The role of multiple modes of cell death in the pathogenesis of acute respiratory distress syndrome
Yongxin Zheng
1,2,3

,
Nanshan Zhong
1
,
Ling Sang
1,*
,
Yongbo Huang
1,*
*Correspondence to:
Ling Sang, Department of Respiratory and Critical Care Medicine, the First Affiliated Hospital of Guangzhou Medical University, Guangzhou 510120, Guangdong, China.
E-mail: sonysang999@vip.163.com
Yongbo Huang, Department of Respiratory and Critical Care Medicine, the First Affiliated Hospital of Guangzhou Medical University, Guangzhou 510120, Guangdong, China. E-mail: yongbo2046@163.com
Yongbo Huang, Department of Respiratory and Critical Care Medicine, the First Affiliated Hospital of Guangzhou Medical University, Guangzhou 510120, Guangdong, China. E-mail: yongbo2046@163.com
Ferroptosis Oxid Stress. 2026;2:202602. 10.70401/fos.2026.0024
Received: January 18, 2026Accepted: April 09, 2026Published: April 13, 2026
Abstract
Acute respiratory distress syndrome (ARDS) is a life-threatening form of acute respiratory failure characterized by diffuse lung inflammation and edema. Despite increased understanding of the molecular biology underlying ARDS, the complex pathogenesis still limits the development of targeted pharmacologic therapies. Cell death plays a vital role in defending against pathogen infections and triggering tissue inflammation, which can damage the alveolar-capillary barrier and ultimately lead to the development of ARDS. Thus, targeting various cell death pathways may be an attractive entry point for therapeutic intervention in ARDS. Intriguingly, recent genetic and biochemical studies have emphasized the importance of revealing the crosstalk among various cell death pathways and indicated that this connectivity exhibits a considerable degree of plasticity in the molecular regulation of potential therapeutic targets in ARDS. In this review, we summarize the mechanisms of the different types of regulated cell death (RCD) and describe the physiological and pathological processes that contribute to ARDS pathogenesis. We also discuss the emerging crosstalk among various RCD modalities, and highlight that targeting cell death pathways is an effective therapeutic strategy for ARDS.
Keywords
Acute respiratory distress syndrome, acute lung injury, cell death, pathogenesis, crosstalk
1. Introduction
Acute respiratory distress syndrome (ARDS) is a common clinical syndrome of acute respiratory failure and is defined by the acute onset of noncardiogenic pulmonary edema, hypoxemia, and the need for mechanical ventilation[1,2]. Despite representing syndromes with heterogeneous etiologies, acute lung injury (ALI) and ARDS exhibit comparable lung injury pathophysiology, similar clinical features, and respond to analogous therapeutic interventions, justifying their treatment as a single clinical entity in research contexts[1,3,4]. ARDS can be induced by a variety of causes, including pneumonia, sepsis, trauma, ischemia-reperfusion, and aspiration of gastric contents[5-7]. Despite improvements in supportive care for ARDS, ARDS is identified in 10.4% of intensive care unit admissions, and mortality remains high at 30-40%[8]. The complex pathophysiological mechanisms of ARDS and inherent challenges of mechanistic studies in humans have significantly impeded the development of effective therapies for ARDS, especially pharmacological treatments[9]. Currently, the classic pathological finding in the lung is diffuse alveolar damage, which is characterized by neutrophilic alveolitis and the formation of hyaline membranes. Ultrastructural analysis has further demonstrated that disruption of the alveolar-capillary barrier directly contributes to the characteristic physiological abnormalities, indicating that alveolar epithelial and endothelial cell death plays a role in the development of ARDS[10].
