Programmable DNA as stereocontrol ligand for asymmetric catalysis in aqueous media
Shanmei Xu

Zixiao Wang
Mengyao Wang
Yashao Chen
Changhao Wang
*
*Correspondence to:
Changhao Wang, Key Laboratory of Applied Surface and Colloid Chemistry (MOE), School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi’an 710119, Shaanxi, China.
E-mail: changhaowang@snnu.edu.cn
Chiral Chem. 2026;2:202611. 10.70401/cc.2026.0022
Received: February 24, 2026Accepted: April 09, 2026Published: April 10, 2026
Abstract
Owing to its intrinsic chirality, programmability and structural diversity, DNA functions as an effective chiral ligand for asymmetric catalysis in water. DNA hybrid catalysts are constructed by anchoring metal species into DNA scaffolds through covalent or noncovalent strategies. These DNA-based catalytic systems exhibit potent reactivity and high enantioselectivity in various C–C, C–N, C–O, and C–F bond formation reactions in aqueous media. Importantly, their catalytic activity and product stereochemistry can be precisely directed by modulating DNA sequences, tertiary structures, metal species, and host-guest interaction. This review highlights recent advances in DNA-mediated asymmetric catalysis and discusses future prospects toward tailored stereocontrol under aqueous conditions.
Graphical Abstract
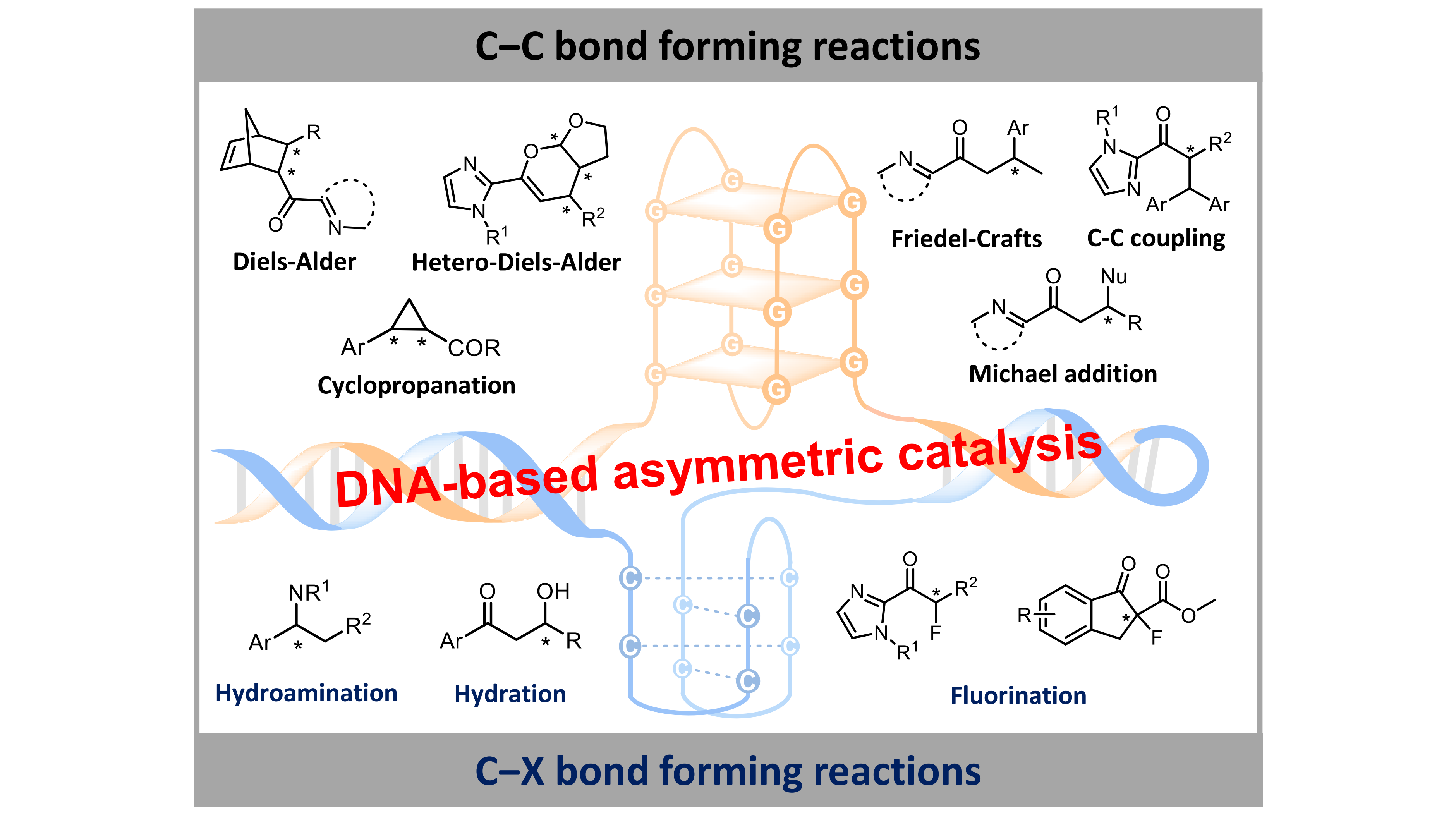
Keywords
Asymmetric catalysis, aqueous medium, chiral inversion, i-motif DNA, G-quadruplex DNA
1. Introduction
In recent years, deoxyribonucleic acid (DNA) has emerged as an effective chiral ligand for asymmetric catalysis in water due to its unique properties[1-3]. As a naturally occurring chiral biomacromolecule, DNA possesses not only inherent chirality and sequence programmability, but also adjustable secondary structures such as duplex, triplex, and quadruplex. These properties provide a versatile platform to construct tunable DNA hybrid catalysts by anchoring metal species through noncovalent or covalent strategies[4,5]. These DNA-metal hybrid catalysts effectively transfer the inherent helical chirality of DNA to the metal catalytic center, thereby enabling precise stereochemical control of the products by finely tuning the DNA structures. Unlike traditional synthetic small-molecule or metal-complex chiral ligands, DNA is innate water-compatible, enabling DNA-based asymmetric catalysis in aqueous media. Its accessibility via modern solid-phase synthesis makes it highly designable, cost-effective, and circumvents the need for complex multistep organic synthesis. Notably, the programmable structure of DNA endows precise stereochemical control of DNA hybrid catalysts for directly produce chiral products with desired enantiomers[6].
Owing to its biocompatible and structural tunability, DNA hybrid catalyst represents a new class of biohybrid catalysts for the sustainable synthesis of chiral compounds. To date, duplex DNA (ds-DNA) hybrid catalysts have been successfully applied to a range of asymmetric reactions in aqueous media, primarily centered on carbon-carbon bond formation reactions including Diels-Alder reactions, Michael addition reactions, Friedel-Crafts alkylations, and cyclopropanations[7]. Beyond C–C bond formation reactions, these DNA catalytic systems have encompassed a broad range of C–X (X = N, O, F) bond formation transformations[8-10]. Most importantly, the G-quadruplex DNA (G4-DNA) and i-motif DNA (im-DNA) hybrid catalysts possess inherently structural tunability, resulting in switchable enantioselectivity via changing the DNA sequences, tertiary structures, metal species, or host-guest interaction[11,12]. Compared with DNA hybrid catalysts, the inferior asymmetric catalytic performance of RNA hybrid catalysts is likely associated with their structural stability and capacity for binding metal complexes[4,13]. In this review (Scheme 1), we summarize the recent advances in DNA-based asymmetric reactions, analyze the intrinsic structure-activity relationship, and prospect future directions toward precise stereochemical control.
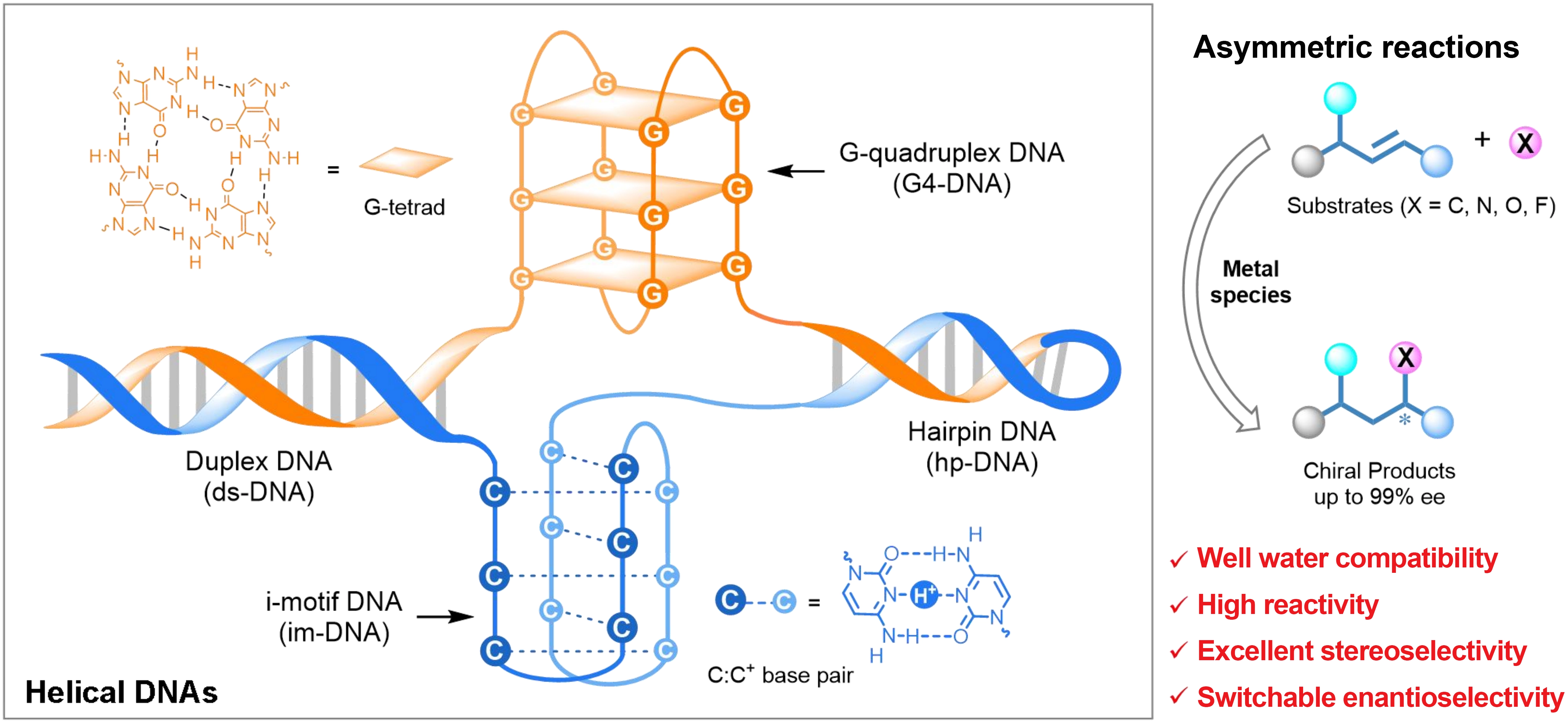
Scheme 1. Schematic representation of DNA-based asymmetric catalysis using different topological DNAs as stereocontrol ligands. DNAs: deoxyribonucleic acids.
2. DNA-Based Asymmetric C–C Bond Formation Reactions
To date, DNA-metal hybrid catalysts have enabled a broad spectrum of C-C bond-forming reactions, including Diels-Alder reactions, Michael addition reactions, Friedel-Crafts alkylations, and carbon-carbon coupling reactions. In these systems, ds-DNA, G4-DNA, and im-DNA have been employed as chiral scaffolds. These DNA scaffolds not only stabilize the active metal species in water, but also orient substrates through π-π stacking, groove binding, and electrostatic interactions. Importantly, tuning the DNA sequence, topology, and metal-binding mode enables systematic modulation of both catalytic activity and enantioselectivity.
2.1 Diels-Alder reactions
Roelfes et al.[4] first introduced the concept of anchoring an achiral metal complex onto natural ds-DNA to construct a DNA hybrid catalyst. By noncovalently associating a copper complex with salmon testes DNA, the resulted ds-DNA hybrid catalyst enabled efficient catalysis of an asymmetric Diels–Alder reaction between (E)-3-(4-methoxyphenyl)-1-(pyridin-2-yl)prop-2-en-1-one (1) and cyclopentadiene (2). Subsequent ligand screening revealed that when Cu(II) forms a complex with 4,4'-dimethyl-2,2'-bipyridyl ligand (L1), the resulting ds-DNA-1 hybrid catalyst dramatically increased the enantiomeric excess (ee) of product 3 to 99% in the same reaction, highlighting the decisive influence of ligand structure on the efficiency of chiral transfer from DNA[14] (Scheme 2a). Later studies further elucidated the critical role of ligand denticity in stereochemical control within DNA-based catalytic systems. The bidentate ligand L1 tends to induce a trigonal bipyramidal coordination environment around Cu(II) and the substrate, favoring the formation of the 2S,3S-configured product 5 at 99% ee. In contrast, the tridentate ligand L2 promotes an octahedral coordination geometry at the metal center, resulting in an inversion of the absolute configuration to the product (2R,3R)-5 at -92% ee (Scheme 2b)[15].
