Catalytic atroposelective synthesis of C–N axially chiral 5-pyrazolyl indoles via multicomponent tandem cycloaddition
Yu-Yu Chen
1
,
Jia Xue
1
,
Yu-Chen Zhang
1,*

,
Feng Shi
1,2,*
*Correspondence to:
Yu-Chen Zhang, Research Center of Chiral Functional Heterocycles, School of Chemistry and Materials Science, Jiangsu Normal University, Xuzhou 221116, Jiangsu, China.
E-mail: zhangyc@jsnu.edu.cn
Feng Shi, Research Center of Chiral Functional Heterocycles, School of Chemistry and Materials Science, Jiangsu Normal University, Xuzhou 221116, Jiangsu, China; School of Petrochemical Engineering, Changzhou University, Changzhou 213164, Jiangsu, China. E-mail: fshi@jsnu.edu.cn; fshi@cczu.edu.cn
Feng Shi, Research Center of Chiral Functional Heterocycles, School of Chemistry and Materials Science, Jiangsu Normal University, Xuzhou 221116, Jiangsu, China; School of Petrochemical Engineering, Changzhou University, Changzhou 213164, Jiangsu, China. E-mail: fshi@jsnu.edu.cn; fshi@cczu.edu.cn
Chiral Chem. 2026;2:202604. 10.70401/cc.2026.0018
Received: January 29, 2026Accepted: April 01, 2026Published: April 02, 2026
Abstract
An organocatalytic asymmetric multicomponent tandem cycloaddition has been established for the construction of C–N axially chiral 5-pyrazolyl indole frameworks, which represent a novel class of five-five-membered indole scaffolds. Utilizing 5-aminopyrazoles, cyclohexanediones, and 2,3-diketoester precursors, this methodology constructs the indole ring de novo in a single step, demonstrating high atom- and step-economy with water as the exclusive byproduct. The protocol enabled the efficient and high-enantioselective synthesis of structurally diverse C–N axially chiral 5-pyrazolyl indoles. This achievement not only introduces a new member to the family of axially chiral pyrazolyl indoles but also provides an efficient and novel strategy for their construction.
Graphical Abstract
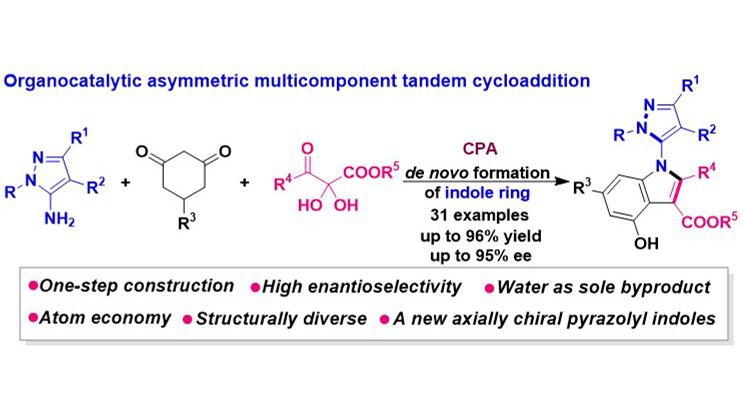
Keywords
Heterocycle, atropisomer, organocatalysis, cycloaddition, axial chirality
1. Introduction
Atropisomers[1-8] serve as indispensable components in medicinal chemistry[9-11], functional materials[12,13], and diverse industrial chemical applications[14,15]. Among these, axially chiral indoles constitute a significant class of atropisomers, frequently encountered in numerous natural products, biologically active molecules, and as valuable chiral catalysts and ligands (Figure 1a)[16-22]. In recent years, the catalytic asymmetric synthesis of axially chiral indoles has evolved into a vibrant research area within the synthetic chemistry community (Figure 1b)[23-28]. Nevertheless, current methodologies are largely restricted to the construction of six-five-membered axially chiral indoles (spanning C–C[29-36], C–N[37-45], and N–N atropisomers[46-49]). In contrast, the catalytic atroposelective assembly of five-five-membered axially chiral indoles remains underdeveloped[50-62]. This disparity originates from the inherent low rotational barriers and limited configurational stability characteristic of five-five-membered frameworks, rendering the catalytic asymmetric construction of such scaffolds significantly more challenging[63-67].
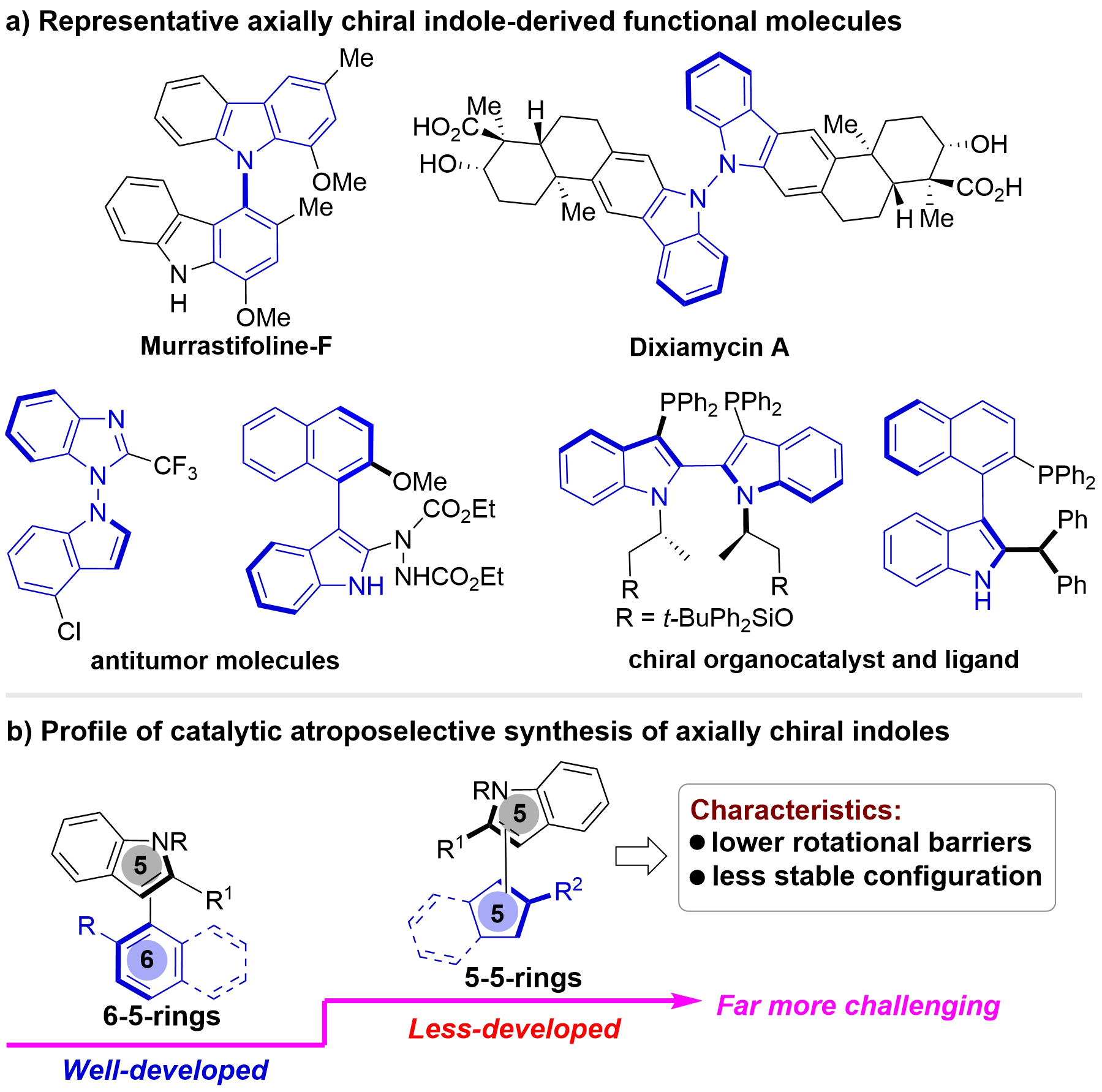
Figure 1. (a) Representative axially chiral indole-derived functional molecules; (b) Profile of catalytic atroposelective synthesis of axially chiral indoles.
Pyrazolyl indoles represent a distinctive class of five-five-membered indole frameworks, serving as the core structure for numerous biologically active molecules (Figure 2a)[68-71]. Despite their prevalence and significance, the catalytic asymmetric construction of axially chiral pyrazolyl indoles has only been realized very recently, with merely two reports documented to date (Figure 2b). In 2022, Huang and their co-workers achieved the highly enantioselective synthesis of C–C axially chiral 4-pyrazolyl indoles utilizing a dynamic kinetic resolution strategy[50]. More recently, in 2025, Feng and their collaborators accomplished the construction of C–N axially chiral 4-pyrazolyl indoles via a central-to-axial chirality transfer strategy[54]. While these two elegant contributions mark significant progress, there remains an urgent need to design novel axially chiral pyrazolyl indole frameworks and to develop highly efficient strategies for their catalytic asymmetric construction.
