Harnessing molecular motion for asymmetry – nitrogen inversion as a springboard for stereoselective C–H functionalization
*Correspondence to:
Guang-Jian Mei, College of Chemistry, Zhengzhou University, Zhengzhou 450001, Henan, China; State Key Laboratory of Green Pesticide, Guizhou University, Guiyang 550025, Guizhou, China; Pingyuan Laboratory (Zhengzhou University), Zhengzhou 450001, Henan, China.
E-mail: meigj@zzu.edu.cn
Chiral Chem. 2026;2:202605. 10.70401/cc.2026.0015
Received: January 31, 2026Accepted: March 05, 2026Published: March 11, 2026
Abstract
The asymmetric construction of nitrogen stereocenters is notoriously difficult due to rapid nitrogen inversion. Zhang and co-workers now showcase a Pd-catalyzed enantioselective C-H activation strategy that turns this inversion into an advantage, delivering stable chiral azepines with high enantioselectivity. Coincidentally, almost at the same time, Shi and co-workers reported a similar C-H alkylation reaction. Beyond methodology, the products serve as promising scaffolds for asymmetric catalysis and chiroptical materials, bridging synthesis and function. This Perspective discusses how their works not only provide a powerful synthetic method but also open a new avenue for constructing heteroatom stereocenters by harnessing, rather than suppressing, molecular dynamics. Here, nitrogen inversion is deliberately exploited as a dynamic feature that enables enantioselective C-H functionalization, rather than being suppressed as a stereochemical liability.
Graphical Abstract
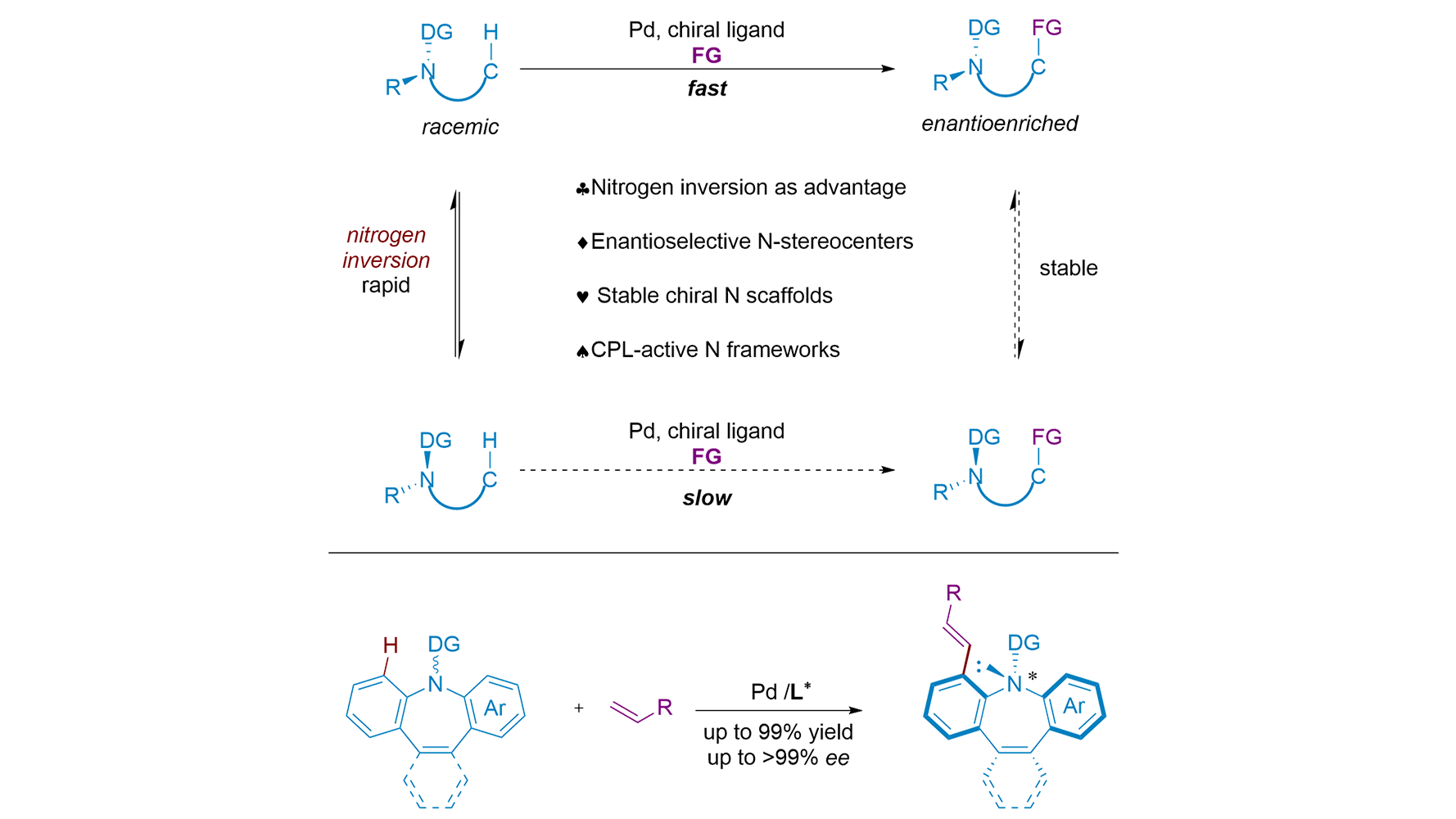
Keywords
Nitrogen stereocenters, Pd-catalyzed, enantioselective C-H activation
Dynamic chirality in organic molecules encompasses diverse stereochemical manifestations, including atropisomerism, helical chirality, conformational chirality, and nitrogen inversion. Although all of these stereochemical elements originate from molecular dynamics, their configurational stability varies substantially. In atropisomeric, helical, and conformational systems, stereochemical integrity is