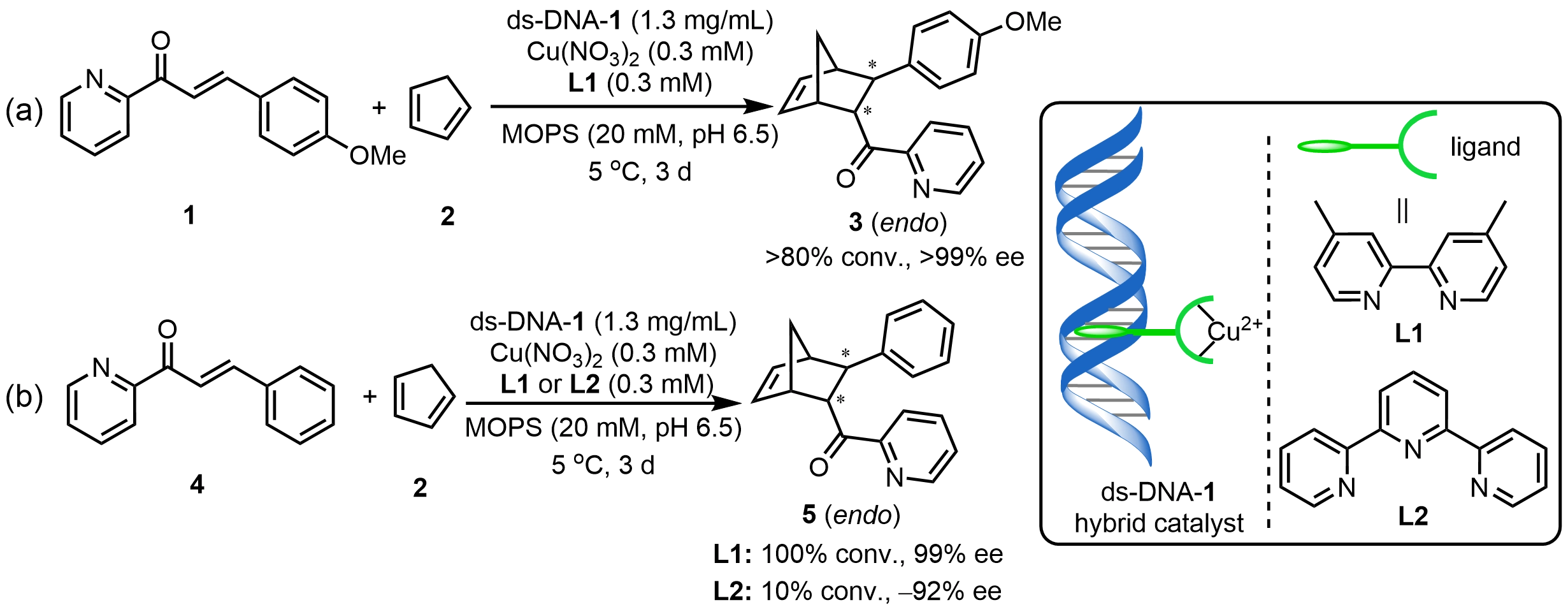
Scheme 2. Asymmetric Diels–Alder reactions catalyzed by ds-DNA hybrid catalysts. MOPS: 2-hydroxy-4-morpholinepropanesulphonic acid; ds-DNA: duplex deoxyribonucleic acid.
To address the issue of catalyst instability in noncovalent systems, Roelfes et al.[5] introduced linkers at specific sites of ds-DNA to covalently attach bipyridyl ligands, thereby constructing a bipyridine-modified ds-DNA-2 hybrid catalyst. The length of the linker was found to significantly affect asymmetric catalytic performance. Shortening the linker to three carbon atoms enhanced the ee of product 5 to 93%, demonstrating that close proximity between the metal complex and the DNA scaffold is crucial for efficient chiral transfer. Subsequently, another class of covalent ds-DNA catalysts was designed by anchoring ligands onto DNA via the strong coordination of platinum, which not only effectively catalyzed Diels–Alder reactions but also proved applicable to Friedel–Crafts reactions[16]. Sugiyama et al.[17] further incorporated histidine residues into the ds-DNA scaffold and coordinated them with Cu2+ to construct an imidazole-modified ds-DNA-3 hybrid catalyst for asymmetric Diels-Alder reactions, achieving up to 90% conversion and 97% ee for product 5 (Scheme 3).
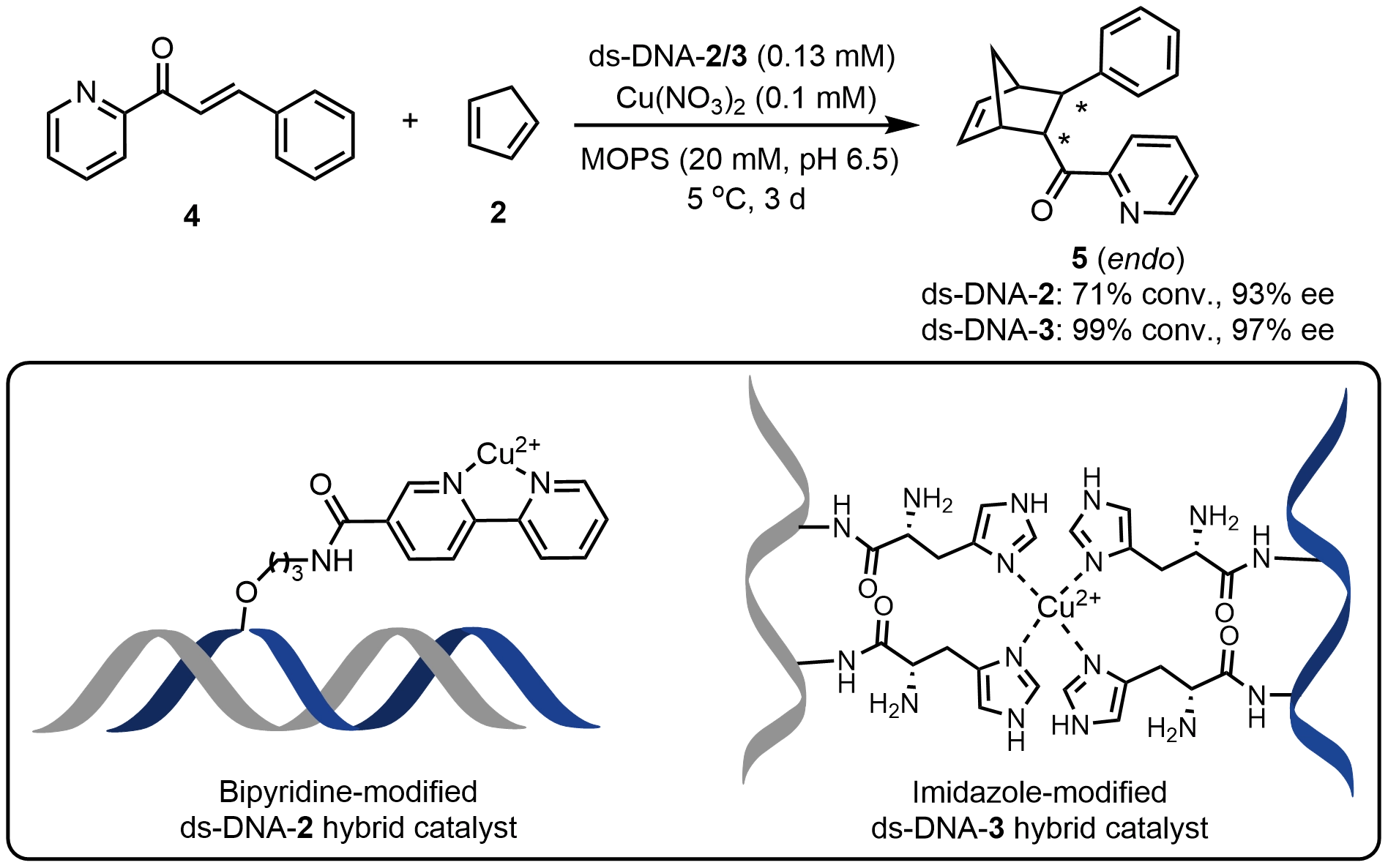
Scheme 3. An asymmetric Diels–Alder reaction catalyzed by bipyridine- and imidazole-modified ds-DNA hybrid catalysts. ds-DNA: duplex deoxyribonucleic acid.
Hennecke et al.[18] reported a hairpin DNA (hp-DNA) scaffold containing only 14 nucleotides, which could assemble with Cu(II)-L1 to form a highly efficient hp-DNA-1 hybrid catalyst for the Diels–Alder reaction between (E)-3-phenyl-1-(pyridin-2-yl)prop-2-en-1-one (4) and cyclopentadiene 2. The reaction proceeded with nearly complete substrate conversion, delivering the product 5 with an ee of 96%. The asymmetric performance of this catalytic system was found to be strongly influenced by the size and nucleotide composition of the hairpin loop, the sequence and length of the stem region, as well as the terminal bases (Scheme 4). Kokkoli and colleagues[19] demonstrated that Cu(II)-L1 can effectively bind to a short ds-DNA comprising only eight base pairs and successfully catalyze the asymmetric Diels-Alder reaction in aqueous media, achieving a product with up to 95% ee. These results demonstrate that the rational design of truncated DNA frameworks can establish well-defined chiral microenvironments through nucleic acid-metal interactions, enabling highly efficient asymmetric catalysis.
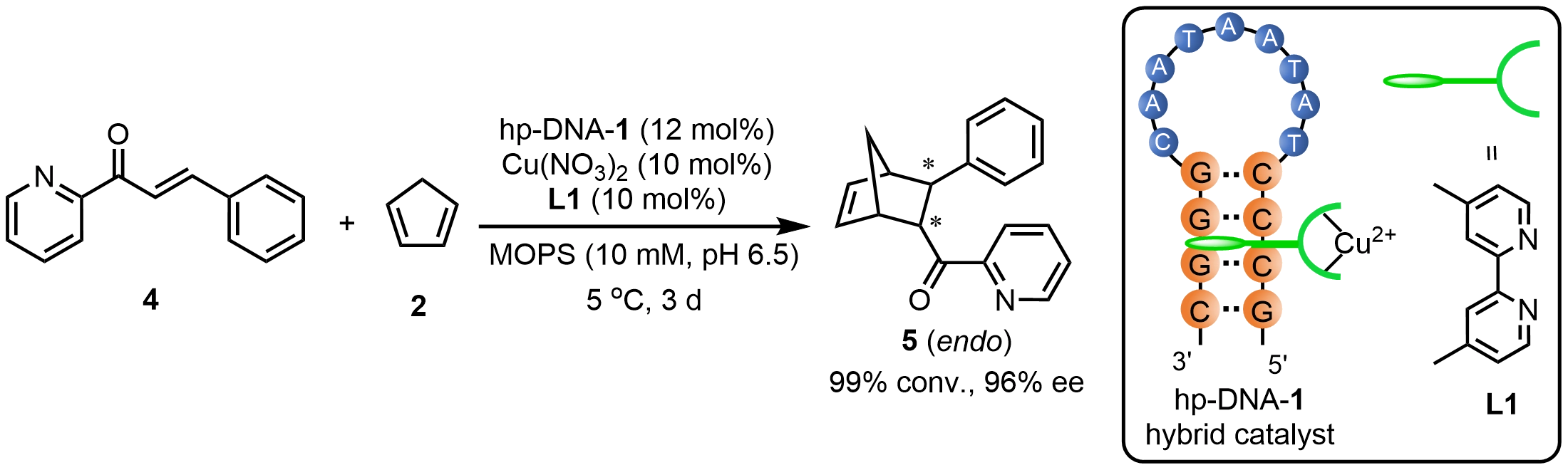
Scheme 4. An asymmetric Diels–Alder reaction catalyzed by a hp-DNA hybrid catalyst. Republished with permission from[18]. hp-DNA: hairpin deoxyribonucleic acid.
Compared with the relatively rigid ds-DNA, single-stranded DNA folding into non-canonical structures such as G-quadruplex or i-motif offers greater structural adaptability, providing new avenues for the development of DNA hybrid catalysts. G4-DNA is formed by a guanine-rich single-stranded DNA via Hoogsteen hydrogen bonding into stacked G-tetrads. The G-quadruplex structure could dynamically transform to parallel, antiparallel, and hybrid topologies[11,20], by varying monovalent cations[17] or manipulating crowded environments[21]. This structural tunability provides a critical basis for designing controllable DNA hybrid catalysts. In related studies, metal-terpyridine complexes have been shown to exhibit strong affinity and selectivity toward human telomeric G4-DNA. Li et al.[22] constructed hybrid catalysts G4-DNA-1 by combining a series of Cu(II)-terpyridine complexes with a 21-mer human telomeric G4-DNA, enabling asymmetric Diels–Alder reactions with up to 99% conversion and 94% ee. The interaction between G4-DNA and the Cu(II) complexes was attributed to a combination of electrostatic interactions and π-π stacking. Notably, replacement of the natural D-form G4-DNA with its enantiomeric L-form resulted in an inversion of the product’s absolute configuration, demonstrating that the stereochemical topology of G4-DNA plays a dominant role in enantioselective control (Scheme 5).
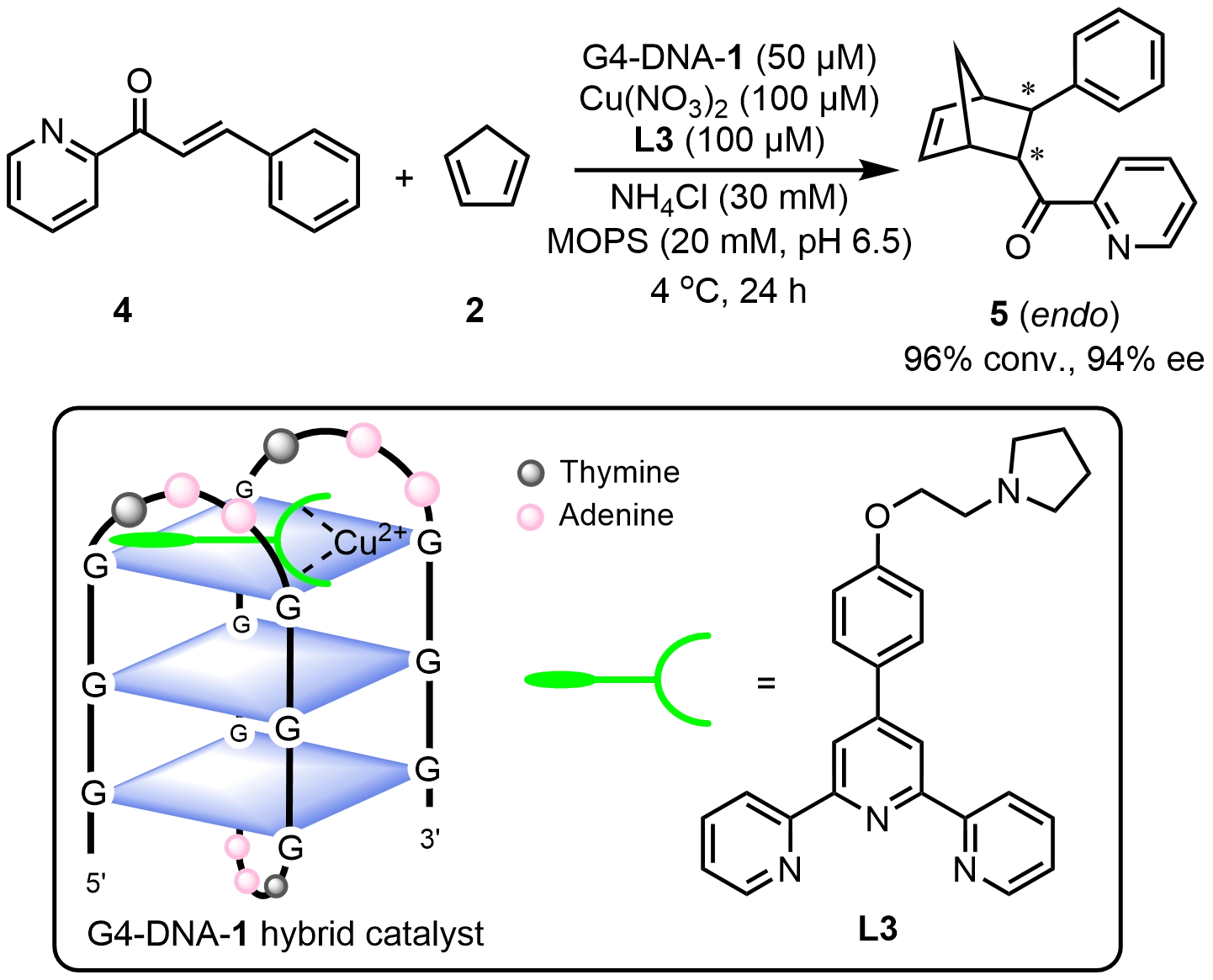
Scheme 5. An asymmetric Diels–Alder reaction catalyzed by a G4-DNA hybrid catalyst. Republished with permission from[11]. DNA: deoxyribonucleic acid.
