Ferroptosis and oxidative stress in glaucoma
*Correspondence to:
Hanhan Liu, Department of Ophthalmology, Faculty of Medicine and University Hospital of Cologne, Cologne 50937, Germany.
E-mail: hanhan.liu@uk-koeln.de
Ferroptosis Oxid Stress. 2026;2:202521. 10.70401/fos.2026.0020
Received: December 05, 2025Accepted: February 14, 2026Published: March 02, 2026
This manuscript is made available in its unedited form to allow early access to the
reported findings. Further editing will be completed before final publication. As such,
the content may include errors, and standard legal disclaimers are applicable.
Abstract
Glaucoma is an ocular disease and a leading cause of irreversible blindness, driven by progressive retinal ganglion cell (RGC) loss. While elevated intraocular pressure (IOP) is a major risk factor, RGC degeneration often persists despite effective IOP-lowering therapy. This persistence suggests the involvement of pressure-independent pathogenic mechanisms. Growing evidence implicates ferroptosis – an
Keywords
Glaucoma, ferroptosis, retinal ganglion cells, mitochondria, oxidative stress
1. Introduction
Glaucoma is a leading cause of irreversible blindness worldwide, characterized by progressive degeneration of retinal ganglion cell (RGC) and their axons. While elevated intraocular pressure (IOP) remains the primary risk factor, IOP reduction alone does not completely halt RGC loss in many patients, indicating that additional pathogenic mechanisms contribute to neurodegeneration in this disease. Clinically, glaucoma encompasses a heterogeneous group of optic neuropathies, including primary open-angle glaucoma (POAG), primary angle-closure glaucoma (PACG), and normal-tension glaucoma (NTG). The existence of NTG unequivocally underscores the necessity to investigate pressure-independent mechanisms, with compelling evidence now positioning ferroptosis as a pivotal driver of RGC loss in glaucoma[1]. Patients typically present with progressive peripheral visual field loss, which may remain asymptomatic in early stages. As the disease advances, it leads to irreversible tunnel vision and eventual blindness if left untreated. POAG, the most common form, progresses gradually and asymptomatically, while PACG can present acutely with severe ocular pain, headache, nausea, and rapid vision loss. The existence of normal-tension glaucoma highlights that RGC degeneration can occur in the absence of markedly elevated IOP, underscoring the importance of pressure-independent neurodegenerative mechanisms.
Recent evidence has identified ferroptosis, an iron-dependent form of regulated cell death distinct from apoptosis, as a critical mechanism underlying glaucomatous neurodegeneration. This review summarizes findings from peer-reviewed literature published through 2025, examining ferroptosis mechanisms in glaucoma, oxidative stress pathways, and therapeutic strategies targeting iron metabolism, antioxidant defenses, and programmed cell death pathways in retinal neurodegeneration. Current evidence for ferroptosis in glaucoma is predominantly preclinical, derived from experimental models including optic nerve crush (ONC), microbead-induced ocular hypertension, and glutamate excitotoxicity models[2-4]. To date, no completed clinical trials have explicitly targeted ferroptosis as a primary therapeutic strategy in glaucoma, though preclinical evidence strongly supports its therapeutic potential.
2. Overview of Ferroptosis
Ferroptosis, first defined by Dixon et al. in 2012[5], represents a unique, iron-dependent form of regulated cell death, mechanistically distinct from apoptosis, necrosis, and pyroptosis. Ferroptotic cell death is precipitated by the uncontrolled accumulation of lipid peroxides within membrane phospholipids, driven by iron-catalyzed oxidative reactions[6,7]. At the biochemical level, ferroptosis is defined by the collapse of glutathione (GSH)-dependent antioxidant defenses and inactivation of glutathione peroxidase 4 (GPX4), leading to unchecked lipid peroxide buildup. Three mechanistic axes underpin ferroptotic cell death: (1) reprogramming of amino acid and lipid metabolism—specifically via the cystine/glutamate antiporter system Xc⁻[5,8]; (2) amplification of reactive oxygen species (ROS) through Fenton chemistry and lipoxygenase activity[9]; and (3) dysregulation of iron homeostasis through transferrin receptor-mediated uptake and ferritinophagy[10]. The triggers and axes form a cohesive model of ferroptosis-driven RGC death are summarized in Figure 1.
