
21. Zhang J, Chen T, Wang Y, Zhou F, Zhang Z, Gridnev ID, et al. Asymmetric hydrogenation of γ-branched allylamines for the efficient synthesis of γ-chirogenic amines. Nat Sci. 2021;1(2):e10021.[DOI]
-
22. Xu Y, Liu D, Deng Y, Zhou Y, Zhang W. Rhodium‐catalyzed asymmetric hydrogenation of 3-benzoylaminocoumarins for the synthesis of chiral 3-amino dihydrocoumarins. Angew Chem Int Ed. 2021;60(44):23602-23607.[DOI]
-
23. Li M, Zhang J, Zou Y, Zhou F, Zhang Z, Zhang W. Asymmetric hydrogenation for the synthesis of 2-substituted chiral morpholines. Chem Sci. 2021;12(45):15061-15066.[DOI]
-
24. Jin Y, Zou Y, Hu Y, Han Y, Zhang Z, Zhang W. Azole‐directed cobalt‐catalyzed asymmetric hydrogenation of alkenes. Chem Eur J. 2022;28(44):e202201517.[DOI]
-
25. Li B, Chen J, Liu D, Gridnev ID, Zhang W. Nickel-catalysed asymmetric hydrogenation of oximes. Nat Chem. 2022;14:920-927.[DOI]
-
26. Wei H, Chen H, Chen J, Gridnev ID, Zhang W. Nickel-catalyzed asymmetric hydrogenation of α-substituted vinylphosphonates and diarylvinylphosphine oxides. Angew Chem Int Ed. 2023;62(6):e202214990.[DOI]
-
27. Hu Y, Zou Y, Yang H, Ji H, Jin Y, Zhang Z, et al. Precise synthesis of chiral z-allylamides by cobalt-catalyzed asymmetric sequential hydrogenations. Angew Chem Int Ed. 2023;62(15):e202217871.[DOI]
-
28. Chen T, Zou Y, Hu Y, Zhang Z, Wei H, Wei L, et al. Cobalt-catalyzed efficient convergent asymmetric hydrogenation of E/Z-enamides. Angew Chem Int Ed. 2023;62(26):e202303488.[DOI]
-
29. Guan J, Chen J, Luo Y, Guo L, Zhang W. Copper-catalyzed chemoselective asymmetric hydrogenation of C=O bonds of exocyclic α,β-unsaturated pentanones. Angew Chem Int Ed. 2023;62(35):e202306380.[DOI]
-
30. Yang H, Hu Y, Zou Y, Zhang Z, Zhang W. Cobalt-catalyzed efficient asymmetric hydrogenation of α-primary amino ketones. JACS Au. 2023;3(11):2981-2986.[DOI]
-
31. Chen T, Hu Y, Tang X, Zou Y, Wei L, Zhang Z, et al. Cobalt-catalyzed enantioselective reductive amination of ketones with hydrazides. Org Lett. 2024;26(4):769-774.[DOI]
-
32. He J, Li Z, Li R, Kou X, Liu D, Zhang W. Bimetallic Ru/Ru-catalyzed asymmetric one-pot sequential hydrogenations for the stereodivergent synthesis of chiral lactones. Adv Sci. 2024;11(23):2400621.[DOI]
-
33. Li B, Wang Z, Luo Y, Wei H, Chen J, Liu D, et al. Nickel-catalyzed asymmetric hydrogenation for the preparation of α-substituted propionic acids. Nat Commun. 2024;15(1):5482.[DOI]
-
34. Ding Z, Luo Y, Yuan Q, Wang G, Yu Z, Zhao M, et al. Ru-catalyzed asymmetric hydrogenation of α,β-unsaturated γ-lactams. J Am Chem Soc. 2024;146(36):25312-25320.[DOI]
-
35. Guan J, Luo Y, Wang Q, Chen J, Zhang W. Copper-catalyzed asymmetric hydrogenation of unsymmetrical ortho-Br substituted benzophenones. Angew Chem Int Ed. 2025;64(4):e202416313.[DOI]
-
36. Wang M, Liu S, Liu H, Wang Y, Lan Y, Liu Q. Asymmetric hydrogenation of ketimines with minimally different alkyl groups. Nature. 2024;631(8021):556-562.[DOI]