Li group[23] selected K+ and NH4+, which have similar ionic radii, as stabilizers to construct higher-order human telomeric G4-DNA hybrid catalysts. In the presence of K+, the resulting G4-DNA-2 hybrid catalyst promoted the asymmetric Diels–Alder reaction with an ee of 90% and a 5-6-fold increase in the reaction rate. When changing K+ to NH4+, the G4-DNA-2 hybrid catalyst adopted a hybrid topology, resulting in a reversal of the absolute configuration of the Diels–Alder product with -82% ee (Scheme 6). The K+/NH4+ swiched ennatioselectivity is mainly ascribed to the distinct G-quadruplex topologies[24,25]. Compared with Na+-stabilized G-quadruplexes, K+ and NH4+ exhibit higher binding affinity via coordinating at the center of two G-tetrads with distinct octahedral-like geometries. Although K+ and NH4+ have approximately equal ionic radius, their difference in binding site preference and exchange kinetics result in distinct chiral cavities for metal species, leading to switchable stereoinduction. These results demonstrate that the enantioselective catalytic performance of DNA hybrid catalysts highly depends on the DNA topologies and the tunable G4-DNA hybrid catalyst achieves precise control over the configuration of target enantiomers.
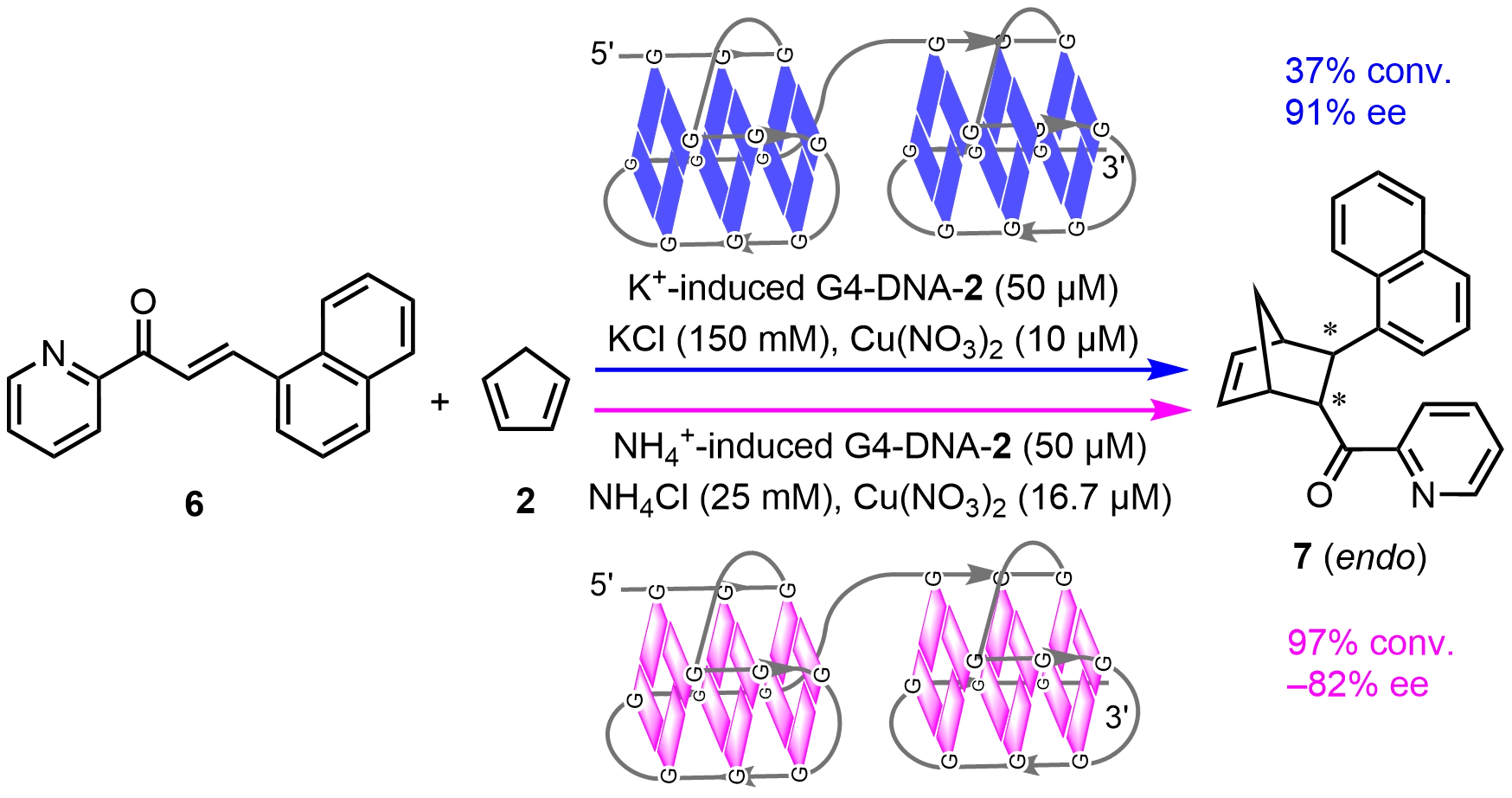
Scheme 6. Chiral inversion in asymmetric Diels–Alder reactions catalyzed by G4-DNA hybrid catalysts. DNA: deoxyribonucleic acid.
Besides G-quadruplex, another non-canonical quadruplex DNA called i-motif has entered the fray to construct DNA hybrid catalysts. Cytosine-rich single-stranded DNA can fold into an i-motif structure under slightly acidic conditions via hemi-protonated cytosine-cytosine base pairs (C:C+). Under near neutral conditions, Ag+ ions can stabilize an i-motif structure through formation of Ag+-mediated cytosine-cytosine base pairs (C-Ag+-C). Recently, our group[26] reported a human telomeric im-DNA containing C-Ag+-C base pairs interacting with Cu2+ to form an im-DNA-1 hybrid catalyst, which highly catalyzes a Diels-Alder reaction with 90% conversion and 95% ee (Scheme 7). The structural analysis revealed that Cu2+ locates in a specific loop region through coordinating with one N7 atom from adenine and two phosphate oxygen atoms. Taken together with G4-DNA and im-DNA hybrid catalysts, it is clear that the unpaired bases in loop regions provide direct binding sites to metal ions. Based on the integrated catalytic data, structural characterizations, and theoretical calculations, a plausible catalytic mechanism for the Diels-Alder reaction catalyzed by im-DNA-1(Ag+)/Cu2+ has been proposed. Theoretical calculations indicate that the Cu2+ ion forms a tetra-coordinated species, im-DNA-1(Ag+)/Cu2+, by coordinating to the N7 atom of the A18 base in the third loop region as well as to the phosphate oxygen atoms of nucleotides T16 and A17. This species then activates substrate 1a via Cu-N and Cu-O bonds to form a hexa-coordinated intermediate, im-DNA-1(Ag+)/Cu2+-8. Attack of cyclopentadiene 2 preferentially occurs from the Re face in this intermediate, as the Si face is sterically shielded by nearby nucleotide residues, leading to the formation of the (2R,3R)-configured enantiomer 9 (Re-endo).
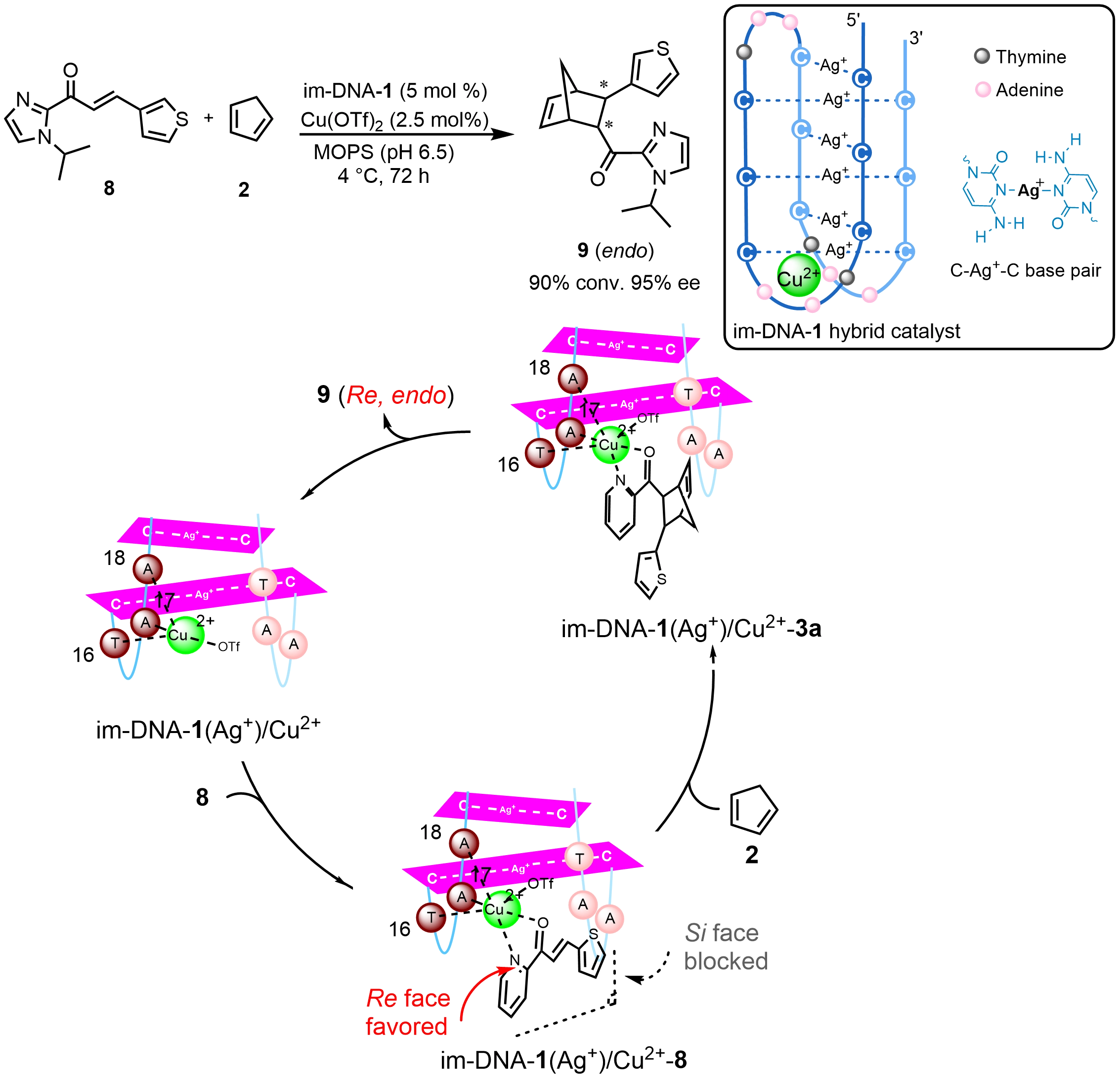
Scheme 7. An asymmetric Diels-Alder reaction catalyzed by an im-DNA hybrid catalyst and the proposed mechanism for stereoinduction. Republished with permission from[26]. DNA: deoxyribonucleic acid.
In addition, Smietana et al.[27] achieved an inverse electron-demand asymmetric hetero-Diels-Alder reaction catalyzed by a ds-DNA hybrid catalyst. Using a ds-DNA/Cu(II)-L1 hybrid catalytic system, the reaction between α,β-unsaturated 2-acyl imidazoles (10) and 2,3-dihydrofuran (11) proceeded smoothly, affording the desired products in 99% yield with a diastereomeric ratio of 97:3 and an ee of 95% (Scheme 8). This methodology was applicable to a broad range of α,β-unsaturated 2-acyl imidazole substrates, exhibiting good substrate generality and amenability to preparative-scale synthesis. Importantly, these findings demonstrate that the concept of DNA-based asymmetric catalysis can be extended to emerging synthetic transformations, providing an efficient, sustainable, and highly selective platform for the construction of chiral structural motifs.
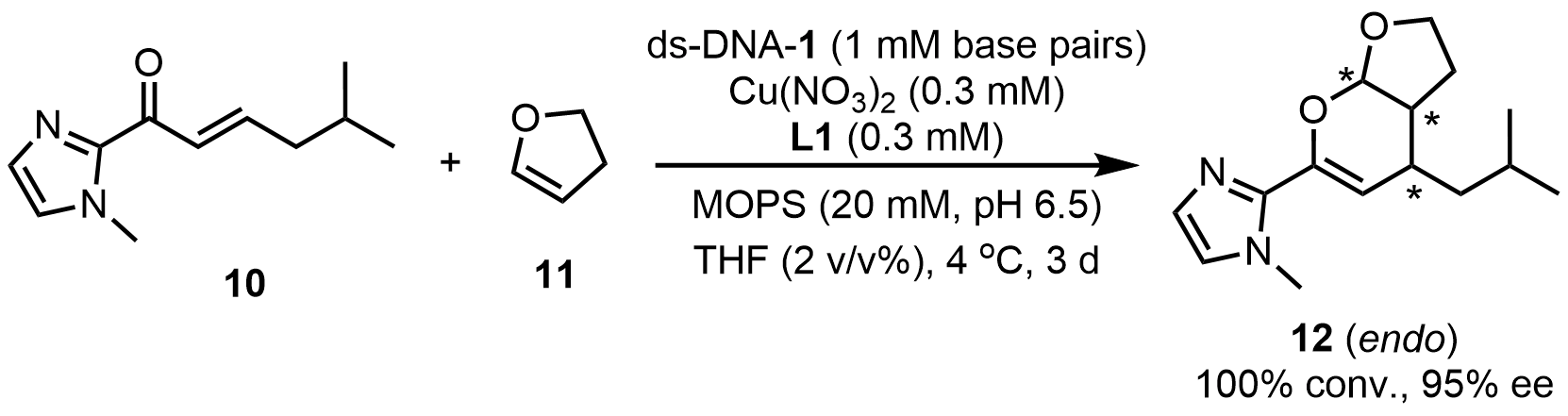
Scheme 8. An inverse electron-demand hetero-Diels-Alder reaction catalyzed by a ds-DNA hybrid catalyst. ds-DNA: duplex deoxyribonucleic acid.