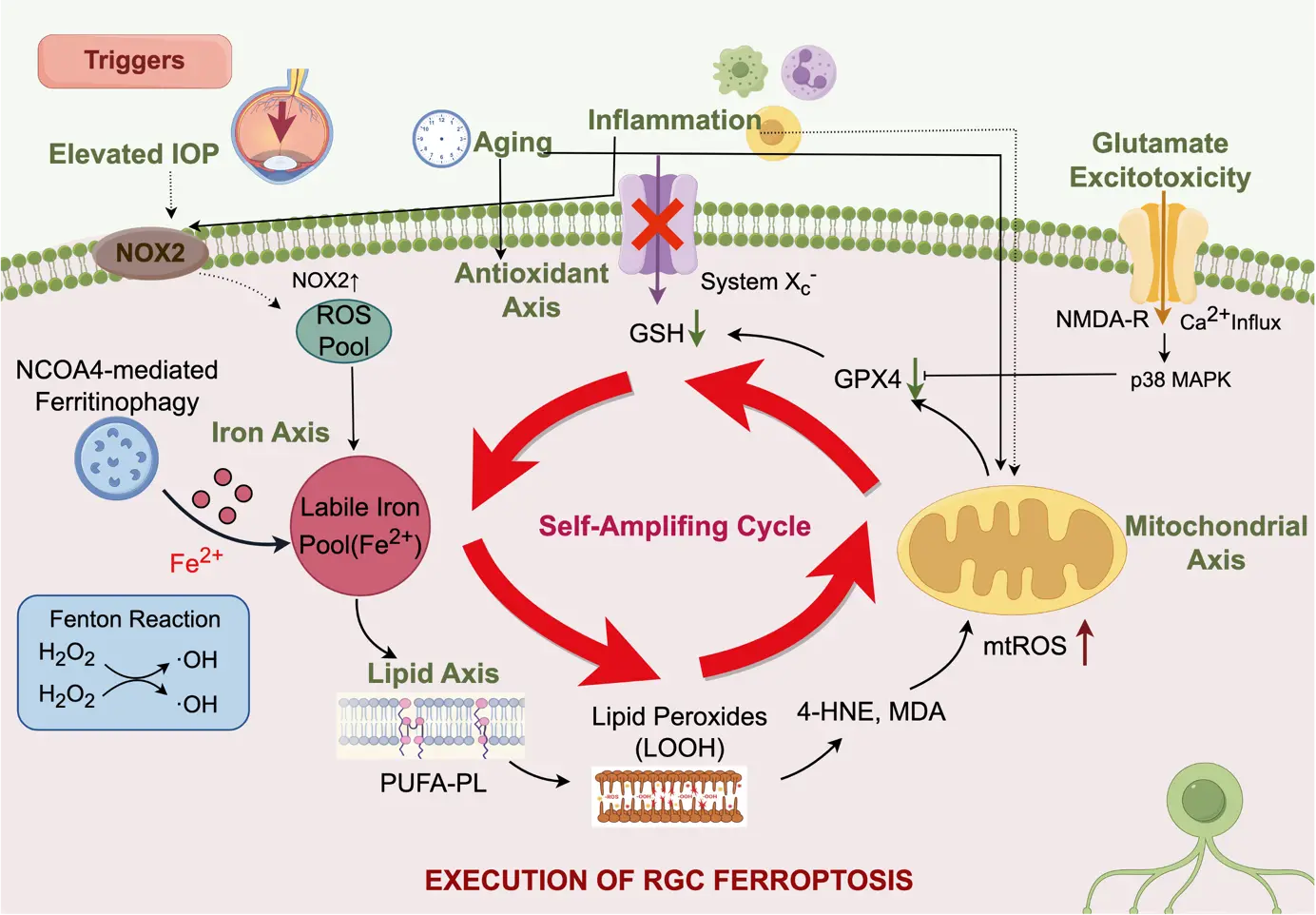
Figure 1. Mechanisms of ferroptosis in glaucomatous RGC injury. Created with FigDraw. RGC: retinal ganglion cell; ROS: reactive oxygen species; GSH: glutathione;
3. Mechanistic Basis and Propagation of Ferroptosis
Ferroptosis is executed through iron-dependent peroxidation of polyunsaturated fatty acids (PUFAs) within membrane phospholipids, propagating a self-sustaining radical chain reaction[6,11]. In Fenton chemistry, ferrous iron (Fe2+) reacts with hydrogen peroxide (H2O2), generating hydroxyl radicals (OH-) that abstract bis-allylic hydrogens from PUFAs, initiating lipid radical formation[12]. Subsequent reactions produce lipid hydroperoxides (LOOH), which, through further iron-mediated reduction, yield lipid alkoxyl (LO-) and alkyl (L-) radicals that perpetuate membrane damage. Enzymatic lipid peroxidation, catalyzed by arachidonate lipoxygenases (ALOXs) and cytochrome P450 oxidoreductase, further accelerates ferroptosis[13,14].