A growing body of work has suggested that cell death is not merely a consequence of lung injury but rather a central driving mechanism in the pathogenesis of ARDS. While ARDS was historically viewed as primarily an inflammatory disorder with secondary cell death, emerging evidence reveals that regulated cell death (RCD) of specific lung cell populations, particularly alveolar epithelial cells, pulmonary endothelial cells and immune cells, actively orchestrates the hallmark pathophysiologic features of ARDS, including alveolar-capillary barrier disruption, inflammatory dysregulation, and impaired tissue repair. Cell death occurs not only through unregulated necrosis, such as that which occurs in trauma, but also as a result of various types of RCD[11]. Main RCD patterns, including apoptosis, necroptosis, pyroptosis, ferroptosis, and NETosis, have been recognized as critical pathogenic mechanisms that actively propagate and amplify lung injury rather than simply resulting from it. RCD has been considered a double-edged sword in ARDS. On the one hand, uncontrolled or excessive cell death of parenchymal cells (epithelial and endothelial) directly activates proinflammatory and procoagulant pathways, increases paracellular permeability, and ultimately contributes to pulmonary edema formation[12,13]. During ARDS, tissue damage and inflammation are tightly linked and are both associated with the release of damage-associated molecular patterns (DAMPs) from dying cells[14-16]. RCD and inflammation mutually induce each other, driving a local auto-amplification loop that leads to exacerbated cell death and inflammation[17]. Paradoxically, RCD also contributes to immune suppression by causing excessive loss of immune cells. This excessive loss of immune cells compromises the immune response, diminishing the body’s capacity to effectively combat infections and repair damaged lung tissue[18]. On the other hand, cell death at moderate levels has been recognized as a defense mechanism against microbial infection. Moderate cell death can directly impede the spread of pathogens by clearing replicative vehicles and further promoting the removal of pathogens[19-21]. Consequently, various RCD modalities play a pivotal role in the pathogenesis of ARDS and may represent one of the most important therapeutic targets for ARDS.
Multiple cell death modalities were once believed to be distinct and independent processes. However, recent genetic and biochemical studies have elucidated the remarkable flexibility and connectivity between various cell death pathways[22]. For example, despite their important role of triggering pyroptosis, inflammatory caspases (CASP) can also induce apoptosis, and conversely, apoptotic stimuli can also initiate pyroptosis[23-26]. Moreover, the induction of excessive lipid peroxidation by deleting glutathione peroxidase 4 (GPX4) not only triggers ferroptosis but also promotes pyroptosis and necroptosis, indicating the complex connections among ferroptosis, necroptosis, and pyroptosis[27,28]. Thus, RCD modalities do not operate as separate entities but act in parallel with various human pathologies.