2.2 Cyclopropanation reactions
Roelfes et al. demonstrated that ds-DNA can serve not only as a chiral scaffold in Lewis acid catalysis, but also as a coordination environment for regulating metal-carbene intermediates in cyclopropanation reactions. By constructing DNA-metal hybrid catalysts, asymmetric cyclopropanation of alkenes was achieved in aqueous media, with DNA acting as the sole source of chirality[28-30]. The introduction of DNA not only significantly accelerated the reaction rate, but also markedly improved the stereoselectivity of the cyclopropanation products, highlighting the unique capability of DNA to modulate highly reactive metal-carbene pathways. Based on thrombin-binding aptamer (TBA) G-quadruplexes as chiral scaffolds, Li et al.[31] employed site-directed mutagenesis to identify optimized sequences such as mG7A9C (5'-GGTTGGGGAGGTTGGC-3') and TmC4T (5'-TGGTCGGTGTGGTTGGT-3'). Assembly of these sequences with an Fe(II)-L4 complex generated the G4-DNA-3 and G4-DNA-4 hybrid catalysts, which enabled asymmetric cyclopropanation reactions, affording enantiomeric excesses of 81% and -79%, respectively (Scheme 9a). Notably, modulation of the DNA sequence alone allowed inversion of the absolute configuration of the products. Furthermore, using another mutated TBA sequence (mA9A: 5'-GGTTGGTGAGGTTGGA-3'), the authors constructed G4-DNA-5 hybrid catalysts in combination with either Fe(II)-L4 or Fe(II)-L5[32]. Remarkably, simply altering the N-methyl substitution pattern on the iron-porphyrin ligand enabled a dramatic switch in enantioselectivity within the same G4-DNA framework, with ee values ranging from 91% to -72% (Scheme 9b). In contrast to Fe(II)-L4, Fe(II)-L5 lacks planar symmetry and is therefore capable of synergistic interactions with the deoxynucleobase residues in the G4-DNA-5 scaffold. This cooperative effect allows the metal complex to participate more directly in chiral induction during cyclopropane formation, ultimately affording products with the opposite absolute configuration compared to those obtained from the G4-DNA-5 catalytic system. These studies have enabled the rational design of DNA-based biocatalysts through cofactor modification, providing an efficient strategy to fine-tune and regulate the catalytic performance of DNA-based catalytic systems.
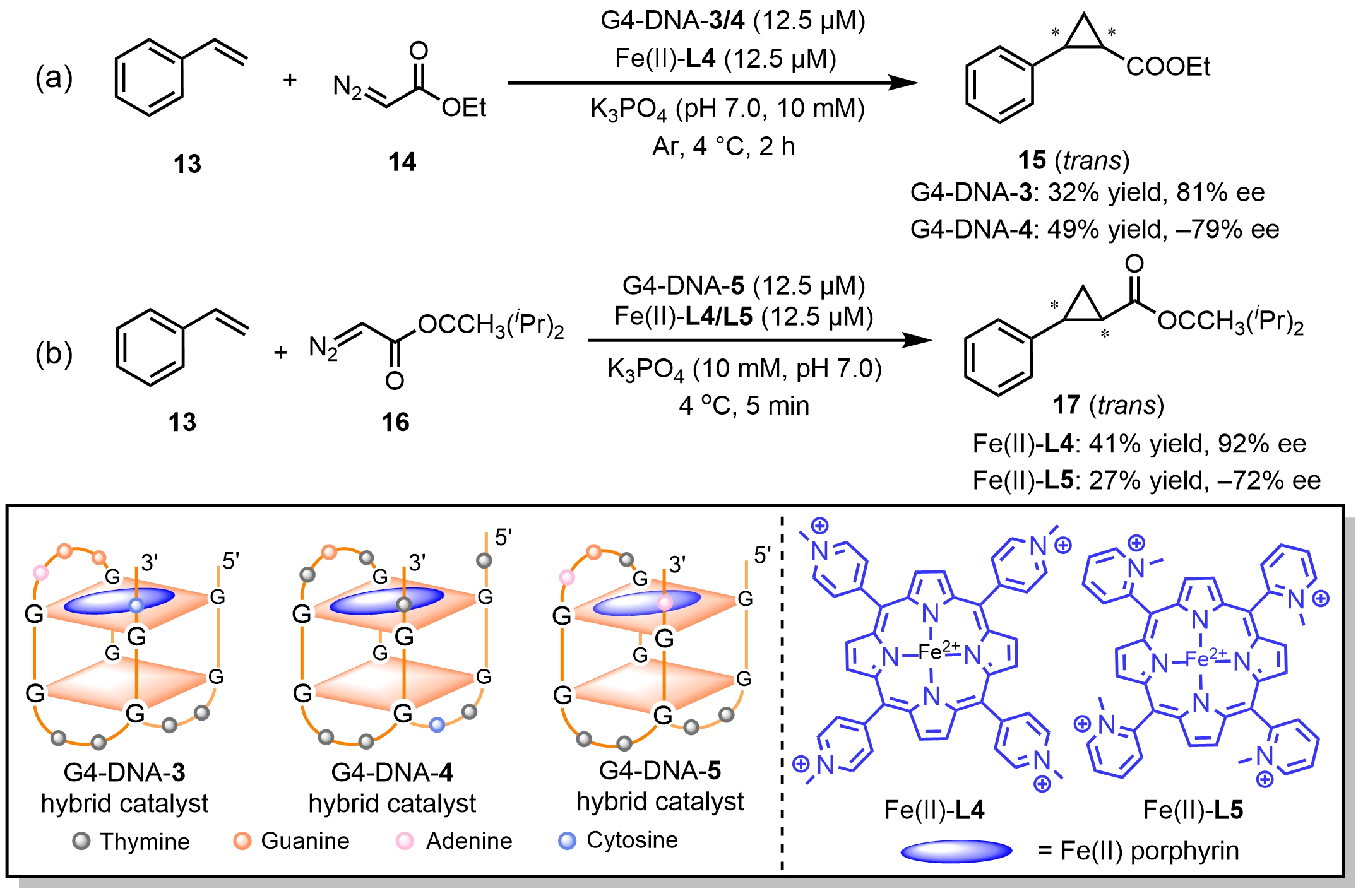
Scheme 9. Chiral inversion in asymmetric cyclopropanation reactions catalyzed by G4-DNA/Fe(II) porphyrin hybrid catalysts. DNA: deoxyribonucleic acid.
2.3 Friedel-Crafts reactions
Roelfes et al.[33] achieved an asymmetric Friedel–Crafts alkylation of (E)-1-(1-methyl-1H-imidazol-2-yl)but-2-en-1-one (18) with 5-methoxyindole(19) in aqueous media, employing a ds-DNA-1/Cu(II)-L1 hybrid catalytic system to obtain the product with 83% ee. By fine-tuning the DNA sequence, they found that using the duplex d(TCAGGGCCCTGA)2 led to nearly quantitative conversion and a further increase in ee to 93%, highlighting the pivotal role of DNA sequences in constructing a well-defined chiral microenvironment (Scheme 10a). The same group[34] subsequently extended this strategy to more challenging tandem Friedel-Crafts/protonation reactions with 84% ee (Scheme 10b). The rate acceleration arises from the high effective molarity of the reactants, as DNA binds and concentrates all components within a cavity-like microenvironment. The observed enantioselectivity indicates the contribution of secondary coordination sphere interactions. These results demonstrate that DNA-based catalysis provides an effective strategy for challenging reactions in aqueous media, benefiting from its rate-enhancing effect.
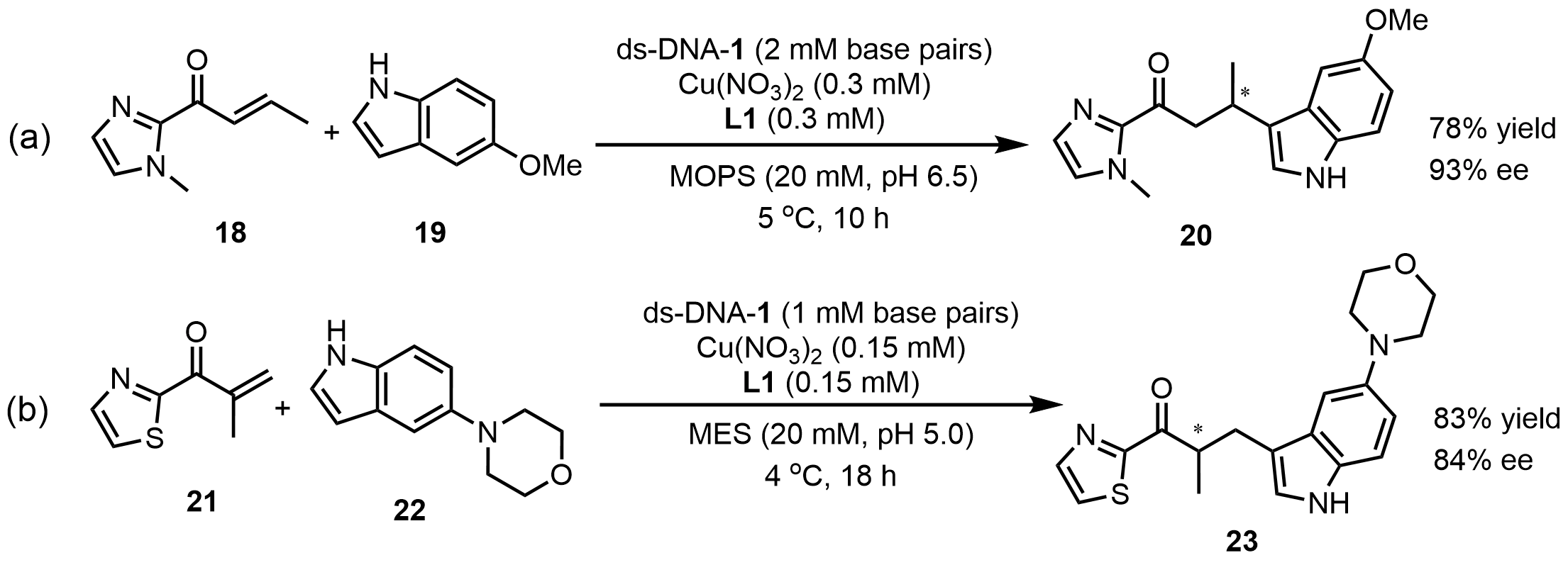
Scheme 10. (a) Asymmetric Friedel–Crafts alkylation and (b) Friedel-Crafts/protonation reactions catalyzed by ds-DNA hybrid catalysts. MES: 2-(N-Morpholino)ethanesulfonic acid; ds-DNA: duplex deoxyribonucleic acid.
In addition to supramolecular anchoring strategies, covalent linkage allows precise positioning of the metal cofactor within the DNA framework, facilitating mechanistic interpretation of experimental results[35,36] Arseniyadis et al.[37] employed sequence screening to develop the bipyridine-modified DNA/RNA (ds-DNA-4) hybrid catalytic system (Scheme 11). This catalytic system exhibited excellent selectivity, achieving up to 99% ee in Friedel-Crafts reactions, significantly outperforming conventional ds-DNA or RNA catalysts. Molecular dynamics simulations revealed that the observed stereoselectivity arises from the combined effects of hydrogen bonding and π-π interactions among the substrate, ligand, and nucleobases, providing a mechanistic explanation for the sequence-dependent differences.
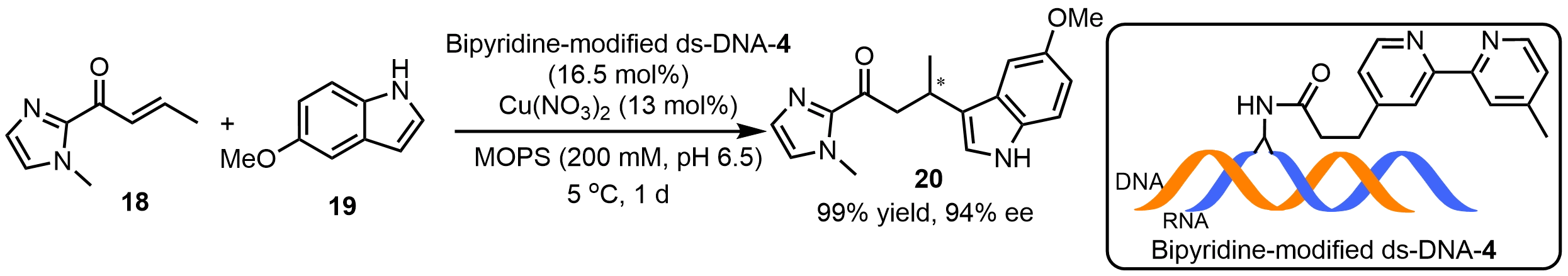
Scheme 11. An asymmetric Friedel-Crafts reactions catalyzed by a bipyridine-modified ds-DNA hybrid catalyst. ds-DNA: duplex deoxyribonucleic acid.
In recent years, Zhu et al.[38] reported a novel design strategy integrating DNA base excision repair with bioorthogonal conjugation. This strategy overcomes the key limitations of conventional constructions for DNA hybrid catalysts, such as poor functional group compatibility and difficulties in high-throughput optimization. In this approach, uracil-DNA glycosylase recognizes and excises deoxyuridine from DNA, generating abasic sites containing free aldehyde groups. These sites are then efficiently coupled with various small-molecule catalysts via oxime or hydrazone bioorthogonal reactions, enabling the facile construction of a diverse library of DNA hybrid catalysts. This strategy allows high-throughput optimization of DNA catalysts, and the screened bipyridine-modified hp-DNA-2 hybrid catalyst exhibited excellent activity and enantioselectivity in Friedel-Crafts alkylation reactions with up to 96% ee (Scheme 12a). Moreover, this strategy created another bipyridine-modified hp-DNA-3 hybrid catalyst, which achieved an atroposelective reaction between α,β-unsaturated acyl imidazole (27) and N-aryl pyrrole (28) with 96% ee (Scheme 12b). Additionally, this strategy enables the anchoring of multiple catalytic moieties onto DNA strands, thereby enriching the diversity of DNA hybrid catalysts and holding promise for advancing the field of DNA-based asymmetric catalysis.