Central to ferroptosis suppression is GPX4, which reduces lipid hydroperoxides to non-toxic alcohols using GSH as a cofactor[15]. Loss of GPX4 activity disrupts this protective barrier, allowing uncontrolled lipid peroxide accumulation and irreversible cell death[3,16,17].
4. Assessing Ferroptosis in Glaucoma and RGC Injury
Determining the extent of ferroptosis in vivo remains challenging due to the absence of specific biomarkers[18-20]. Typical surrogate indicators include elevated levels of malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), along with reduced GSH levels[21,22]. However, these markers are not exclusive to ferroptosis and overlap with other oxidative processes. Definitive evidence relies on demonstrating reversal of cell death phenotypes by ferroptosis inhibitors, such as iron chelators (e.g., deferiprone, deferoxamine) or radical-trapping antioxidants (e.g., ferrostatin-1, liproxstatin-1)[5,23]. Nonetheless, clinical studies using iron chelation in AD and PD have yielded mixed results[24,25], with some trials showing disease progression rather than improvement. These unexpected outcomes reveal the complexity of brain iron regulation. While radical-trapping agents show promise in preclinical models, their clinical efficacy remains to be determined. The mixed findings likely reflect disease-specific differences in iron dependency. Iron plays essential roles in mitochondrial respiration and physiological redox signaling, and non-selective chelation may therefore impair cellular bioenergetics in tissues without overt iron overload[26]. Unlike the brain, the retina is protected by a distinct blood–retinal barrier and allows for localized ocular drug delivery, enabling higher local drug concentrations with minimal systemic exposure. Consequently, targeting ferroptosis through iron regulation may represent a more feasible and controllable therapeutic strategy in glaucoma than in diffuse brain neurodegeneration. Accordingly, these observations highlight the need for disease- and
While ferroptosis can be experimentally induced through genetic or chemical inhibition of GPX4, the physiological triggers driving its activation in human pathology remain poorly defined[27]. Unlike apoptosis or necroptosis, ferroptosis does not rely on a single initiating signal; rather, it arises when antioxidant systems fail to restrain ongoing lipid peroxidation[6]. In glaucoma, multiple stress factors act together to increase the susceptibility of RGCs to ferroptosis. These include chronic inflammation of the optic nerve and retina, dysregulated iron metabolism due to IOP fluctuations or other causes, mitochondrial dysfunction in RGCs and their axons, abnormal lipid metabolism potentially linked to aging or disease, and the possible accumulation of pathological proteins[28]. Understanding how these converging stressors progressively impair the ferroptosis defense mechanisms in RGCs is crucial for clarifying the specific role of this cell death pathway in glaucoma progression and for developing targeted therapeutic strategies.
Importantly, chronic inflammation, iron dyshomeostasis, mitochondrial dysfunction, and excessive oxidative stress, which are all known promoters of ferroptosis, are also well-established core pathogenic features of glaucomatous optic neuropathy. This strongly suggests that ferroptosis may serve as a critical effector pathway linking these disparate pathological processes to the ultimate loss of RGCs. Given that RGCs share with central neurons a high vulnerability to iron-driven lipid peroxidation and antioxidant failure, targeting ferroptosis emerges as a promising new direction for neuroprotection in glaucoma.