typically maintained through restricted bond rotation or conformational bias. In contrast, nitrogen stereocenters interconvert via pyramidal inversion, a process generally associated with comparatively low activation barriers. A tertiary amine bearing three distinct substituents is, in principle, inherently chiral. However, this chirality is often transient[1]. The low activation barrier for nitrogen inversion (typically 5-10 kcal/mol)[2] allows the two enantiomeric conformers to interconvert rapidly at room temperature, rendering them unresolvable under standard conditions. Consequently, controlling nitrogen stereocenters remains a longstanding challenge in asymmetric synthesis[3]. Traditional strategies to access enantioenriched nitrogen compounds have relied on circumventing this inversion (Figure 1A), either by converting it into a configurationally stable tetraalkylammonium cation[4-7] (Strategy A), as demonstrated by Yamamoto and co-workers[3] through a bimetallic titanium-catalyzed asymmetric N-oxidation of tertiary amines, or by locking the nitrogen in a rigid polycyclic framework[8-11] (Strategy B), as exemplified by the enantioselective construction of conformationally stable N-stereogenic centers in ethano- and propano-Tröger’s bases via Pd-catalyzed annulative allylic alkylation[9]. Alternatively, N-chirality can be stabilized by introducing electronegative substituents or incorporating strained three-membered rings. A significant breakthrough in this area was reported by Tan, Houk, and co-workers[12] in a recent Nature article, where they achieved the first catalytic asymmetric control of simple pyramidal N-chirality. Around the same time, List and colleagues[13] developed an imidodiphosphorimidate (IDPi)-catalyzed addition of silyl ketene acetals, enabling the asymmetric synthesis of acyclic N-stereogenic amines. These two approaches complement the strategies discussed in contributing to the broader understanding of nitrogen stereochemistry and expanding the toolbox for the selective synthesis of nitrogen stereoisomers.