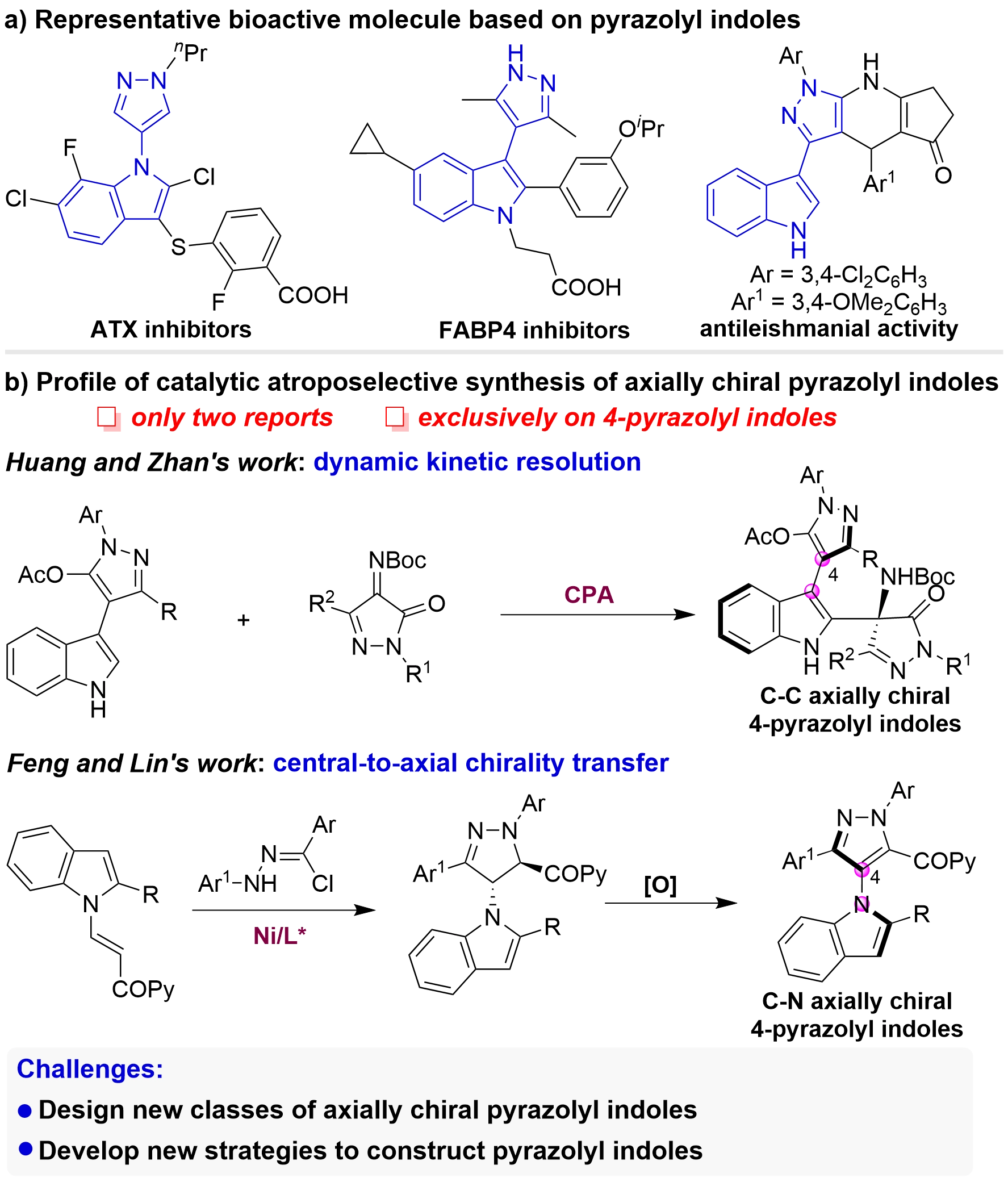
Figure 2. (a) Representative bioactive molecule based on pyrazolyl indoles; (b) Profile of catalytic atroposelective synthesis of axially chiral pyrazolyl indoles.
To address the aforementioned challenges, and drawing upon our extensive experience in chiral indole chemistry[72-75], we designed a novel class of axially chiral pyrazolyl indole frameworks: the C–N axially chiral 5-pyrazolyl indole scaffolds (Figure 3a)[76-79]. Concurrently, we devise the de novo ring formation strategy as a novel approach for synthesizing these C-N axially chiral pyrazolyl indoles (Figure 3b). Recently, our group achieved the construction of C–N axially chiral pyrazolyl pyrroles through an organocatalyzed asymmetric Paal–Knorr reaction involving 5-aminopyrazoles (Figure 3c)[80]. Encouraged by these results, we sought to further utilize 5-aminopyrazole as a platform molecule, aiming to construct our newly designed frameworks through the de novo formation of the indole ring[81-86].
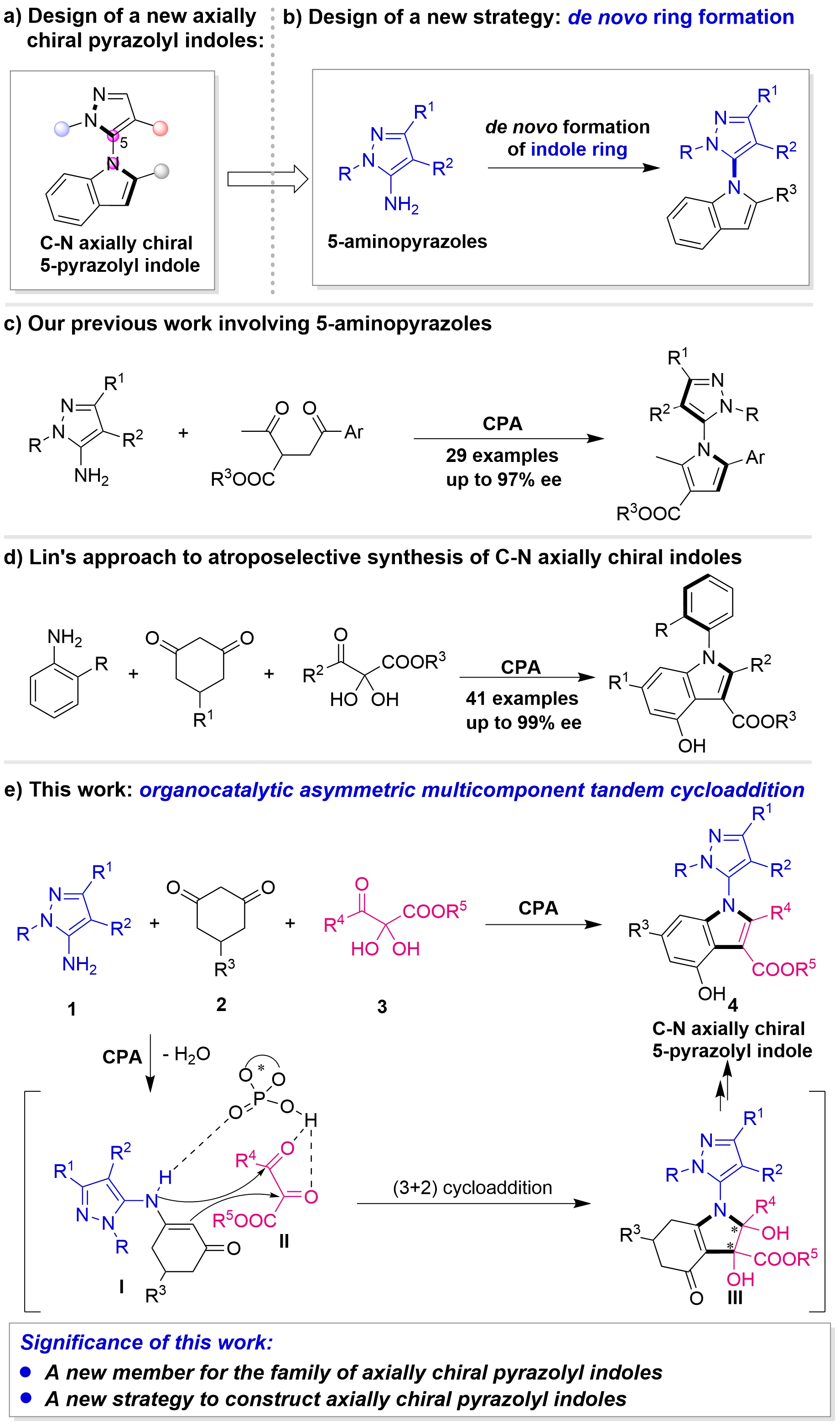
Figure 3. (a) Design of a new axially chiral pyrazolyl indoles; (b) Design of a new strategy; (c) Our previous work involving 5-aminopyrazoles; (d) Lin’s approach to atroposelective synthesis of C–N axially chiral indoles; (e) This work.
Notably, in 2019, Lin and co-workers reported an elegant approach to the enantioselective synthesis of C–N axially chiral aryl indoles via the de novo construction of the indole ring, which involved an organocatalyzed asymmetric tandem cycloaddition of anilines (Figure 3d)[84]. Inspired by this work, we developed an organocatalyzed asymmetric multicomponent tandem cycloaddition for the efficient construction of C–N axially chiral 5-pyrazolyl indole scaffolds (Figure 3e). In this design, 5-aminopyrazoles 1 and cyclohexanediones 2 are envisioned to initially condense in the presence of the chiral phosphoric acid (CPA)[87-93] to form enamine intermediates I. Simultaneously, 2,3-diketoester precursors 3 undergo dehydration to afford intermediates II. Intermediates I and II then participate in this asymmetric (3+2) cycloaddition to yield intermediates III, which subsequently undergo dehydration and aromatization processes, ultimately furnishing the C–N axially chiral 5-pyrazolyl indoles 4. This work will not only introduce a new member to the family of axially chiral pyrazolyl indoles, but also provide an efficient and novel strategy for their construction.