5. Ferroptosis as a New Type of Programmed Cell Death in Glaucoma
Against this backdrop, ferroptosis emerges as a compelling candidate mechanism for retinal ganglion cell loss in glaucoma. Ferroptosis is defined by the iron-dependent buildup of lipid peroxides, representing a regulated cell death pathway that is morphologically, biochemically, and genetically distinct from apoptosis and other forms of cell death[29]. This type of cell death features unique characteristics, including intracellular iron accumulation, reduction of cellular antioxidants, extensive lipid peroxidation, and specific ultrastructural changes in mitochondria, such as shrunken mitochondria with increased membrane density and decreased cristae[3].
Growing evidence suggests that ferroptosis is a significant contributor to RGC death in experimental glaucoma models. Studies using ONC and microbead-induced glaucoma models showed that the ferroptosis inhibitor Ferrostatin-1 (Fer-1) significantly promoted RGC survival and preserved retinal function more effectively than apoptosis inhibitors, indicating ferroptosis rather than apoptosis as the predominant RGC death mechanism in these models[3].
6. Ferroptosis Activation in Glaucomatous RGC Injury
6.1 Distinguishing ferroptosis from other forms of cell death
Ferroptosis is mechanistically and morphologically distinct from other cell death modalities such as apoptosis, necrosis, and autophagy. Unlike apoptosis, ferroptotic cells do not involve nuclear condensation, DNA fragmentation, or caspase activation[30]; instead, they display intact nuclei with shrunken mitochondria, increased membrane density, and reduced cristae. Unlike necrosis, ferroptosis is a regulated process that can be prevented by specific inhibitors (ferrostatin-1, liproxstatin-1, iron chelators)[31]. Although ferroptosis is classified as a form of autophagy-dependent cell death, the terminal cell death event is driven by
6.2 Elevated IOP and iron dysregulation
A paradigm shift in understanding glaucoma pathogenesis emerged from the discovery that pathologically high intraocular pressure directly disturbs iron metabolism in the retina. Clinical analysis of patients with acute primary angle-closure glaucoma revealed elevated free iron levels in peripheral serum compared to control populations[33]. Experimental models confirmed that high IOP rapidly induces abnormal accumulation of ferrous iron (Fe2+) in retinal cells within 1-8 hours post-injury, progressing to elevated ferric iron (Fe3+) in serum within 8 hours[1].
6.3 NCOA4-mediated ferritinophagy and iron release
The upstream mechanism driving iron accumulation involves NCOA4-mediated ferritin autophagy (ferritinophagy). Ferritin, the primary iron storage protein, can sequester up to 4,500 iron atoms within a single molecule. Under IOP stress, NCOA4 acts as a selective autophagy cargo receptor, recognizing and delivering iron-bound ferritin to autophagosomes for lysosomal degradation. This process releases massive quantities of previously sequestered iron into the cytoplasm, creating a pathological iron overload state. Research utilizing NCOA4 knockdown approaches demonstrated that suppressing ferritinophagy prevents iron accumulation in the retina following IOP elevation, establishing NCOA4-FTH1 axis disruption as a critical upstream mechanism of glaucomatous iron dysregulation[1].
6.4 The glutathione-GPX4 antioxidant axis
The system xc⁻/GSH/GPX4 axis represents the central regulatory mechanism controlling ferroptosis susceptibility in RGCs[34]. This pathway operates as follows: cystine imported through the xCT transporter (system xc⁻ cystine/glutamate antiporter) is reduced to cysteine, which is then utilized for GSH synthesis. GSH, a tripeptide containing a redox-active cysteine residue, serves as the essential cofactor enabling GPX4 to reduce lipid hydroperoxides to non-toxic lipid alcohols, thereby preventing lipid peroxidation-induced cell death[35].
