Beyond ferroptosis: Role of GPX4 in osteoarthritis and its therapeutic implications
Junchen He

Xiong Zhang
Liangcai Hou
Haigang Liu
Fengjing Guo
1,*
,
Kai Sun
1,*
*Correspondence to:
Fengjing Guo, Department of Orthopedics, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei, China.
E-mail: guofjdoc@163.com
Kai Sun, Department of Orthopedics, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei, China. E-mail: 1085844308@qq.com
Kai Sun, Department of Orthopedics, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei, China. E-mail: 1085844308@qq.com
Ferroptosis Oxid Stress. 2026;2:202514. 10.70401/fos.2026.0013
Received: November 05, 2025Accepted: January 04, 2026Published: January 09, 2026
Abstract
Osteoarthritis (OA) is increasingly regarded as a whole-organ disease that includes different subsets of joint pathological conditions with variable genetic, biochemical and clinical characteristics. The pathogenesis of OA is perplexing, and disease-modifying drugs are still lacking. Glutathione peroxidase 4 (GPX4), recently best known as the key regulator of ferroptosis implicated in many diseases, including OA, actually has long been identified and reportedly possesses multiple significant biological functions. However, the relationship between GPX4 and OA remains to be elucidated. In this review, we first summarize the current knowledge of GPX4 as a selenoenzyme and the regulation of its expression. Then we scrutinize various possible patterns of involvement of GPX4 in OA. Finally, we also underscore the potential implications and prospects of GPX4-based therapeutic regimens for OA.
Keywords
Osteoarthritis, cartilage, ferroptosis, GPX4
1. Introduction
As the most prevalent chronic joint disorder, osteoarthritis (OA) is a leading cause of impaired mobility among older adults worldwide, contributing to a huge health and socioeconomic burden[1]. OA is a heterogeneous disorder. The risk factors include aging, sex, trauma, obesity, mechanical injury, and genetic and epigenetic traits[2]. The concept of the pathophysiology of OA is continuously advancing, transitioning from being viewed as only cartilage-limited to a multifactorial complex disease that affects the entire joint, including articular cartilage, subchondral bone, and synovium[3]. As for the treatment, disease-modifying drugs are still under development, with joint replacement often considered the only effective approach for symptomatic end-stage osteoarthritis in clinical practice[4]. Hence, a more comprehensive understanding of the molecular mechanisms implicated in the onset and progression of OA will be of paramount importance in developing proactive and preventative strategies.
Articular cartilage is a highly organized structure and primarily contains only one type of specialized cell known as the chondrocyte. As the sole cellular component, chondrocytes play a pivotal role in preserving the structural integrity of articular cartilage, which is achieved through a delicate equilibrium between the synthesis and breakdown of the extracellular matrix (ECM)[5,6]. However, this balance can be disrupted due to the presence of some osteoarthritic pathogenic factors, such as the inflammatory milieu or mechanical aberrations in the joint space. This disruption can result in widespread chondrocyte dysfunction (represented by abnormal ECM metabolism) or death, eventually contributing to cartilage degeneration in OA. Previous research has indicated that various forms of cell death, including necrosis and apoptosis, can induce detrimental effects on chondrocytes during OA[7]. In 2012, Dixon et al. first proposed the epochal concept of ‘ferroptosis’, a novel form of regulated cell death caused by oxidative disturbance of the intracellular microenvironment that is constitutively controlled by Glutathione peroxidase 4 (GPX4)[8,9], which can catalyze the reduction of lipid peroxides at the expense of reduced glutathione and protect cells against oxidative stress. Recent studies have further reinforced the notion that GPX4 is a master regulator in ferroptosis[10]. Besides, in 2020, our research first reported that chondrocyte ferroptosis could contribute to the progression of osteoarthritis. To the best of our knowledge, it’s the first time, in the literature, that the pathophysiology of OA had been associated with cell ferroptosis[11]. Altogether, it seems obvious that GPX4 could regulate the osteoarthritic development through chondrocyte ferroptosis.
However, up to now, the exact function and regulation of GPX4 during the development of OA have not been fully investigated, although GPX4 has been discovered for decades and has been reported to engage in multiple physiological processes including inflammation and cell death and to be related to diverse cancers and nonneoplastic diseases such as inflammatory bowel disease and Alzheimer’s disease[12-15]. Therefore, this review aims to present a concise overview of GPX4 and delve into its regulation and various possible roles in OA. Furthermore, we will investigate and discuss the regulators of GPX4, shedding light on their potential implications for the treatment of OA.
2. A Brief Overview of GPX4
2.1 GPX4 as a selenoenzyme
GPX4 is a member of the glutathione peroxidase family, which consists of eight oxidoreductases that are closely phylogenetically related and found in a wide range of living organisms. The most unique feature of the mammalian GPX family is that many of them, including GPX1-4 and GPX6, are selenoenzymes with the catalytically essential amino acid selenocysteine (Sec) in their active site, whereas GPX-5, GPX-7 and GPX-8 contain a cysteine (Cys) instead of Sec[16]. Sec or Cys, along with glutamine, tryptophan, and asparagine, make up the conserved catalytic tetrad of these enzymes[16]. The catalytic cycle of all GPX proteins occurs in two distinct stages, following a ping-pong mechanism whereby the active site of these enzymes shuttles between an oxidized and reduced state[17,18]. In the first stage, the active site selenocysteine or cysteine of GPX proteins reduces a peroxide species, while undergoing concomitant oxidation. In the second stage, the active site residue is regenerated through the utilization of reducing substrates such as GSH, whereby the active site residue is reduced and the reducing substrate is oxidized simultaneously[19] (Figure 1).
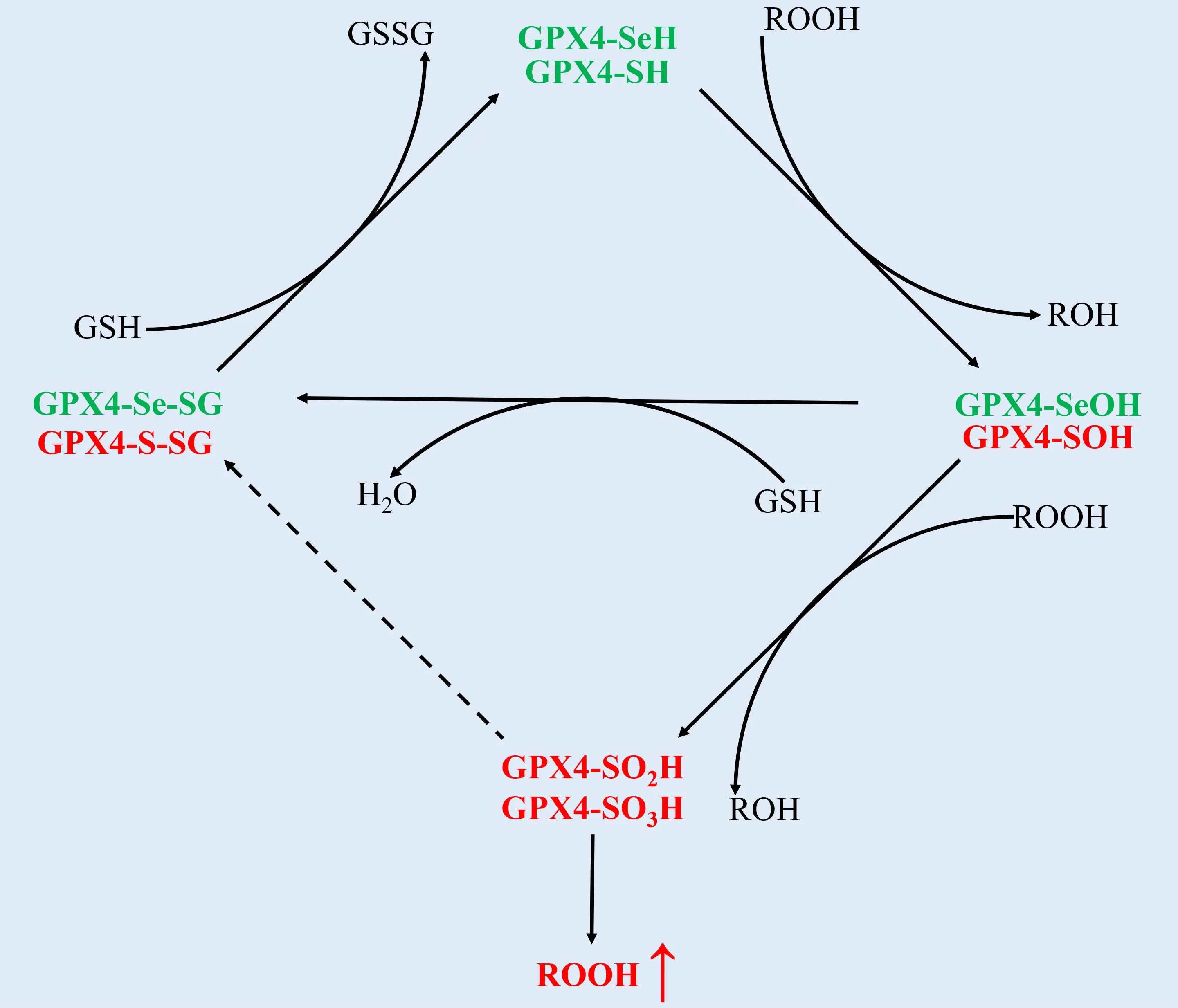
Figure 1. The catalytic cycle of GPX4. The catalytic cycle conforms to a ping-pong mechanism and can be divided into two phases, the oxidative part and reductive part respectively. In the first (oxidative) phase, the selenol in selenium-containing GPX4 (green) becomes oxidized to selenenic acid by a hydroperoxide (ROOH). In the second (reductive) phase, the selenenic acid within oxidized GPX4 interacts with the reductant GSH to form a selenodisulfide. This intermediate would be further resolved by a second GSH molecule back into the original selenol, which marks the regeneration of GPX4 and the end of a cycle. However, the replacement of selenium with sulfur in the GPX4 variant (red) inclines to facilitate the irreversible overoxidation and resultant inactivation of the enzyme, which could trigger a premature termination of the catalytic cycle before it proceeds into the second (reductive) phase. GPX4: glutathione peroxidase 4; GSH: glutathione.
Of note, for the selenoenzymes like GPX4 among GPXs, the substitution of Cys with Sec is reported to improve the sustainability of the catalytic cycle, although Sec differs from Cys only by a single atom, namely selenium replacing sulfur[13]. Several studies have proposed that compared with cysteine-containing variants of GPX4, which generally tend toward the undesired irreversible overoxidation and inactivation during catalysis, the Sec-containing GPX4 is intrinsically resistant to this process due to the enhanced stability of the oxidized intermediate throughout the cycle[13,20]. A possible chemical-biological rationale underlying the aforementioned phenomenon is the specific capacity of selenium to undergo rapid oxidation and then immediate reduction, which is commonly referred to as the “selenium paradox”[21]. Another distinct characteristic of GPX4’s structure-function relationships emerging from comparison among GPX family members is that unlike the other four selenoenzymes which are homotetramers, GPX4 is the sole monomeric member of the vertebrate GPX family bearing Sec in the redox center, which is suggested to be potentially associated with its substrate accessibility[22,23]. The monomer GPX4 operates with substrates generally at an interface, not via a simple collision as the tetramers do[23-25]. This interfacial interaction allows GPX4 to be specially involved in the reduction of more complex lipid hydroperoxides, even those embedded in the biological membrane[25]. This function is of particular therapeutic interest as it suggests GPX4’s key role in maintaining the integrity of biological membranes[19].