-
37. Wei H, Luo Y, Li J, Chen J, Gridnev ID, Zhang W. Enantioselective synthesis of chiral β2-amino phosphorus derivatives via nickel-catalyzed asymmetric hydrogenation. J Am Chem Soc. 2025;147(1):342-352.[DOI]
-
38. Song Y, Zou Y, Chen T, Zhang Z, Zhang W. Cobalt-catalyzed asymmetric hydrogenation of α-hydroxy ketones enabled by a carboxylic acid additive promotion strategy. Angew Chem Int Ed. 2025;64(27):e202504159.[DOI]
-
39. Ye J, Luo Y, Xu Y, Liu D, Zhang W. Rh-catalyzed asymmetric hydrogenation of 3-amino-4-alkyl/aryl disubstituted maleimides. J Am Chem Soc. 2025;147(25):22121-22131.[DOI]
-
40. Cai X, Luo Y, Chen J, Zhang W. Enantioselective nickel-catalyzed hydrogenation of α-alkylidene succinimides enabled by weak noncovalent interactions. Angew Chem Int Ed. 2025;64(34):e202510401.[DOI]
-
41. Jones M, Harris D, Struble J, Hayes M, Koeller K, Chepiga Özgün K, et al. Development of a practical process for the large-scale preparation of the chiral pyridyl-backbone for the crabtree/pfaltz-type iridium complex used in the industrial production of the novel fungicide inpyrfluxam. Org Process Res Dev. 2022;26(8):2407-2414.[DOI]
-
42. Xu WX, Peng Z, Gu QX, Zhu Y, Zhao LH, Leng F, et al. Cyclolignan synthesis streamlined by enantioselective hydrogenation of tetrasubstituted olefins. Nat Synth. 2024;3(8):986-997.[DOI]
-
43. Dong B, Wang T, Gao G, Zhang X, He Y. Development of a practical and scalable manufacturing process for ciprofol. Org Process Res Dev. 2025;29(5):1291-1298.[DOI]
-
44. Huang YH, Gu QX, Chao QC, Xiao HZ, Lu HH. Enantioselective divergent total syntheses of cycloaurenones and dysiherbols. Angew Chem Int Ed. 2025;64(30):e202507638.[DOI]
-
45. Massaro L, Zheng J, Margarita C, Andersson P.G. Enantioconvergent and enantiodivergent catalytic hydrogenation of isomeric olefins. Chem Soc Rev. 2020;49:2504-2522.[DOI]
-
46. Chauhan P, Mahajan S, Enders D. Organocatalytic carbon–sulfur bond-forming reactions. Chem Rev. 2014;114(18):8807-8864.[DOI]
-
47. Ge Y, Ke J, He C. Catalytic asymmetric dehydrogenative Si−H/X−H coupling toward Si-stereogenic silanes. Acc Chem Res. 2025;58(3):375-398.[DOI]
-
48. Bull JA. Catalytic enantioselective synthesis of secondary alkylboronate building blocks with and without metals. Angew Chem Int Ed. 2012;51(36):8930-8932.[DOI]
-
49. Xie JH, Zhu SF, Zhou QL. Transition metal-catalyzed enantioselective hydrogenation of enamines and imines. Chem Rev. 2011;111(3):1713-1760.[DOI]
-
50. Charvillat T, Bernardelli P, Daumas M, Pannecoucke X, Ferey V, Besset T. Hydrogenation of fluorinated molecules: An overview. Chem Soc Rev. 2021;50:8178-8192.[DOI]
-
51. Luo S, Zhu H, Ngai KY, Wang J. Recent advances in stereoselective synthesis of amino-phosphine oxides. ChemCatChem. 2025;17(11):e202500037.[DOI]
-
52. Wu Y, Wang P. Silicon-stereogenic monohydrosilane: Synthesis and applications. Angew Chem Int Ed. 2022;61(36):e202205382.[DOI]