In experimental glaucoma models, downregulation of both xCT and GPX4 expression occurs in RGCs following ONC or IOP elevation, accompanied by increased lipid peroxide accumulation and iron elevation. Laser-capture microdissection combined with PCR specifically confirmed GPX4 downregulation within isolated RGCs, establishing these cells as the primary site of ferroptosis induction in the retina. Importantly, GPX4 expression is reduced predominantly in the mitochondrial compartment, where mitochondrial ROS accumulation and lipid peroxidation drive the ferroptotic death of RGCs[3].
6.5 Oxidative stress and lipid peroxidation
Oxidative stress represents both a cause and consequence of ferroptosis in glaucoma[36]. Elevated intraocular pressure induces prominent generation of ROS, with the retina demonstrating increased lipid peroxidation markers detected through both elevated MDA levels and protein carbonyl formation. This oxidative protein modification impairs cellular homeostasis and glial cell function, potentially contributing to secondary neuronal damage through paracrine mechanisms[3].
The intersection between oxidative stress and ferroptosis creates a pathological feedback loop: iron accumulation catalyzes Fenton reactions that generate ROS, which intensifies lipid peroxidation and further depletes antioxidant defenses. This reciprocal amplification of oxidative stress and iron-mediated lipid peroxidation creates a self-perpetuating cycle driving RGC death[3,33].
6.6 Mitochondrial dysfunction at the interface of ferroptosis and RGC death
Mitochondrial impairment represents a central event in ferroptosis-associated RGC death. Transmission electron microscopy reveals that ONC or IOP elevation induces characteristic ferroptotic alterations in RGC mitochondria, including shrinkage, cristae disruption, and increased membrane density, accompanied by excessive mitochondrial ROS and lipid peroxidation[3].
The mitochondria-targeted antioxidant MitoTEMPO markedly attenuates RGC loss, underscoring the pivotal role of mitochondrial oxidative stress in ferroptotic injury. Consistently, pharmacological inhibition of ferroptosis with Fer-1 preserves ATP production and the expression of mitochondrial genes (mt-Cytb, MT-ATP6), highlighting the importance of maintaining mitochondrial integrity and bioenergetic capacity for RGC survival[3].
6.7 Glutamate as a trigger
Elevated vitreous glutamate concentrations have been documented in both POAG and PACG patients, implicating glutamatergic excitotoxicity in glaucomatous RGC death. Importantly, glutamate functions as a classical ferroptosis inducer: excessive glutamate activates N-methyl-D-aspartate (NMDA) receptors, triggering calcium overload that initiates multiple downstream cell injury pathways, including ferroptosis[35].
In glutamate-induced glaucoma models, ferroptosis is activated through characteristic pathological changes: iron accumulation, lipid peroxide generation, decreased intracellular glutathione, diminished ferritin light chain expression, reduced SLC7A11 (xCT) expression, and downregulated GPX4 levels. These findings establish ferroptosis as a major cell death mechanism in glutamatergic models of glaucoma, complementing the role of ferroptosis in pressure-induced RGC injury[35].
6.8 p38 mitogen-activated protein kinase (MAPK) signaling
The p38 MAPK pathway has emerged as a critical regulator of glutamate-induced ferroptosis in RGCs. The p38 MAPK inhibitor SB202190 protects RGCs from glutamate excitotoxicity by suppressing ferroptosis through regulation of ferritin light chain (FTL), spermidine/spermine N1-acetyltransferase 1 (SAT1), and the SLC7A11/GPX4 pathway. These findings position p38 MAPK inhibition as a potential therapeutic strategy for glutamate-related RGC injury in glaucoma[35].
7. Therapeutic Strategies Targeting Ferroptosis
7.1 Ferroptosis inhibitors
Multiple pharmacological approaches targeting ferroptosis have demonstrated efficacy in experimental glaucoma models, first and foremost being the lipid peroxidation inhibitors and iron chelators. Figure 1 illustrates one such approach (iron chelation) in the context of IOP-induced ferroptosis. Here, we summarize key strategies, which include direct ferroptosis inhibitors, iron modulators, and antioxidant pathway activators.
