Transition metal-catalyzed remote asymmetric C–H activation of arenes
Lili Chen

Senmiao Xu
*
*Correspondence to:
Senmiao Xu, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China.
E-mail: smxu@chem.ecnu.edu.cn
Chiral Chem. 2026;2:202509. 10.70401/cc.2025.0006
Received: November 07, 2025Accepted: December 10, 2025Published: December 16, 2025
Abstract
Transition metal-catalyzed asymmetric C–H activation is vital for chiral molecule synthesis but faces challenges in remote C–H functionalization due to traditional metallacycle constraints and difficulties in long-range chiral recognition. This review summarizes three core strategies to address these issues: template-assisted chiral ligand control, norbornene-mediated palladium catalysis, and bifunctional catalyst control. These strategies achieve high enantioselectivity for diverse chiral architectures. Future directions include expanding to para-C–H bonds of arenes and aliphatic C–H bonds, developing robust chiral mediators/ligands, and applying the methodology to natural products and complex materials.
Graphical Abstract
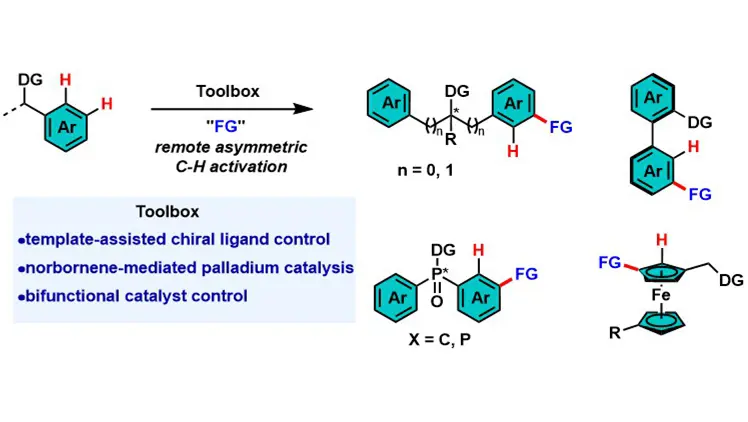
Keywords
Asymmetric catalysis, C–H activation, remote selectivity
1. Introduction
Transition metal-catalyzed asymmetric C–H activation has emerged as a powerful tool for constructing complex chiral molecules, with significant applications in organic synthesis, pharmaceuticals, and materials science[1-12]. Traditional approaches to asymmetric C–H activation typically involve the formation of four-, five-, or six-membered chiral metallacycles, which limits the functionalization of remote C–H bonds distal to existing functional groups. Although numerous achiral protocols for remote C–H activation have been documented[13], spatial constraints pose a significant challenge to achieving remote stereoselectivity. This is because the stereocenter of the chiral ligand or mediator must be efficiently transferred to the remote prochiral center of the substrate. Despite these hurdles, recent advances have demonstrated the feasibility of remote C–H activation of arenes through innovative strategies (Figure 1), including template-assisted chiral ligand-controlled enantioselective meta-C–H activation, norbornene (NBE)-mediated enantioselective meta-C–H activation, and bifunctional chiral catalyst-controlled enantioselective meta-C–H activation. The first strategy requires a template to direct the metal center toward the remote C–H bond, with the chiral ligand governing enantioselective C–H activation (Figure 1A). The second strategy comprises two pathways (Figure 1B): in pathway A, enantioselectivity is controlled by the enantioselective 1,2-migratory insertion of an ortho-metallated intermediate into a chiral NBE, while regioselectivity is determined by subsequent C–H activation; in Pathway B, enantioselectivity is regulated by chiral ligand-enabled ortho-C–H activation, and regioselectivity is controlled by subsequent 1,2-migratory insertion/second C–H activation. In the third strategy, the bifunctional catalyst interacts with the substrate via hydrogen bonding or Lewis acid–base interactions. The distance between the two functionalities can orient the metal center in proximity to the meta-C–H bonds, and the chiral scaffold dictates enantioselectivity (Figure 1C). These approaches have been pivotal in overcoming the limitations of traditional methods and have opened new avenues for the synthesis of chiral compounds. This review aims to comprehensively summarize the aforementioned strategies for enantioselective remote C–H activation of arenes. It is anticipated that this review will not only facilitate a deeper understanding of the current state-of-the-art but also inspire the development of more efficient, practical, and general methodologies for addressing the long-standing challenges in remote stereoselective C–H activation.
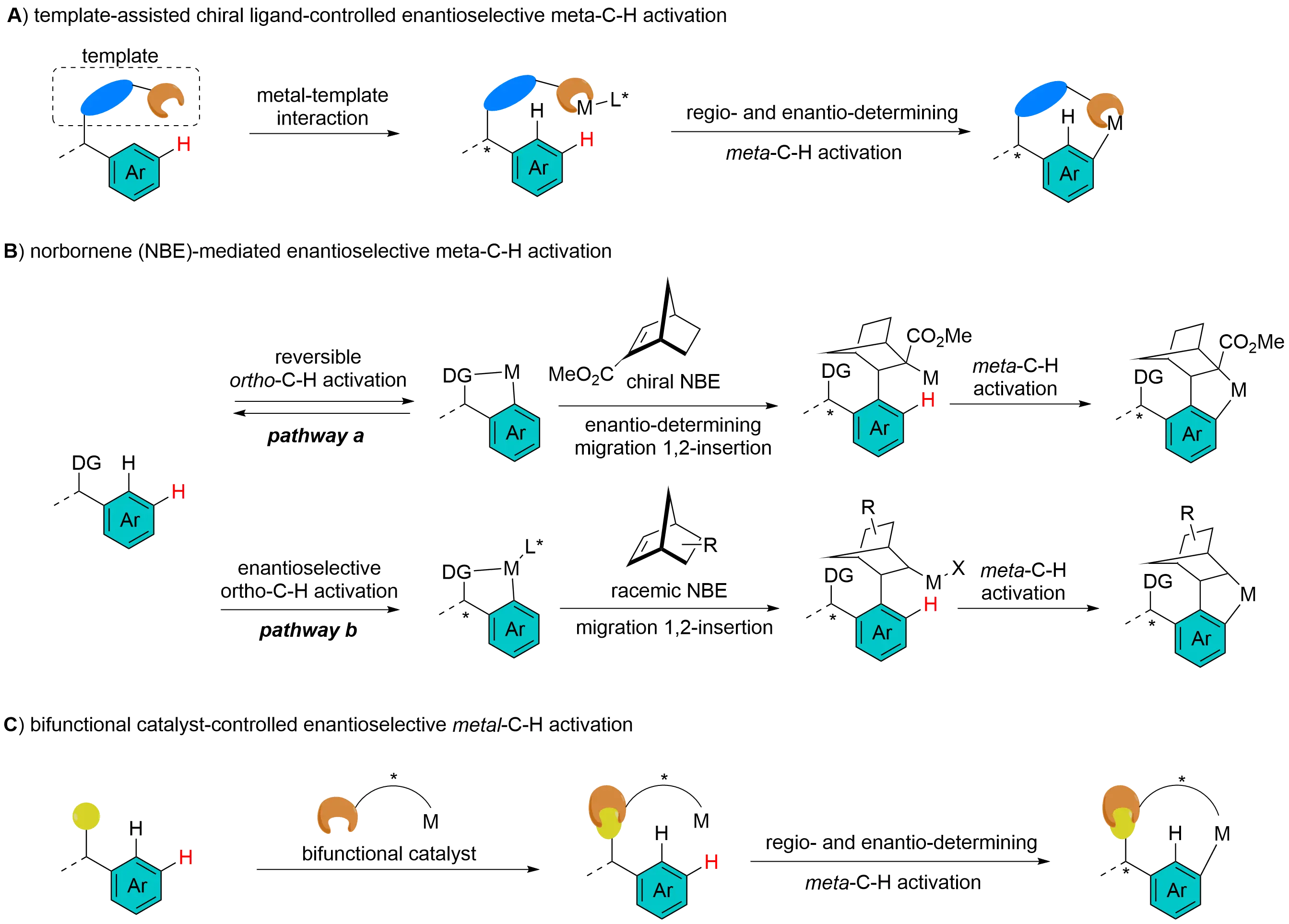
Figure 1. Current strategies for transition metal-catalyzed remote enantioselective C–H activation.