2. Experimental
1H NMR spectra (400 MHz), 13C NMR spectra (100 MHz), 31P NMR spectra (162 MHz) and 19F NMR spectra (376 MHz) were recorded on commercial instruments. High resolution mass spectra (HRMS) were performed on an AB SCIEX TripleTOF 4600. Enantiomeric excesses (ee) were determined by chiral high-performance liquid chromatography (chiral HPLC). The chiral columns used for the determination of enantiomeric excesses by chiral HPLC were Chiralpak columns. Optical rotation values were measured with instruments operating at λ = 589 nm, corresponding to the sodium D line at the temperatures indicated. The X-ray source used for the single crystal X-ray diffraction analysis of compound 4aag was GaKα (λ = 1.34139), and the thermal ellipsoid was drawn at the 50% probability level. Analytical grade solvents for the column chromatography were distilled before use. Substrates 2 commercially available were used directly. Chiral catalysts, substrates 1 and 3 are known compounds and were synthesized according to the literature procedures (see the Supplementary materials for details).
2.1 Procedure for the synthesis of products 4
In a sealed tube were added 5-aminopyrazole 1 (0.1 mmol), cyclohexanedione 2 (0.1 mmol), 2,3-diketoester precursor 3 (0.1 mmol), chiral phosphoric acid catalyst (R)-5d (5 mol%), and Na₂SO₄ (50 mg). Subsequently, CHCl3 (1 mL) was added, and the reaction mixture was stirred at 100 oC for 15 h. The reaction progress was monitored by thin-layer chromatography (TLC). Upon completion of the reaction, the crude mixture was purified by flash column chromatography on silica gel to afford the pure product 4.
2.2 Synthetic procedure of product 7
Under argon atmosphere, CH3MgBr (0.2 mL, 3 mol/mL) was added to a Schlenk tube. Then, the solution of 4aab (51.4 mg, 0.1 mmol) in anhydrous THF (1 mL) was added dropwise to the Schlenk tube at 0 oC. Subsequently, the reaction mixture was stirred at 70 oC for 4 h. After the completion of the reaction indicated by TLC, the reaction mixture was quenched by a saturated ammonium chloride aqueous solution and extracted with ethyl acetate for three times. The combined organic layers were dried and concentrated under reduced pressure to give a residue. Finally, the residue was purified by preparative thin layer chromatography (PE:EA = 4:1) on silica gel to afford pure product 7 in 85% yield (42.5 mg) as a white solid.
2.3 Synthetic procedure of product 8
NaH (60% in oil, 12 mg, 0.3 mmol) and THF (2 mL) were added to a reaction tube at 0 oC. A solution of compound 4aab (102.7 mg, 0.2 mmol) in THF (1 mL) was then added to the reaction mixture, followed by stirring at room temperature for 30 minutes. Afterward, CH3I (18.7 μL, 0.3 mmol) was added to the solution at 0 oC, and the reaction mixture was stirred at 0 oC for 12 hours. After the completion of the reaction indicated by TLC, the reaction mixture was quenched by water and extracted with ethyl acetate for three times. The combined organic layers were dried over anhydrous Na2SO4 and then concentrated under reduced pressure. The residue was purified through flash column chromatography on silica gel (petroleum ether/ethyl acetate = 4/1) to afford pure product 8 in 86% yield (90.3 mg) as a white solid.
2.4 Synthetic procedure of product 9
LiAlH4 (75.9 mg, 2 mmol) and 1.5 mL of THF were added to a reaction tube. Then, the solution of compound 8 (105.5 mg, 0.2 mmol) in 0.5 mL of THF was added to the reaction mixture, which was stirred at 0 oC for 3 h. After the completion of the reaction which was indicated by TLC, the reaction mixture was quenched with 10 mL of H2O and the aqueous layer was extracted with EtOAc (3 × 5 mL). The combined organic layers were dried over anhydrous Na2SO4 and then concentrated under reduced pressure. The residue was purified through flash column chromatography on silica gel (petroleum ether/ethyl acetate = 2/1) to afford pure product 9 in 93% yield (90.3 mg) as a white solid.
2.5 Synthetic procedure of product 10
Compound 9 (48.6 mg, 0.1 mmol) and EtOAc (2.0 mL) were added to a reaction tube. Then, 2-iodoxybenzoic acid (IBX) (56 mg, 0.2 mmol) was added to the reaction mixture, which was stirred at 80 °C for 7 h. After the completion of the reaction, which was indicated by TLC, the reaction mixture was filtered and concentrated under reduced pressure. The residue was purified through preparative thin layer chromatography on silica gel (petroleum ether/ethyl acetate = 2/1) to afford pure product 10 in 99% yield (48.0 mg) as a white solid.
3. Results and Discussion
To validate the feasibility of our designed organocatalyzed asymmetric multicomponent tandem cycloaddition, we initially attempted the reaction using CPA (R)-5a as the catalyst and ClCH2CH2Cl (DCE) as the solvent (Table 1, entry 1). Encouragingly, we successfully obtained the target C–N axially chiral 5-pyrazolyl indole 4aaa, albeit with a low yield and moderate enantioselectivity (17% yield, 47% ee). Subsequently, we screened a series of CPAs with different substituents (entries 2–6). The results indicated that CPA (R)-5d was the most effective (entry 4), delivering excellent enantioselectivity (91% ee). Following this, we evaluated various types of solvents (entries 7–11). It was observed that the reaction exhibited poor solvent compatibility, proceeding efficiently only in DCE and toluene (entries 4 and 7). Moreover, DCE remained the superior solvent for the