Lipid peroxidation inhibitors, such as Fer-1, the prototype ferroptosis inhibitor, significantly promote RGC survival and preserve retinal function in both ONC and microbead-induced glaucoma models, demonstrating superior efficacy compared to apoptosis inhibitors. Liproxstatin-1, another potent ferroptosis inhibitor operating at nanomolar concentrations, exhibits superior potency compared to other ferroptosis suppressors and effectively restores expression of GSH, GPX4, and FSP1 while suppressing mitochondrial lipid peroxidation[3,37].
Iron chelation: Iron chelators represent a mechanistic approach to ferroptosis suppression by reducing intracellular iron availability. Deferoxamine (DFO), a classical iron chelator, reduces intracellular iron accumulation and mitigates ferroptosis. However, clinical translation faces challenges, as excessive iron chelation can impair mitochondrial function. Deferiprone, an oral iron chelator with improved blood-retinal barrier penetration, effectively chelates abnormally elevated ferrous iron in the retina following IOP elevation, inhibiting RGC ferroptosis and protecting visual function without compromising mitochondrial bioenergetics[1,38].
7.2 NRF2 activation
NRF2 pathway activation represents a transcriptional approach to ferroptosis suppression. Dimethyl fumarate (DMF), a clinically available NRF2 activator, ameliorates neurological dysfunction and reduces ferroptosis through coordinated activation of multiple antioxidant response elements (AREs) upstream of ferroptosis-suppressive genes, including SLC7A11, GPX4, HO-1, and FSP1[39].
7.3 MAPK inhibition
The p38 MAPK pathway inhibitor SB202190 protects RGCs from glutamate excitotoxicity-induced ferroptosis in experimental models, positioning p38 MAPK as a druggable target for ferroptosis-related glaucomatous RGC death[35].
7.4 Hydrogen sulfide-based therapies
Emerging evidence demonstrates that hydrogen sulfide (H2S) provides neuroprotection against pressure-induced ferroptosis through multiple mechanisms: iron chelation, iron metabolism regulation, oxidative stress reduction, and ferroptosis pathway modulation. Importantly, H2S exerts these effects partially through NOX2 inhibition, suggesting that slow-releasing H2S donors represent a promising multi-target therapeutic approach for glaucoma, with NOX2 emerging as a key ferroptosis regulator[40].
7.5 Natural Compounds and Antioxidants
Several natural compounds exhibit ferroptosis-inhibitory activity through complementary antioxidant and regulatory mechanisms. Epigallocatechin-3-gallate (EGCG), the major catechin in green tea, mitigates ferroptosis via iron chelation, GSH preservation, inhibition of lipid peroxidation, and activation of the NRF2 pathway[41]. Vitamin E (α-tocopherol) directly scavenges lipid radicals and, through FSP1-mediated recycling, prevents ferroptotic damage in neurodegenerative models[41,42]. Flavonoids such as baicalin and quercetin suppress ferroptosis by reducing oxidative stress, chelating iron, and modulating the GSH/GPX4 and NRF2 pathways[43]. Similarly, proanthocyanidins from grape seeds inhibit iron accumulation, maintain GSH/GPX4 levels, and prevent lipid peroxidation, thereby supporting neuronal survival[42] (Table 1).
Table 1. Key therapeutic strategies targeting ferroptosis in glaucoma.