2.2 The expression and regulation of GPX4
The genomic structure of GPX4 locus is conserved among mammals. In humans, it is located on chromosome 19p13.3 and spans a length of 2.8 kb with eight exons, two of which are alternative, i.e., exons 1A and 1B[26,27]. Alternative splicing yields three different mRNA and protein isoforms: mitochondrial GPX4 (mGPX4), cytosolic GPX4 (cGPX4), and nuclear GPX4 (nGPX4)[28]. More specifically, the production of mGPX4 (long form) and cGPX4 (short form) is attributed to two slightly different transcription start sites at the same exon 1A, whereas the transcription initiation of nGPX4 takes place at the alternative exon 1B[29]. Despite adherence to the same fundamental biochemical rules of enzymatic catalysis described above, these isoforms would eventually enter different subcellular compartments with the guidance of dissimilar organelle import N-terminal sequences, thus serving different biological functions. The cytosolic GPX4 is now widely recognized as the dominant isoform in somatic cells and plays a central role in suppressing lipid peroxidation and ferroptosis. By contrast, the mitochondrial and nuclear isoforms of GPX4 seem to have more restricted expression patterns, and the research on these two variants focuses more on their roles in germ cells including sperm: mGPX4 is linked with mitochondrial apoptosis and male infertility[30-33], while nGPX4 focuses on protecting chromatin condensation and genetic integrity in sperm[34,35]. While mGPX4 and nGPX4 are recognized as the prevailing isoforms in male germ cells and play essential roles in reproductive biology, their functions beyond the realm of reproduction remain largely unknown.
As a TATA-less gene, GPX4 is deemed to own a feature commonly shared by housekeeping genes, which are often defined as being not highly regulated[23]. Indeed, the precise regulatory mechanisms governing GPX4 expression remain undeciphered, although some fragmentary knowledge of its regulation has been gained.
Epigenetic research has revealed the presence of CpG islands in the vicinity of the transcription start site (TSS) of GPX4, where the methylation process controls the expression of GPX4[36]. Under conditions of certain cancer types, chronic mild stress, or metabolic disorders of amino acids, including glycine, homocysteine, and S-adenosylmethionine, there is an observed increase in methylation of the GPX4 promoter, which leads to GPX4 inhibition and resultant ROS accumulation, thus inducing ferroptosis[37-41]. In addition to DNA methylation, some histone modifications (e.g., methylation like H3K4me3, acetylation like H3K27ac) have been reported to play an important role in the epigenetic regulation of GPX4[42,43].
In the field of transcriptional regulation, it is noteworthy that Uefer and Imai et al. have systematically addressed the functional characterization of the positive regulatory and distinct core regions of GPX4 promoters in mouse various tissues and cells by site-directed mutagenesis and electrophoretic mobility shift analysis (EMSA), successively[44,45]. Some promoter regions and binding sites of transcription factors, including stimulating protein 1 (SP1), activator protein 2 (AP2), cAMP-response element-binding protein (CREB), CCAAT/enhancer-binding protein (C/EBP-ε), and so on, have been disclosed in their studies.
At the post-transcriptional and translational level, diverse mechanisms have been investigated to regulate GPX4 synthesis. For instance, GPX4 mRNA stability could be augmented in a METTL14-mediated m6A-HuR-dependent manner to inhibit osteoclastogenesis and bone resorption for the amelioration of osteoporosis[46]. Some microRNAs, including miR-15a, miR-1656, miR-214-3p, miR-1290 and miR-324-3p, are also worth noting since they predominantly target the 3’ UTR of GPX4 mRNAs as translational suppressors, decreasing GPX4 level and promoting ferroptosis[47-51]. Predictably, with selenocysteine in the catalytic center, selenoenzyme GPX4 expression relies on Se availability partly, although some studies have found that in selenium-deficient rats GPX4 activity decreased very slowly[52,53]. This phenomenon is tentatively attributed to GPX4 mRNA stability and selenocysteine insertion sequence efficiency, which rank GPX4 high in the hierarchy of selenoprotein expression[54]. Apart from that, GPX4 translation has been reportedly coupled with intracellular cysteine levels partly through the Rag-mTORC1-4EBP signaling axis, in which mTORC1 is the master sensor monitoring responses to nutrient availability and growth stimuli and could be inactivated upon cysteine starvation[55].
Several post-translational modifications have been reviewed to regulate GPX4[56]. To name a few, the succination of GPX4 by intracellular fumarate aggregation could sensitize cancer cells to ferroptosis inducers (FINs), whereas the alkylation of GPX4 could serve as a route for class II FINs to induce ferroptosis[57,58]. The ubiquitination of GPX4 has been reported as an important mechanism of regulating ferroptosis in various diseases[59,61]. Recently our research has demonstrated that the senescence indicator P21 could also regulate GPX4 protein stability in chondrocytes by affecting GPX4 recruitment to the linear ubiquitin chain assembly complex (LUBAC) and subsequent M1-linked ubiquitination on GPX4 during OA progression[62]. Apart from ubiquitin-proteasome mediated degradation, another major system to decompose GPX4 is also noteworthy: chaperone-mediated autophagy (CAM). Heat shock protein family A (Hsp70) member 8 (HSPA8) and HSP90 have been confirmed to participate in the CAM-dependent GPX4 hydrolysis[63] (Figure 2).
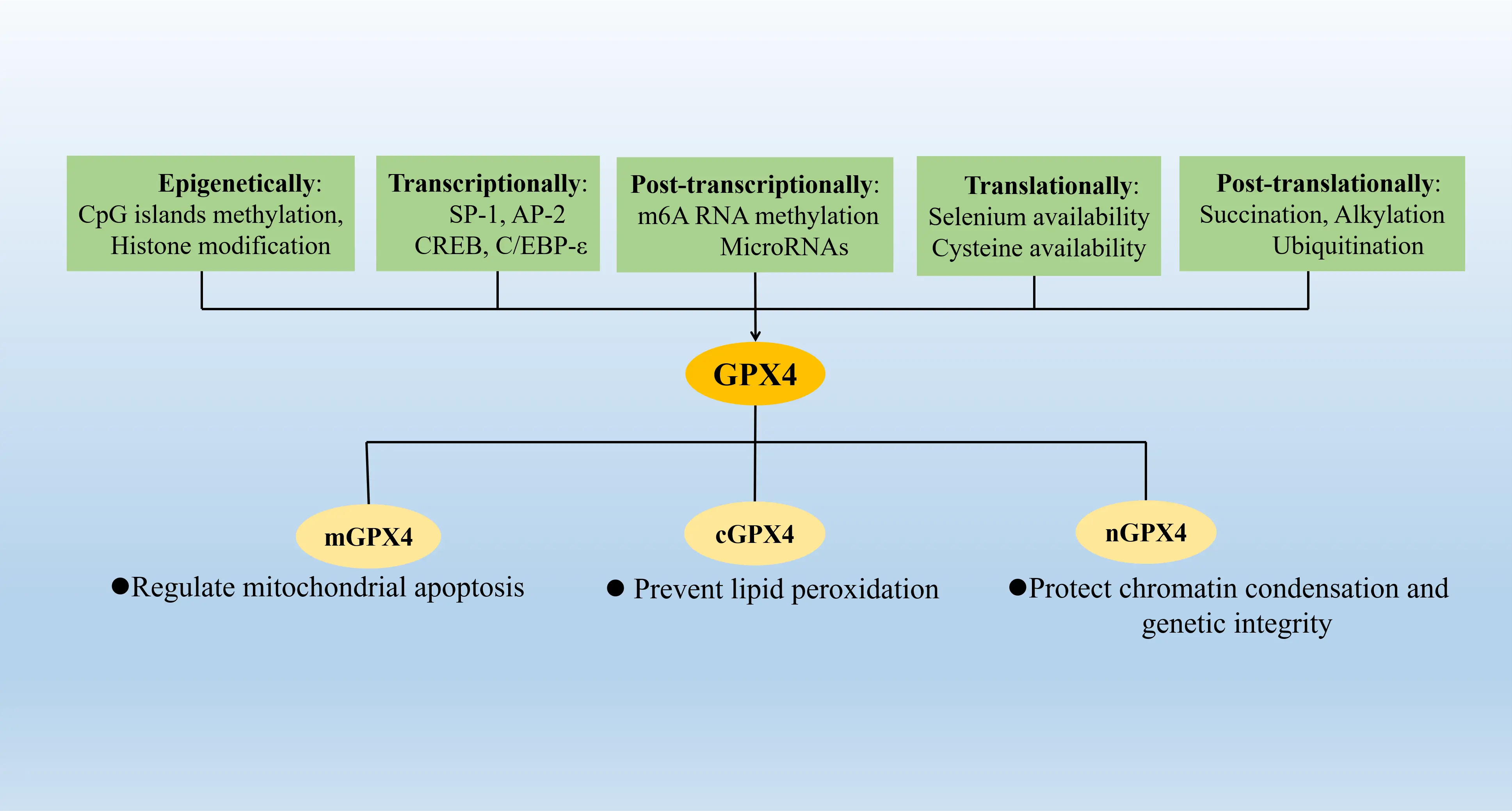
Figure 2. The expression and regulation of GPX4. The expression of GPX4 is regulated in multiple ways (epigenetically by DNA CpG islands methylation and histone modification, transcriptionally by SP1, AP2, CREB and C/EBP-ε, post-transcriptionally by RNA methylation and microRNA silencing, translationally by selenium and cysteine availability, post-translationally by succination, alkylation, ubiquitination, etc.). The products of GPX4 expression include three protein isoforms which bear distinct biological functions in different cellular compartments. SP1, stimulating protein 1. AP2, activator protein 2. CREB, cAMP-response element-binding protein. C/EBP-ε, CCAAT/enhancer-binding protein. GPX4: glutathione peroxidase 4; SP1: stimulating protein 1; AP2: activator protein 2; CREB: cAMP-response element-binding protein; C/EBP-ε: CCAAT/enhancer-binding protein.
3. The Expression and Regulation of GPX4 in OA
When it comes to the specific disease, the expression and regulation of GPX4 in osteoarthritic condition remain not fully elucidated, but indeed, they have also garnered increasing attention in recent years.
First, regarding the expression profile of GPX4 isoforms in the musculoskeletal system, the majority of current OA studies do not deliberately distinguish or detect the expression levels of individual GPX4 isoforms. Given that most OA research focuses on the role of GPX4 in suppressing lipid peroxidation and ferroptosis, the cytosolic isoform of GPX4 is generally presumed to be the specific variant under investigation. In contrast, the expression and regulation of the other two GPX4 isoforms have not been reported in the context of OA research.
The phenomenon of decreased GPX4 expression has been observed in both OA patients and animal models of OA[11,64]. Pro-inflammatory cytokines (such as IL-1β) and oxidative stress factors (such as the oxidant TBHP) are frequently utilized as inducers to establish OA models in chondrocytes, and a reduction in GPX4 expression is typically observed in these treated cells[11,65]. Although there is currently no dedicated study focusing on why GPX4 is downregulated in OA, some regulatory mechanisms underlying the reduced levels of GPX4 in OA have been reported.
Pan et al. found that IL-1β can promote methylation within the GPX4 promoter region at the epigenetic level, thereby reducing GPX4 mRNA and protein levels in chondrocytes[66]. Chen et al. reported that TPX2 may decrease GPX4 expression levels in LPS-induced C28/I2 chondrocytes via the NF-κB p65 signaling pathway[67]. It is well established that inflammatory cytokines could activate the classical NF-κB pathway to reduce the transcription of antioxidant molecules[68]. Consequently, phosphorylated p65 is also highly likely to act as the key transcriptional factor reducing the transcriptional levels of GPX4, another antioxidant molecule, in IL-1β-treated OA chondrocytes.
Selenium deficiency is receiving growing attention in the pathogenesis of musculoskeletal disorders, including OA and Kashin-Beck disease[69,70]. Therefore, abnormal selenium metabolism may partially explain the reduced expression level of GPX4 in OA pathogenesis.
Lv et al. found that low expression of TRPV1 is associated with decreased GPX4 levels in OA, and that pharmacological activation of TRPV1 could upregulate GPX4 to ameliorate OA progression[71]. Interestingly, similar to an ion channel, the Piezo1 channel may play a different role in regulating GPX4. Reportedly, the Piezo1 channel could be activated under conditions of excessive mechanical stress to decrease GPX4 expression and promote chondrocyte ferroptosis in OA[72]. This study could also partially explain, from a molecular mechanistic perspective, why joint mechanical disorders can promote OA progression.