2. Transition Metal-Catalyzed Remote Asymmetric C-H Activation
2.1 Template-assisted chiral ligand controlled enantioselective meta-C-H activation
To address the challenge of activating remote meta-C–H bonds, which is hindered by proximity-driven reactivity in traditional directed C–H activation, and to achieve high positional selectivity overriding the intrinsic electronic/steric biases of arenes, Leow et al. designed easily removable nitrile-containing templates that guide Pd(II) catalysts to distal meta-C–H bonds via weak “end-on” coordination of the nitrile group, relieving the strain of cyclophane-like pre-transition states, hence achieving highly meta-selective C–H olefination of toluene derivatives and hydrocinnamic acids[14]. Li et al. designed a carboxyl group-containing template for the meta-C–H olefination of hydrocinnamic acids with a Pd catalyst via possible κ² coordination[15]. Later, by utilizing the same template for long-range site recognition, the Li group achieved enantioselective meta–C–H olefination and arylation of prochiral hydrocinnamic acid-derived amides 1 using the mono-protected amino acid (MPAA) Ac-L-Phe-OH through desymmetrization (Figure 2). This protocol features a broad substrate scope, delivering the corresponding olefinated and arylated products (2 and 3) with moderate to good enantioselectivities[16]. Preliminary computational studies revealed that the origin of the enantioselectivity was controlled by the distortion energy of the substrate moiety imparted by the formation of the large 12-membered palladacycle.
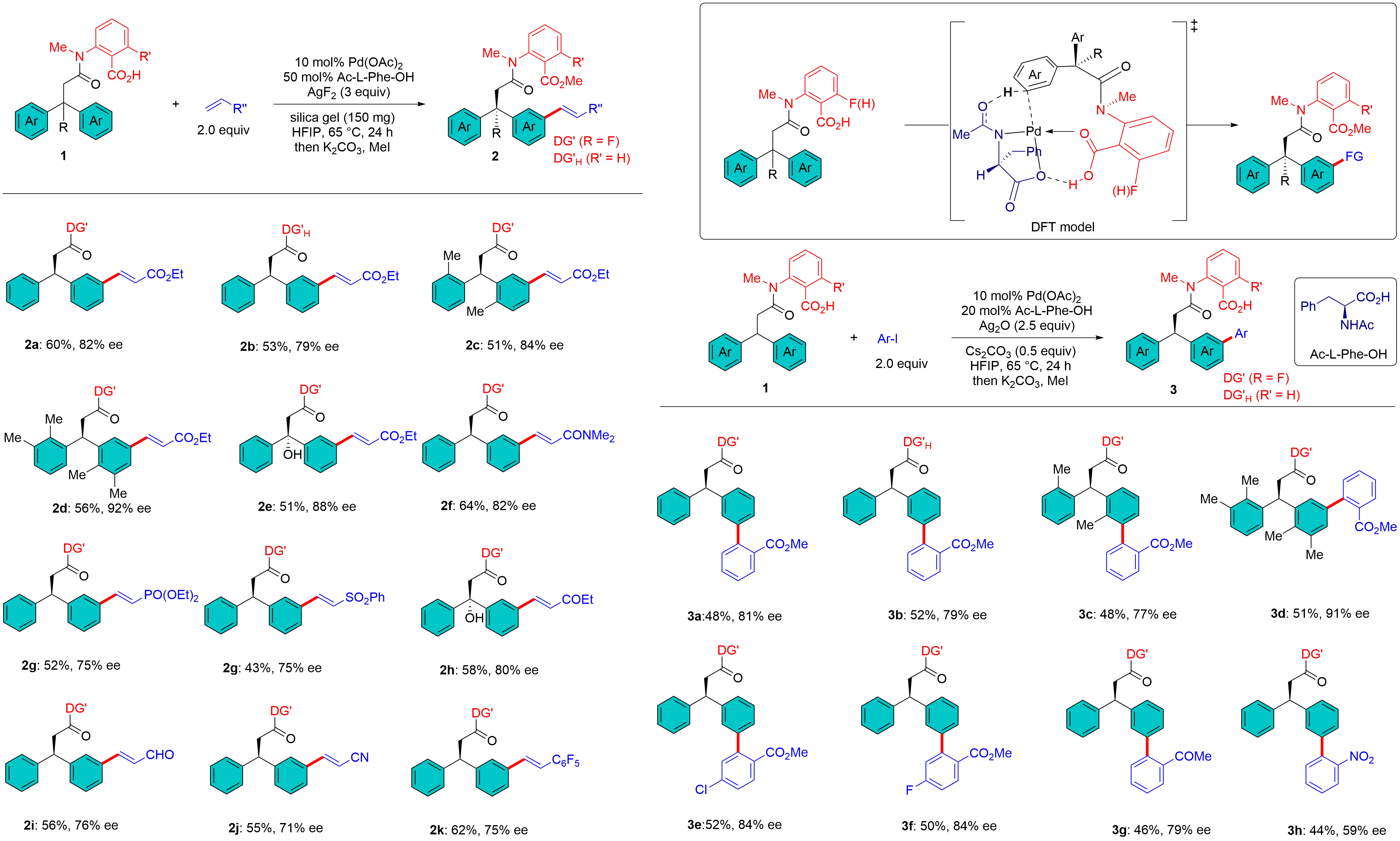
Figure 2. Template-assisted MMPA controlled enantioselective meta-C–H activation. MMPA: mono-protected amino acid.
2.2 Norbornene-mediated asymmetric meta-C–H activation of arenes
The Catellani reaction[17], which employs Pd/NBE catalysis, enables convenient functionalization or dual functionalization of ortho C–H bonds in aryl iodides. This efficient regioselectivity offers potential for directing-group-enabled remote C–H bond functionalization. In 2015, Wang et al. demonstrated that Pd-catalyzed meta-C–H activation of arenes could be achieved using NBE as a transient mediator[18]. A newly developed pyridine-based ligand was found to be crucial for relaying the palladium catalyst to the meta position via NBE after the initial ortho C–H activation. In the same year, Dong et al. reported a meta-selective C–H arylation reaction[19]. This transformation utilized a simple tertiary amine as the directing group and was enabled by Pd/NBE catalysis. The reaction was facilitated by a commercially available ligand, AsPh₃, and a unique “acetate cocktail”. These two pioneering studies opened up avenues for achieving asymmetric remote C–H functionalization via Pd/NBE catalysis, as the NBE could affect the step of meta-C–H activation.