reaction. We further explored other halogenated solvents (entries 12–13), finding that employing CHCl3 as the solvent (entry 13) significantly improved the yield to 82% while maintaining excellent enantioselectivity (91% ee). Next, we investigated the effect of reaction temperature (entries 14–15). It was determined that 100 °C (entry 13) still provided the best overall results. Screening of various additives (entries 16–19) revealed that Na2SO4 (entry 18) further enhanced both the yield and enantioselectivity (92% yield, 92% ee). We also found that adjusting the stoichiometric ratio of reactants to 1:1:1 (entry 20) led to a marginal improvement in both yield and enantioselectivity (96% yield, 93% ee). Furthermore, the influence of catalyst loading was investigated. Reducing the catalyst loading to 2 mol% (entry 21) led to a significant decrease in yield (66%) and a slight drop in enantioselectivity (91% ee). In contrast, increasing the catalyst amount to 10 mol% showed no impact on either yield or enantioselectivity (entry 22). Finally, the effect of water on the reaction was examined (entries 23-24). Performing the reaction in the absence of Na2SO4 resulted in a substantial decrease in yield and a slight reduction in enantioselectivity (entry 23). Besides, the further addition of 10 equiv. of water showed no further impact on either the yield or the enantioselectivity (entry 24). Based on these studies, the reaction conditions outlined in entry 20 were identified as the optimal conditions.
Table 1. Optimization of reaction conditionsa.
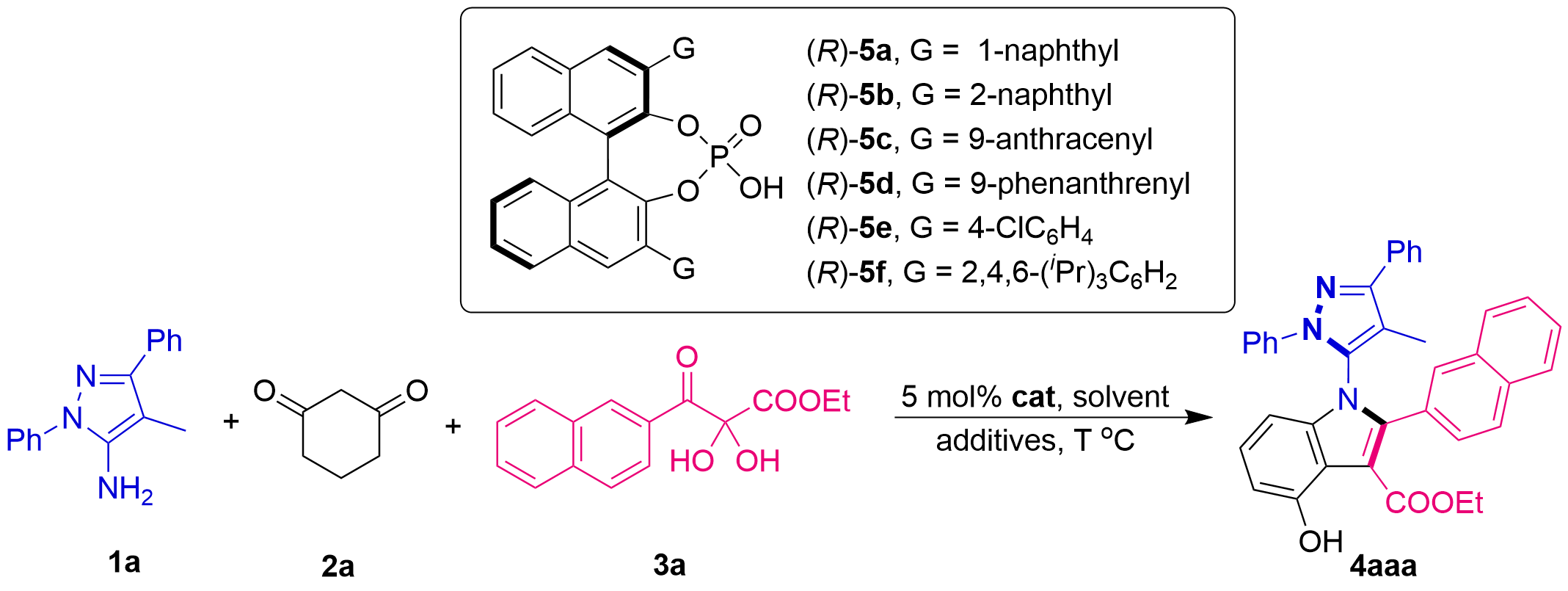
| entry | Cat. | solvent | T (oC) | additives | 1a:2a:3a | yield (%)b | ee (%)c |
| 1 | (R)-5a | DCE | 100 | 5 Å MS | 1:1:2 | 17 | 47 |
| 2 | (R)-5b | DCE | 100 | 5 Å MS | 1:1:2 | 8 | 15 |
| 3 | (R)-5c | DCE | 100 | 5 Å MS | 1:1:2 | 32 | 86 |
| 4 | (R)-5d | DCE | 100 | 5 Å MS | 1:1:2 | 59 | 91 |
| 5 | (R)-5e | DCE | 100 | 5 Å MS | 1:1:2 | trace | / |
| 6 | (R)-5f | DCE | 100 | 5 Å MS | 1:1:2 | 25 | 78 |
| 7 | (R)-5d | toluene | 100 | 5 Å MS | 1:1:2 | 25 | 64 |
| 8 | (R)-5d | THF | 100 | 5 Å MS | 1:1:2 | N.R. | / |
| 9 | (R)-5d | CH3CN | 100 | 5 Å MS | 1:1:2 | N.R. | / |
| 10 | (R)-5d | acetone | 100 | 5 Å MS | 1:1:2 | N.R. | / |
| 11 | (R)-5d | EtOAc | 100 | 5 Å MS | 1:1:2 | N.R. | / |
| 12 | (R)-5d | CCl4 | 100 | 5 Å MS | 1:1:2 | 29 | 85 |
| 13 | (R)-5d | CHCl3 | 100 | 5 Å MS | 1:1:2 | 82 | 91 |
| 14 | (R)-5d | CHCl3 | 80 | 5 Å MS | 1:1:2 | 41 | 91 |
| 15 | (R)-5d | CHCl3 | 120 | 5 Å MS | 1:1:2 | 81 | 91 |
| 16 | (R)-5d | CHCl3 | 100 | 3 Å MS | 1:1:2 | 53 | 90 |
| 17 | (R)-5d | CHCl3 | 100 | 4 Å MS | 1:1:2 | 11 | 61 |
| 18 | (R)-5d | CHCl3 | 100 | Na2SO4 | 1:1:2 | 92 | 92 |
| 19 | (R)-5d | CHCl3 | 100 | MgSO4 | 1:1:2 | 89 | 92 |
| 20 | (R)-5d | CHCl3 | 100 | Na2SO4 | 1:1:1 | 96 | 93 |
| 21d | (R)-5d | CHCl3 | 100 | Na2SO4 | 1:1:1 | 66 | 91 |
| 22e | (R)-5d | CHCl3 | 100 | Na2SO4 | 1:1:1 | 95 | 93 |
| 23 | (R)-5d | CHCl3 | 100 | / | 1:1:1 | 69 | 91 |
| 24f | (R)-5d | CHCl3 | 100 | / | 1:1:1 | 69 | 90 |
a: Unless indicated otherwise, the reaction was carried out at 0.1 mmol scale and catalyzed by 5 mol% Cat. in CHCl3 (1 mL) at T oC with addictives (50 mg) for 15 h; b: Isolated yield; c: The ee values were determined by HPLC; d: 2 mol% Cat; e: 10 mol% Cat; f: With water (10.0 equiv.); DCE: ClCH2CH2Cl.
With the optimal conditions established, we proceeded to investigate the substrate scope of this catalytic asymmetric multicomponent tandem cycloaddition. We initially focused on exploring the scope of 2,3-diketoester precursors 3 (Figure 4). It was found that 2,3-diketoester precursors 3 bearing various Ar/R substituents were all amenable to this reaction, affording the products 4 in generally high yields (up to 96%) and excellent enantioselectivities (up to 94% ee). Specifically, a phenyl group proved to be a suitable Ar substituent of substrate 3b, affording the corresponding product 4aab with 80% yield and 93% ee. Substrates 3c–3i, bearing either electron-withdrawing or electron-donating groups at the meta- or para-positions of the phenyl ring, underwent the reaction smoothly to furnish the respective C–N axially chiral 5-pyrazolyl indoles 4aac–4aai in both high yields and excellent enantioselectivities. Heteroaryl-substituted substrate 3j also proved to be a suitable substrate for this reaction; however, its performance was marginally lower than that of aryl-substituted substrates 3a–3i, furnishing product 4aaj in a moderate yield of 52% and a good enantioselectivity of 85% ee. Furthermore, when the ethyl ester group of substrate 3b was replaced with a methyl ester group (substrate 3k), the reaction also proceeded smoothly, yielding the corresponding product 4aak (57% yield, 91% ee). The moderate yields observed in these cases were attributed to the formation of inseparable mixtures during the reaction. In addition, the absolute configuration of product 4aag was determined to be (Ra) by single-crystal X-ray diffraction analysis (Supplementary materials).
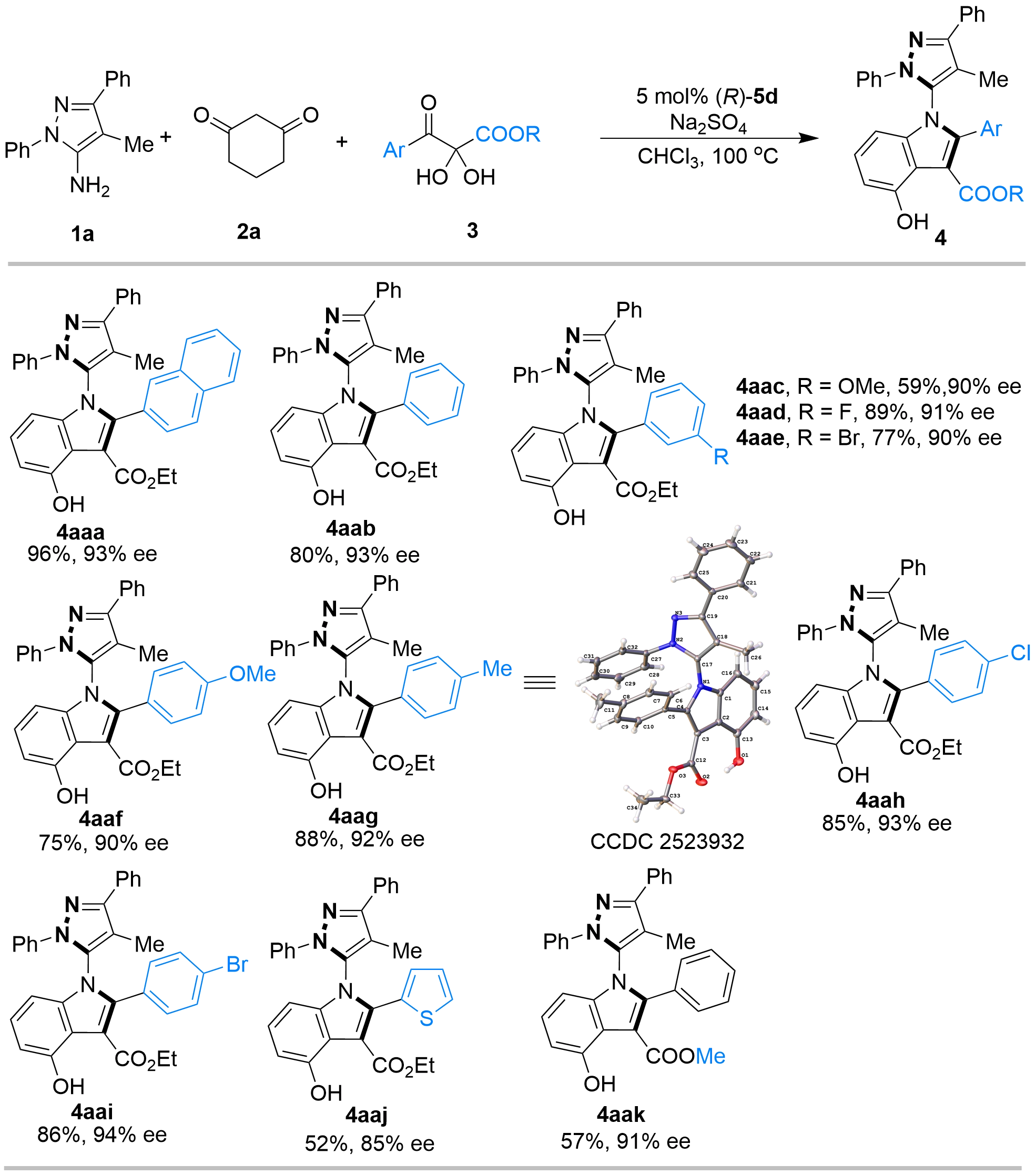
Figure 4. Substrate scope of 2,3-diketoester precursors 3a. a: Unless indicated otherwise, the reaction was carried out at 0.1 mmol scale and catalyzed by 5 mol% (R)-5d in CHCl3 (1 mL) at 100 oC with Na2SO4 (50 mg) as additives for 15 h, and the molar ratio of 1a:2a:3 was 1:1:1. Isolated yield. The ee values were determined by HPLC. HPLC: high-performance liquid chromatography.