In the field of post-translational modification, Zhou et al. have provided evidence for the possible existence of OGT-dependent O-GlcNAcylation of the GPX4 protein and demonstrated that reduced levels of O-GlcNAcylation in OA cartilage tissue may be associated with decreased GPX4 expression[73]. HSPA5 has been reported to inhibit the degradation of GPX4 in cancer cells[74]. This phenomenon may be interpreted as HSPA5 exerting a negative regulatory effect on the chaperone-mediated autophagy of GPX4[75]. In the context of OA, Lv et al. have demonstrated that the RNA-binding protein SND1 could downregulate HSPA5 to promote the degradation of GPX4 and chondrocyte ferroptosis[76].
Collectively, these reported regulatory mechanisms could partially explain why decreased expression level of GPX4 is typically observed during OA.
4. The Function of GPX4 in OA
OA is a severe and complex condition that is now recognized as a whole-organ disease characterized by structural alterations of different joint tissues, including cartilage degeneration, subchondral bone sclerosis, osteophyte formation, and synovial inflammation[77]. Emerging research demonstrates that a series of cellular events, including chondrocyte ferroptosis and apoptosis, aberrant ECM metabolism, inflammatory responses of synovial cells, and imbalanced activity of osteoblasts and osteoclasts, can contribute to the pathogenesis of OA[78]. According to the available evidence, GPX4 is proposed to be involved in these cellular events underlying the pathophysiology of osteoarthritis to some extent, which will be the focus of our next discussion in this review below.
4.1 GPX4 in articular cartilage
4.1.1 GPX4 in chondrocyte
Since chondrocytes are the only cell type present in cartilage, the balance between chondrocyte proliferation and death plays a crucial role in cartilage homeostasis. Due to the avascular and aneural nature of articular cartilage, the chondrocytes in adult human cartilage, which are literally imprisoned in scattered lacunas within the matrix and restricted from migrating, are normally quiescent and not actively proliferative. Consequently, it is usually compromised when cartilage self-repair is considered[79,80]. Yet on the other side of the coin, this also implies that shifting the focus of treatment strategies to how to appropriately inhibit detrimental chondrocyte death during OA development may prove to be a more effective approach in maintaining cartilage integrity and health.
Regulated cell death can be classified into apoptosis and non-apoptotic processes, including necroptosis, pyroptosis and ferroptosis[81,82]. Recent studies have indicated a possible association between ferroptosis and OA, as supported by examinations of OA patients’ specimens and experimental models, including TBHP-induced OA chondrocyte models, Erastin-induced OA chondrocyte models, ACLT-induced OA mouse models and DMM-induced OA mouse models[11,62,64,65,83-87]. These studies show that chondrocytes undergo ferroptosis during OA and that chondrocyte ferroptosis can be regulated multidimensionally to contribute to the progression of OA.
The role of ferroptosis-related pathways in osteoarthritis has attracted increasing attention in recent years[88,89]. Abnormal intracellular iron homeostasis (e.g., altered expression of Tfr1 and FTH1) and dysregulated lipid metabolism (e.g., upregulation of Acsl4 and Lpcat3) promote the occurrence of cellular ferroptosis[90]. Conversely, the classical defense mechanisms against ferroptosis comprise four major axes: the cysteine/GSH/GPX4 axis, the NADPH/FSP1/CoQ10 axis, the GCH1/BH4 axis, and the DHODH/CoQH2 axis[91,92]. The balance or imbalance between the ferroptosis execution system and the ferroptosis defense system ultimately determines whether a cell undergoes ferroptosis[91]. Among these ferroptosis-related pathways, GPX4, serving as a key regulator of ferroptosis and a “star molecule” as well, has received the majority of research focus in the context of osteoarthritis. It has also been reported to decrease significantly at both mRNA and protein levels in chondrocytes of OA animal models and human patients[11,64,72,76,93].
Although the more detailed mechanism of GPX4’s enzymatic action in osteoarthritic chondrocyte ferroptosis remains to be further investigated, yet the eventual effect may be relatively explicit. During the process of OA development, the reduction of GPX4 expression may not directly induce chondrocyte ferroptosis as it usually does in other cells, such as cancer cells, but it does make chondrocytes more sensitive to oxidative stress and mechanical overloading[64,72,94]. Meanwhile, under conditions of only GPX4-knockdown with no TBHP treatment, there is also an observed increase of the substrate GSH, which is opposite to the results from OA cartilage and may partly counteract the detriment from decreased GPX4, thus accounting for the survival of GPX4-knockdown cells[64].
In addition to mediating ferroptosis, GPX4 may also confer a role in chondrocyte apoptosis during OA development. The three canonical apoptotic pathways, the mitochondrial pathway, the death receptor pathway, and the endoplasmic reticulum (ER) stress pathway, can be all implicated in chondrocyte apoptosis during OA[95,96]. Although it remains uncertain in which pathway the downregulation of GPX4 induces chondrocyte apoptosis in OA, both decreased GPX4 expression and the occurrence of apoptosis (detected by flow cytometry) have been concurrently observed in IL-1β-treated chondrocytes[97]. During the process of a specific disease such as OA, different types of regulated cell death usually coexist, sharing similar initial signals and molecular regulators[7,98,99]. GPX4, essentially as an antioxidant, initially gained attention for its anti-apoptotic function through multiple mechanisms before it was connected to ferroptosis[100,101]. For instance, downregulation of GPX4 could upregulate EGR1 to induce mitochondria-mediated apoptosis in TNBC cells[102]. GPX4 could inhibit apoptosis by preventing the release of cytochrome c through detoxification of cardiolipin hydroperoxide in the mitochondrial membrane[103]. This mechanism may also occur in chondrocytes from OA cartilage, where a significant decrease in the mitochondrial membrane potential and alteration in proteins associated with the integrity of the mitochondrial membrane and apoptosis were observed[104-106]. Collectively, these studies suggest GPX4 could be implicated in ROS-induced mitochondrial-mediated apoptosis, besides ferroptosis during OA.
4.1.2 GPX4 in ECM homeostasis
The ECM in cartilage is a sophisticated assembly of macromolecules, consisting primarily of two components that define its mechano-physical properties: the collagenous network, responsible for the tensile strength of the ECM, and the proteoglycans, responsible for the elastic properties of ECM and maintaining osmotic balance[107]. Beyond its structural function as a cellular scaffold, ECM also acts as a dynamic repository for cytokines and other growth factors that modulate chondrocyte behavior and turnover[108].
The imbalance of chondrocyte anabolism and catabolism could lead to abnormal ECM homeostasis and thus OA development[109]. Available studies, including Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis, gene set enrichment analysis (GSEA) and cell experiments, have indicated GPX4 downregulation in osteoarthritic chondrocytes may promote ECM degradation through the MAPK/NF-kB pathway, which could activate cartilage catabolism-relate proteins, including MMP3 and MMP13, while exerting limited regulatory influence on anabolism-related genes such as COL2A1 and aggrecan (ACAN)[64,110,111]. Noteworthily, the upregulation of matrix metalloproteinases (MMPs), particularly MMP13, is usually considered as one of the signs of early-stage OA and a point of no return for the disease progression[112-115]. This insight is further reflective of the role of GPX4 in the OA process from the viewpoint of MMP-induced ECM degradation.
4.2 GPX4 in subchondral bone and osteophyte
Although articular cartilage degeneration is the primary concern in osteoarthritis, the integrity of articular cartilage also relies on its biochemical and biomechanical interplay with subchondral bone[116,117]. Subchondral bone can provide the mechanical support and nutrition supply for overlying articular cartilage during the movement of joints and undergo continuous adaptation in response to changes in the mechanical environment through bone formation and resorption[117,118].
Available research has shown that GPX4 suppression could affect bone metabolism and cause bone disorder by prompting osteoblast ferroptosis in osteoporosis[119]. During OA progression, knockdown of GPX4 has been demonstrated to increase subchondral bone plate thickness (SBP) and bone volume fraction of subchondral trabecular bone (STB) significantly in mice[64]. A decrease in GPX4 with concomitant cellular ferroptosis has been observed during mouse osteoclast differentiation in recent research[120]. In this study, upregulation of FOXO3 could inhibit ferroptosis and differentiation of subchondral osteoclasts to attenuate abnormal bone remodeling in obesity-induced OA by partially restoring GPX4. Despite these aforementioned phenomena, the exact molecular mechanism underlying subchondral bone remodeling during OA remains largely unknown.
The role of TGF–β/SMAD pathway in the pathogenesis of osteoarthritis has drawn more and more attention in recent years[121]. Not only is TGF–β essential for maintenance of articular cartilage metabolic and structural homeostasis through regulating the expression of collagen type II and MMP13, the TGF–β signaling also seems to mediate Smad2/3 to facilitate osteoclastic differentiation and bone resorption, recruit nestin-positive MSCs and osteoprogenitors to resorption pits in subchondral bone, and promote osteoblast terminal maturation, depositing extracellular matrix components Col1 and osteocalcin in bone[117,122-124]. These alterations eventually result in an imbalance between osteoblasts and osteoclasts and consequent aberrant bone remodeling that may initiate and worsen osteoarthritis[125,126]. Intriguingly, emerging research has suggested that GPX4 may block the phosphorylation of Smad2/3 and thus exhibit antifibrotic activities in certain fibrotic diseases like lung fibrosis[127]. This intriguing inference about the relationship between GPX4 and SMAD implies a potential involvement of GPX4 in abnormal subchondral bone remodeling during OA through regulating TGF–β/SMAD2/3 pathway.
Osteophyte formation is predominantly characterized by the neo-chondrogenesis of mesenchymal stem cells located in the periosteum at the bone-cartilage junction. This process is followed by chondrocyte hypertrophy and subsequent endochondral ossification. A mature osteophyte typically becomes integrated with the original subchondral bone upon completion[128]. Despite the details remaining obscure, the TGF–β/SMAD pathway may play a role in most of these sections of osteophyte formation according to previous studies[128]. Based on the aforementioned putative link between GPX4 and SMAD, GPX4 may also be involved in osteophyte formation by mediating regulation of TGF–β/SMAD pathway. Moreover, available research has verified that GPX4 downregulation can indeed increase osteophyte maturity and size significantly[64].
4.3 GPX4 in synovial inflammation
Nowadays inflammation is gaining increasing recognition as a central player in the pathogenesis of OA. The chronic low-grade inflammation in OA, following joint trauma or overuse, can frequently evolve into a vicious, self-sustaining cycle of local joint damage, inflammation and failed tissue repair[129,130]. The synovium, which mainly consists of immunocytes and fibroblast-like synoviocytes (FLS), despite not being the sole tissue implicated in OA-related inflammation, represents a major site of gross and microscopic inflammatory alterations[129].
Previous investigations have shown that GPX4 overexpression could inhibit the activities of LOXs and COXs, which require basal levels of lipid hydroperoxides for activation, and thus inhibit the synthesis of proinflammatory eicosanoids from arachidonic acids[131,132]. Moreover, GPX4 overexpression may inhibit the production of a wide variety of inflammatory genes, including TNF-α, IL-1β, IL-6, IL-17A, and IL-22[133-136]. Not surprisingly, these inflammatory mediators can also be induced in OA synovial inflammation as well[137-139]. Taken together, these observations suggest a potential association between GPX4 and inflammatory signaling pathways in OA synovitis.
Recently, Hu et al. demonstrated a decrease of GPX4 level in the synovium with increasing KL grade in OA. In this study, lipoxin A4 reportedly activated the ESR2/LPAR3/Nrf2 axis to reverse the downregulation of GPX4 in OA FLSs, thus antagonizing FLS ferroptosis and mitigating the pain and progression of KOA[140]. Additionally, it’s noteworthy that another interesting research examining the alteration of GPX4 level in the rheumatoid arthritis (RA) synovium reached a different conclusion[39]. An increase of GPX4 in the RA synovium and FLSs was observed in this study, accompanied by a decrease in FLS ferroptosis. This study demonstrated that although both oxidation and antioxidation levels were elevated in RA FLS compared to healthy FLS, there was a reduction in the ratio of oxidation/antioxidation, suggesting that the aberrant ‘tumor-like’ growth of RA FLS displayed resistance to ferroptosis.