The present work focuses on summarizing the mechanisms of different types of RCD and describing the pathophysiological processes that contribute to ARDS pathogenesis. We discuss the intriguing notion that different RCD modalities could be recognized as a single, coordinated system in which individual pathways are highly interconnected and collaboratively lead to tissue damage and lung edema. In addition, we thoroughly discuss the emerging therapeutic applications of targeting cell death in ARDS.
2. Apoptosis in ARDS
2.1 Molecular mechanism of apoptosis
Apoptosis, as the classic RCD form, commonly occurs due to cell damage or infection[11,29]. While the molecular mechanisms regulating apoptosis have been extensively investigated, it is now established that apoptosis is classified as two major pathways, namely intrinsic and extrinsic apoptosis (Figure 1). When cells are exposed to DNA damage, oxidative stress, or nutrient deprivation, the intrinsic apoptosis pathway can be activated. Intrinsic apoptosis is initiated by mitochondrial outer membrane permeabilization (MOMP), which leads to the release of cytochrome c (CYCS) and subsequent activation of Caspase 9 and inhibition of antiapoptotic protease activating factor-1 (Apaf-1)[30]. The release of CYCS into cytosol is promoted by pro-apoptotic proteins of the B-cell lymphoma-2 (BCL2) family members, such as BCL2-associated X protein (BAX), Bcl2-antagonist/killer (BAK) and p53 upregulated modulator of apoptosis (PUMA). These proteins can be further divided into activators (e.g., BIM, BID and PUMA), and sensitizers (e.g., NOXA and BIK). In response to cellular stress, both transcriptional and post-translational activation of the activator proteins bind directly to the main effectors (BAX and BAK) by undergoing oligomerization. Oligomerized BAX and BAK further form pores in the mitochondrial membrane, leading to MOMP. Sensitizer proteins contribute to MOMP by promoting the cellular localization of BAX and BAK or inhibiting the activation of anti-apoptotic proteins. Interestingly, other BCL2 family members, such as BCL2, BCL2-xl, BCL-W, and MCL-1, inhibit apoptosis. These anti-apoptotic proteins sequester activators or sensitizer BCL-2 proteins or the BAX and BAK complexes by utilizing two BH3 domains to form a binding groove. The fine balance of binding affinities and expression of BCL-2 family proteins regulates the activation and termination of apoptosis. Next, the rupture of the mitochondrial membrane leads to the release of CYCS and second mitochondrial-derived activator of CASP (SMAC, also known as DIABLO) to activate downstream caspases[31]. CYCS induces the formation of the apoptosome, comprising Apaf-1 and procaspase9, to promote the activation of caspase9, which in turn mediates the cleavage of other downstream caspases, such as caspase3/7. Similarly, SMAC, which functions as an inhibitor of the inhibitor of apoptosis (IAP) family (IAP1/2 and XIAP), can also promote apoptosis by activating downstream caspase9/3/7. Revolutionarily, knockout of caspase 3 or caspase7 also delay mitochondrial damage and cell death, indicating that the view of a simple upstream and downstream cascade of apoptosis should be modified[32]. Apoptosis may be a circular cascade which can achieve self-amplification.