In 2018, the Shi et al. group reported an enantioselective version of remote C–H arylation/alkylation of diarylmethylamines and homobenzyl amines using Pd/NBE catalysis for the construction of central chirality[20]. They designed the chiral NBE methyl (1S, 4R)-bicyclo[2.2.1]-hept-2-ene-2-carboxylate ((+)-NBE-CO2Me) as the catalytic chiral transient mediator to control enantioselectivity. In the meta-C-H arylation of diarylmethylamines (4), the combination of (+)-NBE-CO2Me and a chiral CPA was important for achieving high chiral induction. This protocol could tolerate various diarylmethylamines and aryl iodides, including heteroaryl iodides, affording corresponding products 5a-5l with good to excellent enantioselectivities (Figure 3A). Further kinetic studies revealed that a pyridone ligand could improve the product selectivity and the catalyst robustness for the enantio-determining insertion of an arylpalladium into (+)-NBE-CO2Me[21]. Importantly, the chiral transient mediator (+)-NBE-CO2Me was also effective for the asymmetric meta-C–H activation of homobenzyl amines (6), including, desymmetrization and kinetic resolution. After screening several chiral mediators, (+)-NBE-CO2Me was identified as the most effective. Additionally, a chiral phosphoric acid (CPA) was not required for this transformation. Consequently, nosyl (Ns)-protected homobenzyl amines underwent meta-C–H arylation and alkylation smoothly, yielding the corresponding chiral amines 7a-4l with good enantioselectivities (Figure 3B). Building on kinetic studies for arylation of diarylmethylamine 4m, a proposed mechanism is illustrated in Figure 3C[21]. The reaction begins with the coordination of substrate 4m to the Pd species, forming intermediate 9. Subsequent reversible ortho-C–H activation generates palladacycle 10, which is identified as a catalytic resting state. The rate- and enantio-determining step is the 1,2-migratory insertion of 10 into (+)-NBE-CO2Me, leading to intermediate 11. This is followed by oxidative addition of an aryl iodide, ultimately affording the final product 5m.
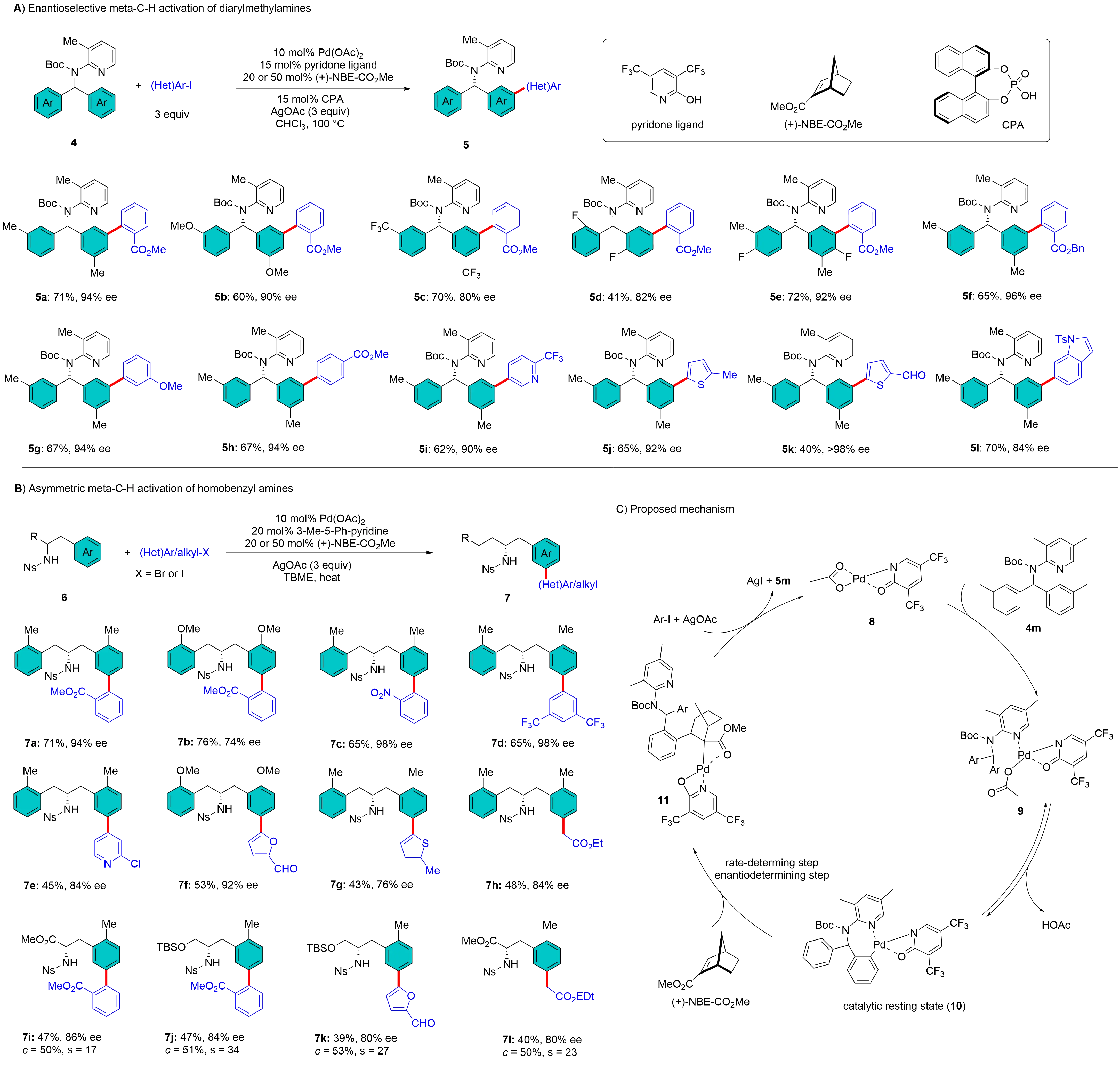
Figure 3. Pd/Chiral NBE catalysis for constructing central chirality. NBE: norbornene.
Beyond the construction of central chirality, the Pd/NBE catalytic system has also proven to be an effective approach in generating planar chirality. In 2023, Zhou et al. reported an enantioselective remote C–H arylation of ferrocenes, which led to the synthesis of 1,3-disubstituted chiral ferrocenes (Figure 4). This achievement was facilitated by employing a simple tertiary amine as the directing group, an MPAA Boc-L-Val-OH as the chiral ligand, and racemic bridgehead alkyl substituted NBE (NBE-alkyl: NBE-C6H13 or NBE-C7H15) as the transient mediator. This combination allowed for the incorporation of a wide range of functionalities, resulting in the synthesis of chiral ferrocenes 13a-l with outstanding enantioselectivities (Figure 4A)[22]. Through control experiments, the authors demonstrated that the chirality of Boc-L-Val-OH is the primary determinant of the enantioselectivity, rather than the chirality of the NBE mediator, indicating that the initial ortho-C–H activation is the enantio-determining step (Figure 4B). An initial mechanistic investigation, utilizing deuterium-labeling experiments, suggested that the proton source for the final C–Pd protonation step is derived from the adventitious water (or other exchangeable protic species) present in the reaction system. The authors also demonstrated synthetic applications, particularly in the preparation of chiral bidentate phosphine ligands and chiral amino alcohols (Figure 4C). The latter could serve as ligands, exhibiting high enantioselectivity in the Zn-catalyzed nucleophilic addition of benzaldehyde with Et2Zn.