4.4 GPX4 in cell-cell interaction during OA
The joint is generally regarded as a single functional unit. In OA, articular cartilage degeneration, subchondral bone remodeling/sclerosis, and synovitis are inextricably linked and reciprocally influence one another.
Regarding the role of GPX4 in OA, current research has predominantly focused on cartilage. When pathogenic factors induce a reduction in GPX4 levels within chondrocytes, these cells become more susceptible to ferroptosis. Prior to cell death, chondrocytes often undergo functional dysregulation characterized by decreased anabolism (e.g., downregulation of type II collagen) and increased catabolism (e.g., upregulation of MMP3 and MMP13), consequently leading to the depletion of the ECM. As cartilage is primarily composed of chondrocytes and the ECM, it deteriorates progressively.
GPX4-regulated chondrocyte death-derived cell debris and ECM degradation products can act as damage-associated molecular patterns (DAMPs), activating pattern recognition receptors on macrophages in the synovium[141,142]. This promotes M1 macrophage polarization, triggering the release of pro-inflammatory cytokines and contributing to synovitis. The downregulation of GPX4 in fibroblast-like synoviocytes also promotes the secretion of inflammatory mediators. These inflammatory mediators from M1 macrophages and FLSs, in turn, can activate the NF-κB or MAPK pathways in chondrocytes, thereby exacerbating chondrocyte inflammation and further accelerating ECM degradation.
As articular cartilage becomes progressively worn and thinned, the unprotected subchondral bone is more vulnerable to aberrant joint stress. A decrease in GPX4 levels in osteoclasts within the subchondral bone renders them more prone to ferroptosis, thereby disrupting the balance between osteoclasts and osteoblasts. This imbalance further promotes aberrant subchondral bone remodeling.
Collectively, the downregulation of GPX4 in joint would contribute to the progression of OA (Figure 3).
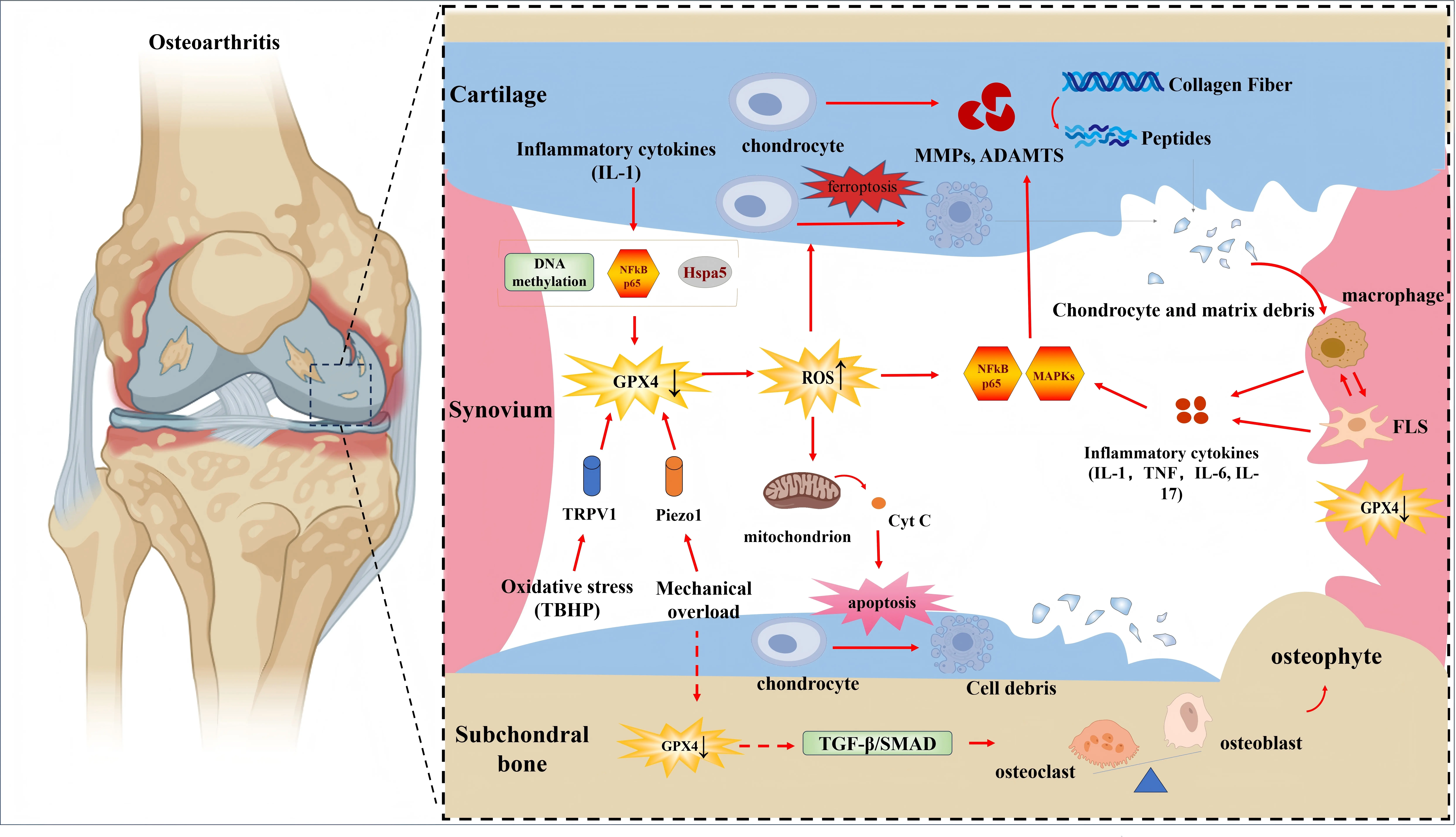
Figure 3. GPX4 in the pathogenesis of OA. OA is a whole joint disease involving cartilage, subchondral bone and synovium. Many pathogenetic factors, including inflammatory chemokines, oxidative stress and mechanical overload, could cause a decrease of GPX4 in chondrocytes. The downregulation of GPX4 could contribute to not only cartilage degradation by facilitating chondrocyte death such as ferroptosis and apoptosis and ECM breakdown, but also synovitis by eliciting the release of inflammatory mediators. Cartilage degradation-derived products including cell and ECM debris could activate PPRs in synovium macrophages as DAMPs to induce inflammatory mediators and aggravate synovitis, ultimately causing a vicious cycle of local joint damage, inflammation and failed tissue repair. In addition, decreased GPX4 may modulate TGF-β/SMAD signaling pathway to mediate abnormal bone remodeling and osteophyte formation, where the exact role of GPX4 remains ambiguous and need more validation yet, as labeled by dotted lines. DAMPs, damage associated molecular patterns. PPRs, pattern recognition receptors. GPX4: glutathione peroxidase 4; OA: osteoarthritis; ECM: extracellular matrix; PPRs: pattern recognition receptors; DAMPs: damage-associated molecular patterns; MMPs: matrix metalloproteinases; ROS: reactive oxygen species; MAPKs: mitogen activated protein kinases.
5. Therapeutic Implications and Prospects
5.1 GPX4-regulated ferroptosis: A central driver of OA initiation or a downstream amplifying pathway
Growing evidence indicates that lipid peroxidation contributes to chondrocyte dysfunction and oxidative stress in OA. However, it remains unclear whether ferroptosis represents a central driver of disease initiation or acts as one of several downstream amplifying mechanisms. Here, we speculate that ferroptosis predominantly functions as a core downstream amplifying signaling pathway in the current landscape of OA patients.
First, it is well established that aging is one of the primary risk factors for OA[2]. Aging has been characterized as a state of systemic chronic inflammation (‘inflammaging’), wherein senescent cells secrete the senescence-associated secretory phenotype (SASP), which contains numerous inflammatory factors that promote chronic inflammation[143]. The canonical inflammatory cytokine IL-1β is the most commonly used reagent to treat chondrocyte and simulate OA in vitro. In these models, a decrease in GPX4 is frequently observed. However, it is noteworthy that p21, a marker of chondrocyte senescence, is also upregulated and could reportedly increase resistance to chondrocyte ferroptosis by inhibiting the ubiquitination of GPX4[62]. Indeed, the resistance of senescent cells to ferroptosis has been reported in other studies[144-146]. Therefore, for aging-related OA patients, ferroptosis does not appear to be the central driver of OA initiation. Nevertheless, the occurrence of ferroptosis has been reported to exacerbate inflammation in chronic inflammatory diseases[147].
For patients with OA induced by joint mechanical derangement, ferroptosis may play a crucial role in both the initiation and progression of the disease. Mechanical stress has been reported to mechanically downregulate GPX4 and induce ferroptosis in chondrocytes via upregulation of the mechanosensor Piezo1 channel[72]. Furthermore, animal experiments have reported that conditional knockout of GPX4 in the articular cartilage of sham-operated mice can induce an OA phenotype[64]. Collectively, mechanical stress-induced downregulation of GPX4 and ferroptosis has likely already served as a central driver in the initiation stage of mechanically induced OA or post-traumatic OA. In addition, it is noteworthy that GPX4-regulated ferroptosis could aggravate OA progression, as evidenced by the exacerbation of OA phenotypes in the ACLT mouse model following GPX4 deletion[64].
Obesity is another significant risk factor for the increasing prevalence of OA, aside from aging[2]. Similar to aging, obesity is viewed as a state of chronic, low-grade inflammation. A unifying factor in obesity-related diseases is immune dysregulation, particularly inflammation, in response to metabolic overload[148]. Moreover, obesity-related OA patients are often prone to joint mechanical disorder due to excess body weight. Therefore, for obesity-related OA patients, targeting ferroptosis is important for both the initiation and development stages of the disease.
Furthermore, selenium deficiency has been increasingly found to be highly correlated with radiographic OA severity[69]. Selenium deficiency can mechanistically lead to decreased expression of the selenoprotein GPX4, thereby increasing susceptibility to ferroptosis. For this subset of OA patients, targeting GPX4 or ferroptosis appears to be a more specific approach and should arguably be regarded as a disease-modifying strategy.
Collectively, whether ferroptosis acts more as the initiator or a downstream amplifier may depend on the specific etiology of OA. However, regardless of the subtype, targeting GPX4-regulated ferroptosis holds an indispensable position in the treatment of OA.
5.2 Finely tuning GPX4 level in selectively targeted tissues or cells is of significance
In addition to preventing lipid peroxidation and ferroptosis, GPX4 also fulfills other physiological functions, including modulating inflammation, immunity, autophagy and differentiation[15,23]. Yet, admittedly, the exact role of GPX4 in these physiological processes and related molecular pathways remains largely elusive until now. Thus, it is actually difficult but also important to figure out how to properly regulate GPX4 expression levels in specific tissues while avoiding the potential side effect of disturbing normal physiological conditions.
To be more specific, as one of the lipid antioxidants, GPX4 serves as both a guardian against oxidative damage as well as a key regulator of physiologically versatile signaling lipids, such as oxygenated PUFAs-derived arachidonic acid (AA) and its metabolites[100,135,149,150]. Undoubtedly, this functional duality of GPX4 must be carefully balanced to sustain homeostasis. One evolutionarily plausible reason why such complex post-translational modification of GPX4 exists is that it enables GPX4 level and activity to be tuned in a way that avoids excessive lipid peroxidation while permitting the generation of bioactive signaling lipids engaged in physiologically fundamental redox signaling[135,151,152].