| Therapeutic Target | Representative molecules | Mechanism | Evidence level | Key Considerations |
| Iron overload | DFO, DFP | Chelation of labile Fe2+, inhibition of Fenton reaction | Preclinical (Animal) | Systemic iron depletion risk, ocular delivery challenges, mixed results in neurodegenerative diseases |
| Lipid peroxidation | Fer-1, Lip-1 | Scavenging lipid radicals, inhibition of lipid peroxidation | Preclinical (In vitro and Animal) | Poor chemical stability,limited ocular pharmacokinetic data,not clinically available |
| SystemXc⁻–GSH–GPX4 axis | N-acetylcysteine, GPX4 activators | Restoration of antioxidant capacity, detoxification of lipid peroxides | Preclinical (Animal) | GPX4 modulation in retina requires further validation |
| NRF2 signaling | DMF | Transcriptional induction of antioxidant and iron-handling genes | Animal models, FDA-approved for multiple sclerosis | No glaucoma-specific clinical trials |
| Mitochondrial dysfunction | MitoTEMPO | Reduction of mtROS, preservation of ATP production | Preclinical (In vitro and Animal) | Mitochondrial targeting efficiency varies,long-term safety unknown |
| p38 MAPK pathway | SB202190 | Modulation of ferroptosis-related gene expression (FTL, SAT1, SLC7A11/GPX4) | Preclinical (In vitro and Animal) | Indirect effect on ferroptosis |
| H₂S signaling | H2S donors (e.g., GYY4137) | Iron chelation, antioxidant activity, regulation of iron metabolism | Preclinical (In vitro and Animal) | Mechanisms in glaucoma still under investigation,controlled release needed |
| Natural antioxidants | EGCG, Quercetin, Baicalein | Multi-target antioxidant effects, iron chelation, NRF2 activation, anti-inflammatory | Preclinical (In vitro and Animal) | Favorable safety profile,variable bioavailability, limited potency vs synthetic inhibitors |
DFO: deferoxamine; DFP: deferiprone; Fer-1: Ferrostatin-1; Lip-1: Liproxstatin-1; GPX4: glutathione peroxidase 4; GSH: glutathione; DMF: dimethyl fumarate;
8. Therapeutic Implications and Future Directions
Recognizing ferroptosis as a key driver of RGC death in glaucoma highlights novel therapeutic opportunities beyond IOP reduction. Promising strategies include combinatorial targeting of multiple ferroptosis pathways (GPX4-GSH-xCT, FSP1-CoQ10-NADPH,
Despite this progress, moving these approaches into the clinic faces several critical research gaps:
(1) Development of ferroptosis-specific biomarkers. In glaucoma research, aqueous humor samples obtained during ocular surgery provide a direct source for investigating disease-related biomarkers[44]. Meanwhile, non-invasive retinal imaging techniques such as optical coherence tomography (OCT) and OCT angiography (OCTA) offer valuable tools for long-term disease monitoring and patient stratification[45]. However, a critical question remains: can current imaging or fluid-based indicators specifically reflect ferroptosis, rather than general neurodegenerative changes or oxidative damage? Therefore, establishing specific signatures of ferroptosis still requires rigorous experimental validation.
(2) Systematic evaluation of combination therapies. Preclinical data suggest that targeting multiple ferroptotic pathways (e.g., iron chelation plus NRF2 activation) enhances neuroprotection. However, optimal combinations, dosing strategies, and potential interactions remain undefined.
(3) Advanced drug delivery systems. Effective translation of iron chelators and synthetic ferroptosis inhibitors requires delivery platforms capable of crossing the blood-retinal barrier while minimizing systemic exposure. Sustained-release formulations that maintain therapeutic concentrations in the posterior segment are essential.
(4) Long-term safety assessment. While natural antioxidants like flavonoids are generally safe, their therapeutic potency may be limited[46]. In contrast, potent synthetic inhibitors such as Lip-1 must be thoroughly assessed to ensure that sustained suppression of ferroptosis does not interfere with essential iron signaling or redox balance[23].
(5) Translational clinical trial design. Future trials should incorporate ferroptosis-relevant endpoints, define appropriate patient populations (e.g., normal-tension glaucoma or IOP-refractory cases), and establish proof-of-concept efficacy before advancing to larger studies.
9. Conclusion
Ferroptosis is a key mechanism of RGC death in glaucoma, driven by iron accumulation, lipid peroxidation, and antioxidant depletion. Its regulation through iron metabolism, mitochondrial function, and antioxidant pathways offers multiple therapeutic targets. Preclinical studies support ferroptosis inhibitors, iron chelators, NRF2 activators, and natural compounds as neuroprotective strategies. Translating these approaches alongside IOP-lowering therapies could enhance vision preservation, and future work should focus on optimizing combinations, assessing safety, and validating efficacy in clinical trials.
Acknowledgements
AI tools were used solely for language editing and polishing of the manuscript. The authors take full responsibility for the integrity, accuracy, and originality of the content.