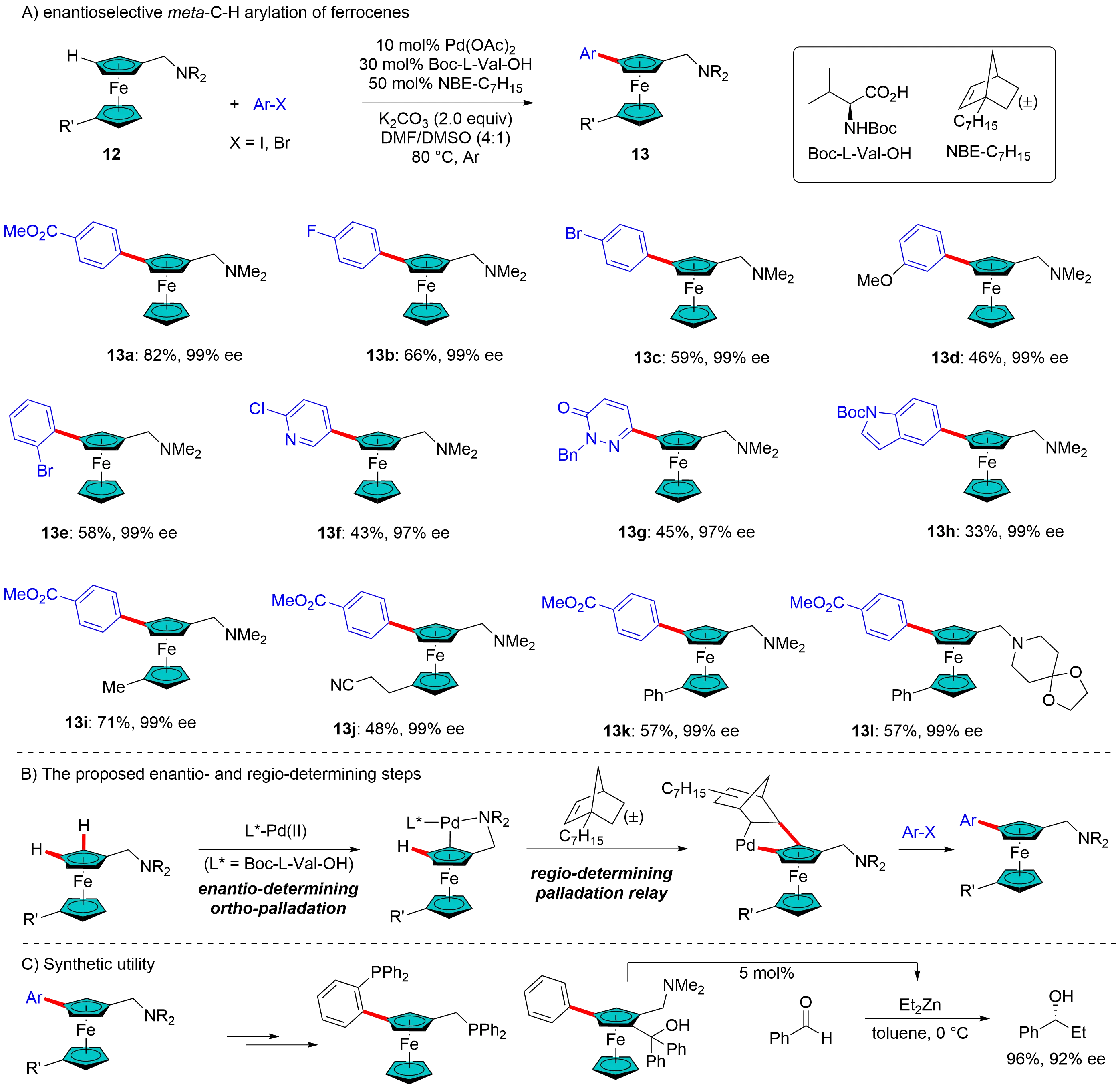
Figure 4. Pd/Racemic NBE catalysis for enantioselective meta-C–H activation to ferrocenes. NBE: norbornene.
In 2023, the Yu group further extended the capabilities of Pd/NBE catalysis to the construction of axial chirality through atroposelective remote meta-C–H activation, which was recently highlighted by Dai and Lu[23]. Only a few examples were recorded for distal atroposelective C–H bromination of biaryls via organocatalysis[24-26]. This elegant approach involved the use of racemic primary biaryl amines, which, in the presence of a catalytic amount of Pd(OAc)2 and the chiral transient mediator (+)-NBE-CO2Me, underwent kinetic resolution (KR) smoothly using aryl bromides as the limiting reagent (Figure 5). This protocol effectively produced both meta-C–H arylated chiral biaryl amines 15a-o and recovered starting materials 14a-o with high enantioselectivities (up to an s factor of 167), demonstrating exceptional KR performance[27]. This method features a broad spectrum of substrate scope and aryl bromides. One of the arylated products obtained was successfully converted into a chiral thiourea and a chiral monodentate phosphine ligand. Notably, the latter exhibited excellent enantiocontrol in Pd-catalyzed asymmetric allylic alkylation reactions. Control experiments confirmed the indispensable roles of both Pd and (+)-NBE-CO2Me in facilitating the meta-C–H arylation. Furthermore, the inclusion of AgOAc was shown to significantly enhance the reactivity. The authors further elaborated on a proposed mechanism that involves a Pd(II)/Pd(IV)/Pd(II) catalytic cycle, where (+)-NBE-CO2Me acts as the chiral transient mediator, enabling the stereoselective transfer of the Pd center to the remote meta-position.