Subsequently, we extended our investigation to evaluate the versatility of 5-aminopyrazoles 1 and cyclohexanediones 2 (Figure 5). In general, a broad array of 5-aminopyrazoles 1 bearing diverse R and Ar groups proved to be compatible with this catalytic asymmetric multicomponent tandem cycloaddition, providing the corresponding C–N axially chiral 5-pyrazolyl indoles 4 in moderate to good yields (67-95%) with high enantioselectivities (85-95% ee). Specifically, 5-aminopyrazoles 1b–1i, featuring ortho-, meta-, or para-substituted phenyl rings as the R substituent, served as suitable substrates to afford products 4bab–4iab with high enantioselectivities. Notably, alkyl substituents, such as benzyl, cyclohexyl, and tert-butyl groups, also served as effective R substituents for substrates 1j–1l, undergoing the tandem cyclization to generate the respective products 4jab–4lab with good enantioselectivity. Furthermore, substrates 1m–1s, possessing phenyl Ar groups with varying electronic properties at different positions, proceeded smoothly to furnish the corresponding products 4mab–4sab in good yields and enantioselectivities. To our delight, heteroaromatic motifs, such as the thienyl group (1t), were also accommodated, successfully delivering the desired product 4tab. Regarding the cyclohexanediones 2, substrate 2b substituted with an alkyl group served as an effective substrate, delivering the corresponding product 4abb efficiently.
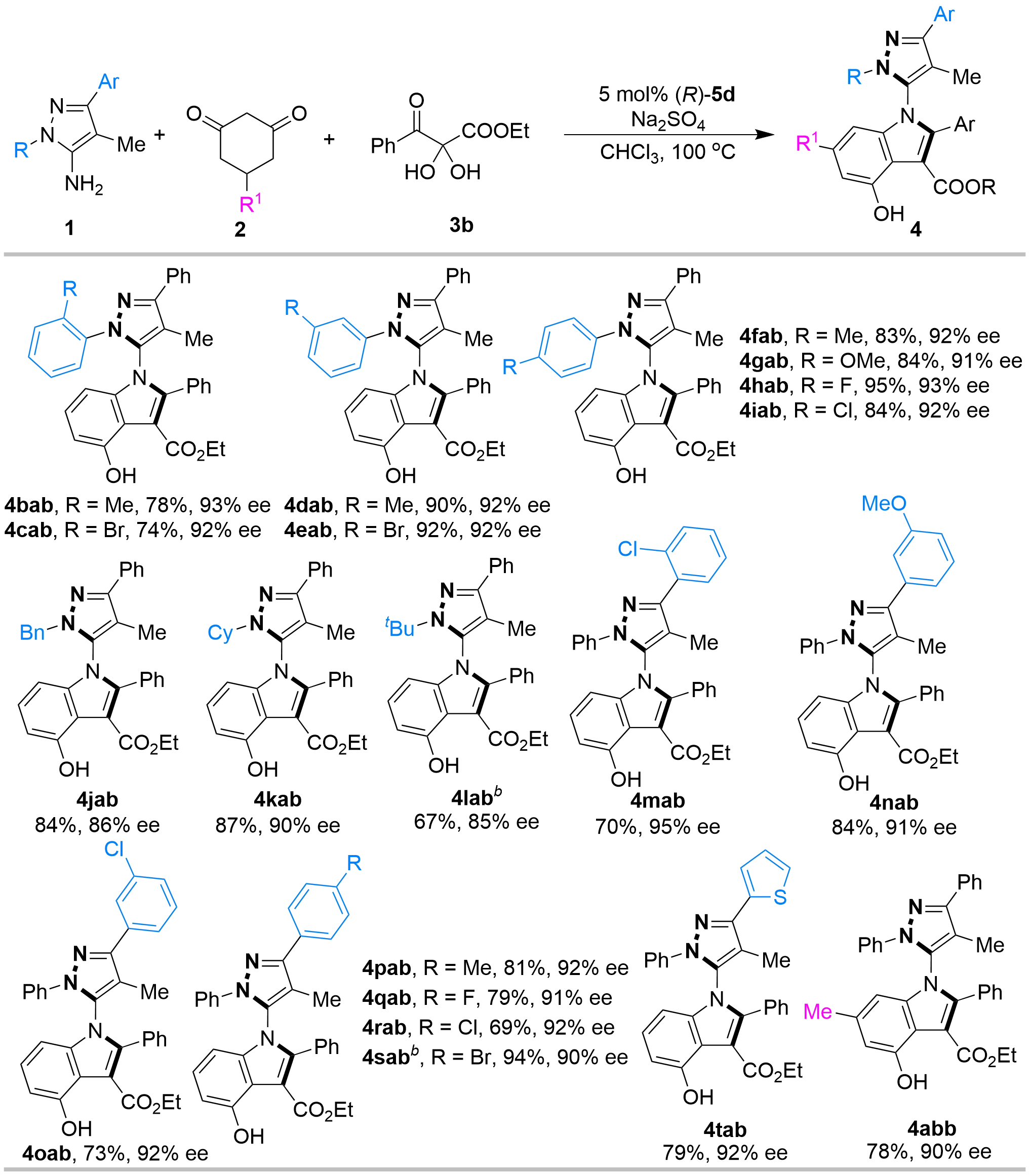
Figure 5. Substrate scope of 5-aminopyrazoles 1 and cyclohexanediones 2a. a: Unless indicated otherwise, the reaction was carried out at 0.1 mmol scale and catalyzed by 5 mol% (R)-5d in CHCl3 (1 mL) at 100 oC with Na2SO4 (50 mg) as additives for 15 h, and the molar ratio of 1:2:3b was 1:1:1. Isolated yield. The ee values were determined by HPLC; b: 30 mol% (R)-5d; HPLC: high-performance liquid chromatography.
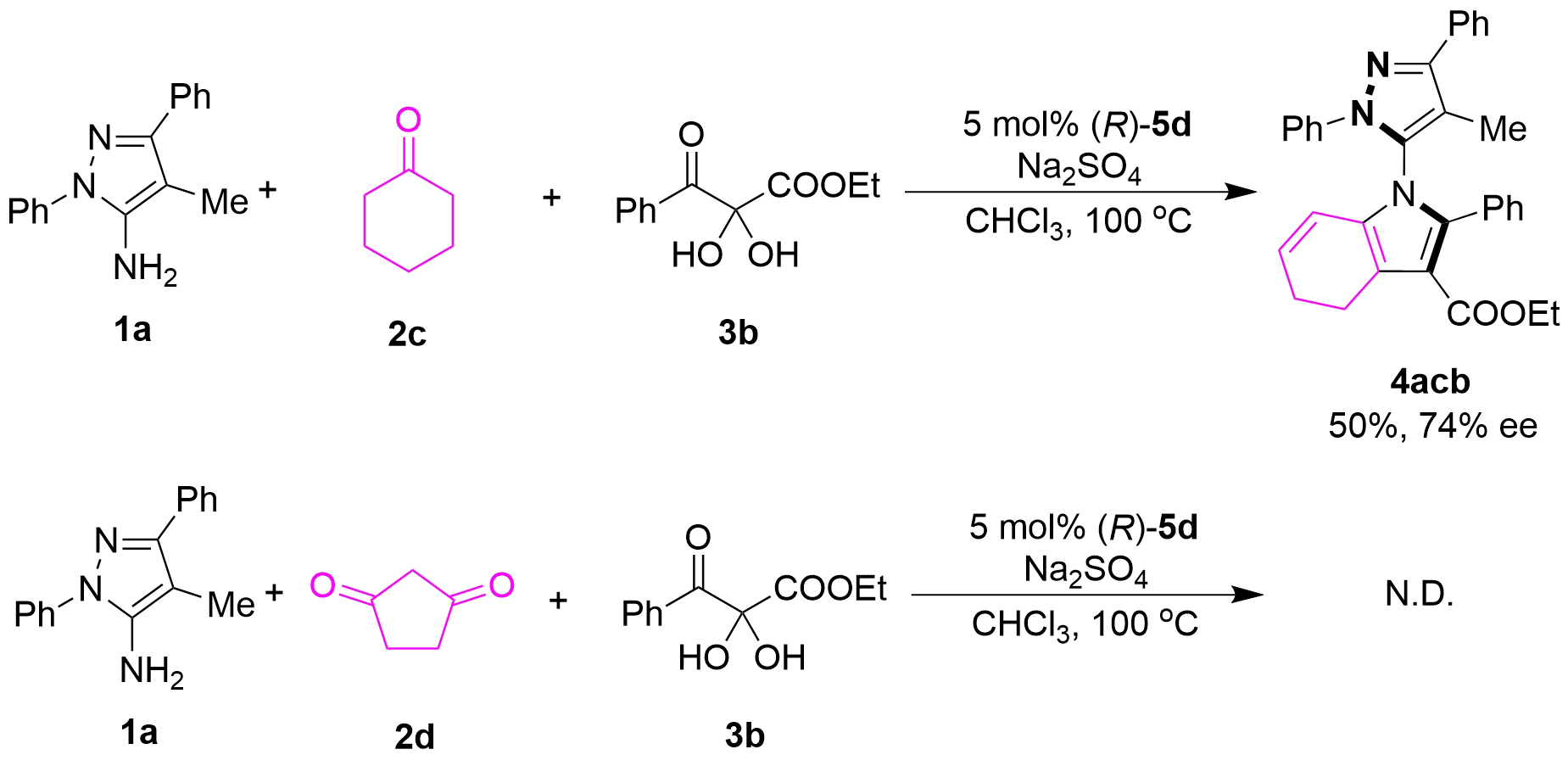
We further explored the scope of different cyclic ketones as substrates 2 (Figure 6). Cyclohexanone (2c) was found to be a competent substrate for this catalytic asymmetric multicomponent tandem cycloaddition, providing the corresponding product 4acb in 50% yield with an acceptable enantioselectivity of 74% ee. However, when 1,3-cyclopentanedione (2d) was employed as the substrate, the desired product could not be obtained.