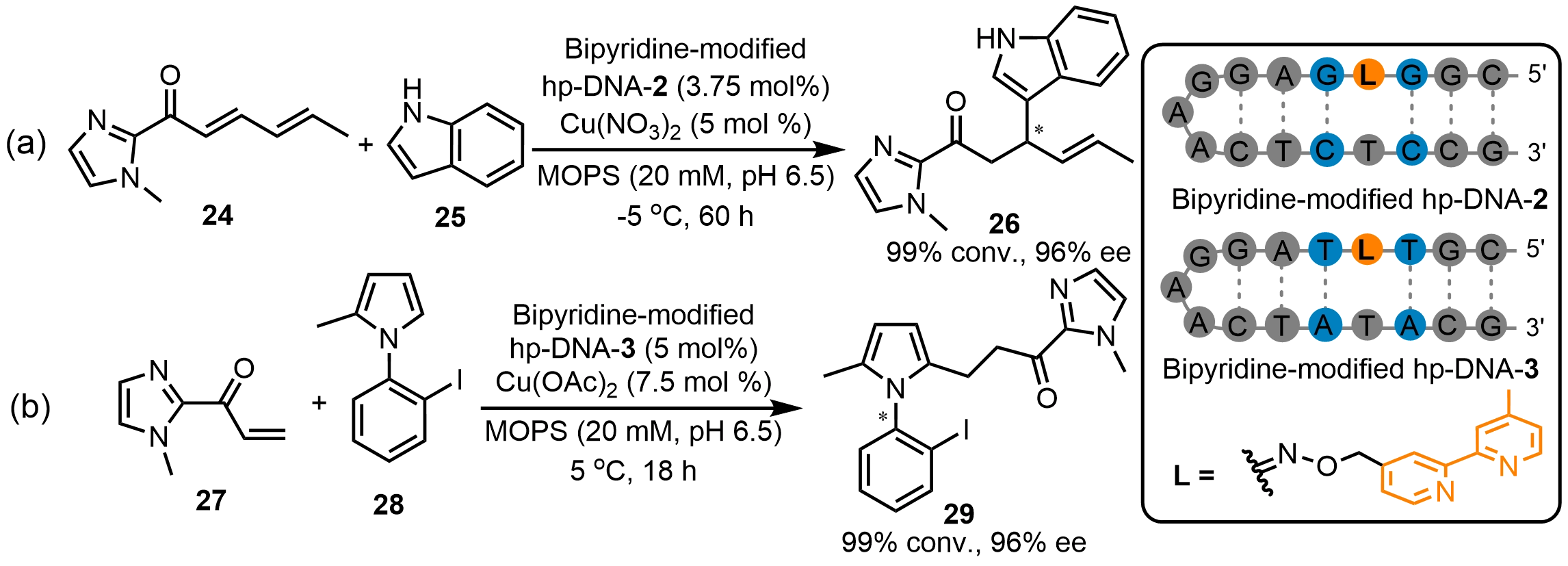
Scheme 12. (a) Asymmetric Friedel-Crafts alkylation and (b) atroposelective reactions catalyzed by bipyridine-modified hp-DNA hybrid catalysts. hp-DNA: hairpin deoxyribonucleic acid.
Very recently, our group[12] has introduced a tetrad-capping strategy to construct a potent im-DNA-2 hybrid catalyst featuring C:C+ base pairs (Scheme 13). For the Friedel-Crafts reaction of (E)-1-(1-propyl-1H-imidazol-2-yl)but-2-en-1-one (30) with 1-methyl-1H-pyrrole (31), this H+-mediated im-DNA-2/Cu2+ catalytic system achieved quantitative conversion and up to 97% ee. Notably, the introduction of Ag+ triggered the structural reorganization of im-DNA(H+) into a near-mirror symmetric conformation of im-DNA(Ag+). In the presence of the Cu(II)-L1, Ag+-mediated im-DNA-2 enabled switchable enantioselectivity of Friedel-Crafts reaction with -93% ee. This chiral inversion arises from the distinct i-motif topologies selectively regulated by H+ and Ag+, respectively. Based on spectroscopic and experimental data, tentative structural models were proposed for H+/Ag+-mediated im-DNA-2 hybrid catalysts. In the H+-mediated im-DNA-2/Cu2+ system, three Cu2+ ions reside in the three loop regions, interacting with deoxythymidine monophosphate residues via coordination or electrostatic interactions. The substrate 31 coordinates to Cu2+ to generate a hexacoordinated intermediate showing a favorable Re face attack, while the Si face is sterically shielded by the G:C:G:C tetrad or adjacent nucleotide residues. In the Ag+-mediated im-DNA-2/Cu(II)-L1 system, two Cu(II)-L1 complexes are primarily positioned at the terminal regions through π-π stacking interactions with G-Ag+-G base pairs. The substrate 31 coordinates to Cu(II)-L1 showing a preferential Si-face attack, while the Re face is sterically hindered by nucleotide residues.
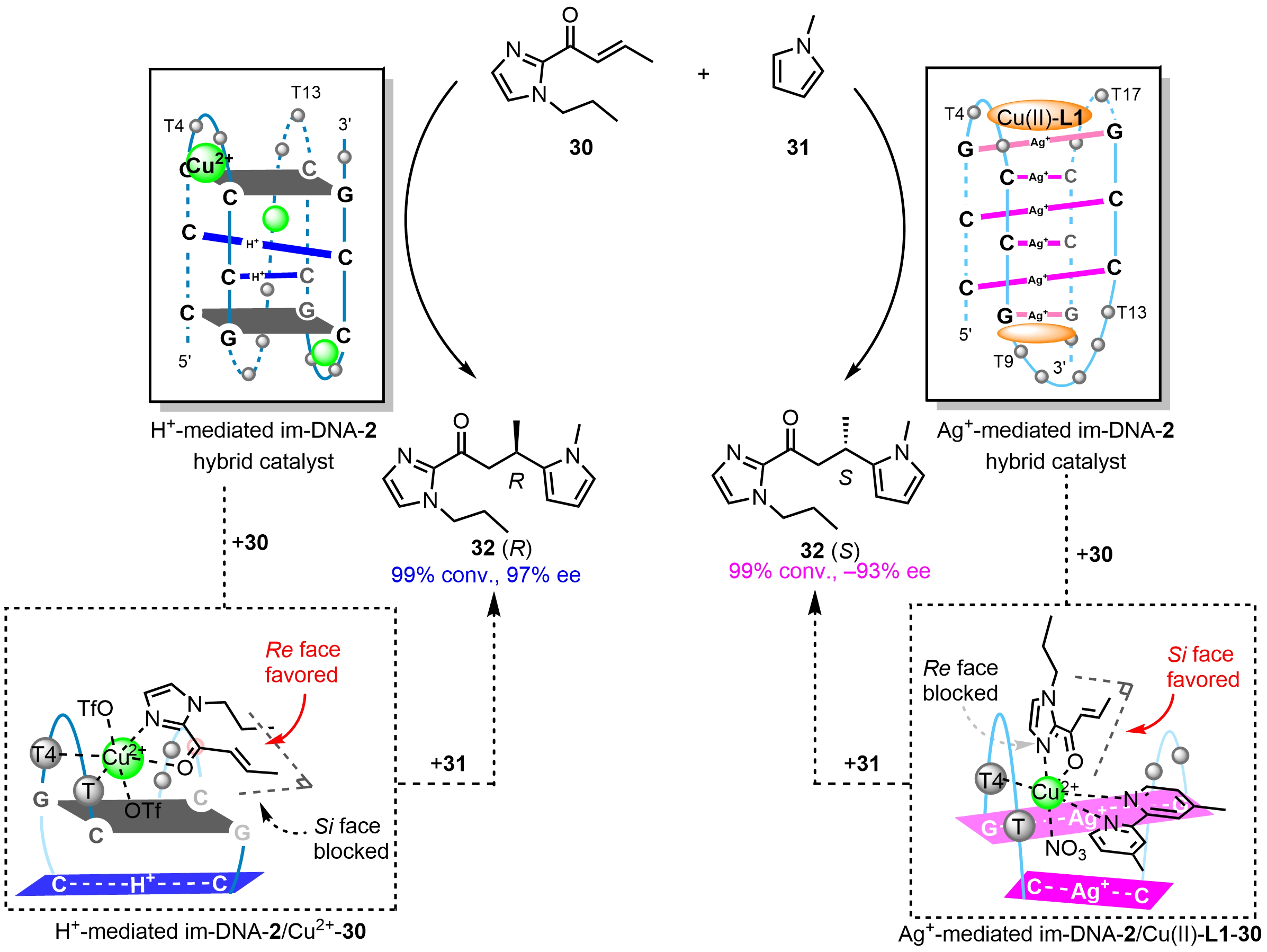
Scheme 13. H+/Ag+ switch of enantioselectivity in i-motif DNA-based Friedel-Crafts reactions and the proposed chiral inversion mechanism. Republished with permission from[12]. DNA: deoxyribonucleic acid.
Besides DNA, our group[39] reported that cyclic diadenosine monophosphate (c-di-AMP) coordinates with Cu2+ ions to generate a c-di-AMP·Cu2+ catalyst, enabling asymmetric Friedel-Crafts reactions with up to 97% ee (Scheme 14). Control experiments demonstrated that both the twelve-membered cyclic framework and the two adenine moieties of c-di-AMP are indispensable for its asymmetric catalytic performance. Spectroscopic characterizations and theoretical calculations supported a dimeric structure of the c-di-AMP·Cu2+ catalyst, in which the Cu2+ ion resides in the center of an adenine-adenine plane via binding to two N7 atoms and one phosphate-oxygen atom. An α,β-unsaturated ketone (33) coordinates to the c-di-AMP·Cu2+ dimer, showing a favorable Si face attack, while the Re face is blocked by the nearby nucleotide residues.
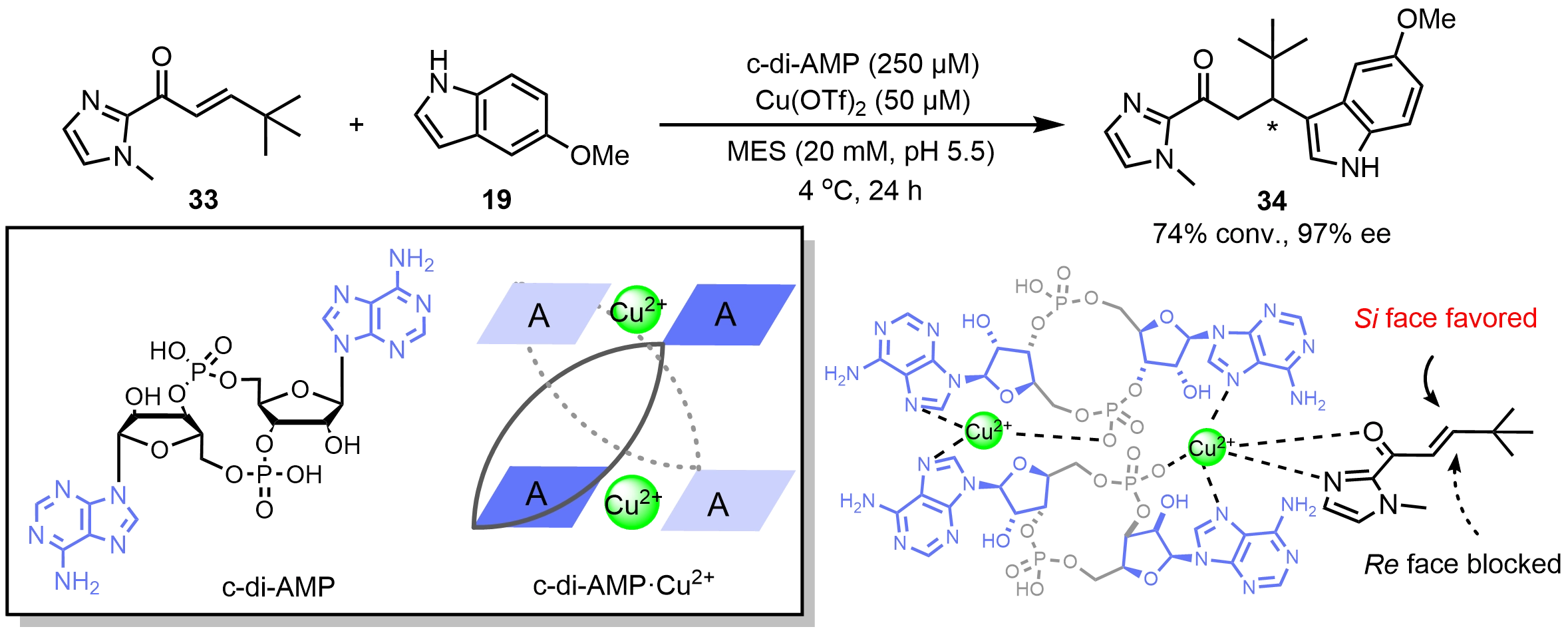
Scheme 14. An asymmetric Friedel-Crafts reaction catalyzed by a c-di-AMP·Cu2+ catalyst. Republished with permission from[39]. c-di-AMP: cyclic diadenosine monophosphate.
2.4 Michael addition reactions
Roelfes et al.[40] demonstrated that the ds-DNA-1/Cu(II)-L1 hybrid catalyst could efficiently catalyze asymmetric Michael addition reactions between α,β-unsaturated 2-acyl imidazoles (35) and dimethyl malonate (36) or nitroalkanes (37), delivering the corresponding products with up to 99% ee (Scheme 15). Mechanistic studies showed that this catalyst interacts with 35 to generate a favorable Si face attacked by nucleophiles. This mode of approach is consistent with the related DNA-based Diels-Alder reaction, suggesting that the DNA scaffold effectively shields the Re face of the coordinated α,β-unsaturated 2-acylimidazole substrates[41]. Subsequently, Li et al.[42] expanded the substrate scope of this catalytic platform to include Michael additions between 2-acyl imidazoles and cyano ester nucleophiles. Although a decrease in enantioselectivity was observed in some cases, these studies clearly validated the notable substrate compatibility of such non-covalently assembled DNA hybrid catalytic systems.