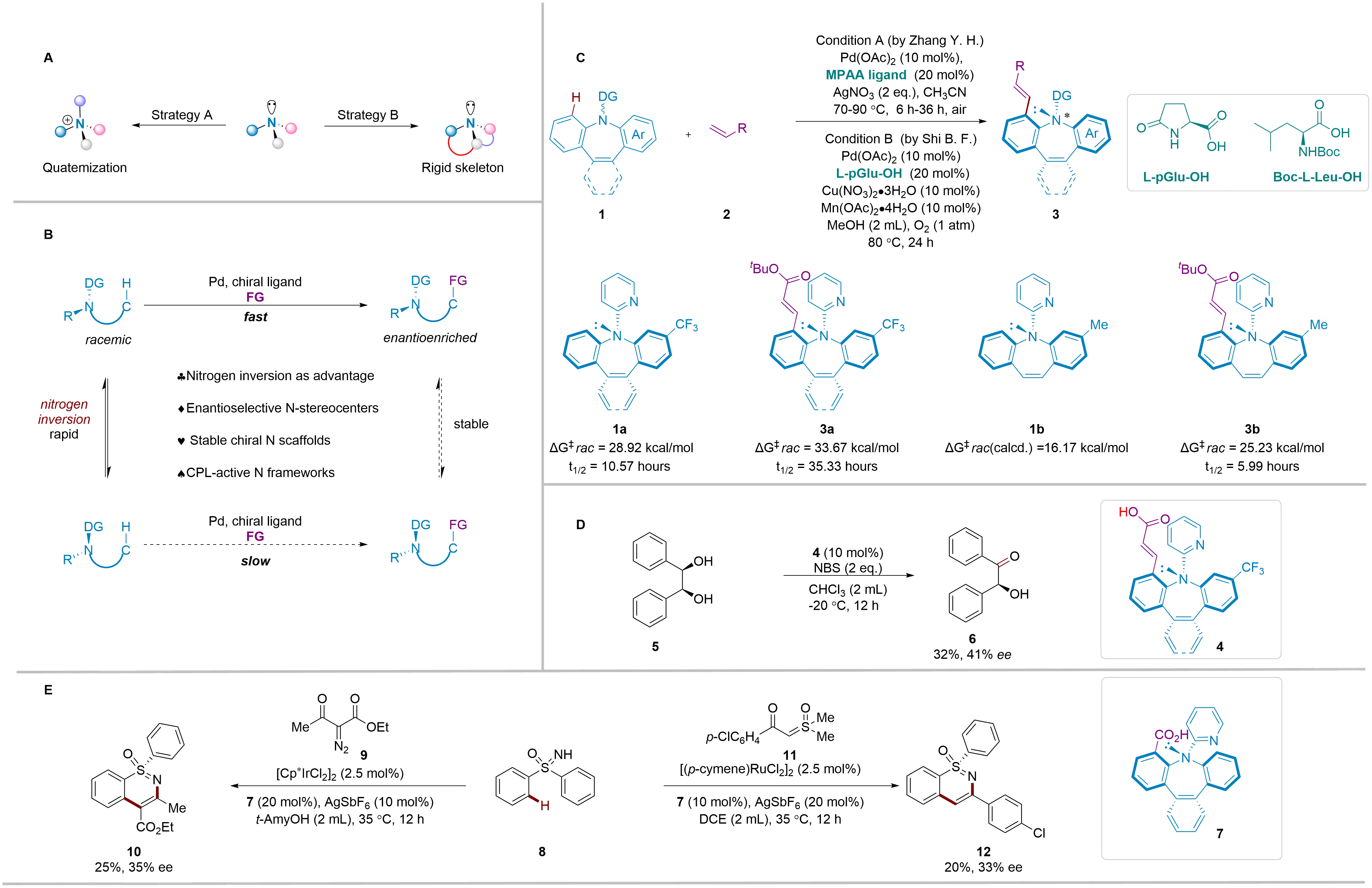
Figure 1. Construction of chiral nitrogen stereocenters via enantioselective C-H activation. (A) Stabilizing N-stereocenters by quaternization or rigid skeleton; (B) Strategies for construction of chiral nitrogen stereocenters; (C) Established reaction and racemization barrier; (D) Application as a chiral catalyst; (E) Application as an efficient ligand.