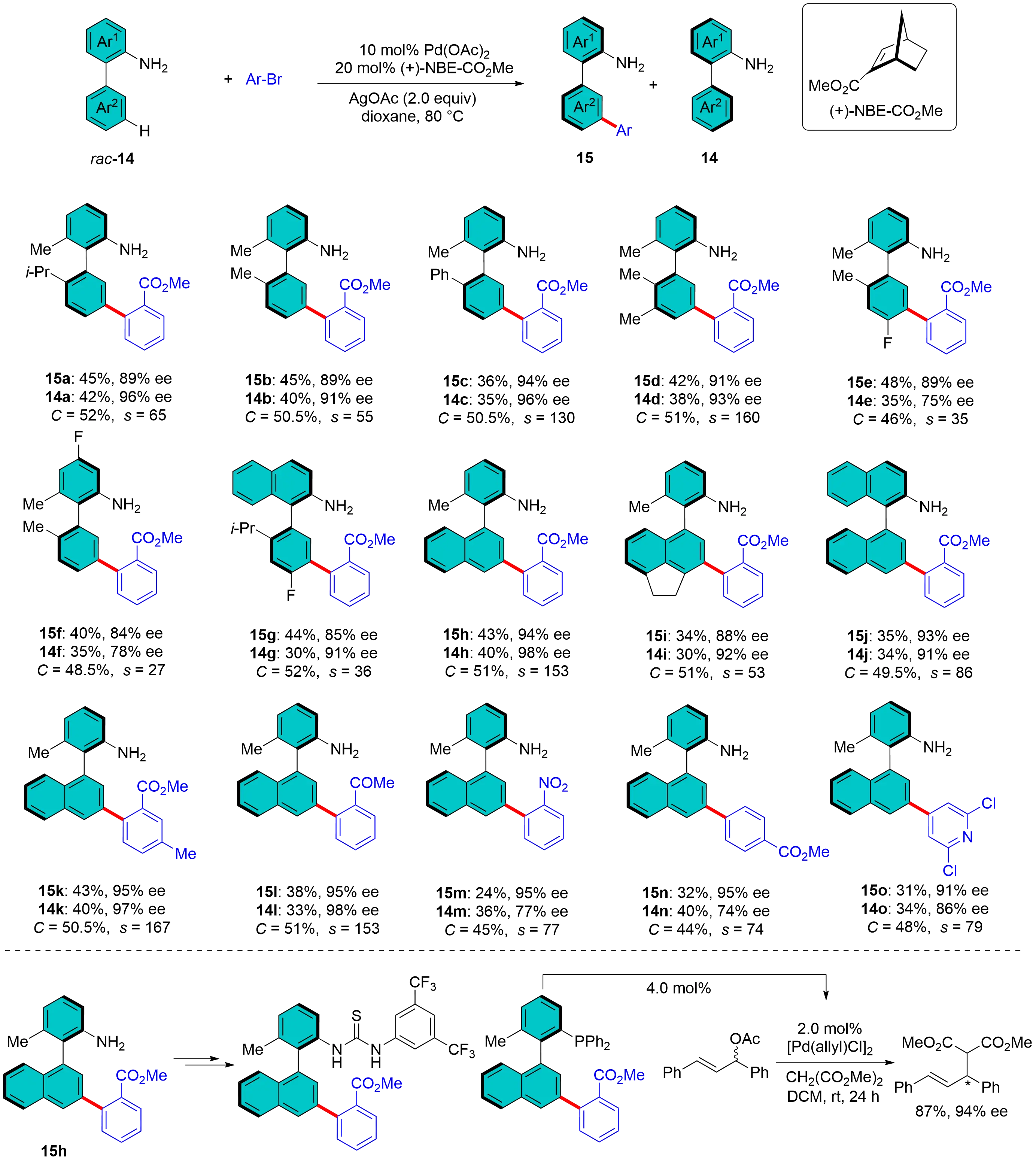
Figure 5. Pd/Chiral NBE catalysis for meta-C–H activation to biaryls via kinetic resolution. NBE: norbornene.
An alternative strategy for atroposelective C–H functionalization involves the combination of a chiral MPAA ligand and a racemic NBE. This approach initiates with a stereoselective ortho-C–H activation achieved through KR, followed by a subsequent Catellani relay step. In 2025, Li et al. demonstrated the effectiveness of this method in the Pd-catalyzed KR of racemic quinoline-based biaryls via remote C–H arylation in the presence of a chiral MPAA ligand L-Pyr-OH and racemic NBE-CO2Me (Figure 6). This protocol efficiently produced a diverse array of chiral meta-functionalized biaryl products. Both the products 17a-o and the recovered starting materials 16a-o exhibited outstanding enantioselectivities (with s factors up to 458), indicating the highly effective performance of the KR process[28]. The chiral biaryls functionalized with meta-C–H bonds, along with the recovered starting materials, were successfully transformed into various useful molecules. Notably, these transformations led to the creation of a novel P,N-ligand and a chiroptical functional material, highlighting the practical applications of the atroposelective remote C-H functionalization strategy.
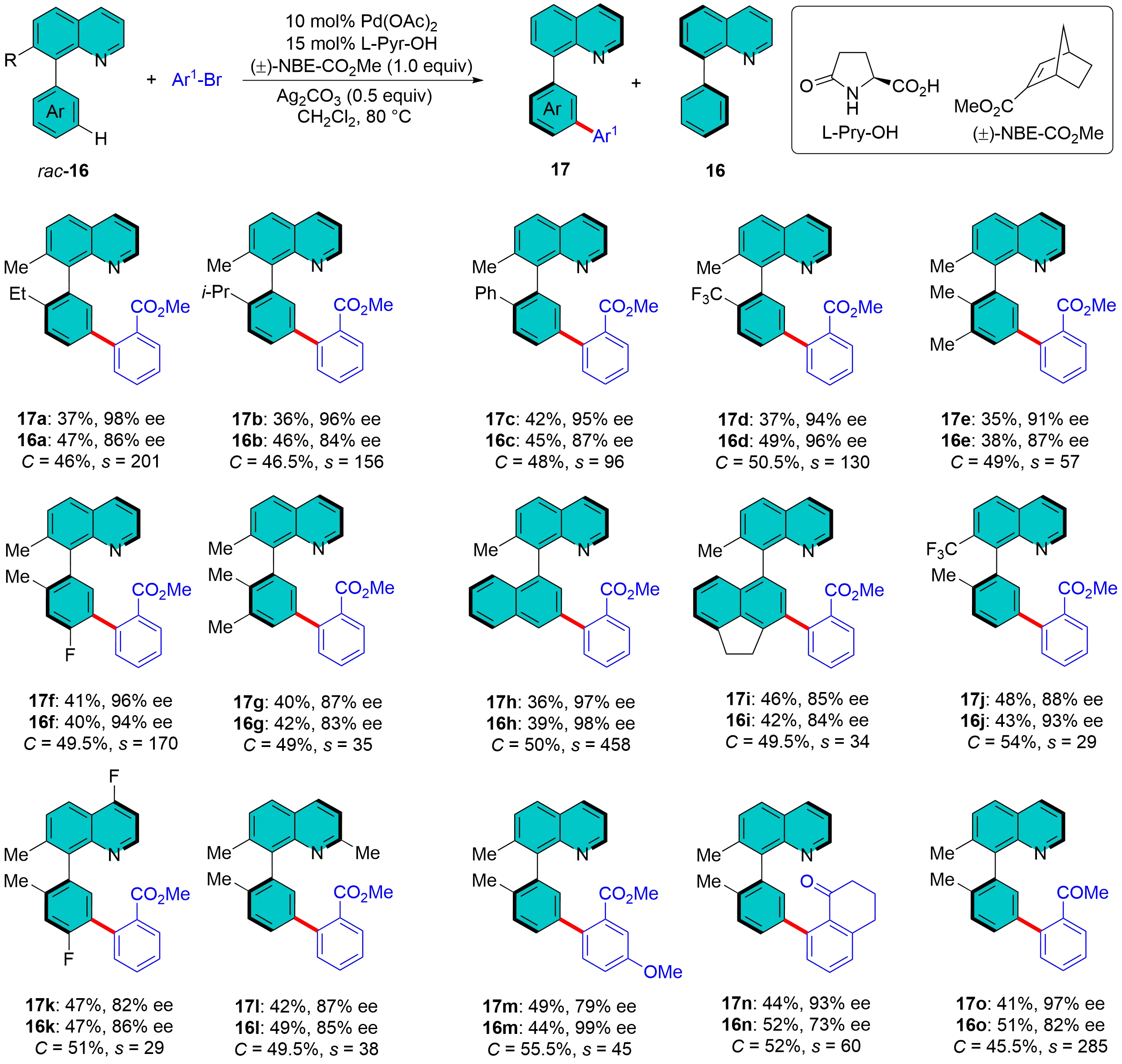
Figure 6. Pd/Racemic NBE catalysis for meta-C–H activation to biaryls via kinetic resolution. NBE: norbornene.