-
53. Franz AK, Wilson SO. Organosilicon molecules with medicinal applications. J Med Chem. 2013;56(2):388-405.[DOI]
-
54. Hayashi Y. Time and pot economy in total synthesis. Acc Chem Res. 2021;54(6):1385-1398.[DOI]
-
55. Jagannathan JR, Fettinger JC, Shaw JT, Franz AK. Enantioselective Si−H insertion reactions of diarylcarbenes for the synthesis of silicon-stereogenic silanes. J Am Chem Soc. 2020;142(27):11674-11679.[DOI]
-
56. Wu Y, Zheng L, Wang Y, Wang P. Catalytic asymmetric synthesis of silicon-stereogenic organosilanes. Chem. 2023;9(12):3461-3514.[DOI]
-
57. Liu MM, Xu Y, He C. Catalytic asymmetric dehydrogenative Si−H/N−H coupling: Synthesis of silicon-stereogenic silazanes. J Am Chem Soc. 2023;145(21):11727-11734.[DOI]
-
58. Wu L, Zhang L, Guo J, Gao J, Ding Y, Ke J, et al. Catalytic asymmetric construction of C- and Si-stereogenic silacyclopentanes via hydrosilylation of arylmethylenecyclopropanes. Angew Chem Int Ed. 2024;63(48):e202413753.[DOI]
-
59. Panella L, Feringa BL, de Vries JG, Minnaard AJ. Enantioselective Rh-catalyzed hydrogenation of enol acetates and enol carbamates with monodentate phosphoramidites. Org Lett. 2005;7(19):4177-4180.[DOI]
-
60. Zhang Z, Han Z, Gu G, Dong XQ, Zhang X. Enantioselective synthesis of chiral 3-substituted-3-silylpropionic esters via rhodium/bisphosphine-thiourea-catalyzed asymmetric hydrogenation. Adv Synth Catal. 2017;359(15):2585-2589.[DOI]
-
61. Källström K, Munslow IJ, Hedberg C, Andersson PG. Iridium-catalysed asymmetric hydrogenation of vinylsilanes as a route to optically active silanes. Adv Synth Catal. 2006;348(18):2575-2578.[DOI]
-
62. Mazuela J, Paptchikhine A, Pàmies O, Andersson PG, Diéguez M. Adaptative biaryl phosphite−oxazole and phosphite−thiazole ligands for asymmetric Ir-catalyzed hydrogenation of alkenes. Chem Eur J. 2010;16(15):4567-4576.[DOI]
-
63. Mazuela J, Norrby PO, Andersson PG, Pàmies O, Diéguez M. Pyranoside phosphite−oxazoline ligands for the highly versatile and enantioselective Ir-catalyzed hydrogenation of minimally functionalized olefins. A combined theoretical and experimental study. J Am Chem Soc. 2011;133(34):13634-13645.[DOI]
-
64. Coll M, Pàmies O, Diéguez M. A modular furanoside thioether-phosphite/phosphinite/phosphine ligand library for asymmetric iridium-catalyzed hydrogenation of minimally functionalized olefins: Scope and limitations. Adv Synth Catal. 2013;355(1):143-160.[DOI]
-
65. Ma DD, Gu P, Li R. Asymmetric hydrogenation of 1-silyl-1-substituted alkenes for preparation of optically active silanes. Tetrahedron Lett. 2016;57(50):5666-5668.[DOI]
-
66. Wang A, Bernasconi M, Pfaltz A. Iridium-catalyzed enantioselective hydrogenation of vinylsilanes. Adv Synth Catal. 2017;359(15):2523-2529.[DOI]
-
67. Guo J, Shen X, Lu Z. Regio- and enantioselective cobalt-catalyzed sequential hydrosilylation/hydrogenation of terminal alkynes. Angew Chem Int Ed. 2017;56(2):615-618.[DOI]