Although there have been several studies on the catalytic asymmetric synthesis of nitrogen chirality, these approaches, while effective, are often substrate-specific and can limit structural diversity. The direct catalytic asymmetric synthesis of chiral amines from simple, racemizing precursors has thus remained a “holy grail” in the field of stereoselective synthesis.
The work by Zhang et al. represents a conceptual leap. Instead of viewing nitrogen inversion as an obstacle to be overcome, the authors ingeniously exploit it as the fundamental engine for a Dynamic Kinetic Resolution (DKR)[14]. The principle is elegant in its simplicity: if a racemic substrate racemizes rapidly under the reaction conditions, and a catalyst can selectively transform one enantiomer into a configurationally stable product, then theoretically, a 100% yield of a single enantiomer can be achieved.
Zhang and co-workers demonstrated this principle with remarkable efficacy[14]. They selected azepine scaffolds, molecules of significant interest in materials science and pharmacology, as their platform. The key to their success lies in the subtle interplay between substrate and product stability (Figure 1B). The starting benzazepines, while chiral, undergo rapid nitrogen inversion, ensuring a constant supply of both enantiomers. The C-H olefination reaction, catalyzed by a chiral Pd/amino acid complex, selectively functionalizes one enantiomer. Critically, the resulting olefinated product possesses a significantly higher barrier to nitrogen inversion, “freezing” the chirality and preventing racemization. This perfect storm of rapid substrate racemization and high product stability, coupled with an exceptionally enantioselective C-H activation step, enables a highly efficient DKR process. Leveraging a combination of chiral catalysis and selective C-H activation, this method provides a powerful approach to forming nitrogen-containing stereocenters with high enantioselectivity from readily available and often simple substrates. The process is highly versatile, demonstrating excellent functional group tolerance, scalability, and the ability to access a wide range of structurally diverse nitrogen-containing compounds. Coincidentally, a similar approach to constructing N-chirality was explored by the research group of Shi[15], who also employed C-H olefination reactions to build stable nitrogen stereocenters (Figure 1C). Their work utilized analogous substrates and palladium-catalyzed C-H activation to selectively functionalize nitrogen-containing heterocycles, ultimately introducing stable nitrogen stereocenters. Notably, their methodology was demonstrated in 41 examples, achieving yields of up to 99% and enantioselectivities of up to > 99%. Shi’s group demonstrated the broad applicability of this methodology in creating a variety of nitrogen-containing chiral molecules, offering further support for the potential of Pd-catalyzed C-H olefination in the synthesis of chiral nitrogen centers.
The energy barrier analysis (Figure 1C) underscores this transformation. The parent substrate (1b) exhibits a low inversion barrier (ΔG‡rac ≈ 16 kcal/mol), which means it racemizes rapidly. By contrast, the olefinated products (e.g., 3a and 3b) show markedly higher barriers (ΔG‡rac up to 33.7 kcal/mol) and half-lives exceeding 35 h, confirming that the catalytic transformation stabilizes previously labile stereocenters. This observation provides a clear mechanistic insight: C-H functionalization not only constructs a nitrogen stereocenter but also preserves it. Supporting this, kinetic isotope effect experiments and DFT calculations show that C-H bond cleavage is both the rate-determining and enantioselectivity-determining step. These findings offer guiding principles for the design of future catalysts.
Building on these mechanistic insights, the resulting chiral tribenzo[b,d,f]azepines also exhibit intriguing functional properties. First, they serve as scaffolds for chiral organocatalysis: hydrolyzed derivatives were shown to promote enantioselective diol oxidation (Figure 1D), offering a glimpse of new bifunctional catalyst frameworks. Second, their rigid, saddle-shaped architecture imparts strong circularly polarized luminescence (CPL), with glum values up to 10-2, unusually high for small organic molecules. This positions them as promising candidates for next-generation chiroptical materials.