To elucidate the reaction mechanism of this catalytic asymmetric multicomponent tandem cycloaddition, we conducted a series of control experiments (Figure 7a). We directly employed pre-synthesized enamine 6 as a substrate to react with 2,3-diketoester precursor 3b under optimal conditions. This reaction proceeded smoothly, affording product 4aab in high yield and excellent enantioselectivity (eq 1). Furthermore, when 5-aminopyrazole 1a and cyclohexanedione 2a were reacted under optimal conditions for 8 hours, TLC monitoring indicated complete conversion of both starting materials to enamine 6. Subsequently, the addition of substrate 3b and continuation of the reaction for an additional 7 hours afforded product 4aab in 79% yield and 92% ee (eq 2). These results unequivocally demonstrated that enamine 6 is a crucial intermediate in this reaction. Moreover, as we initially envisioned, in this catalytic asymmetric multicomponent tandem cycloaddition, substrates 1 and 2 first condensed to form the enamine intermediates, which then underwent a cycloaddition reaction with substrates 3.
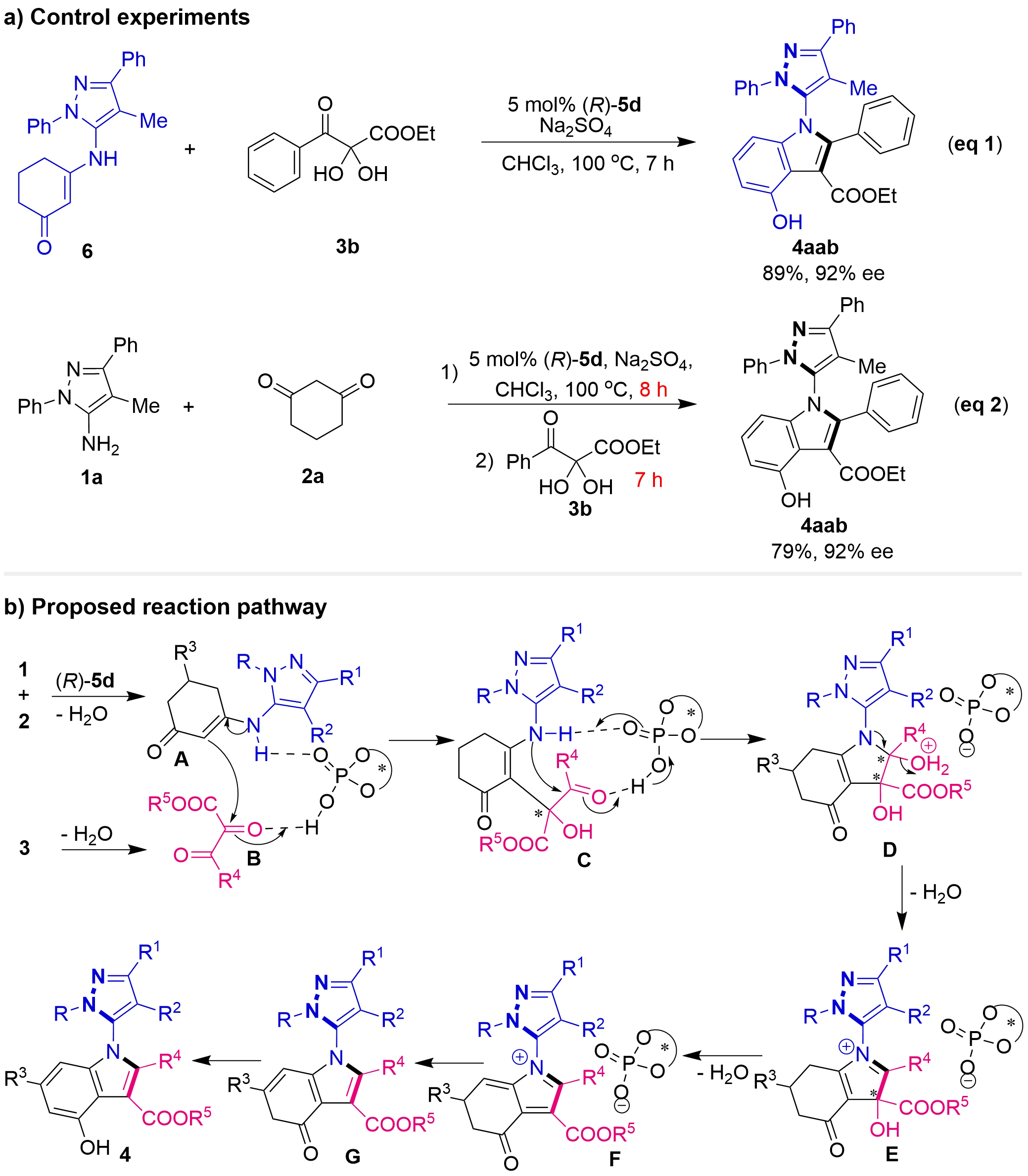
Based on these experimental results, we proposed a plausible mechanism and the activation mode for this reaction (Figure 7b). Under the action of CPA (R)-5d, 5-aminopyrazoles 1 and cyclohexanediones 2 underwent dehydration–condensation to form enamine intermediates A. Concurrently, 2,3-diketoester precursors 3 underwent dehydration to form 2,3-diketoester intermediates B. CPA (R)-5d then activated both intermediates A and intermediates B simultaneously through hydrogen bonding, promoting the asymmetric addition to generate intermediates C with a newly formed stereocenter. Subsequently, intermediates C, activated by CPA (R)-5d, underwent intramolecular cycloaddition via amine attack on the carbonyl group, leading to intermediates D containing two stereocenters. During the subsequent dehydration and aromatization steps involving intermediates E, F, and G, the established central chirality was efficiently transferred to axial chirality. Finally, a subsequent isomerization process delivered the C–N axially chiral 5-pyrazolyl indoles 4.
To assess the configurational stability of the newly designed C–N axially chiral 5-pyrazolyl indoles, a racemization experiment was conducted (Figure 8a). Product 4aab, after stirring at 120 °C for 8 hours, maintained excellent chemical and configurational stability, being recovered with quantitative recovery (99% recovery) and retaining enantioselectivity (94% ee). In addition, the rotational barrier of product 4aab was theoretically calculated as 35.8 kcal mol-1, further supporting the high configurational stability of these C–N axially chiral 5-pyrazolyl indoles. To demonstrate the synthetic utility of this methodology, a 1 mmol scale-up reaction was performed (Figure 8b). The transformation proved to be highly robust upon scaling, delivering product 4aab in 86% yield with 94% ee, which was consistent with the results obtained on a 0.1 mmol scale. Furthermore, several synthetic transformations of the obtained products were also explored (Figure 8c). Product 4aab underwent nucleophilic addition with a Grignard reagent to afford the corresponding tertiary alcohol derivative 7 with preservation of enantiopurity. Additionally, the phenolic hydroxyl group of product 4aab could be methylated to afford compound 8 in 86% yield. Subsequent reduction of the ester group in compound 8 with LiAlH4 efficiently furnished the C–N axially chiral benzylic alcohol derivative 9, which was further oxidized to access the axially chiral indole-3-carboxaldehyde derivative 10. Notably, the enantioselectivity was well-maintained throughout this sequence of transformations.
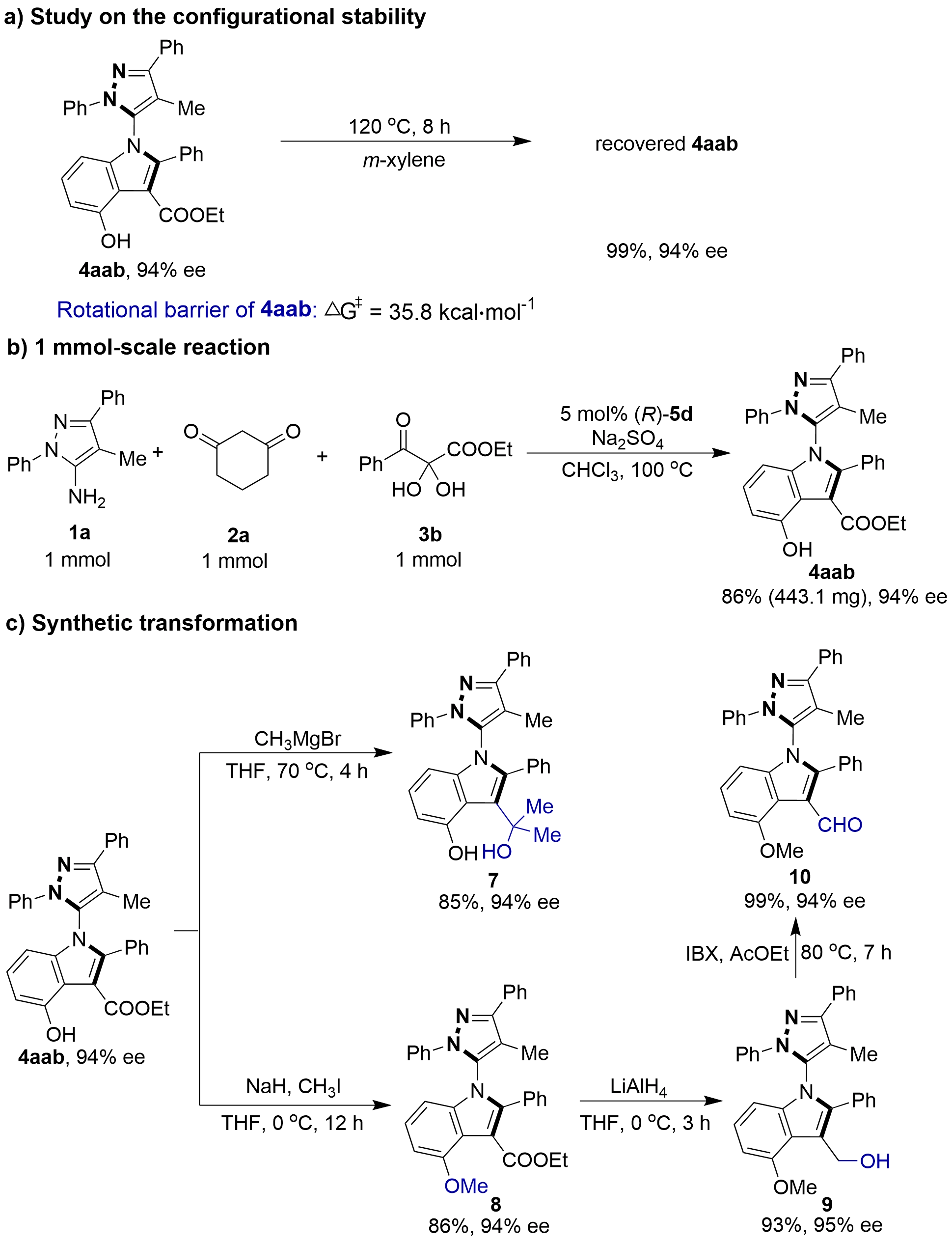
Figure 8. (a) Study on the configurational stability; (b) One mmol scale reaction; (c) Synthetic transformation.