-
68. Zuo Z, Xu S, Zhang L, Gan L, Fang H, Liu G, et al. Cobalt-catalyzed asymmetric hydrogenation of vinylsilanes with a phosphine−pyridine−oxazoline ligand: Synthesis of optically active organosilanes and silacycles. Organometallics. 2019;38(20):3906-3911.[DOI]
-
69. Leeuwen PWNMv, Kamer PCJ, Claver C, Pàmies O, Diéguez M. Phosphite-containing ligands for asymmetric catalysis. Chem Rev. 2011;111(3):2077-2118.[DOI]
-
70. Shen C, Zhang P, Sun Q, Bai S, Hor TSA, Liu X. Recent advances in C–S bond formation via C–H bond functionalization and decarboxylation. Chem Soc Rev. 2015;44(1):291-314.[DOI]
-
71. Tokat AO, Akbulut A, Billur D, Koca G, Bayram P, Kuru S, et al. Montelukast attenuates radioactive I131-induced pulmonary damage on rats. Int J Radiat Biol. 2018;94(6):542-550.[DOI]
-
72. Peters BK, Zhou T, Rujirawanich J, Cadu A, Singh T, Rabten W, et al. An enantioselective approach to the preparation of chiral sulfones by ir-catalyzed asymmetric hydrogenation. J Am Chem Soc. 2014;136(47):16557-16562.[DOI]
-
73. Jiang J, Wang Y, Zhang X. Rhodium-catalyzed asymmetric hydrogenation of β-acetylamino acrylosulfones: A practical approach to chiral β-amido sulfones. ACS Catal. 2014;4(5):1570-1573.[DOI]
-
74. Tosatti P, Pfaltz A. Iridium-catalyzed asymmetric hydrogenation of benzo[b]thiophene 1,1-dioxides. Angew Chem Int Ed. 2017;56(16):4579-4582.[DOI]
-
75. Liu G, Zhang H, Huang Y, Han Z, Liu G, Liu Y, et al. Efficient synthesis of chiral 2,3-dihydro-benzo[b]thiophene 1,1-dioxides via rh-catalyzed hydrogenation. Chem Sci. 2019;10(8):2507-2512.[DOI]
-
76. Yan Q, Xiao G, Wang Y, Zi G, Zhang Z, Hou G. Highly efficient enantioselective synthesis of chiral sulfones by Rh-catalyzed asymmetric hydrogenation. J Am Chem Soc. 2019;141(4):1749-1756.[DOI]
-
77. Kuroki Y, Asada D, Sakamaki Y, Iseki K. Enantioselective synthesis of 1,1,1-trifluoroalkan-2-ols by ruthenium-catalyzed hydrogenation. Tetrahedron Lett. 2000;41(23):4603-4607.[DOI]
-
78. Urban S, Beiring B, Ortega N, Paul D, Glorius F. Asymmetric hydrogenation of thiophenes and benzothiophenes. J Am Chem Soc. 2012;134(37):15241-15244.[DOI]
-
79. Li W, Schlepphorst C, Daniliuc C, Glorius F. Asymmetric hydrogenation of vinylthioethers: Access to optically active 1,5-benzothiazepine derivatives. Angew Chem Int Ed. 2016;55(10):3300-3303.[DOI]
-
80. Paul D, Beiring B, Plois M, Ortega N, Kock S, Schlüns D, et al. A cyclometalated ruthenium-NHC precatalyst for the asymmetric hydrogenation of (hetero)arenes and its activation pathway. Organometallics. 2016;35(20):3641-3646.[DOI]
-
81. Meindertsma AF, Pollard MM, Feringa BL, de Vries JG, Minnaard AJ. Asymmetric hydrogenation of alkyl (vinyl) thioethers: a promising approach to α-chiral thioethers. Tetrahedron: Asymmetry. 2007;18(24):2849-2858.[DOI]
-
82. Gao W, Lv H, Zhang X. Rh/duanphos-catalyzed asymmetric hydrogenation of β-acetylamino vinylsulfides: An approach to chiral β-acetylamino sulfides. Org Lett. 2017;19(11):2877-2880.[DOI]