One inevitable concern about the impact of an imbalanced regulation of GPX4 on osteoarthritis is whether overexpression of GPX4 in specific tissues, such as the synovium in OA, would lead to FLS insensitivity to ferroptosis and resultant synovium hyperplasia and fibrosis, which occurs in RA synovium as mentioned earlier[39]. Another more relevant and thought-provoking instance is that Zi et al. have recently demonstrated that senescent chondrocytes could exhibit resistance to ferroptosis and thus subsist, probably through upregulating EAAT1-GSH-GPX4 signaling axis[153]. This mechanism may be evolutionarily advantageous in maintaining unicellular redox homeostasis but may exert a counterproductive effect on the whole organism when applied to undesired senescent chondrocytes producing detrimental SASPs and inducing age-related OA. Similarly, Ran et al. indicated that lifelong reduction in GPX4 actually increased the lifespan for GPX4+/-mice and retarded age-related pathology, most likely through alterations in the sensitivity of tissues to apoptosis[154]. Intriguingly, this phenomenon concerning reduced GPX4 and longevity may be also potentially associated with the controversial function of the PUFAs-derived AA mentioned above, which may delay the aging process along with its anti-inflammatory metabolites, including resolvins and lipoxins, through mitigating inflammation, facilitating stem cell proliferation, and augmenting autophagy and apoptosis[100,149,155]. Collectively, these studies highlight the necessity of finely manipulating GPX4 levels in specifically targeted tissues for safer clinical translation.
5.3 OA treatment-oriented GPX4-targeted therapeutics deserve more research attention
Overall, the panorama of GPX4 regulation mechanism remains to be delineated in more detail, which would be favorable to GPX4-based drug development. Due to the huge therapeutic potential to target ferroptosis in cancer cells, inhibitors of GPX4 have been well developed and are generally divided into GPX4 activity inhibitors (e.g., RSL3, ML210, ML162) and protein degraders (e.g., FIN56 and some miRNAs), while the regulators of GPX4 deserve further investigation[15,156-160]. Particularly, to the best of our knowledge, no selective activator of GPX4 has so far been reportedly evaluated or applied in OA treatment in published literature, although some GPX4 activators have been tested in other conditions (Table 1). Recent studies utilizing virtual screening methodologies have identified several promising compounds that function as allosteric enzymatic activators of GPX4, effectively suppressing ferroptosis[161-163]. Selenium-based nutritional supplementation has shown efficacy in boosting GPX4 function in cardiomyopathy models, probably through upregulating GPX4 expression[164]. In the context of OA, some pharmacologically active compounds have been found to upregulate the levels of GPX4 to alleviate OA progression. However, it is noteworthy, in these studies, the reported regulatory patterns are indirect, mostly via the Nrf2/SLC7A11/GPX4 signaling pathway[66,111,165-175] (Table 2). Among these compounds, some are generally considered as classical lipophilic antioxidants. Compared with GPX4-targeted approaches, these lipophilic antioxidants offer greater accessibility and a more rapid onset of action in treatment. However, they are also prone to aberrant biodistribution within the body, leading to potential off-target effects. Thus, GPX4-targeted therapeutics appropriate for OA need further development and assessment in order to serve as a safer precision therapeutic strategy. In addition, a one-size-fits-all approach may not be suitable for OA due to the confusing heterogeneity of the disease. In the future, further investigations are warranted to develop effective GPX4 regulators specifically targeting distinct OA subtypes. Current research relies heavily on DMM or ACLT-induced murine models, which predominantly simulate post-traumatic OA. However, the translational relevance of these models to the most prevalent OA subtypes affecting patients, particularly aging-related and obesity-related OA, remains unclear[176].
Table 1. GPX4-targeted compounds reported in non-OA context.
| Compound | Target/Mechanism | Experimental models | Ref. |
| Compound 1 and Analogues | GPX4 Allosteric Activator | HT-1080 Cells, Human PMNs | [162] |
| Compound 102 | GPX4 Allosteric Activator | HT-1080 Cells, Human PMNs | [161] |
| Compound A9 and C3 | GPX4 Allosteric Activator | HT-1080 Cells, DOX-Induced Myocardial Injury (Mice) | [163] |
| Selenomethionine (SeMet) | Selenium supplementation (upregulate GPX4 expression) | H9C2 Cells, DOX-Induced Cardiomyopathy (Mice) | [164] |
GPX4: glutathione peroxidase 4; OA: osteoarthritis; PMNs: polymorphonuclear neutrophils.
Table 2. Potential pharmacologically active regulators of GPX4 in OA treatment.
| Regulators | Target cell/tissue | Target pathway | Main findings | Ref. |
| Lipophilic antioxidants | ||||
| Mitoquinone | Murine chondrocytes | Nrf2/GPX4↑ | inhibits chondrocyte ferroptosis, inflammation and ECM degradation | [172] |
| Theaflavin-3,3'-digallate | Human chondrocytes | Nrf2/GPX4↑ | protects chondrocytes against Erastin-induced ferroptosis | [174] |
| Astaxanthin | Rat chondrocytes | SLC7A11/ GPX4↑ | defenses against IL-1β-mediated mitochondrial damage and chondrocyte ferroptosis | [165] |
| Icariin | Human cell line SW1353 | SLC7A11/GPX4↑ | mitigates chondrocyte ferroptosis, ECM degradation and the production of inflammatory mediators during OA | [169] |
| Ruscogenin | Murine chondrocytes | Nrf2/SLC7A11/GPX4↑ | inhibits IL-1β-induced inflammation and ferroptosis in chondrocytes | [173] |
| Vitamin k2 | Rat chondrocytes | SLC7A11/GPX4↑ | inhibits chondrocyte ferroptosis and MAPK/NFκB signaling pathway to decrease ECM degradation | [111] |
| Baicalin | Human chondrocytes | Nrf2/GPX4↑ | inhibits IL-1β-induced chondrocyte ferroptosis and ECM degradation | [166] |
| Melatonin | Murine chondrocytes, Mouse cell line ATDC5 | NOX4/GRP78/GPX4↑ | mitigates mitochondrial dysfunction and chondrocyte ferroptosis | [171] |
| Hydrophilic/Amphiphilic compounds | ||||
| Cardamonin | Rat chondrocytes | P53/SLC7A11/GPX4↑ | mitigates IL-1β-induced chondrocyte ferroptosis | [168] |
| Biochanin A | Murine chondrocytes | Nrf2/ SLC7A11/GPX4↑ | scavenges free radicals and prevents lipid peroxidation | [167] |
| Kukoamine A | Mouse cell line ATDC5 | Sirt1/Nrf2/GPX4↑ | inhibits chondrocyte inflammation and ferroptosis | [170] |
| Vinpocetine | Murine chondrocytes | Nrf2/GPX4↑ | inhibits TBHP-induced chondrocyte ferroptosis and ECM degradation | [175] |
| 4-octyl itaconate | Rat chondrocytes | GPX4 methylation↓ | protects chondrocytes against IL-1β-induced oxidative stress and ferroptosis | [66] |
GPX4: glutathione peroxidase 4; OA: osteoarthritis; ECM: extracellular matrix; ↑: increase; ↓: decrease.
6. Conclusion
In summary, the selenoenzyme GPX4, as a key antioxidant in cellular redox homeostasis, is not only most familiar as the 'star molecule’ in lipid peroxidation and ferroptosis, but also may mediate OA development in multiple aspects due to its possible involvement in the pathophysiological alterations of different joint tissues. OA is no longer regarded solely as a degenerative process ascribed to aging-related “wear and tear”, but appears to more resemble an umbrella term encompassing heterogenous pathological conditions, which partly accounts for the current lack of coherent therapeutic strategies[176]. In order to optimize the palliative effects of current treatments and develop efficacious disease-modifying anti-osteoarthritic drugs, therapies based on the regulation of GPX4 may need to be tailored to the individual or a subset of osteoarthritic joints.
Authors contribution
Sun K, Guo F: Conceptualization, funding acquisition.
He J: Writing-original draft, visualization.
Zhang X, Hou L, Liu H: Writing-review & editing.
All authors read and approved the final version of the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China [Grant Nos. 82172498 and 82302768].
Copyright
© The Author(s) 2026.
References
-
1. Dell’Isola A, Recenti F, Giardulli B, Lawford BJ, Kiadaliri A. Osteoarthritis year in review 2025: Epidemiology and therapy. Osteoarthr Cartil. 2025;33(11):1300-1306.[DOI]
-
2. Kloppenburg M, Namane M, Cicuttini F. Osteoarthritis. Lancet. 2025;405(10472):71-85.[DOI]
-
3. Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, et al. Osteoarthritis. Nat Rev Dis Primers. 2016;2(1):16072.[DOI]
-
4. Peng X, Chen X, Zhang Y, Tian Z, Wang M, Chen Z. Advances in the pathology and treatment of osteoarthritis. J Adv Res. 2025;78:257-283.[DOI]
-
5. Akkiraju H, Nohe A. Role of chondrocytes in cartilage formation, progression of osteoarthritis and cartilage regeneration. J Dev Biol. 2015;3(4):177-192.[DOI]
-
6. Chen H, Tan XN, Hu S, Liu RQ, Peng LH, Li YM, et al. Molecular mechanisms of chondrocyte proliferation and differentiation. Front Cell Dev Biol. 2021;9:664168.[DOI]
-
7. Charlier E, Relic B, Deroyer C, Malaise O, Neuville S, Collée J, et al. Insights on molecular mechanisms of chondrocytes death in osteoarthritis. Int J Mol Sci. 2016;17(12):2146.[DOI]
-
8. Dixon Scott J, Lemberg Kathryn M, Lamprecht Michael R, Skouta R, Zaitsev Eleina M, Gleason Caroline E, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-1072.[DOI]
-
9. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486-541.[DOI]
-
10. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144-152.[DOI]
-
11. Yao X, Sun K, Yu S, Luo J, Guo J, Lin J, et al. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J Orthop Translat. 2021;27:33-43.[DOI]
-
12. Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. 1982;710(2):197-211.[DOI]
-
13. Nishida Xavier da Silva T, Friedmann Angeli JP, Ingold I. GPX4: Old lessons, new features. Biochem Soc Trans. 2022;50(3):1205-1213.[DOI]
-
14. Liu Y, Wan Y, Jiang Y, Zhang L, Cheng W. GPX4: The hub of lipid oxidation, ferroptosis, disease and treatment. Biochim Biophys Acta Rev Cancer. 2023;1878(3):188890.[DOI]
-
15. Xie Y, Kang R, Klionsky DJ, Tang D. GPX4 in cell death, autophagy, and disease. Autophagy. 2023;19(10):2621-2638.[DOI]
-
16. Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830(5):3289-3303.[DOI]