4. Conclusion
In summary, we have established an organocatalytic asymmetric multicomponent tandem cycloaddition for the construction of C–N axially chiral 5-pyrazolyl indole frameworks, representing a novel class of five-five-membered indole scaffolds. Utilizing 5-aminopyrazoles, cyclohexanediones, and 2,3-diketoester precursors, this methodology constructs the indole ring de novo in a single step, demonstrating high atom- and step-economy with water as the exclusive byproduct. The protocol enabled the efficient and high-enantioselective synthesis of structurally diverse C–N axially chiral 5-pyrazolyl indoles (up to 96% yield, 95% ee). Mechanistic insights were gained through control experiments, and the resulting products demonstrated outstanding configurational stability. Further validation of the practical utility of this reaction was achieved through scale-up experiments and exploration of synthetic transformations. This achievement not only introduces a new member to the family of axially chiral pyrazolyl indoles but also provides an efficient and novel strategy for their construction.
Supplementary materials
The supplementary material for this article is available at: Supplementary materials.
Authors contribution
Chen YY, Xue J: Investigation, formal analysis.
Zhang YC, Shi F: Conceptualization, methodology, writing-original draft, writing-review & editing.
Conflicts of interest
Feng Shi is an Editorial Board Member of Chiral Chemistry. Other authors declared that there are no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
The data reported in this paper are available in the main text or Supplementary materials, including methods, NMR data, HRMS data, HPLC spectra Crystal data and NMR spectra. Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2523932 (4aag).
Funding
This work was supported by the National Science Foundation of China (Grant No. 22125104) and the Natural Science Foundation of Jiangsu Province (Grant No. BK20240052), Project for Excellent Scientific and Technological Innovation Team of Jiangsu Province.
Copyright
© The Author(s) 2026.
References
-
1. Wang YB, Tan B. Construction of axially chiral compounds via asymmetric organocatalysis. Acc Chem Res. 2018;51(2):534-547.[DOI]
-
3. Metrano AJ, Miller SJ. Peptide-based catalysts reach the outer sphere through remote desymmetrization and atroposelectivity. Acc Chem Res. 2019;52(1):199-215.[DOI]
-
4. Qin W, Liu Y, Yan H. Enantioselective synthesis of atropisomers via vinylidene ortho-quinone methides (VQMs). Acc Chem Res. 2022;55(19):2780-2795.[DOI]
-
5. Cheng JK, Xiang SH, Tan B. Organocatalytic enantioselective synthesis of axially chiral molecules: Development of strategies and skeletons. Acc Chem Res. 2022;55(20):2920-2937.[DOI]
-
6. Kozlowski MC, Miller SJ, Perreault S. Atropisomers: Synthesis, analysis, and applications. Acc Chem Res. 2023;56(3):187-188.[DOI]
-
8. Gaucherand A, Yen-Pon E, Domain A, Bourhis A, Rodriguez J, Bonne D. Enantioselective synthesis of molecules with multiple stereogenic elements. Chem Soc Rev. 2024;53(22):11165-11206.[DOI]
-
9. LaPlante SR, Fader LD, Fandrick KR, Fandrick DR, Hucke O, Kemper R, et al. Assessing atropisomer axial chirality in drug discovery and development. J Med Chem. 2011;54(20):7005-7022.[DOI]
-
11. Basilaia M, Chen MH, Secka J, Gustafson JL. Atropisomerism in the pharmaceutically relevant realm. Acc Chem Res. 2022;55(20):2904-2919.[DOI]
-
12. Li Q, Green L, Venkataraman N, Shiyanovskaya I, Khan A, Urbas A, et al. Reversible photoswitchable axially chiral dopants with high helical twisting power. J Am Chem Soc. 2007;129(43):12908-12909.[DOI]
-
13. Takaishi K, Yasui M, Ema T. Binaphthyl–bipyridyl cyclic dyads as a chiroptical switch. J Am Chem Soc. 2018;140(16):5334-5338.[DOI]
-
14. Zhou, QL. Privileged chiral ligands and catalysts. Hoboken: John Wiley & Sons; 2011.
-
15. Tan B. Axially chiral compounds: Asymmetric synthesis and applications. New York: John Wiley & Sons; 2021.[DOI]
-
16. Ito C, Thoyama Y, Omura M, Kajiura I, Furukawa H. Alkaloidal constituents of murraya koenigii. isolation and structural elucidation of novel binary carbazolequinones and carbazole alkaloids. Chem Pharm Bull. 1993;41(12):2096-2100.[DOI]
-
17. Zhang Q, Mándi A, Li S, Chen Y, Zhang W, Tian X, et al. N–N-coupled indolo-sesquiterpene atropo-diastereomers from a marine-derived actinomycete. Eur J Org Chem. 2012;2012(27):5256-5262.[DOI]
-
18. Ge FB, Yin QK, Lu CJ, Xuan X, Feng J, Liu RR. Enantioselective synthesis of benzimidazole atropisomers featuring a N-N axis. Chin J Chem. 2024;42(7):711-718.[DOI]
-
22. Mino T, Komatsu S, Wakui K, Yamada H, Saotome H, Sakamoto M, et al. N-Aryl indole-derived C–N bond axially chiral phosphine ligands: Synthesis and application in palladium-catalyzed asymmetric allylic alkylation. Tetrahedron Asymmetry. 2010;21(6):711-718.[DOI]
-
24. Zhang HH, Shi F. Organocatalytic atroposelective synthesis of indole derivatives bearing axial chirality: Strategies and applications. Acc Chem Res. 2022;55(18):2562-2580.[DOI]
-
25. Kee Cheng J, Tan B. Chiral phosphoric acid-catalyzed enantioselective synthesis of axially chiral compounds involving indole derivatives. Chem Rec. 2023;23(11):e202300147.[DOI]
-
26. Wei J, Zhu M, Zhang B, Li K, Zhang X. Recent advances in atroposelective synthesis of axially chiral heterobiaryl featuring indole frameworks. Tetrahedron. 2023;149:133716.[DOI]
-
27. Feng J, Liu RR. Catalytic asymmetric synthesis of N–N biaryl atropisomers. Chemistry A European J. 2024;30(2):e202303165.[DOI]
-
28. Wang YD, Luan WY, Ma C, Shi F. Catalytic asymmetric reactions of indolylmethanols for the synthesis of chiral indole derivatives. Chem Commun. 2026;62(4):1109-1127.[DOI]
-
29. Hang QQ, Wu SF, Yang S, Wang X, Zhong Z, Zhang YC, et al. Design and catalytic atroposelective synthesis of axially chiral isochromenone-indoles. Sci China Chem. 2022;65(10):1929-1937.[DOI]
-
31. Xu D, Huang S, Hu F, Peng L, Jia S, Mao H, et al. Diversity-oriented enantioselective construction of atropisomeric heterobiaryls andN-aryl indoles via VinylideneOrtho-quinone methides. CCS Chem. 2022;4(8):2686-2697.[DOI]
-
33. Yang H, Sun HR, He RQ, Yu L, Hu W, Chen J, et al. Organocatalytic cycloaddition of alkynylindoles with azonaphthalenes for atroposelective construction of indole-based biaryls. Nat Commun. 2022;13:632.[DOI]
-
36. Liu M, Zhang Y, Ke XY, Ni SF, Li P. Asymmetric organocatalytic 1, 6-conjugate addition of alkynyl 8-methylenenaphthalen-2(8H)-one formed in situ: Synergistic construction of axial and central chirality. Org Lett. 2025;27(5):1271-1275.[DOI]
-
37. Wang Y, Zhou X, Shan W, Liao R, Deng Y, Peng F, et al. Construction of axially chiral indoles by cycloaddition–isomerization via atroposelective phosphoric acid and silver sequential catalysis. ACS Catal. 2022;12(13):8094-8103.[DOI]
-
39. Xie ZY, Tang C, Li L, Zhou Z, Zou J, Qian PC. Rhodium-catalyzed regioselective [4 + 2] cycloaddition of ynamines and 2-(cyanomethyl)phenylboronates. Org Lett. 2024;26(31):6586-6590.[DOI]
-
40. Zhang G, Yang B, Yang J, Zhang J. Pd-catalyzed asymmetric larock indole synthesis to access axially chiral N-arylindoles. J Am Chem Soc. 2024;146(8):5493-5501.[DOI]
-
41. Guo Y, Liu X, Hu L, Xia J, Li Y, Zhu L, et al. Visible-light-driven cobalt-catalyzed assembly of indole-alcohols bearing concurrent axial and central chiralities via dynamic kinetic resolution. ACS Catal. 2025;15(21):18305-18314.[DOI]
-