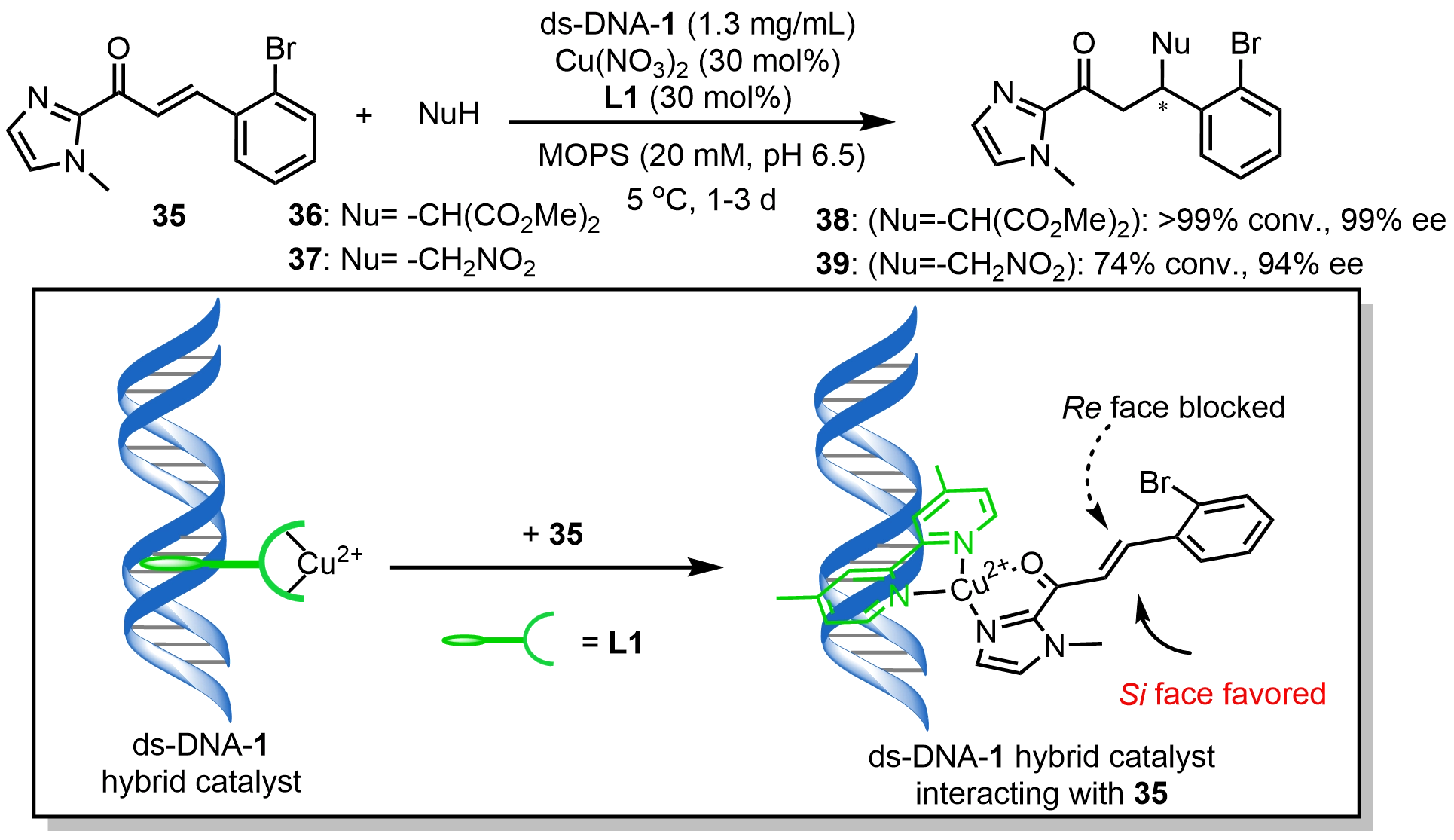
Scheme 15. Asymmetric Michael addition reactions catalyzed by a ds-DNA hybrid catalyst. ds-DNA: duplex deoxyribonucleic acid.
Zhu et al.[43] recently reported a bimetallic hp-DNA hybrid catalyst bearing two covalently appended bipyridine ligands that coordinate Cu(II) to form a bimetallic center (Scheme 16). This catalyst promoted an asymmetric Michael addition between (E)-1-(1-(tert-butyl)-1H-imidazol-2-yl)but-2-en-1-one (40) and 1-(1-isopropyl-1H-imidazol-2-yl)propan-1-one (41) to provide a chiral 1,5-dicarbonyl compound (42). Notably, modulating the spacing between the bipyridyl ligands inverted the absolute configuration of the products, enabling access to the desired enantiomers. Computational analysis of the “adjacent” and “intervening” bimetallic DNA catalysts showed that stereocontrol originates from the spatial arrangement of the two Cu(II)-bipyridine centers along the DNA scaffold. In both systems, each substrate can adopt multiple coordination geometries, and the bipyridine-Cu(II) units may orient toward either the major or minor groove. The vertical positioning of substrates and the rotational orientation of the bipyridine ligands determine the distance between the electrophilic enone β-carbon and the nucleophilic ketone α-carbon, directly influencing enantioselectivity.
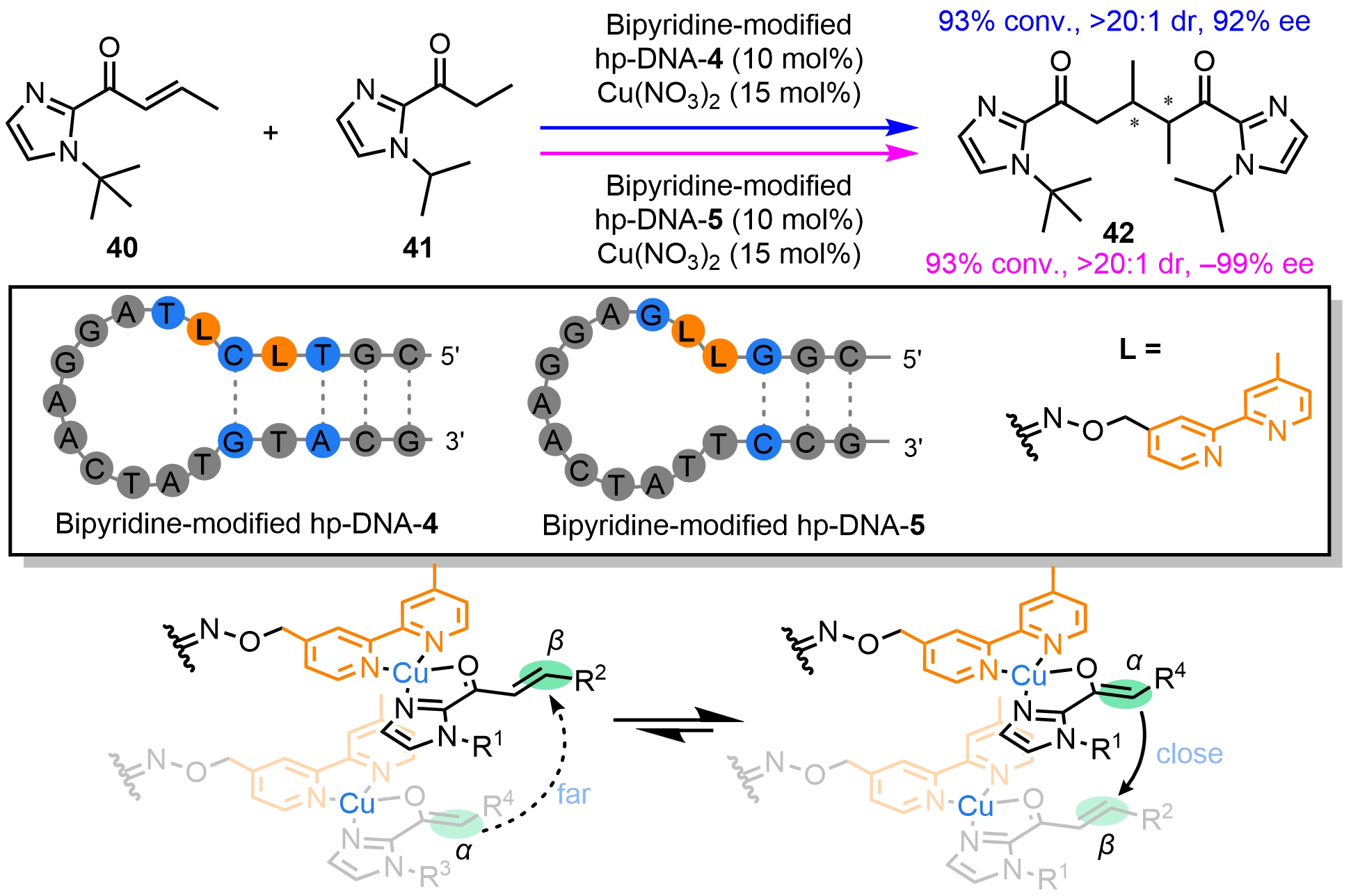
Scheme 16. Chiral inversion in asymmetric Michael addition reactions by bipyridine-modified hp-DNA hybrid catalysts by tuning the position of two metal centers. hp-DNA: hairpin deoxyribonucleic acid.
Clever et al.[44] covalently anchored R- or S-configured imidazole ligands to the loop regions of human telomeric G4-DNA. The resulting G4-DNA hybrid catalysts enabled asymmetric Michael additions with tunable enantioselectivity by adjusting imidazole numbers and locations, as well as the G-quadruplex topologies (Scheme 17). The G4-DNA-6 hybrid catalyst bearing one and two R-configured imidazole ligands at the first and third loop regions adopted an antiparallel structure in the presence of K+, which catalyzed the Michael addition with 99% ee. In contrast, the G4-DNA-7 hybrid catalyst functionalized with two R-configured imidazole ligands at the first loop region folded into an antiparallel G4 topology in the presence of Na+ and delivered the product with -90% ee. The chirality of the appended ligands had little effect on product configuration, underscoring the dominant role of the G4-DNA scaffold topology in governing stereoselectivity.
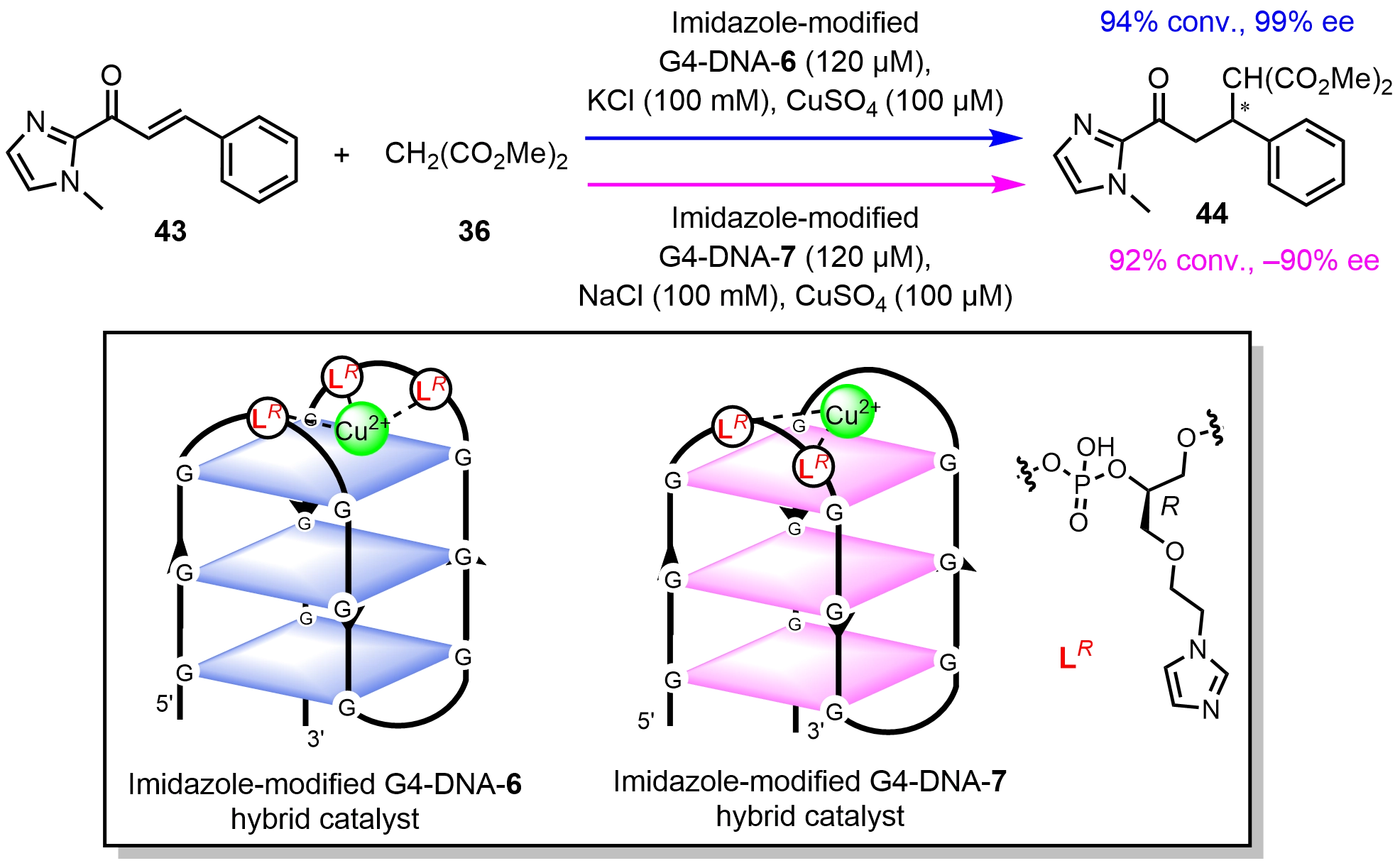
Scheme 17. Chiral inversion in asymmetric Michael addition reactions catalyzed imidazole-modified G4-DNA hybrid catalysts by tuning the position and number of covalently attached imidazole groups. Republished with permission from[44]. DNA: deoxyribonucleic acid.