-
17. Brigelius-Flohé R, Flohé L. Regulatory phenomena in the glutathione peroxidase superfamily. Antioxid Redox Signal. 2020;33(7):498-516.[DOI]
-
18. Labrecque CL, Fuglestad B. Electrostatic drivers of GPx4 interactions with membrane, lipids, and DNA. Biochemistry. 2021;60(37):2761-2772.[DOI]
-
19. Weaver K, Skouta R. The selenoprotein glutathione peroxidase 4: From molecular mechanisms to novel therapeutic opportunities. Biomedicines. 2022;10(4):891.[DOI]
-
20. Maiorino M, Aumann KD, Brigelius-Flohé R, Doria D, van den Heuvel J, McCarthy J, et al. Probing the presumed catalytic triad of selenium-containing peroxidases by mutational analysis of phospholipid hydroperoxide glutathione peroxidase (PHGPx). Biol Chem Hoppe Seyler. 1995;376(11):651-660.[DOI]
-
21. Hondal RJ, Ruggles EL. Differing views of the role of selenium in thioredoxin reductase. Amino Acids. 2011;41(1):73-89.[DOI]
-
22. Flohé L, Toppo S, Orian L. The glutathione peroxidase family: Discoveries and mechanism. Free Radic Biol Med. 2022;187:113-122.[DOI]
-
23. Ursini F, Bosello Travain V, Cozza G, Miotto G, Roveri A, Toppo S, et al. A white paper on Phospholipid Hydroperoxide Glutathione Peroxidase (GPx4) forty years later. Free Radic Biol Med. 2022;188:117-133.[DOI]
-
24. Maiorino FM, Brigelius-Flohé R, Aumann KD, Roveri A, Schomburg D, Flohé L. Diversity of glutathione peroxidases. Methods Enzymol. 1995;252:38-53.[DOI]
-
25. Toppo S, Flohé L, Ursini F, Vanin S, Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: Variations of a basic scheme. Biochim Biophys Acta. 2009;1790(11):1486-1500.[DOI]
-
26. Chu FF. The human glutathione peroxidase genes GPX2, GPX3, and GPX4 map to chromosomes 14, 5, and 19, respectively. Cytogenet Cell Genet. 2008;66(2):96-98.[DOI]
-
27. Kelner MJ, Montoya MA. Montoya, Structural organization of the human selenium-dependent phospholipid hydroperoxide glutathione peroxidase gene (GPX4): Chromosomal localization to 19p13.3. Biochem Biophys Res Commun. 1998;249(1):53-55.[DOI]
-
28. Maiorino M, Scapin M, Ursini F, Biasolo M, Bosello V, Flohé L. Distinct promoters determine alternative transcription of gpx-4 into phospholipid-hydroperoxide glutathione peroxidase variants. J Biol Chem. 2003;278(36):34286-34290.[DOI]
-
29. Brigelius-Flohe R, Aumann KD, Blöcker H, Gross G, Kiess M, Klöppel KD, et al. Phospholipid-hydroperoxide glutathione peroxidase. Genomic DNA, cDNA, and deduced amino acid sequence. J Biol Chem. 1994;269(10):7342-7348.[DOI]
-
30. Nomura K, Imai H, Koumura T, Arai M, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase suppresses apoptosis mediated by a mitochondrial death pathway. J Biol Chem. 1999;274(41):29294-29302.[DOI]
-
31. Puglisi R, Bevilacqua A, Carlomagno G, Lenzi A, Gandini L, Stefanini M, et al. Mice overexpressing the mitochondrial phospholipid hydroperoxide glutathione peroxidase in male germ cells show abnormal spermatogenesis and reduced fertility. Endocrinology. 2007;148(9):4302-4309.[DOI]
-
32. Schneider M, Forster H, Boersma A, Seiler A, Wehnes H, Sinowatz F, et al. Mitochondrial glutathione peroxidase 4 disruption causes male infertility. Faseb J. 2009;23(9):3233-3242.[DOI]
-
33. Imai H, Hakkaku N, Iwamoto R, Suzuki J, Suzuki T, Tajima Y, et al. Depletion of selenoprotein GPx4 in spermatocytes causes male infertility in mice. J Biol Chem. 2009;284(47):32522-32532.[DOI]
-
34. Puglisi R, Maccari I, Pipolo S, Conrad M, Mangia F, Boitani C. The nuclear form of glutathione peroxidase 4 is associated with sperm nuclear matrix and is required for proper paternal chromatin decondensation at fertilization. J Cell Physiol. 2012;227(4):1420-1427.[DOI]
-
35. Pipolo S, Puglisi R, Mularoni V, Esposito V, Fuso A, Lucarelli M, et al. Involvement of sperm acetylated histones and the nuclear isoform of Glutathione peroxidase 4 in fertilization. J Cell Physiol. 2018;233(4):3093-3104.[DOI]
-
36. Liu Y, Cheng D, Wang Y, Xi S, Wang T, Sun W, et al. UHRF1-mediated ferroptosis promotes pulmonary fibrosis via epigenetic repression of GPX4 and FSP1 genes. Cell Death Dis. 2022;13(12):1070.[DOI]
-
37. Wigner P, Synowiec E, Jóźwiak P, Czarny P, Bijak M, Barszczewska G, et al. The changes of expression and methylation of genes involved in oxidative stress in course of chronic mild stress and antidepressant therapy with agomelatine. Genes. 2020;11(6):644.[DOI]
-
38. Zhang X, Huang Z, Xie Z, Chen Y, Zheng Z, Wei X, et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic Biol Med. 2020;160:552-565.[DOI]
-
39. Ling H, Li M, Yang C, Sun S, Zhang W, Zhao L, et al. Glycine increased ferroptosis via SAM-mediated GPX4 promoter methylation in rheumatoid arthritis. Rheumatology. 2022;61(11):4521-4534.[DOI]
-
40. Deng L, Di Y, Chen C, Xia J, Lei B, Li N, et al. Depletion of the N6-Methyladenosine (m6A) reader protein IGF2BP3 induces ferroptosis in glioma by modulating the expression of GPX4. Cell Death Dis. 2024;15(3):181.[DOI]
-
41. Meng X, Wang Z, Yang Q, Liu Y, Gao Y, Chen H, et al. Intracellular C5aR1 inhibits ferroptosis in glioblastoma through METTL3-dependent m6A methylation of GPX4. Cell Death Dis. 2024;15(10):729.[DOI]
-
42. Wu Y, Zhang S, Gong X, Tam S, Xiao D, Liu S, et al. The epigenetic regulators and metabolic changes in ferroptosis-associated cancer progression. Mol Cancer. 2020;19(1):39.[DOI]
-
43. Wang Z, Zhang X, Tian X, Yang Y, Ma L, Wang J, et al. CREB stimulates GPX4 transcription to inhibit ferroptosis in lung adenocarcinoma. Oncol Rep. 2021;45(6):88.[DOI]
-
44. Ufer C, Borchert A, Kuhn H. Functional characterization of cis- and trans-regulatory elements involved in expression of phospholipid hydroperoxide glutathione peroxidase. Nucleic Acids Res. 2003;31(15):4293-4303.[DOI]
-
45. Imai H, Saito M, Kirai N, Hasegawa J, Konishi K, Hattori H, et al. Identification of the positive regulatory and distinct core regions of promoters, and transcriptional regulation in three types of mouse phospholipid hydroperoxide glutathione peroxidase. J Biochem. 2006;140(4):573-590.[DOI]
-
46. Deng M, Luo J, Cao H, Li Y, Chen L, Liu G. METTL14 represses osteoclast formation to ameliorate osteoporosis via enhancing GPX4 mRNA stability. Environ Toxicol. 2023;38(9):2057-2068.[DOI]
-
47. Hou Y, Cai S, Yu S, Lin H. Metformin induces ferroptosis by targeting miR-324-3p/GPX4 axis in breast cancer. Acta Biochim Biophys Sin. 2021;53(3):333-341.[DOI]
-
48. Gu X, Wang Y, He Y, Zhao B, Zhang Q, Li S. MiR-1656 targets GPX4 to trigger pyroptosis in broilers kidney tissues by activating NLRP3 inflammasome under Se deficiency. J Nutr Biochem. 2022;105:109001.[DOI]
-
49. Xu P, Wang Y, Deng Z, Tan Z, Pei X. MicroRNA-15a promotes prostate cancer cell ferroptosis by inhibiting GPX4 expression. Oncol Lett. 2022;23(2):67.[DOI]
-
50. Zhou J, Xiao C, Zheng S, Wang Q, Zhu H, Zhang Y, et al. MicroRNA-214-3p aggravates ferroptosis by targeting GPX4 in cisplatin-induced acute kidney injury. Cell Stress Chaperones. 2022;27(4):325-336.[DOI]
-
51. Cao W, He Y, Lan J, Luo S, Sun B, Xiao C, et al. FOXP3 promote the progression of glioblastoma via inhibiting ferroptosis mediated by linc00857/miR-1290/GPX4 axis. Cell Death Dis. 2024;15(4):239.[DOI]
-
52. Weitzel F, Ursini F, Wendel A. Phospholipid hydroperoxide glutathione peroxidase in various mouse organs during selenium deficiency and repletion. Biochim Biophys Acta. 1990;1036(2):88-94.[DOI]
-
53. Yao Y, Chen Z, Zhang H, Chen C, Zeng M, Yunis J, et al. Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat Immunol. 2021;22(9):1127-1139.[DOI]
-
54. Wingler K, Böcher M, Flohé L, Kollmus H, Brigelius-Flohé R. mRNA stability and selenocysteine insertion sequence efficiency rank gastrointestinal glutathione peroxidase high in the hierarchy of selenoproteins. Eur J Biochem. 1999;259(1-2):149-157.[DOI]
-
55. Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. 2021;12(1):1589.[DOI]
-
56. Cui C, Yang F, Li Q. Post-translational modification of GPX4 is a promising target for treating ferroptosis-related diseases. Front Mol Biosci. 2022;9:901565.[DOI]
-
57. Kerins MJ, Milligan J, Wohlschlegel JA, Ooi A. Fumarate hydratase inactivation in hereditary leiomyomatosis and renal cell cancer is synthetic lethal with ferroptosis induction. Cancer Sci. 2018;109(9):2757-2766.[DOI]
-
58. Vučković AM, Bosello Travain V, Bordin L, Cozza G, Miotto G, Rossetto M, et al. Inactivation of the glutathione peroxidase GPx4 by the ferroptosis-inducing molecule RSL3 requires the adaptor protein 14-3-3ε. FEBS Lett. 2020;594(4):611-624.[DOI]
-
59. Chu LK, Cao X, Wan L, Diao Q, Zhu Y, Kan Y, et al. Autophagy of OTUD5 destabilizes GPX4 to confer ferroptosis-dependent kidney injury. Nat Commun. 2023;14(1):8393.[DOI]
-
60. Wang Z, Xia Y, Wang Y, Zhu R, Li H, Liu Y, et al. The E3 ligase TRIM26 suppresses ferroptosis through catalyzing K63-linked ubiquitination of GPX4 in glioma. Cell Death Dis. 2023;14(10):695.[DOI]
-
61. Liu J, Wei X, Xie Y, Yan Y, Xue S, Wang X, et al. MDM4 inhibits ferroptosis in p53 mutant colon cancer via regulating TRIM21/GPX4 expression. Cell Death Dis. 2024;15:825.[DOI]
-
62. Zheng Z, Shang X, Sun K, Hou Y, Zhang X, Xu J, et al. P21 resists ferroptosis in osteoarthritic chondrocytes by regulating GPX4 protein stability. Free Radic Biol Med. 2024;212:336-348.[DOI]
-
63. Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U. A. 2019;116(8):2996-3005.[DOI]
-
64. Miao Y, Chen Y, Xue F, Liu K, Zhu B, Gao J, et al. Contribution of ferroptosis and GPX4’s dual functions to osteoarthritis progression. EBioMedicine. 2022;76[DOI]
-
65. Sun W, Lv Z, Li W, Lu J, Xie Y, Wang P, et al. XJB-5-131 protects chondrocytes from ferroptosis to alleviate osteoarthritis progression via restoring Pebp1 expression. J Orthop Translat. 2024;44:114-124.[DOI]
-
66. Pan X, Kong X, Feng Z, Jin Z, Wang M, Lu H, et al. 4-Octyl itaconate protects chondrocytes against IL-1β-induced oxidative stress and ferroptosis by inhibiting GPX4 methylation in osteoarthritis. Int Immunopharmacol. 2024;137:112531.[DOI]
-
67. Chen B, Liu H, Xie Y, Yu J. TPX2 promotes ferroptosis in LPS-induced C28/I2 chondrocytes via NF-κB p65-mediated downregulation of GPX4 and SLC7A11. Mol Biol Rep. 2025;53(1):171.[DOI]
-
68. Chen Y, Fang ZM, Yi X, Wei X, Jiang DS. The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis. 2023;14(3):205.[DOI]