42. Jin S, Wang Y, Yan JX, Xu T, Ning M, Yuan Q, et al. P(=O)R2-directed asymmetric catalytic C–H olefination leading to C–N axially chiral targets. Org Lett. 2025;27(12):2838-2844.[DOI]
-
43. Zhang X, Teng Q, Tung CH, Xu Z. Synthesis of axially and centrally chiral N-alkenyl indoles by asymmetric Rh-catalyzed [2 + 2 + 2] cycloaddition. ChemCatChem. 2025;17:e202401143.[DOI]
-
44. Liu Q, Gu J, Zhuang HF, He Y. Stereospecific positional alkene isomerization enables bidirectional central-to-axial chirality transfer. Nat Commun. 2025;16:6782.[DOI]
-
46. Zhu X, Wu H, Wang Y, Huang G, Wang F, Li X. Rhodium-catalyzed annulative approach to N–N axially chiral biaryls via C–H activation and dynamic kinetic transformation. Chem Sci. 2023;14(32):8564-8569.[DOI]
-
47. Qu H, Huo C, Ge J, Xue X, Gu Z, Deng R. Symmetric anion mediated dynamic kinetic asymmetric Knoevenagel reaction for N–C and N–N atropisomers synthesis. Angew Chem Int Ed. 2024;63(39):e202410012.[DOI]
-
48. Ranganathappa SS, Dehury BS, Singh GK, Shee S, Biju AT. Atroposelective synthesis of N–N axially chiral indoles and pyrroles via NHC-catalyzed diastereoselective (3 + 3) annulation strategy. ACS Catal. 2024;14(9):6965-6972.[DOI]
-
49. Wang NY, Gao S, Shu ZD, Cheng BB, Ma C, Zhang YC, et al. Catalytic atroposelective synthesis of indolyl quinazolinones bearing N–N/C–C diaxes. Sci China Chem. 2025;68(7):3130-3137.[DOI]
-
50. Li C, Zuo WF, Zhou J, Zhou WJ, Wang M, Li X, et al. Catalytic asymmetric synthesis of 3,4′-indole–pyrazole derivatives featuring axially chiral bis-pentatomic heteroaryls. Org Chem Front. 2022;9(7):1808-1813.[DOI]
-
53. Gao C, Li Y, Luo G, Ni Q. Chiral phosphoric acid-catalyzed asymmetric Doyle indolization: Enantiodivergent access to indolizinylindoles. Org Lett. 2025;27(35):9576-9581.[DOI]
-
57. Sparr C, Hutskalova V. Control over stereogenic N–N axes by Pd-catalyzed 5-endo-hydroaminocyclizations. Synthesis. 2023;55(11):1770-1782.[DOI]
-
60. Wang D, Zong J, Zhang B, Wang J, Wang B, Piao H, et al. Enantioselective synthesis of N–N indole-pyrrole atropisomers via palladium/chiral phosphonic acid relay catalysis. ACS Catal. 2025;15(14):12450-12462.[DOI]
-
61. Li X, Wang XZ, Shen B, Chen QY, Xiang H, Yu P, et al. Organocatalyzed diastereo- and enantioselective synthesis of N–N atropisomeric isoindolinones bearing central chirality. Nat Commun. 2025;16:1662.[DOI]
-
62. Gu J, Zhang LH, Zhuang HF, He Y. Atroposelective [4+1] annulation for the synthesis of isotopic isoindolinones bearing both central and axial chirality. Chem Sci. 2025;16(13):5735-5744.[DOI]
-
64. Bonne D, Rodriguez J. A bird’s eye view of atropisomers featuring a five-membered ring. Eur J Org Chem. 2018;2018(20-21):2417-2431.[DOI]
-
65. Zhang S, Liao G, Shi B. Enantioselective synthesis of atropisomers featuring pentatomic heteroaromatics. Chin J Org Chem. 2019;39(6):1522.[DOI]
-
66. Chen YB, Yang YN, Huo XZ, Ye LW, Zhou B. Recent advances in the construction of axially chiral arylpyrroles. Sci China Chem. 2023;66(9):2480-2491.[DOI]
-
67. Tan W, Wu XY, Shi F. Catalytic atroposelective construction of furan-based axially chiral scaffolds. ChemCatChem. 2024;16(21):e202401022.[DOI]
-
68. Ma D, Tan Z, Li S, Zhao B, Yue L, Wei X, et al. Discovery of novel 4,5,6,7-tetrahydro-7H-pyrazolo[3,4-c]pyridin-7-one derivatives as orally efficacious ATX allosteric inhibitors for the treatment of pulmonary fibrosis. J Med Chem. 2025;68(1):792-818.[DOI]
-
69. Tagami U, Takahashi K, Igarashi S, Ejima C, Yoshida T, Takeshita S, et al. Interaction analysis of FABP4 inhibitors by X-ray crystallography and fragment molecular orbital analysis. ACS Med Chem Lett. 2016;7(4):435-439.[DOI]
-
70. Anand D, Yadav PK, Patel OPS, Parmar N, Maurya RK, Vishwakarma P, et al. Antileishmanial activity of pyrazolopyridine derivatives and their potential as an adjunct therapy with miltefosine. J Med Chem. 2017;60(3):1041-1059.[DOI]
-
72. Zhang YC, Jiang F, Shi F. Organocatalytic asymmetric synthesis of indole-based chiral heterocycles: Strategies, reactions, and outreach. Acc Chem Res. 2020;53(2):425-446.[DOI]
-
73. Wang HQ, Gao S, Miao XH, Zhang YC, Shi F. Condition-controlled catalytic asymmetric chemodivergent reaction of benzofuran-derived azadienes with 2-indolylmethanols. Sci China Chem. 2025.[DOI]
-
74. Liu SJ, Wang X, Yang JX, Ao XS, Ni SF, Zhang YC, et al. Atroposelective construction of axially chiral alkenylindole-fused nine-membered rings via catalytic asymmetric formal (4 + 5) cycloaddition. Nat Commun. 2025;16:6605.[DOI]
-
76. Kitagawa O. Chiral Pd-catalyzed enantioselective syntheses of various N–C axially chiral compounds and their synthetic applications. Acc Chem Res. 2021;54(3):719-730.[DOI]
-
77. Wu YJ, Liao G, Shi BF. Stereoselective construction of atropisomers featuring a C–N chiral axis. Green Synth Catal. 2022;3(2):117-136.[DOI]
-
78. Rodríguez-Salamanca P, Fernández R, Hornillos V, Lassaletta JM. Asymmetric synthesis of axially chiral C–N atropisomers. Chemistry A European J. 2022;28(28):e202104442.[DOI]
-
79. Feng J, Xi LL, Lu CJ, Liu RR. Transition-metal-catalyzed enantioselective C–N cross-coupling. Chem Soc Rev. 2024;53(19):9560-9581.[DOI]
-
80. Chen YY, Xue J, Shi Q, Zhang YC, Shi F. Catalytic atroposelective synthesis of C–N axially chiral pyrazolyl pyrroles via De Novo Construction of a pyrrole ring. Org Lett. 2026;28(4):1481-1486.[DOI]
-
81. Yang BM, Ng XQ, Zhao Y. Enantioselective synthesis of indoles through catalytic indolization. Chem Catal. 2022;2(11):3048-3076.[DOI]
-
82. Sun HR, Sharif A, Chen J, Zhou L. Atroposelective synthesis of heterobiaryls through ring formation. Chemistry A European J. 2023;29(27):e202300183.[DOI]
-
83. Wang LY, Yang L, Chen J, Zhou L. Chiral phosphoric acid catalyzed asymmetric cycloadditions: From alkenes to alkynes. Synlett. 2023;34(11):1200-1214.[DOI]
-
86. Wang LY, Miao J, Zhao Y, Yang BM. Chiral acid-catalyzed atroposelective indolization enables access to 1,1′-indole-pyrroles and bisindoles bearing a chiral N–N axis. Org Lett. 2023;25(9):1553-1557.[DOI]
-
88. Terada M. Chiral phosphoric acids as versatile catalysts for enantioselective transformations. Synthesis. 2010;2010(12):1929-1982.[DOI]
-
89. Parmar D, Sugiono E, Raja S, Rueping M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: History and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem Rev. 2014;114(18):9047-9153.
-
90. Xia ZL, Xu-Xu QF, Zheng C, You SL. Chiral phosphoric acid-catalyzed asymmetric dearomatization reactions. Chem Soc Rev. 2020;49(1):286-300.[DOI]
-
91. Woldegiorgis AG, Lin X. Recent advances in the asymmetric phosphoric acid-catalyzed synthesis of axially chiral compounds. Beilstein J Org Chem. 2021;17:2729-2764.[DOI]
-
92. Guo X, Yu H, Wan H, Lu Y, Tan W, Shi F. Organophosphoric acid catalyzed [3 + 3] cyclization for the synthesis of indenoquinolinedione derivatives. Chin J Org Chem. 2024;44(12):3727-3738. Chinese.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Chen YY, Xue J, Zhang YC, Shi F. Catalytic atroposelective synthesis of C–N axially chiral 5-pyrazolyl indoles via multicomponent tandem cycloaddition. Chiral Chem. 2026;2:202604. https://doi.org/10.70401/cc.2026.0018
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Chen YY, Xue J, Zhang YC, Shi F. Catalytic atroposelective synthesis of C–N axially chiral 5-pyrazolyl indoles via multicomponent tandem cycloaddition. Chiral Chem. 2026;2:202604. https://doi.org/10.70401/cc.2026.0018
copy
Share Link
copy