2.3 Bifunctional catalyst-controlled asymmetric meta-C–H activation of arenes
Although considerable efforts have been directed toward the meta-C–H borylation of arenes, asymmetric variants remain limited[29]. In 2016, Mihai et al. developed anionic sulfonate-tethered bipyridine ligands for ion pair-directed Ir-catalyzed meta-C–H borylation of quaternized phenethylamines and phenylpropylamines[30]. The distal positioning of the sulfonate group in bipyridine ligands is essential for achieving high remote regioselectivity. Subsequently, they reported that the tetra-butylammonium salt of the previously used sulfonate ligand could function as a single anionic bipyridine ligand in hydrogen bonding-directed meta-C–H borylation of amides derived from benzylamine, phenylethylamine, and phenylpropylamine[31]. Building on these results, the same group replaced the achiral tetrabutylammonium counterion of the ligand with a chiral cation derived from N-benzyl-substituted dihydroquinine (DHQ), obtaining a chiral ion pair. In this system, the distal sulfonate group acts as a hydrogen bond acceptor to control regioselectivity, while the DHQ-based counterion provides a chiral environment that governs long-range enantioselectivity[32]. The reaction of prochiral diarylmethyl amides 18a-j with B₂pin₂ achieved highly meta-selective C–H borylation in 85-96% enantioselectivities. Furthermore, this protocol is applicable to the synthesis of optically active P-stereogenic compounds 20k-o through enantioselective meta-C–H borylation of diarylphosphinamides 18k-o (Figure 7). Computational studies identified distinct noncovalent interaction patterns between amide/phosphinamide substrates and cinchona alkaloid-derived chiral cations in iridium-catalyzed enantioselective C–H borylation, with amides relying on cation–dipole interactions and phosphinamides engaging in hydrogen bonds and π–π interactions[33]. These insights confirm the chiral cation’s role in controlling enantioselectivity and expand the methodology’s scope to substrates lacking hydrogen bond donors, promising broader applications in asymmetric transition-metal catalysis.
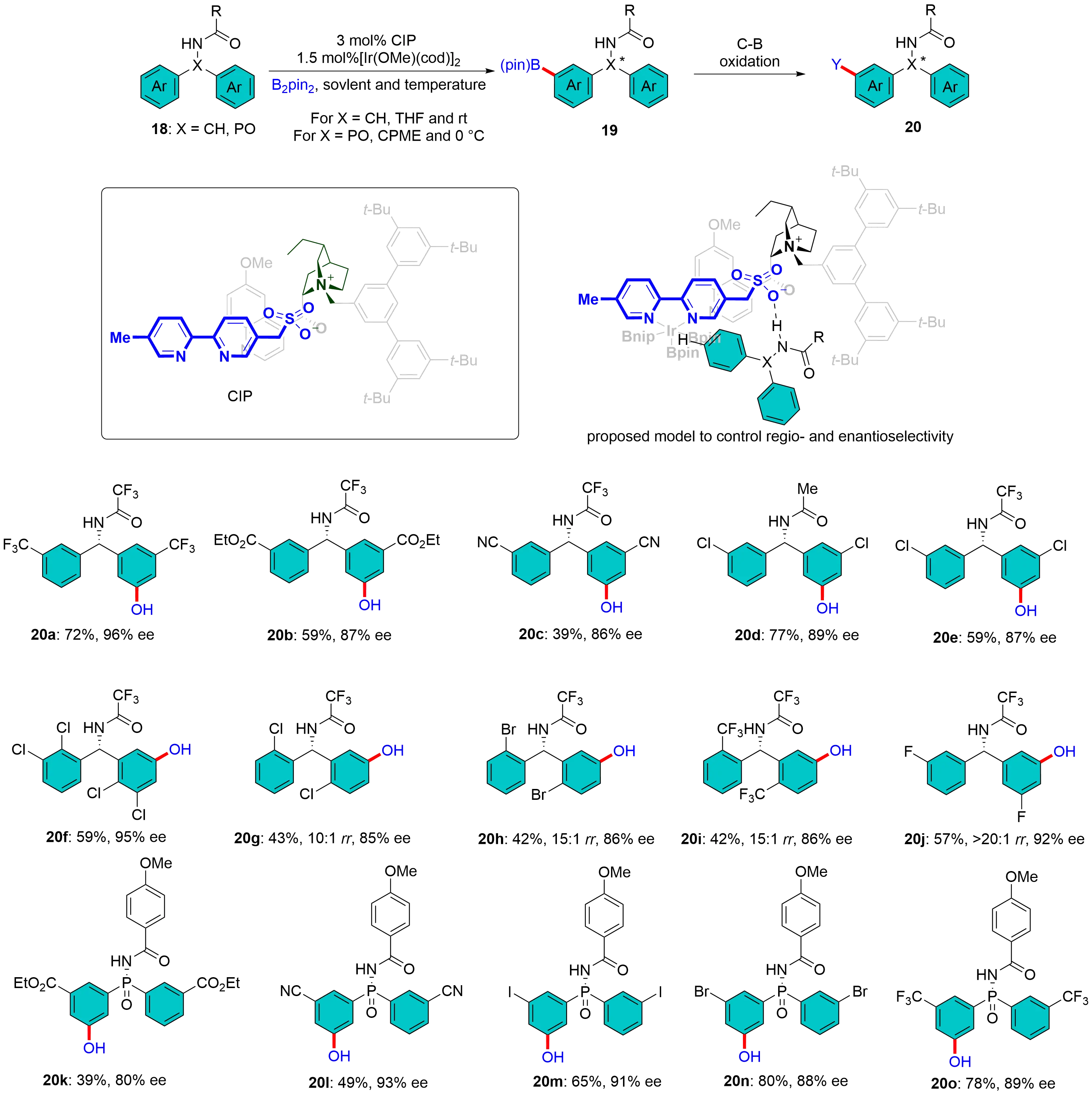
Figure 7. Chiral ion pair enabled Ir-catalyzed enantioselective meta-C–H borylation.
In 2019, Yang et al. reported an iridium-Lewis acid bifunctional catalyst for the meta-selective C–H borylation of (hetero)arenes containing an aminocarbonyl group or an sp2-hybridized nitrogen atom[34]. A bipyridine ligand bearing a remote Lewis acidic alkylaluminum biphenoxide moiety was particularly effective for the Ir-catalyzed meta-C–H borylation benzamides. Inspired by Yang’s work, Aditya et al. developed an iridium-Lewis acid bifunctional catalyst based on an H8-BINOL-derived chiral bipyridine ligand (L), which successfully enabled the remote asymmetric meta-C–H borylation of α,α-diarylcarboxamides (Figure 8)[35]. This reaction overcomes the challenge of long-range asymmetric induction through the close proximity between the chiral scaffold of the ligand and the (pro)stereogenic center of the substrate, achieving desymmetrization of prochiral substrates 21a-j with excellent regioselectivity (up to > 30:1 rr) and enantioselectivity (up to 99% ee), while also realizing both single- and double-fold kinetic resolution of racemic substrates 21k-n for such transformations with moderate to good selectivity (s-factor up to 19.5). The synthetic utility of this protocol is demonstrated by scale-up experiments and subsequent derivatization of the boronate ester products into diverse functional groups (e.g., chiral biaryls, aryl halides), providing an efficient strategy for constructing all-carbon quaternary stereocenters.