-
69. Wang N, Xie M, Lei G, Zeng C, Yang T, Yang Z, et al. A cross-sectional study of association between plasma selenium levels and the prevalence of osteoarthritis: Data from the xiangya osteoarthritis study. J Nutr Health Aging. 2022;26(2):197-202.[DOI]
-
70. Wang L, Yin J, Yang B, Qu C, Lei J, Han J, et al. Serious selenium deficiency in the serum of patients with kashin–beck disease and the effect of nano-selenium on their chondrocytes. Biol Trace Elem Res. 2020;194(1):96-104.[DOI]
-
71. Riegger J. TRPV1 as an anti-ferroptotic target in osteoarthritis. eBioMedicine, 2022;84:104279.[DOI]
-
72. Wang S, Li W, Zhang P, Wang Z, Ma X, Liu C, et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. J Adv Res. 2022;41:63-75.[DOI]
-
73. Zhou X, Yang Y, Qiu X, Deng H, Cao H, Liao T, et al. Antioxidant taurine inhibits chondrocyte ferroptosis through upregulation of OGT/Gpx4 signaling in osteoarthritis induced by anterior cruciate ligament transection. J Adv Res. 2025;77:551-567.[DOI]
-
74. Zhu S, Zhang Q, Sun X, Zeh HJ, III , Lotze MT, Kang R, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017;77(8):2064-2077.[DOI]
-
75. Zhao P, Yin S, Qiu Y, Sun C, Yu H. Ferroptosis and pyroptosis are connected through autophagy: a new perspective of overcoming drug resistance. Mol Cancer. 2025;24(1):23.[DOI]
-
76. Lv M, Cai Y, Hou W, Peng K, Xu K, Lu C, et al. The RNA-binding protein SND1 promotes the degradation of GPX4 by destabilizing the HSPA5 mRNA and suppressing HSPA5 expression, promoting ferroptosis in osteoarthritis chondrocytes. Inflamm Res. 2022;71(4):461-472.[DOI]
-
77. Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697-1707.[DOI]
-
78. Sun K, Guo J, Guo Z, Hou L, Liu H, Hou Y, et al. The roles of the Hippo-YAP signalling pathway in Cartilage and Osteoarthritis. Ageing Res Rev. 2023;90:102015.[DOI]
-
79. Leijten JC, Georgi N, Wu L, van Blitterswijk CA, Karperien M. Cell sources for articular cartilage repair strategies: shifting from monocultures to cocultures. Tissue Eng Part B Rev. 2013;19(1):31-40.[DOI]
-
80. Wei W, Dai H. Articular cartilage and osteochondral tissue engineering techniques: Recent advances and challenges. Bioact Mater. 2021;6(12):4830-4855.[DOI]
-
81. Tait SWG, Ichim G, Green DR. Die another way--non-apoptotic mechanisms of cell death. J Cell Sci. 2014;127(10):2135-2144.[DOI]
-
82. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106-1121.[DOI]
-
83. Cao S, Wei Y, Xu H, Weng J, Qi T, Yu F, et al. Crosstalk between ferroptosis and chondrocytes in osteoarthritis: A systematic review of in vivo and in vitro studies. Front Immunol. 2023;14:1202436.[DOI]
-
84. Xu W, Zhang B, Xi C, Qin Y, Lin X, Wang B, Kong P, Yan J. Ferroptosis plays a role in human chondrocyte of osteoarthritis induced by IL-1β in vitro. Cartilage. 2023;14(4).[DOI]
-
85. Sun K, Hou L, Guo Z, Wang G, Guo J, Xu J, et al. JNK-JUN-NCOA4 axis contributes to chondrocyte ferroptosis and aggravates osteoarthritis via ferritinophagy. Free Radic Biol Med. 2023;200:87-101.[DOI]
-
86. Xue X, Dai T, Chen J, Xu Y, Yang Z, Huang J, et al. PPARγ activation suppresses chondrocyte ferroptosis through mitophagy in osteoarthritis. J Orthop Surg Res. 2023;18(1):620.[DOI]
-
87. Zhang X, Hou L, Guo Z, Wang G, Xu J, Zheng Z, et al. Lipid peroxidation in osteoarthritis: focusing on 4-hydroxynonenal, malondialdehyde, and ferroptosis. Cell Death Discov. 2023;9(1):320.[DOI]
-
88. Liu Y, Zhang Z, Fang Y, Liu C, Zhang H. Ferroptosis in osteoarthritis: Current understanding. J Inflamm Res. 2024;17:8471-8486.[DOI]
-
89. Sheng W, Liao S, Wang D, Liu P, Zeng H. The role of ferroptosis in osteoarthritis: Progress and prospects. Biochem Biophys Res Commun. 2024;733:150683.[DOI]
-
90. Xia S, Li L, Shi Z, Sun N, He Y. Ferroptosis in osteoarthritis: Metabolic reprogramming, immunometabolic crosstalk, and targeted intervention strategies. Front Immunol. 2025;16:1604652.[DOI]
-
91. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185(14):2401-2421.[DOI]
-
92. Cao J, Chen X, Chen L, Lu Y, Wu Y, Deng A, et al. DHODH-mediated mitochondrial redox homeostasis: a novel ferroptosis regulator and promising therapeutic target. Redox Biol. 2025;85:103788.[DOI]
-
93. Lv Z, Han J, Li J, Guo H, Fei Y, Sun Z, et al. Single cell RNA-seq analysis identifies ferroptotic chondrocyte cluster and reveals TRPV1 as an anti-ferroptotic target in osteoarthritis. eBioMedicine. 2022;84:104258.[DOI]
-
94. Lee J, Roh JL. Targeting GPX4 in human cancer: Implications of ferroptosis induction for tackling cancer resilience. Cancer Lett. 2023;559:216119.[DOI]
-
95. Hwang HS, Kim HA. Chondrocyte apoptosis in the pathogenesis of osteoarthritis. Int J Mol Sci. 2015;16(11):26035-26054.[DOI]
-
96. Li R, Sun K. Regulation of chondrocyte apoptosis in osteoarthritis by endoplasmic reticulum stress. Cell Stress Chaperones. 2024;29(6):750-763.[DOI]
-
97. Yang Z, Jiang W, Xiong C, Shang J, Huang Y, Zhou X, et al. Calcipotriol suppresses GPX4-mediated ferroptosis in OA chondrocytes by blocking the TGF-β1 pathway. Cytokine. 2023;171:156382.[DOI]
-
98. Yang J, Hu S, Bian Y, Yao J, Wang D, Liu X, et al. Targeting cell death: Pyroptosis, ferroptosis, apoptosis and necroptosis in osteoarthritis. Front Cell Dev Biol. 2021;9:789948.[DOI]
-
99. Liu S, Pan Y, Li T, Zou M, Liu W, Li Q, et al. The role of regulated programmed cell death in osteoarthritis: From pathogenesis to therapy. Int J Mol Sci. 2023;24(6):5364.[DOI]
-
100. Imai H, Koumura T, Nakajima R, Nomura K, Nakagawa Y. Protection from inactivation of the adenine nucleotide translocator during hypoglycaemia-induced apoptosis by mitochondrial phospholipid hydroperoxide glutathione peroxidase. Biochem J. 2003;371(3):799-809.[DOI]
-
101. Liang H, Ran Q, Jang YC, Holstein D, Lechleiter J, McDonald-Marsh T, et al. Glutathione peroxidase 4 differentially regulates the release of apoptogenic proteins from mitochondria. Free Radic Biol Med. 2009;47(3):312-320.[DOI]
-
102. Ding Y, Chen X, Liu C, Ge W, Wang Q, Hao X, et al. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol. 2021;14(1):19.[DOI]
-
103. Ran Q, Liang H, Gu M, Qi W, Walter CA, Roberts LJ, II , et al. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J Biol Chem. 2004;279(53):55137-55146.[DOI]
-
104. Maneiro E, Martín MA, de Andres MC, López-Armada MJ, Fernández-Sueiro JL, del Hoyo P, et al. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum. 2003;48(3):700-708.[DOI]
-
105. Ruiz-Romero C, Calamia V, Mateos J, Carreira V, Martiénez-Gomariz M, Fernaéndez M, et al. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol Cell Proteomics. 2009;8(1):172-189.[DOI]
-
106. Sun K, Luo J, Jing X, Xiang W, Guo J, Yao X, et al. Hyperoside ameliorates the progression of osteoarthritis: An in vitro and in vivo study. Phytomedicine. 2021;80:153387.[DOI]
-
107. Cancedda R. Cartilage and bone extracellular matrix. Curr Pharm Des. 2009;15(12):1334-1348.[DOI]
-
108. Van der Kraan PM, Buma P, Van Kuppevelt T, Van Den Berg WB. Interaction of chondrocytes, extracellular matrix and growth factors: Relevance for articular cartilage tissue engineering. Osteoarthritis Cartilage. 2002;10(8):631-637.[DOI]
-
109. Peng Z, Sun H, Bunpetch V, Koh Y, Wen Y, Wu D, et al. The regulation of cartilage extracellular matrix homeostasis in joint cartilage degeneration and regeneration. Biomaterials. 2021;268:120555.[DOI]
-
110. Han J, Zhan L, Huang Y, Guo S, Zhou X, Kapilevich L, et al. Moderate mechanical stress suppresses chondrocyte ferroptosis in osteoarthritis by regulating NF-κB p65/GPX4 signaling pathway. Sci Rep. 2024;14(1):5078.[DOI]
-
111. He Q, Lin Y, Chen B, Chen C, Zeng J, Dou X, et al. Vitamin K2 ameliorates osteoarthritis by suppressing ferroptosis and extracellular matrix degradation through activation GPX4’s dual functions. Biomed Pharmacother. 2024;175:116697.[DOI]
-
112. Sato T, Konomi K, Yamasaki S, Aratani S, Tsuchimochi K, Yokouchi M, et al. Comparative analysis of gene expression profiles in intact and damaged regions of human osteoarthritic cartilage. Arthritis Rheum. 2006;54(3):808-817.[DOI]
-
113. Karsdal MA, Madsen SH, Christiansen C, Henriksen K, Fosang AJ, Sondergaard BC. Cartilage degradation is fully reversible in the presence of aggrecanase but not matrix metalloproteinase activity. Arthritis Res Ther. 2008;10(3):R63.[DOI]
-
114. Bay-Jensen AC, Hoegh-Madsen S, Dam E, Henriksen K, Sondergaard BC, Pastoureau P, et al. Which elements are involved in reversible and irreversible cartilage degradation in osteoarthritis? Rheumatol Int. 2010;30(4):435-442.[DOI]
-
115. Li H, Wang D, Yuan Y, Min J. New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res Ther. 2017;19(1):248.[DOI]
-
116. Castañeda S, Vicente EF. Osteoarthritis: More than cartilage degeneration. Clinic Rev Bone Miner Metab. 2017;15(2):69-81.[DOI]
-
117. Zhen G, Wen C, Jia X, Li Y, Crane JL, Mears SC, et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med. 2013;19(6):704-712.[DOI]
-
118. Wen C, Lu WW, Chiu KY. Importance of subchondral bone in the pathogenesis and management of osteoarthritis from bench to bed. J Orthop Transl. 2014;2(1):16-25.[DOI]
-
119. Ruan B, Dong J, Wei F, Huang Z, Yang B, Zhang L, et al. DNMT aberration-incurred GPX4 suppression prompts osteoblast ferroptosis and osteoporosis. Bone Res. 2024;12(1):68.[DOI]
-
120. Zhao C, Kong K, Liu P, Chen X, Rong K, Zhang P, et al. Regulating obesity-induced osteoarthritis by targeting p53-FOXO3, osteoclast ferroptosis, and mesenchymal stem cell adipogenesis. Nat Commun. 2025;16(1):4532.[DOI]
-
121. Zhai G, Doré J, Rahman P. TGF-β signal transduction pathways and osteoarthritis. Rheumatol Int. 2015;35(8):1283-1292.[DOI]
-
122. Lee B, Oh Y, Jo S, Kim TH, Ji JD. A dual role of TGF-β in human osteoclast differentiation mediated by Smad1 versus Smad3 signaling. Immunol Lett. 2019;206:33-40.[DOI]
-
123. Dai G, Xiao H, Liao J, Zhou N, Zhao C, Xu W, et al. Osteocyte TGFβ1‑Smad2/3 is positively associated with bone turnover parameters in subchondral bone of advanced osteoarthritis. Int J Mol Med. 2020;46(1):167-178.[DOI]
-
124. Shen J, Li S, Chen D. TGF-β signaling and the development of osteoarthritis. Bone Res. 2014;2(1):14002.[DOI]
-
125. Donell S. Subchondral bone remodelling in osteoarthritis. EFORT Open Rev, 2019;4(6):221-229.[DOI]
-