2.5 Photoinduced cross-dehydrogenative coupling reaction
Besides addition reactions, Zhu et al.[45] employed DNA phosphate groups as catalysts for asymmetric ion-pairing catalysis. Using a rationally designed hp-DNA-6 hybrid catalyst, they achieved an asymmetric photoinduced cross-dehydrogenative coupling between 1-(1-methyl-1H-imidazol-2-yl)-2-(naphthalen-2-yl)ethan-1-one (45) and 3,6-dimethyl-9H-xanthene (46), giving rise to product 47 at 96% ee (Scheme 18). DNA sequence had little effect on conversion but strongly influenced enantioselectivity. Notably, sequences with cytosine at the 5′-terminus consistently gave lower ee values. Furthermore, inorganic phosphate buffer saline (PBS) proved markedly superior to organic buffering systems, underscoring the decisive role of ion-pair interactions in the catalytic process. This study overcomes the reliance of ion-pair catalysis on nonpolar solvents and offers a biomimetic strategy for sustainable asymmetric catalysis.
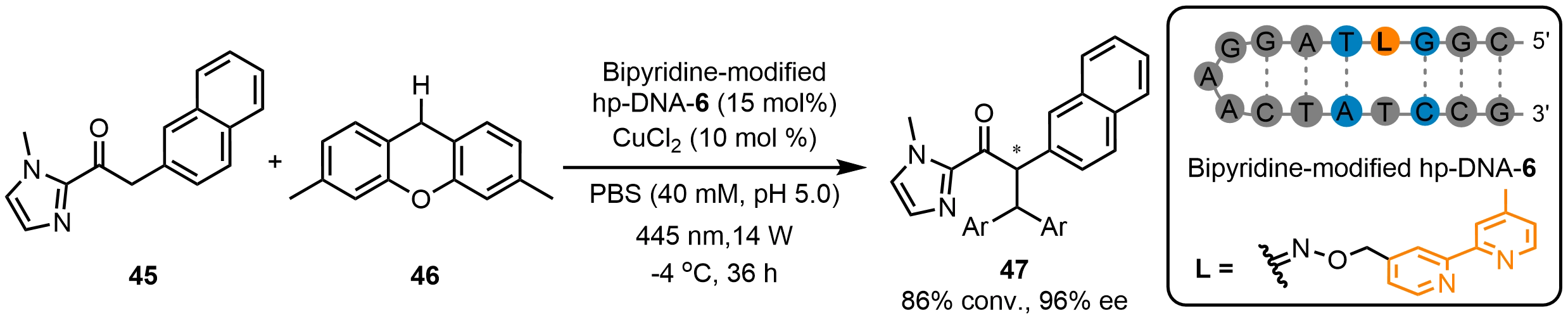
Scheme 18. Asymmetric photoinduced cross-dehydrogenative coupling catalyzed by a bipyridine-modified hp-DNA hybrid catalyst. hp-DNA: hairpin deoxyribonucleic acid.
3. DNA-Based Asymmetric C–X Bond Formation Reactions
Beyond C–C bond formation, DNA hybrid catalysts have been successfully applied to diverse C–X (X = N, O, F) bond formation reactions[8,46-56]. These transformations typically involve stronger substrate-catalyst interactions, intricate proton transfer, and more pronounced electronic effects than C–C bond-forming reactions, thereby imposing greater demands on catalyst design and stereochemical control. In particular, the activation and orientation of heteroatom nucleophiles are highly sensitive to the catalytic microenvironment created by the catalytic system. Thus, precise control over the DNA-substrate binding mode via sequence engineering, secondary structure tuning, and tailored metal coordination is key to high reactivity and enantioselectivity.
For C–N bond formation reactions, Roy et al.[53] reported ds-DNA-based Markovnikov-type hydroamination of β-nitrostyrene (48) and methoxylamine (49) to afford O-methyl-N-(2-nitro-1-phenylethyl)hydroxylamine (50) product with 76% ee (Scheme 19). In this system, ds-DNA served as the chiral scaffold and was noncovalently assembled with Pd(L6)(OAc)2 to generate a DNA hybrid catalyst. Notably, this sustainable and modular strategy enables ready integration of diverse metal complexes into the chiral DNA framework, offering a versatile platform for construction of chiral C–N compounds.
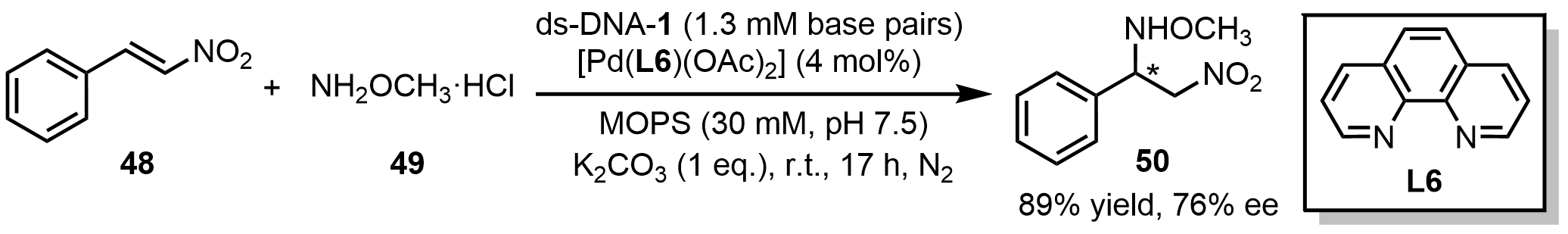
Scheme 19. An asymmetric hydroamination reaction catalyzed by a ds-DNA hybrid catalyst. ds-DNA: duplex deoxyribonucleic acid.
Zhu et al.[45] reported that a bipyridine-modified hp-DNA hybrid catalyst enables asymmetric Mannich reactions affording products with newly formed C-N bonds (Scheme 20). A typical bipyridine-modified hp-DNA-7 hybrid catalyst proceeded the inter-Mannich reaction between 1-(1-isopropyl-1H-imidazol-2-yl)-2-(o-tolyl)ethan-1-one (51) and 1,3,5-triaryl-1,3,5-triazinane (52) with 86% conversion and 94% ee (Scheme 20a). The adjacent nucleotides flanking the modified bipyridyl moiety in hp-DNA-7 play a decisive role in stereochemical control. Meanwhile, variations in pH markedly affect the reaction efficiency. These findings highlight the importance of the local DNA microenvironment in stabilizing and orienting the iminium intermediate. Furthermore, this catalytic system was also applicable to the intra-Mannich reaction of 1-(1-isopropyl-1H-imidazol-2-yl)-2-phenylethan-1-one (54), N-alkyl aniline (55) and paraformaldehyde (56), affording the corresponding product 57 with 92% ee (Scheme 20b). Collectively, these results highlight the ability of DNA phosphate groups to dynamically stabilize cationic intermediates through ion-pair interactions, while the hydrophobic microenvironment of the DNA scaffold effectively mitigates competitive solvation in aqueous media.
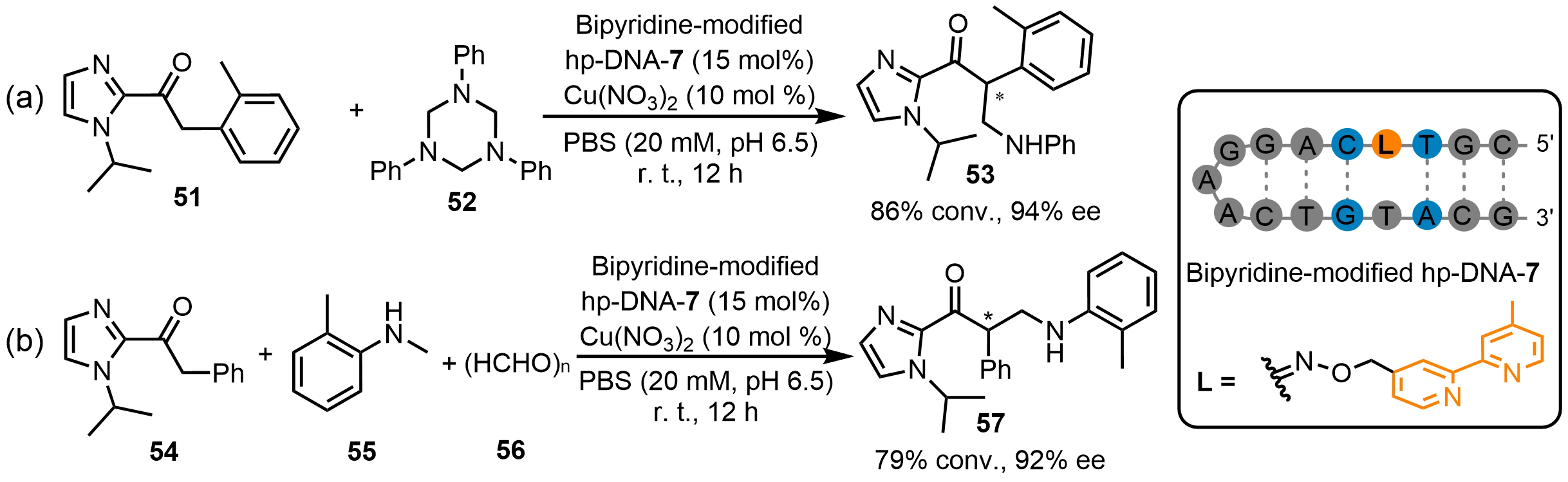
Scheme 20. Asymmetric Mannich reactions catalyzed by a bipyridine-modified hp-DNA hybrid catalyst. hp-DNA: hairpin deoxyribonucleic acid.
For C–O bond formation reactions, Roelfes et al.[9] reported an enantioselective hydration reaction of α,β-unsaturated enones by a ds-DNA hybrid catalyst via noncovalently assembling ds-DNA-1 and a Cu(II)-L7 complex. This system efficiently promoted the Re-face selective hydration of enone 33 with water, affording the corresponding product 59 at 72% ee (Scheme 21a). Building upon this, Sugiyama et al.[55] developed a covalently functionalized DNA (ds-DNA-5) hybrid catalyst by directly embedding bipyridine ligands into the DNA strand. Systematic tuning of linker length, adjacent base pairs, and the steric environment of noncoordinating groups improved the enantioselectivity of hydration product 59 to 87% ee (Scheme 21a). Furthermore, replacing water with its deuterium form further enhanced the enantioselectivity in the hydration of 33 (Scheme 21b). A typical sequence of ds-DNA-6 interacting with Cu(II)-L7 complex catalyzed this hydration with 82% ee, in which the presence of central AT base pairs in the DNA sequence significantly enhanced stereocontrol. In short, this DNA hybrid platform overcomes the intrinsic reversibility of asymmetric hydration and the low nucleophilicity of water under neutral conditions, offering a robust strategy for constructing chiral C–O bonds.
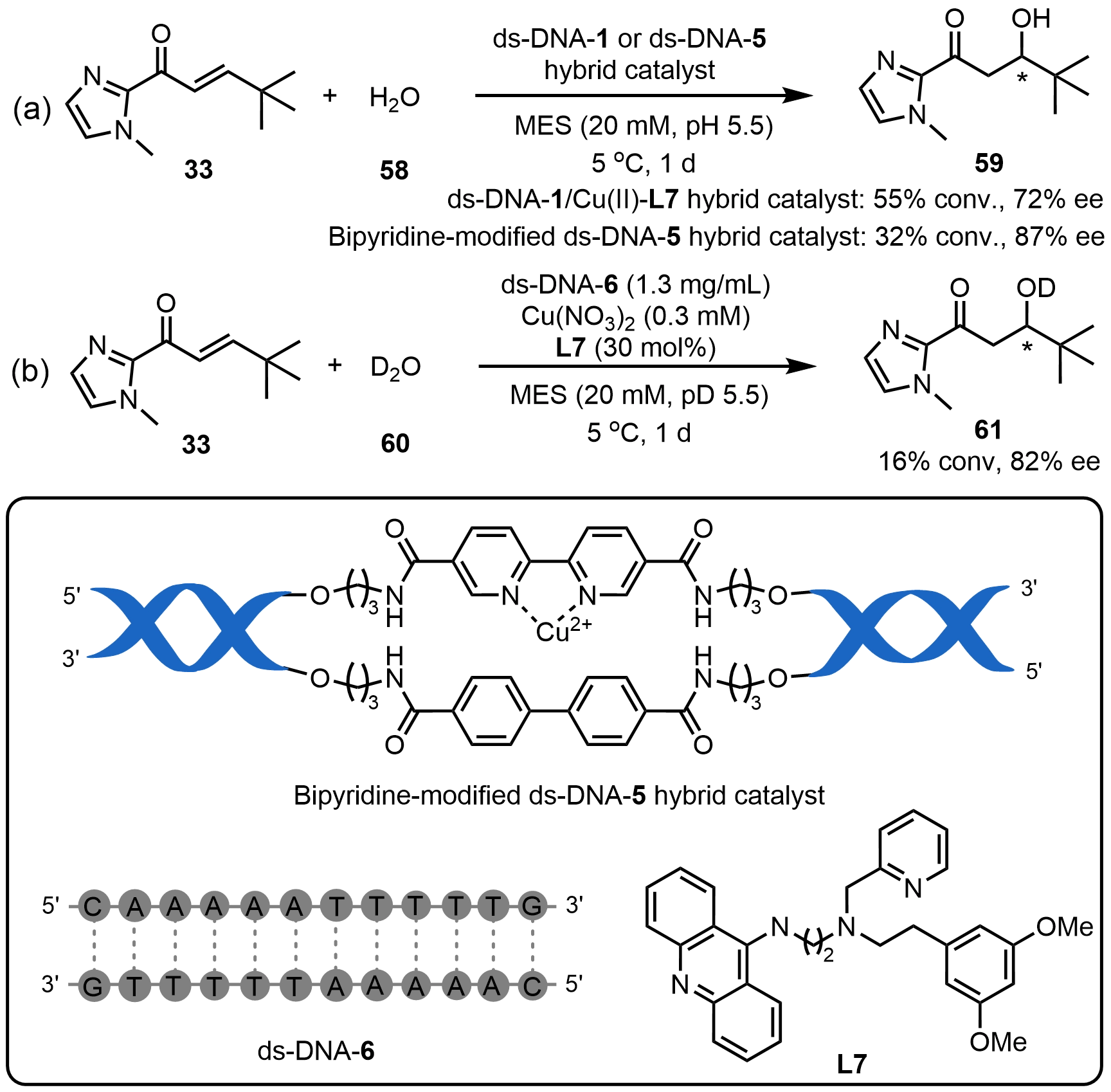
Scheme 21. Asymmetric hydration reactions catalyzed by ds-DNA hybrid catalysts. ds-DNA: duplex deoxyribonucleic acid.