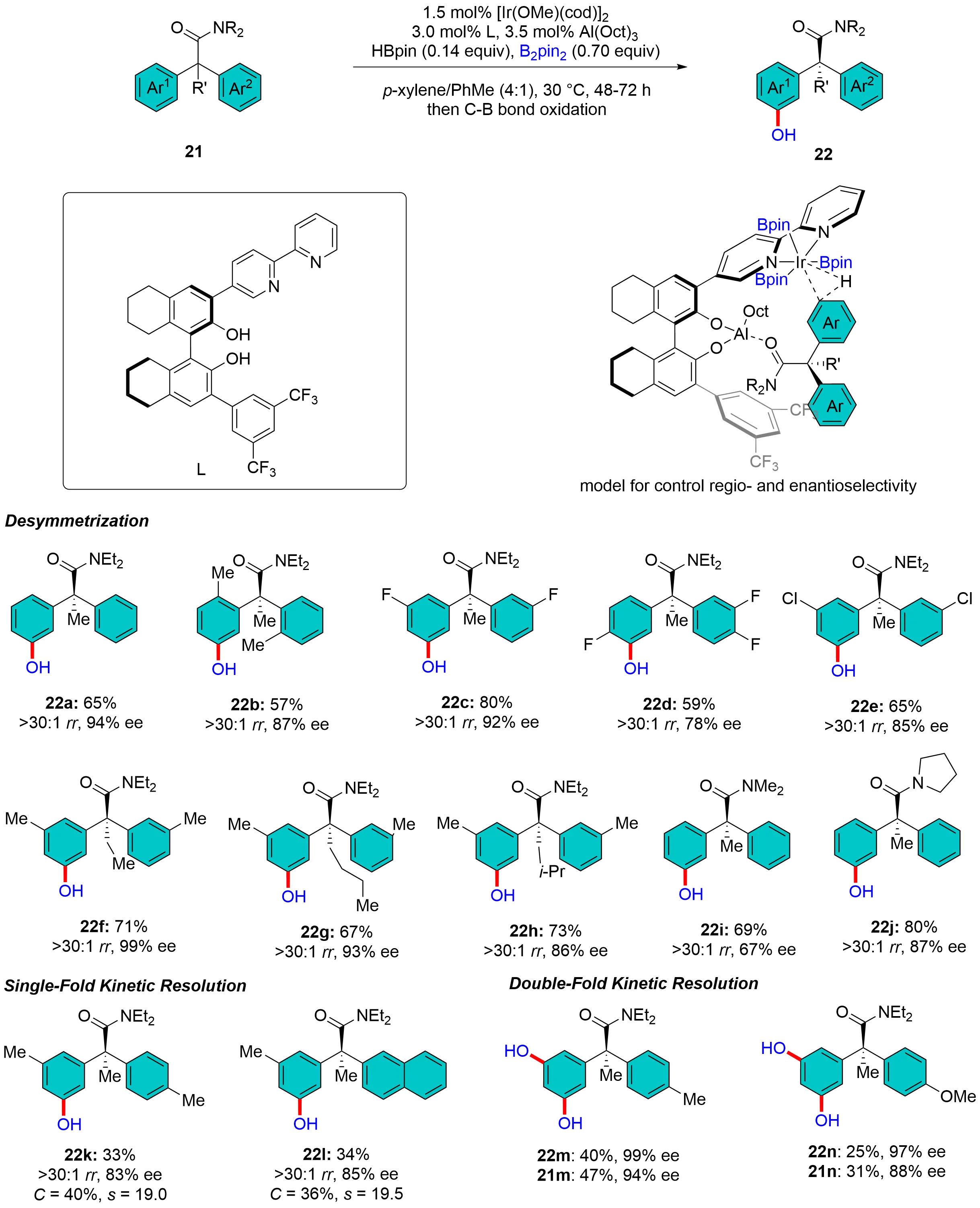
Figure 8. H8-BINOL-derived bipyridine enabled Ir-catalyzed enantioselective meta-C–H borylation.
3. Conclusion and Perspective
The reviewed strategies for asymmetric remote C–H activation demonstrate the growing sophistication of chiral control in organic synthesis. By utilizing template-chiral ligand cooperativity, transient mediators, and bifunctional chiral catalysts, researchers have successfully achieved high enantioselectivities in the functionalization of remote C-H bonds, typically at the meta-position of arenes. These strategies have enabled the synthesis of compounds with central, planar, and axial chiral architectures, which are of great importance in pharmaceuticals, catalysis, and materials science. Mechanistic studies have further elucidated the controlling factors for long-range enantioselectivity, providing deeper insights into the underlying mechanisms and guiding future development. Despite these advancements, key constraints of the three core strategies are obvious, including reliance on custom templates/ligands, narrow substrate scope, and limited reaction types. Additionally, functionalization of para-C–H and aliphatic C–H bonds remains underdeveloped, and the lack of robust chiral mediators limits scalability and applications in complex molecule synthesis. Challenges remain in extending the scope and efficiency of remote C–H activation, particularly for para-C-H bonds of arenes and aliphatic C–H bonds. Future research should focus on developing more robust chiral mediators and ligands, and more powerful strategies that can accommodate a broader range of substrates and functional groups. The more immediate and transformative potential of this strategy lies in its ability to create novel, complex chiral architectures, thereby dramatically expanding chemical space and opening new avenues for discovering bioactive compounds. Furthermore, exploring the application of remote C–H activation in the synthesis of natural products and complex chiral materials will continue to drive innovation in the field. The ongoing development of these technologies promises to unlock new possibilities in asymmetric catalysis and chiral synthesis.
Authors contribution
Chen L: Investigation, writing-original draft.
Xu S: Conceptualization, writing-review & editing.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Key R & D Program of China (No. 2022YFA1504302), the National Natural Science Foundation of China (Nos. 92256302 and 22331011).
Copyright
© The Author(s) 2025.