Authors contribution
Liu H: Conceptualization, writing-original draft.
Zhou Y: Visualization, data curation, writing-review & editing.
Conflicts of interest
Not applicable.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
None.
Copyright
© The Author(s) 2026.
References
-
3. Guo M, Zhu Y, Shi Y, Meng X, Dong X, Zhang H, et al. Inhibition of ferroptosis promotes retina ganglion cell survival in experimental optic neuropathies. Redox Biol. 2022;58:102541.[DOI]
-
6. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73(11-12):2195-2209.[DOI]
-
7. Wei Y, Lin Y, Li Y, Liu J, Yang Y, Chen H, et al. Redefining cell death: Ferroptosis as a game-changer in ophthalmology. Front Immunol. 2025;16:1709354.[DOI]
-
8. Wang J, Shanmugam A, Markand S, Zorrilla E, Ganapathy V, Smith SB. Sigma 1 receptor regulates the oxidative stress response in primary retinal Müller glial cells via NRF2 signaling and system xc−, the Na+-independent glutamate–cystine exchanger. Free Radic Biol Med. 2015;86:25-36.[DOI]
-
15. Zhang W, Liu Y, Liao Y, Zhu C, Zou Z. GPX4, ferroptosis, and diseases. Biomed Pharmacother. 2024;174:116512.[DOI]
-
20. Chen X, Comish PB, Tang D, Kang R. Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol. 2021;9:637162.[DOI]
-
23. Zhang Y, Zhou X, Liang G, Cui M, Qiu Z, Xu J, et al. Iron-chelating and ROS-scavenging polymers with thioketal and thioether bonds delivering ferroptosis inhibitor lip-1 provide a triple therapeutic strategy for retina ganglion cells in acute glaucoma. Adv Mater. 2025;37(39):e2507526.
-
26. Nuñez MT, Chana-Cuevas P. New perspectives in iron chelation therapy for the treatment of neurodegenerative diseases. Pharmaceuticals. 2018;11(4):109.[DOI]
-
28. Huang S, Liu K, Su Y, Wang F, Feng T. Research progress of ferroptosis in glaucoma and optic nerve damage. Mol Cell Biochem. 2023;478(4):721-727.[DOI]
-
30. Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24-49.[DOI]
-
33. Yang Y, Lin Y, Han Z, Wang B, Zheng W, Wei L. Ferroptosis: A novel mechanism of cell death in ophthalmic conditions. Front Immunol. 2024;15:1440309.[DOI]
-
41. Zhang S, Hu R, Geng Y, Chen K, Wang L, Imam MU. The regulatory effects and the signaling pathways of natural bioactive compounds on ferroptosis. Foods. 2021;10(12):2952.[DOI]
-
42. She W, Su J, Ma W, Ma G, Li J, Zhang H, et al. Natural products protect against spinal cord injury by inhibiting ferroptosis: A literature review. Front Pharmacol. 2025;16:1557133.[DOI]
-
43. Li Q, Yang X, Li T. Natural flavonoids from herbs and nutraceuticals as ferroptosis inhibitors in central nervous system diseases: Current preclinical evidence and future perspectives. Front Pharmacol. 2025;16:1570069.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Zhou Y, Liu H. Ferroptosis and oxidative stress in glaucoma. Ferroptosis Oxid Stress. 2026;2:202521. https://doi.org/10.70401/fos.2026.0020
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Overview of Ferroptosis
- 3. Mechanistic Basis and Propagation of Ferroptosis
- 4. Assessing Ferroptosis in Glaucoma and RGC Injury
- 5. Ferroptosis as a New Type of Programmed Cell Death in Glaucoma
- 6. Ferroptosis Activation in Glaucomatous RGC Injury
- 7. Therapeutic Strategies Targeting Ferroptosis
- 8. Therapeutic Implications and Future Directions
- 9. Conclusion
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- Copyright
Science Exploration Style
Zhou Y, Liu H. Ferroptosis and oxidative stress in glaucoma. Ferroptosis Oxid Stress. 2026;2:202521. https://doi.org/10.70401/fos.2026.0020
copy
Share Link
copy