126. Zhu X, Chan YT, Yung PSH, Tuan RS, Jiang Y. Subchondral bone remodeling: A therapeutic target for osteoarthritis. Front Cell Dev Biol. 2020;8:607764.[DOI]
-
127. Li Z, Zhu Z, Liu Y, Liu Y, Zhao H. Function and regulation of GPX4 in the development and progression of fibrotic disease. J Cell Physiol. 2022;237(7):2808-2824.[DOI]
-
128. Blaney Davidson EN, van der Kraan PM, van den Berg WB. TGF-beta and osteoarthritis. Osteoarthritis Cartilage. 2007;15(6):597-604.[DOI]
-
129. Sokolove J, Lepus CM. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther Adv Musculoskelet Dis. 2013;5(2):77-94.[DOI]
-
130. Terkawi MA, Ebata T, Yokota S, Takahashi D, Endo T, Matsumae G, et al. Low-grade inflammation in the pathogenesis of osteoarthritis: Cellular and molecular mechanisms and strategies for future therapeutic intervention. Biomedicines. 2022;10(5):1109.[DOI]
-
131. Huang HS, Chen CJ, Suzuki H, Yamamoto S, Chang WC. Inhibitory effect of phospholipid hydroperoxide glutathione peroxidase on the activity of lipoxygenases and cyclooxygenases. Prostaglandins Other Lipid Mediat. 1999;58(2):65-75.[DOI]
-
132. Heirman I, Ginneberge D, Brigelius-Flohé R, Hendrickx N, Agostinis P, Brouckaert P, et al. Blocking tumor cell eicosanoid synthesis by GP x 4 impedes tumor growth and malignancy. Free Radic Biol Med. 2006;40(2):285-294.[DOI]
-
133. Brigelius-Flohé R, Maurer S, Lötzer K, Böl GF, Kallionpää H, Lehtolainen P, et al. Overexpression of PHGPx inhibits hydroperoxide-induced oxidation, NFkappaB activation and apoptosis and affects oxLDL-mediated proliferation of rabbit aortic smooth muscle cells. Atherosclerosis. 2000;152(2):307-316.[DOI]
-
134. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2(1):17023.[DOI]
-
135. Brigelius-Flohé R, Flohé L. Is there a role of glutathione peroxidases in signaling and differentiation? Biofactors. 2003;17(1-4):93-102.[DOI]
-
136. Li X, He J, Gao X, Zheng G, Chen C, Chen Y, et al. GPX4 restricts ferroptosis of NKp46+ILC3s to control intestinal inflammation. Cell Death Dis. 2024;15(9):687.[DOI]
-
137. Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. 2010;6(11):625-635.[DOI]
-
138. Rahmati M, Mobasheri A, Mozafari M. Inflammatory mediators in osteoarthritis: A critical review of the state-of-the-art, current prospects, and future challenges. Bone. 2016;85:81-90.[DOI]
-
139. Deligne C, Casulli S, Pigenet A, Bougault C, Campillo-Gimenez L, Nourissat G, et al. Differential expression of interleukin-17 and interleukin-22 in inflamed and non-inflamed synovium from osteoarthritis patients. Osteoarthritis Cartilage. 2015;23(11):1843-1852.[DOI]
-
140. Hu Z, Chen L, Zhao J, Zhang W, Jin Z, Sun Y, et al. Lipoxin A(4) ameliorates knee osteoarthritis progression in rats by antagonizing ferroptosis through activation of the ESR2/LPAR3/Nrf2 axis in synovial fibroblast-like synoviocytes. Redox Biol. 2024;73:103143.[DOI]
-
141. Lambert C, Zappia J, Sanchez C, Florin A, Dubuc J-E, Henrotin Y. The damage-associated molecular patterns (DAMPs) as potential targets to treat osteoarthritis: Perspectives from a review of the literature. Front Med. 2020;7:607186.[DOI]
-
142. Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J, et al. The emerging role of ferroptosis in inflammation. Biomed Pharmacother. 2020;127:110108.[DOI]
-
143. Li X, Li C, Zhang W, Wang Y, Qian P, Huang H. Inflammation and aging: signaling pathways and intervention therapies. Signal Transduct Target Ther. 2023;8(1):239.[DOI]
-
144. Masaldan S, Clatworthy SAS, Gamell C, Meggyesy PM, Rigopoulos AT, Haupt S, et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018;14:100-115.[DOI]
-
145. Wei X, Liu M, Zheng Z, Yu S, Huang L, Ma J, et al. Defective NCOA4-dependent ferroptosis in senescent fibroblasts retards diabetic wound healing. Cell Death Discov. 2023;9(1):138.[DOI]
-
146. Loo TM, Zhou X, Tanaka Y, Sugawara S, Yamauchi S, Kawasaki H, et al. Senescence-associated lysosomal dysfunction impairs cystine deprivation-induced lipid peroxidation and ferroptosis. Nat Commun. 2025;16(1):6617.[DOI]
-
147. Shen G, Liu J, Wang Y, Deng Z, Deng F Ferroptosis in cancer and inflammatory diseases: Mechanisms and therapeutic implications. MedComm. 2025;6(9):e70349.[DOI]
-
148. Shaikh SR, Beck MA, Alwarawrah Y, MacIver NJ. Emerging mechanisms of obesity-associated immune dysfunction. Nat Rev Endocrinol. 2024;20(3):136-148.[DOI]
-
149. Das UN. Ageing: Is there a role for arachidonic acid and other bioactive lipids? A review. J Adv Res. 2018;11:67-79.[DOI]
-
150. Zheng Y, Sun J, Luo Z, Li Y, Huang Y. Emerging mechanisms of lipid peroxidation in regulated cell death and its physiological implications. Cell Death Dis. 2024;15(11):859.[DOI]
-
151. Forcina GC, Dixon SJ. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics. 2019;19(18):1800311.[DOI]
-
152. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21(7):363-383.[DOI]
-
153. Wen Z, Xia G, Liang C, Wang X, Huang J, Zhang L, Shan D, Wu S, Cao X. Selective clearance of senescent chondrocytes in osteoarthritis by targeting excitatory amino acid transporter protein 1 to induce ferroptosis. Antioxid Redox Signal. 2023;39(4-6):262-277.[DOI]
-
154. Ran Q, Liang H, Ikeno Y, Qi W, Prolla TA, Roberts LJ, II , et al. Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis. J Gerontol A Biol Sci Med Sci. 2007;62(9):932-942.[DOI]
-
155. Tang DG, La E, Kern J, Kehrer JP. Fatty acid oxidation and signaling in apoptosis. Biol Chem. 2002;383(3-4):425-442.[DOI]
-
156. Qian JY, Lou CY, Chen YL, Ma LF, Hou W, Zhan ZJ. A prospective therapeutic strategy: GPX4-targeted ferroptosis mediators. Eur J Med Chem. 2025;281:117015.[DOI]
-
157. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551(7679):247-250.[DOI]
-
158. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10(1):1617.[DOI]
-
159. Herrera-Abreu MT, Guan J, Khalid U, Ning J, Costa MR, Chan J, et al. Inhibition of GPX4 enhances CDK4/6 inhibitor and endocrine therapy activity in breast cancer. Nat Commun. 2024;15(1):9550.[DOI]
-
160. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22(7):381-396.[DOI]
-
161. Li C, Deng X, Xie X, Liu Y, Friedmann Angeli JP, Lai L. Activation of glutathione peroxidase 4 as a novel anti-inflammatory strategy. Front Pharmacol, 2018;9:1120.[DOI]
-
162. Li C, Deng X, Zhang W, Xie X, Conrad M, Liu Y, et al. Novel allosteric activators for ferroptosis regulator glutathione peroxidase 4. J Med Chem. 2019;62(1):266-275.[DOI]
-
163. Liu X, Guo Y, Huang Y, Wang Q, Huang Y, Lei Y, et al. GPX4 allosteric activators inhibit ferroptosis and exert myocardial protection in doxorubicin-induced myocardial injury mouse model. Eur J Med Chem. 2024;277:116721.[DOI]
-
164. Huang C, Guo Y, Li T, Sun G, Yang J, Wang Y, et al. Pharmacological activation of GPX4 ameliorates doxorubicin-induced cardiomyopathy. Redox Biol. 2024;70:103024.[DOI]
-
165. Wang X, Liu Z, Peng P, Gong Z, Huang J, Peng H. Astaxanthin attenuates osteoarthritis progression via inhibiting ferroptosis and regulating mitochondrial function in chondrocytes. Chem Biol Interact. 2022;366:110148.[DOI]
-
166. Liu J, Zhou H, Chen J, Zuo Q, Liu F. Baicalin inhibits IL-1β-induced ferroptosis in human osteoarthritis chondrocytes by activating Nrf-2 signaling pathway. J Orthop Surg Res. 2024;19(1):23.[DOI]
-
167. He Q, Yang J, Pan Z, Zhang G, Chen B, Li S, et al. Biochanin A protects against iron overload associated knee osteoarthritis via regulating iron levels and NRF2/System xc-/GPX4 axis. Biomed Pharmacother. 2023;157:113915.[DOI]
-
168. Gong Z, Wang Y, Li L, Li X, Qiu B, Hu Y. Cardamonin alleviates chondrocytes inflammation and cartilage degradation of osteoarthritis by inhibiting ferroptosis via p53 pathway. Food Chem Toxicol. 2023;174:113644.[DOI]
-
169. Xiao J, Luo C, Li A, Cai F, Wang Y, Pan X, et al. Icariin inhibits chondrocyte ferroptosis and alleviates osteoarthritis by enhancing the SLC7A11/GPX4 signaling. Int Immunopharmacol. 2024;133:112010.[DOI]
-
170. Sun J, Zhang Y, Wang C, Ruan Q. Kukoamine A protects mice against osteoarthritis by inhibiting chondrocyte inflammation and ferroptosis via SIRT1/GPX4 signaling pathway. Life Sci. 2023;332:122117.[DOI]
-
171. Wang Q, Qi B, Shi S, Jiang W, Li D, Jiang X, et al. Melatonin alleviates osteoarthritis by regulating nadph oxidase 4–induced ferroptosis and mitigating mitochondrial dysfunction. J Pineal Res. 2024;76(6):e12992.[DOI]
-
172. Hou L, Wang G, Zhang X, Lu F, Xu J, Guo Z, et al. Mitoquinone alleviates osteoarthritis progress by activating the NRF2-Parkin axis. iScience. 2023;26(9)[DOI]
-
173. Ruan Q, Wang C, Zhang Y, Sun J. Ruscogenin attenuates cartilage destruction in osteoarthritis through suppressing chondrocyte ferroptosis via Nrf2/SLC7A11/GPX4 signaling pathway. Chem Biol Interact. 2024;388:110835.[DOI]
-
174. Xu C, Ni S, Xu N, Yin G, Yu Y, Zhou B, et al. Theaflavin-3,3'-digallate inhibits erastin-induced chondrocytes ferroptosis via the Nrf2/GPX4 signaling pathway in osteoarthritis. Oxid Med Cell Longev. 2022;2022(1):3531995.[DOI]
-
175. Wang J, Yang J, Fang Y, Lou C, Yu H, Li Y, et al. Vinpocetine protects against osteoarthritis by inhibiting ferroptosis and extracellular matrix degradation via activation of the Nrf2/GPX4 pathway. Phytomedicine. 2024;135:156115.[DOI]
-
176. Hart DA. Osteoarthritis as an umbrella term for different subsets of humans undergoing joint degeneration: The need to address the differences to develop effective conservative treatments and prevention strategies. Int J Mol Sci. 2022;23(23):15365.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
He J, Zhang X, Hou L, Liu H, Guo F, Sun K. Beyond ferroptosis: Role of GPX4 in osteoarthritis and its therapeutic implications. Ferroptosis Oxid Stress. 2026;2:202514. https://doi.org/10.70401/fos.2026.0013
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. A Brief Overview of GPX4
- 3. The Expression and Regulation of GPX4 in OA
- 4. The Function of GPX4 in OA
- 5. Therapeutic Implications and Prospects
- 6. Conclusion
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- Copyright
Science Exploration Style
He J, Zhang X, Hou L, Liu H, Guo F, Sun K. Beyond ferroptosis: Role of GPX4 in osteoarthritis and its therapeutic implications. Ferroptosis Oxid Stress. 2026;2:202514. https://doi.org/10.70401/fos.2026.0013
copy
Share Link
copy