For C–F bond formation reactions, Zhu et al.[45] reported a typical bipyridine-modified hp-DNA-8 hybrid catalyst that efficiently catalyzes the fluorination of α-aryl-1-(1-methylimidazol-2-yl)ethanone (62) with 98% conversion and 92% ee (Scheme 22a). Compared with the previous discussed bipyridine-modified hp-DNA hybrid catalysts, the difference is in the adjacent nucleotides flanking the modified bipyridyl moiety, further confirming its key role in stereocontrol. They systematically investigated the impact of different flanking base-pair sequences on the fluorination reactions by examining all possible combinations within a 17-mer hp-DNA scaffold. The results revealed that the local DNA environment significantly influenced both reactivity and enantioselectivity. For instance, conversions remained below 10% when the catalyst was flanked by AA, TA, or CA, whereas the presence of guanine at the 5′ position (in the GA sequence) restored catalytic activity. In contrast, sequences other than AA, TA, and CA afforded conversions exceeding 80%, indicating a clear detrimental effect of adenine at the 3′-flanking position. Enantioselectivity exhibited even greater sequence-dependent variation, with ee values ranging from 20% to 92%. In addition, our group[50] developed a c-di-AMP hybrid catalyst via assembling it with a Cu(II)-L6 complex (Scheme 22b). This hybrid system enabled the asymmetric fluorination reaction of methyl 6-iodo-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (65) with Selectfluor (63), achieving quantitative conversion and 90% ee. Spectroscopic characterizations indicated the interaction of c-di-AMP and Cu(II)-L6 in a supramolecular mode via π-π stacking. Kinetic studies revealed a distinct rate acceleration for C-F bond formation, highlighting the intrinsic catalytic competence of cyclic dinucleotides.
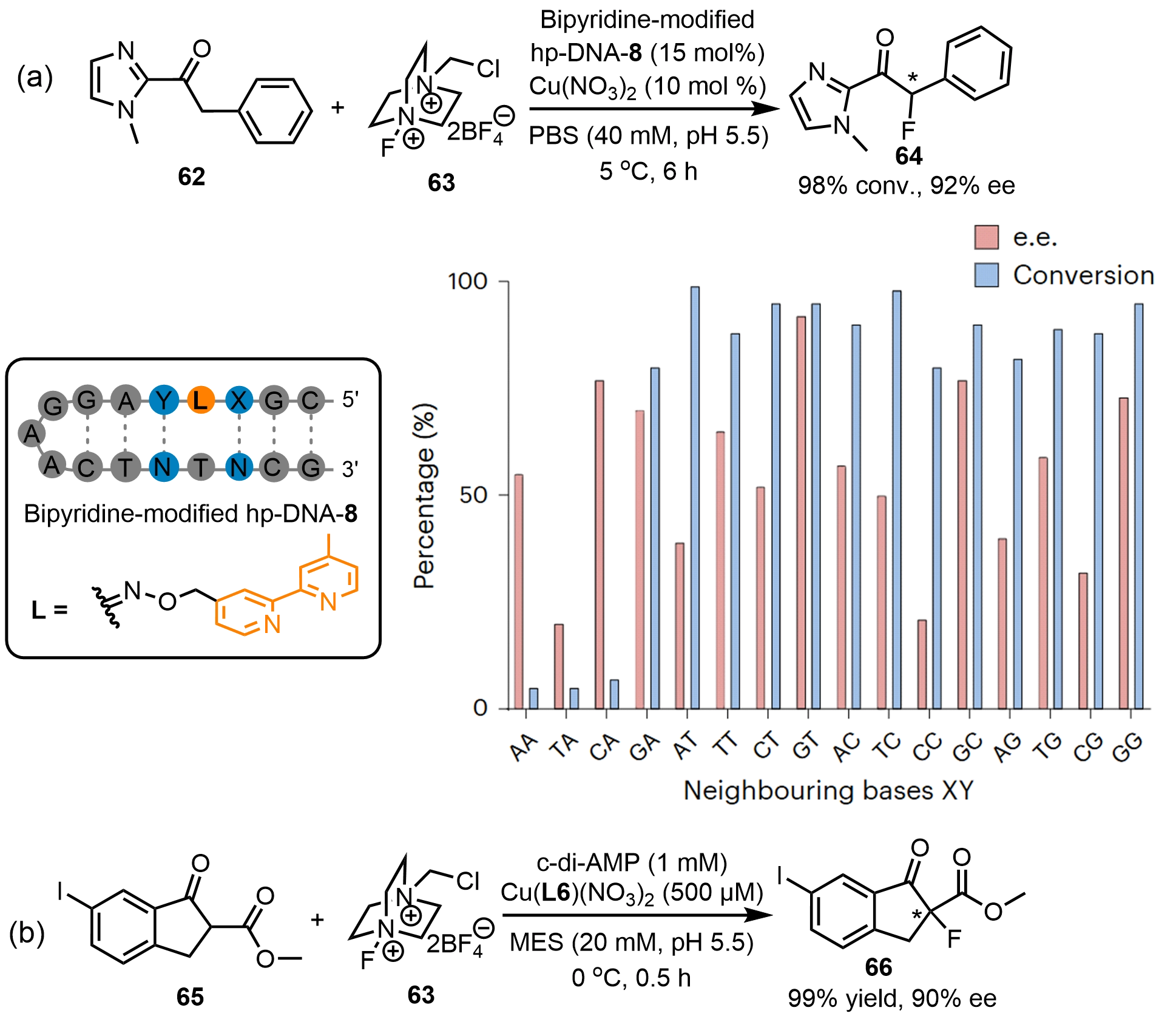
Scheme 22. Asymmetric fluorination reactions catalyzed by (a) bipyridine-modified hp-DNA and (b) c-di-AMP hybrid catalysts. Republished with permission from[45]. hp-DNA: hairpin deoxyribonucleic acid; c-di-AMP: cyclic diadenosine monophosphate.
4. Conclusion and Outlook
DNA hybrid catalytic systems have been successfully applied to a broad spectrum of transformations including C–C and carbon–heteroatom bond formations, offering distinct advantages in aqueous media over conventional small-molecule or metal-ligand systems. Importantly, their catalytic activity and stereochemical outcome can be precisely modulated by adjusting the structural parameters of DNA. In C–C bond-forming reactions, the chiral microenvironment provided by DNA can effectively control substrate conformation through spatial constraints, π-π stacking, and electrostatic interactions. These reactions currently represent the most mature and highly enantioselective class within DNA-based catalysis. For carbon-heteroatom bond formations (C–N, C–O, C–F), DNA hybrid catalysts primarily manifest as fine control over reaction pathways and regioselectivity.
Despite significant advances in DNA-based asymmetric catalysis, several key challenges still remain. First, different reaction types exhibit distinct dependencies on DNA secondary structures and metal active sites, yet their structure-activity relationships remain poorly understood. Second, most highly enantioselective systems are still restricted to specific substrates or reaction types, which limit the overall reaction diversity. Third, the mechanistic understanding of chiral induction is largely empirical, constraining the rational catalyst design.
DNA-based asymmetric catalysis is progressing from proof-of-concept toward programmable and predictable control. On one hand, sequence engineering, secondary structure modulation, and multi-site cooperative design could create tailored chiral microenvironments for specific asymmetric transformations. On the other hand, integrating DNA-based catalysis with organocatalysis, photochemistry, or electrochemistry could expand the repertoire of reaction scope[57]. Furthermore, combined with computational modeling and machine learning, general principles of stereocontrol can be systematically uncovered, providing critical mechanistic insight[58,59]. Overall, the full potential of DNA-based asymmetric catalysis remains untapped but holds great promise for green and sustainable chemistry.
Acknowledgements
We thank Mr. Xingchen Dong and Ms. Weijun Qin for helpful discussions.
Authors contribution
Xu S: Resources, investigation, writing-original draft, writing review & editing.
Wang Z, Wang M: Resources, visualization.
Chen Y: Writing-review & editing.
Wang C: Conceptualization, funding acquisition, writing-review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was financially supported by the National Natural Science Foundation of China (Grant Nos. 22572111, 22222203), the Natural Science Foundation of Shaanxi Province of China (Grant No. 2025JC-QYCX-014) and the Fundamental Research Funds for the Central Universities (Grant No. GK202404001).
Copyright
© The Author(s) 2026.
References
-
1. Qin WJ, Wang ZX, Dong XC, Wang ZH, Chen YS, Wang CH. Research progress of nucleic acids based asymmetric catalysis. Sci Sin Chim. 2024;54(10):1727-1743. Chinese.[DOI]
-
6. Wang Z, Dong X, Chen Y, Wang C. Quadruplex DNA hybrid catalysts for enantioselective reactions. ChemBioChem. 2025;26(1):e202400909.[DOI]
-
7. Bos J, Roelfes G. Artificial metalloenzymes for enantioselective catalysis. Curr Opin Chem Biol. 2014;19:135-143.[DOI]
-
10. Shibata N, Yasui H, Nakamura S, Toru T. DNA-mediated enantioselective carbon-fluorine bond formation. Synlett. 2007;7:1153-1157.[DOI]
-
11. Wang C, Jia G, Zhou J, Li Y, Liu Y, Lu S, et al. Enantioselective Diels–Alder reactions with G-quadruplex DNA-based catalysts. Angew Chem Int Ed. 2012;51(37):9352-9355.[DOI]
-
14. Roelfes G, Boersma AJ, Feringa BL. Highly enantioselective DNA-based catalysis. Chem Commun. 2006;6:635-637.[DOI]
-
15. Boersma AJ, de Bruin B, Feringa BL, Roelfes G. Ligand denticity controls enantiomeric preference in DNA-based asymmetric catalysis. Chem Commun. 2012;48(18):2394.[DOI]
-
16. Gjonaj L, Roelfes G. Novel catalyst design by using cisplatin to covalently anchor catalytically active copper complexes to DNA. ChemCatChem. 2013;5(7):1718-1721.[DOI]
-
25. Hud NV, Schultze P, Feigon J. Ammonium ion as an NMR probe for monovalent cation coordination sites of DNA quadruplexes. J Am Chem Soc. 1998;120(25):6403-6404.[DOI]
-
28. Oelerich J, Roelfes G. DNA-based asymmetric organometallic catalysis in water. Chem Sci. 2013;4(5):2013-2017.[DOI]
-
29. Rioz-Martínez A, Oelerich J, Ségaud N, Roelfes G. DNA-accelerated catalysis of carbene-transfer reactions by a DNA/cationic iron porphyrin hybrid. Angew Chem Int Ed. 2016;55(45):14136-14140.[DOI]
-
30. Miao W, Hao J, Zhou W, Jia G, Li C. Enantioselective induction by G-quadruplex DNA/hemin in intramolecular cyclopropanation. Chin J Catal. 2025;78:138-143.[DOI]
-
31. Hao J, Miao W, Cheng Y, Lu S, Jia G, Li C. Enantioselective olefin cyclopropanation with G-quadruplex DNA-based biocatalysts. ACS Catal. 2020;10(11):6561-6567.[DOI]
-
35. Park S, Zheng L, Kumakiri S, Sakashita S, Otomo H, Ikehata K, et al. Development of DNA-based hybrid catalysts through direct ligand incorporation: Toward understanding of DNA-based asymmetric catalysis. ACS Catal. 2014;4(11):4070-4073.[DOI]
-
36. Mansot J, Aubert S, Duchemin N, Vasseur JJ, Arseniyadis S, Smietana M. A rational quest for selectivity through precise ligand-positioning in tandem DNA-catalysed Friedel–Crafts alkylation/asymmetric protonation. Chem Sci. 2019;10(10):2875-2881.[DOI]
-
41. Boersma AJ, Feringa BL, Roelfes G. α, β-unsaturated 2-acyl imidazoles as a practical class of dienophiles for the DNA-based catalytic asymmetric Diels−Alder reaction in water. Org Lett. 2007;9(18):3647-3650.[DOI]
-
42. Li Y, Wang C, Jia G, Lu S, Li C. Enantioselective Michael addition reactions in water using a DNA-based catalyst. Tetrahedron. 2013;69(32):6585-6590.[DOI]
-
45. Li Z, Zheng Y, Zhao Q, Li Y, Yap A, Zhang X, et al. DNA phosphates are effective catalysts for asymmetric ion-pairing catalysis in water. Nat Catal. 2025;8(11):1220-1231.[DOI]
-
46. Bai J, Sun X, Wang H, Li C, Qiao R. Guanosine-based self-assembly as an enantioselective catalyst scaffold. J Org Chem. 2020;85(4):2010-2018.[DOI]
-
48. Cheng M, Li Y, Zhou J, Jia G, Lu SM, Yang Y, et al. Enantioselective sulfoxidation reaction catalyzed by a G-quadruplex DNA metalloenzyme. Chem Commun. 2016;52(62):9644-9647.[DOI]
-
49. Dijk EW, Feringa BL, Roelfes G. DNA-based hydrolytic kinetic resolution of epoxides. Tetrahedron: Asymmetry. 2008;19(20):2374-2377.[DOI]
-
51. Festa C, Esposito V, Benigno D, De Marino S, Zampella A, Virgilio A, et al. Discovering new G-quadruplex DNA catalysts in enantioselective sulfoxidation reaction. Int J Mol Sci. 2022;23(3):1092.[DOI]
-
52. Megens RP, Roelfes G. DNA-based catalytic enantioselective intermolecular oxa-Michael addition reactions. Chem Commun. 2012;48(51):6366.[DOI]
-
54. Ropartz L, Meeuwenoord NJ, van der Marel GA, van Leeuwen PWNM, Slawin AMZ, Kamer PCJ. Phosphine containing oligonucleotides for the development of metallodeoxyribozymes. Chem Commun. 2007;15:1556.[DOI]
-
59. Gao L, Tong X, Ye T, Gao H, Zhang Q, Yan C, et al. G-quadruplex-based photooxidase driven by visible light. ChemCatChem. 2020;12(1):169-174.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Xu S, Wang Z, Wang M, Chen Y, Wang C. Programmable DNA as stereocontrol ligand for asymmetric catalysis in aqueous media. Chiral Chem. 2026;2:202611. https://doi.org/10.70401/cc.2026.0022
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. DNA-Based Asymmetric C–C Bond Formation Reactions
- 3. DNA-Based Asymmetric C–X Bond Formation Reactions
- 4. Conclusion and Outlook
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Xu S, Wang Z, Wang M, Chen Y, Wang C. Programmable DNA as stereocontrol ligand for asymmetric catalysis in aqueous media. Chiral Chem. 2026;2:202611. https://doi.org/10.70401/cc.2026.0022
copy
Share Link
copy