References
-
1. Giri R, Shi BF, Engle KM, Maugel N, Yu JQ. Transition metal-catalyzed C–H activation reactions: Diastereoselectivity and enantioselectivity. Chem Soc Rev. 2009;38(11):3242-3272.[DOI]
-
2. Engle KM, Mei TS, Wasa M, Yu JQ. Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions. Acc Chem Res. 2012;45(6):788-802.[DOI]
-
3. He J, Wasa M, Chan KSL, Shao Q, Yu JQ. Palladium-catalyzed transformations of alkyl C–H bonds. Chemical Reviews. 2017;117(13):8754-8786.[DOI]
-
4. Newton CG, Wang SG, Oliveira CC, Cramer N. Catalytic enantioselective transformations involving C–H bond cleavage by transition-metal complexes. Chem Rev. 2017;117(13):8908-8976.[DOI]
-
5. Saint-Denis TG, Zhu RY, Chen G, Wu QF, Yu JQ. Enantioselective C(sp3)–H bond activation by chiral transition metal catalysts. Science. 2018;359(6377):eaao4798.[DOI]
-
6. Chen Z, Rong MY, Nie J, Zhu XF, Shi BF, Ma JA. Catalytic alkylation of unactivated C(sp3)–H bonds for C(sp3)–C(sp3) bond formation. Chem Soc Rev. 2019;48(18):4921-4942.[DOI]
-
7. Shao Q, Wu K, Zhuang Z, Qian S, Yu JQ. From to chiral catalysts: the discovery and development of bifunctional mono-N-protected amino acid ligands for diverse C–H functionalization reactions. Acc Chem Res. 2020;53(4):833-851.[DOI]
-
8. Liu B, Romine AM, Rubel CZ, Engle KM, Shi BF. Transition-metal-catalyzed, coordination-assisted functionalization of nonactivated C(sp3)–H bonds. Chem Rev. 2021;121(24):14957-15074.[DOI]
-
9. Su B, Hartwig JF. Development of chiral ligands for the transition-metal-catalyzed enantioselective silylation and borylation of C−H bonds. Angew Chem Int Ed. 2022;61(9):e202113343.[DOI]
-
10. Zhan BB, Jin L, Shi BF. Palladium-catalyzed enantioselective C–H functionalization via C–H palladation. Trends Chem. 2022;4(3):220-235.[DOI]
-
11. Zhang Q, Wu LS, Shi BF. Forging C–heteroatom bonds by transition-metal-catalyzed enantioselective C–H functionalization. Chem. 2022;8(2):384-413.[DOI]
-
12. Yu IF, Wilson JW, Hartwig JF. Transition-metal-catalyzed silylation and borylation of C–H bonds for the synthesis and functionalization of complex molecules. Chem Rev. 2023;123(19):11619-11663.[DOI]
-
13. Dutta U, Maiti S, Bhattacharya T, Maiti D. Arene diversification through distal C(sp2)–H functionalization. Science. 2021;372(6543):eabd5992.[DOI]
-
14. Leow D, Li G, Mei TS, Yu JQ. Activation of remote meta-C–H bonds assisted by an end-on template. Nature. 2012;486(7406):518-522.[DOI]
-
15. Li S, Wang H, Weng Y, Li G. Carboxy group as a remote and selective chelating group for C−H activation of arenes. Angew Chem Int Ed. 2019;58:18502-18507.[DOI]
-
16. Wang H, Li H, Chen X, Zhou C, Li Shang, Yang YF, et al. Asymmetric remote meta-C–H activation controlled by a chiral ligand. ACS Catal. 2022;12(21):13435-13445.[DOI]
-
17. Catellani M. Palladium in organic synthesis. Germany: Springer Berlin Heidelberg; 2005. p. 21-53.
-
18. Wang XC, Gong W, Fang LZ, Zhu RY, Li S, Engle KM, et al. Ligand-enabled meta-C–H activation using a transient mediator. Nature. 2015;519(7543):334-338.[DOI]
-
19. Dong Z, Wang J, Dong G. Simple Amine-directed meta-selective C–H arylation via Pd/Norbornene catalysis. J Am Chem Soc. 2015;137(18):5887-5890.[DOI]
-
20. Shi H, Herron AN, Shao Y, Shao Q, Yu JQ. Enantioselective remote meta-C–H arylation and alkylation via a chiral transient mediator. Nature. 2018;558(7711):581-585.[DOI]
-
21. Hill DE, Yu JQ, Blackmond DG. Insights into the role of transient chiral mediators and pyridone ligands in asymmetric Pd-Catalyzed C–H functionalization. J Org Chem. 2020;85(21):13674-13679.[DOI]
-
22. Zhou L, Cheng HG, Li S, Wu K, Hou J, Jiao C, et al. Synthesis of planar chiral ferrocenes via enantioselective remote C–H activation. Nat Chem. 2023;15(6):815-823.[DOI]
-
23. Dai L, Lu Y. Atroposelective remote meta-C-H arylation of 2-arylanilines. Chem. 2023;9(6):1347-1349.[DOI]
-
24. Gustafson JL, Lim D, Miller SJ. Dynamic kinetic resolution of biaryl atropisomers via peptide-catalyzed asymmetric bromination. Science. 2010;328(5893):1251-1255.[DOI]
-
25. Metrano AJ, Miller SJ. Peptide-based catalysts reach the outer sphere through remote desymmetrization and atroposelectivity. Acc Chem Res. 2019;52(1):199-215.[DOI]
-
26. Mori K, Ichikawa Y, Kobayashi M, Shibata Y, Yamanaka M, Akiyama T. Enantioselective synthesis of multisubstituted biaryl skeleton by chiral phosphoric acid catalyzed desymmetrization/kinetic resolution sequence. J Am Chem Soc. 2013;135(10):3964-3970.[DOI]
-
27. Li JJ, Zhao JH, Shen HC, Wu K, Kuang X, Wang P, et al. Atroposelective remote meta-C–H activation. Chem. 2023;9(6):1452-1463.[DOI]
-
28. Li JJ, Zeng XX, Kuang X, Shen HC, Wang P, Yu JQ. Atroposelective diverse remote meta-C-H functionalization via kinetic resolution. J Am Chem Soc. 2025;147(8):6594-6603.[DOI]
-
29. Zou X, Xu S. Recent progress in iridium-catalyzed remote regioselective C–H borylation of (Hetero)Arenes. Chin J Org Chem. 2021;41(7):2610-2620.[DOI]
-
30. Mihai MT, Davis HJ, Genov GR, Phipps RJ. Ion pair-directed C–H activation on flexible ammonium salts: meta-selective borylation of quaternized phenethylamines and phenylpropylamines. ACS Catal. 2018;8(5):3764-3769.[DOI]
-
31. Davis HJ, Genov GR, Phipps RJ. meta-Selective C−H borylation of benzylamine-, phenethylamine-, and phenylpropylamine-derived amides enabled by a single anionic ligand. angew. Chem Int Ed. 2017;56(43):13351-13355.[DOI]
-
32. Genov GR, Douthwaite JL, Lahdenperä ASK, Gibson DC, Phipps RJ. Enantioselective remote C–H activation directed by a chiral cation. Science. 2020;367(6483):1246-1251.[DOI]
-
33. Ermanis K, Gibson DC, Genov GR, Phipps RJ. Interrogating the crucial interactions at play in the chiral cation-directed enantioselective borylation of arenes. ACS Catal. 2023;13(19):13043-13055.[DOI]
-
34. Yang L, Uemura N, Nakao Y. meta-selective C–H borylation of benzamides and pyridines by an iridium–lewis acid bifunctional catalyst. J Am Chem Soc. 2019;141(19):7972-7979.[DOI]
-
35. Aditya N, Das S, Datta A, Maji B. Iridium-lewis acid bifunctional catalyst-enabled regio- and enantioselective C(sp2)-H meta-borylation of α,α-diarylcarboxamides. J Am Chem Soc. 2025;147(31):27458-27470.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Chen L, Xu S. Transition metal-catalyzed remote asymmetric C–H activation of arenes. Chiral Chem. 2026;2:202509. https://doi.org/10.70401/cc.2025.0006
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Chen L, Xu S. Transition metal-catalyzed remote asymmetric C–H activation of arenes. Chiral Chem. 2026;2:202509. https://doi.org/10.70401/cc.2025.0006
copy
Share Link
copy