Similarly, to demonstrate the scalability and practical utility of their methodology, Shi and co-workers integrated C-H activation with complementary reactions, such as reduction and hydrolysis, thus opening new avenues for the synthesis of more complex molecular architectures. Furthermore, the novel N-chiral carboxylic acids obtained from this method could serve as efficient ligands in the Cp*Ir and (p-cymene)Ru-catalyzed enantioselective C-H annulation of sulfoximine 8, yielding both enantiomers of S stereogenic chiral compounds 10 and 12 with moderate yields and enantioselectivities, showcasing the potential applications of this type of structure as a chiral ligand.
The works by Zhang and Shi are brilliant examples of creative problem-solving in synthetic chemistry. By reframing a fundamental chemical phenomenon, nitrogen inversion, from a problem into a solution, they have opened a powerful and potentially general pathway to enantioenriched nitrogen stereocenters. They firmly place enantioselective C-H activation at the forefront of modern strategies for constructing challenging stereogenic elements. This perspective underscores that their contribution is not merely a new reaction but a conceptual advance that invites us to rethink how we approach the synthesis of dynamically chiral molecules.
Acknowledgements
GPT-5 was used solely for language editing and polishing. The authors take full responsibility for the integrity, accuracy, and originality of the content.
Authors contribution
Guan CY: Investigation, writing-original draft, writing-review & editing.
Mei GJ: Conceptualization, funding acquisition, supervision, writing-review & editing.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
We acknowledge the financial support from the National Natural Science Foundation of China (22371265 and 22208302), Natural Science Foundation of Henan Province (232301420047), and the project of State Key Laboratory of Green Pesticide, Guizhou Medical University (GPLKF202507).
Copyright
© The Author(s) 2026.
References
-
1. Boerner LK. Catching the elusive chiral nitrogen compound. Chem Eng News. 2021;5.[DOI]
-
2. Koeppl GW, Sagatys DS, Krishnamurthy GS, Miller SI. Inversion barriers of pyramidal (XY3) and related planar (=XY) species. J Am Chem Soc. 1967;89(14):3396-3405.[DOI]
-
6. Luo Z, Liao M, Li W, Zhao S, Tang K, Zheng P, et al. Ionic hydrogen bond-assisted catalytic construction of nitrogen stereogenic center via formal desymmetrization of remote diols. Angew Chem. 2024;136(31):e202404979.[DOI]
-
7. Zhang S, Yi D, Li GX, Deng Y, Cui X, Tian Y, et al. Biomimetic dynamic kinetic asymmetric N-oxidation with H2O2 and O2. ACS Catal. 2023;13(18):11954-11962.[DOI]
-
8. Yu T, Cheng S, Luo Y, Li J, Liang Y, Luo S, et al. Immobilizing stereogenic nitrogen center in doubly fused triarylamines through palladium-catalyzed asymmetric C–H activation/seven-membered-ring formation. ACS Catal. 2023;13(14):9688-9694.[DOI]
-
14. Wu Z, Yi D, Tang J, Meng F, Zhu H, Wu K, et al. Construction of chiral nitrogen stereocenters via enantioselective C–H activation. Chem. 2026;12(1):102730.[DOI]
-
15. Jiang YL, Wang BJ, Qian PF, Xu Y, Wang SS, Yao QJ, et al. Efficient construction of nitrogen-stereogenic azepines via Pd(II)-catalyzed enantioselective C–H olefination. ACS Catal. 2026;16(3):2722-2731.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Guan C, Mei G. Harnessing molecular motion for asymmetry – nitrogen inversion as a springboard for stereoselective C–H functionalization. Chiral Chem. 2026;2:202605. https://doi.org/10.70401/cc.2026.0015
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Guan C, Mei G. Harnessing molecular motion for asymmetry – nitrogen inversion as a springboard for stereoselective C–H functionalization. Chiral Chem. 2026;2:202605. https://doi.org/10.70401/cc.2026.0015
copy
Share Link
copy