-
83. Ruchelman AL, Connolly TJ. Enantioselective synthesis of the apremilast aminosulfone using catalytic asymmetric hydrogenation. Tetrahedron: Asymmetry. 2015;26(10-11):553-559.[DOI]
-
84. Liu G, Han Z, Dong XQ, Zhang X. Rh-catalyzed asymmetric hydrogenation of β-substituted-β-thio-α,β-unsaturated esters: Expeditious access to chiral organic sulfides. Org Lett. 2018;20(18):5636-5639.[DOI]
-
85. Yin C, Yang T, Pan Y, Wen J, Zhang X. Rh-catalyzed asymmetric hydrogenation of unsaturated medium-ring NH lactams: Highly enantioselective synthesis of N-unprotected 2,3-dihydro-1,5-benzothiazepinones. Org Lett. 2020;22(3):920-923.[DOI]
-
86. Wu X, Li M, Guo Q, Zi G, Hou G. Rh-catalyzed asymmetric hydrogenation of 2-substituted 4H-(thio)chromenes for synthesis of chiral (thio)chromanes. Org Lett. 2024;26(28):5917-5922.[DOI]
-
87. Valla C, Baeza A, Menges F, Pfaltz A. Enantioselective synthesis of chromanes by iridium-catalyzed asymmetric hydrogenation of 4H-chromenes. Synlett. 2008;2008(20):3167-3171.[DOI]
-
88. Chung W, Vanderwal CD. Stereoselective halogenation in natural product synthesis. Angew Chem Int Ed. 2016;55(14):4396-4434.[DOI]
-
89. Wang CJ, Sun X, Zhang X. Enantioselective hydrogenation of allylphthalimides: An efficient method for the synthesis of β-methyl chiral amines. Angew Chem Int Ed. 2005;44(31):4933-4935.[DOI]
-
90. Sun X, Zhou L, Li W, Zhang X. Convenient divergent strategy for the synthesis of tunephos-type chiral diphosphine ligands and their applications in highly enantioselective ru-catalyzed hydrogenations. J Org Chem. 2008;73(3):1143-1146.[DOI]
-
91. King SM, Calandra NA, Herzon SB. Total syntheses of (-)-acutumine and (-)-dechloroacutumine. Angew Chem Int Ed. 2013;52(13):3642-3645.[DOI]
-
92. Gazić Smilović I, Casas-Arcé E, Roseblade SJ, Nettekoven U, Zanotti-Gerosa A, Kovačevič M, et al. Iridium-catalyzed chemoselective and enantioselective hydrogenation of (1-chloro-1-alkenyl) boronic esters. Angew Chem Int Ed. 2012;51(4):1014-1018.[DOI]
Copyright
© The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Zhang J, Chen Y, Zhang Z, Zhang W. Advances in catalytic asymmetric hydrogenation of third-row heteroatom-substituted alkenes. Chiral Chem. 2025;1:202507. https://doi.org/10.70401/cc.2025.0002
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Asymmetric Hydrogenation of Vinyl Silanes
- 3. Asymmetric Hydrogenation of Vinyl Phosphines
- 4. Asymmetric Hydrogenation of Vinyl Sulfides
- 5. Asymmetric Hydrogenation of Vinyl Chlorides
- 6. Summary and Outlook
- Authors contribution
- Conflict of interests
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- Copyright
Science Exploration Style
Zhang J, Chen Y, Zhang Z, Zhang W. Advances in catalytic asymmetric hydrogenation of third-row heteroatom-substituted alkenes. Chiral Chem. 2025;1:202507. https://doi.org/10.70401/cc.2025.0002
copy
Share Link
copy