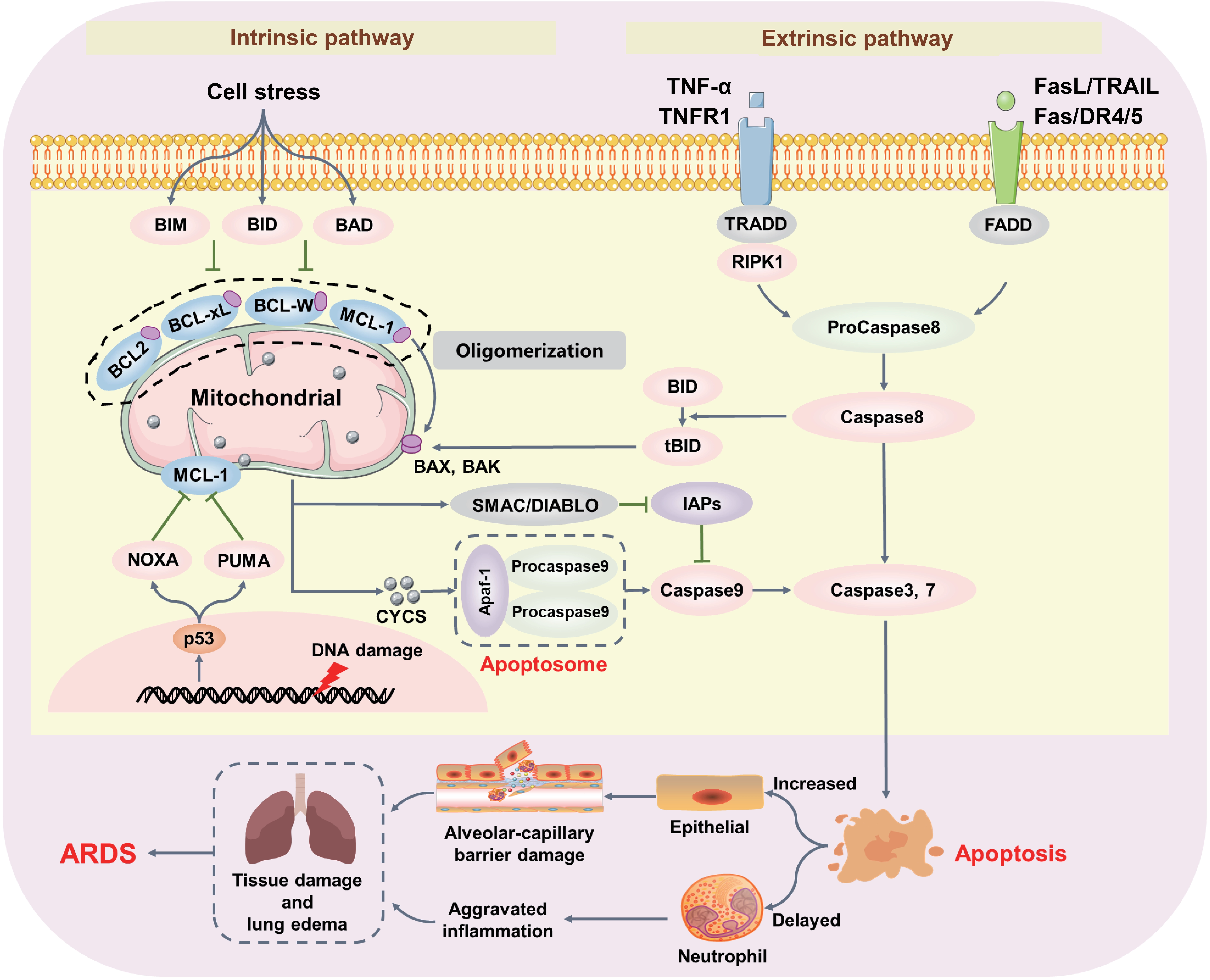
Figure 1. Intrinsic and extrinsic apoptosis pathways in ARDS. Intrinsic apoptosis can be activated by cellular stress, including DNA damage and mitochondrial damage. Inactivation of pro-survival BCL-2 family members and activation of pro-death BCL-2 family members BAK and BAX induce mitochondrial damage and lead to the release of CYCS and other mitochondrial-derived activators of caspases (e.g., SMAC). The release of mitochondrial contents promotes Apaf-1-mediated activation of Caspase9, resulting in downstream caspase cleavage and ultimately apoptosis. The extrinsic apoptosis pathway is mediated by death receptors (e.g., TNFR1 and Fas) binding to their cognate ligands (e.g., TNF and FasL). Activation of death receptors triggers a caspase cascade that induces apoptosis. Activated apoptosis increases lung inflammation and damages the alveolar-capillary barrier, contributing to ARDS pathogenesis. ARDS: acute respiratory distress syndrome; BCL-2: B-cell lymphoma-2; CYCS: cytochrome c; SMAC: second mitochondrial-derived activator of caspase; TNF: tumor necrosis factor; TNFR1: TNF-receptor 1; BAX: BCL2-associated X protein; BAK: Bcl-2 antagonist killer.
Extrinsic apoptosis is initiated by the activation of cell-surface death receptors that are characterized by the presence of an intracellular protein-protein interaction domain known as the death domain, such as TNFR1/2, Fas, and the TNF-related apoptosis-inducing ligand (TRAIL) receptors DR4 and DR5. Upon activation by their cognate ligands (e.g., TNFα, FasL, or TRAIL), death receptors oligomerize to form platforms on the cell surface that recruit adapter proteins (TRADD and FADD), which further activate the apoptotic initiator caspase8, leading to the formation of the death-inducing signaling complex. Interestingly, activation of TNFR1 by TNF may also promote RIPK1-dependent apoptosis. Indeed, before necroptosis was discovered, RIPK1 had been found to interact with TRADD and FADD to form complex IIa, which further mediates the activation of caspase8[33,34]. The activated caspase8 further activates effector caspases3/7 either directly through proteolytic cleavage or indirectly by activating the BCL-2 family member BID, thus generating feedback to initiate intrinsic apoptosis and promote MOMP[35-37].
2.2 Apoptosis in ARDS
Apoptosis is both a consequence of upstream pathogenic mechanisms and a driver of further lung injury, leading to barrier disruption and inflammatory regulation. The first ultrastructural measurements of lungs from patients with ARDS revealed that apoptosis mainly occurred in type I epithelial cells and to a lesser extent in endothelial cells[38,39]. The activation of Fas by soluble FasL in bronchoalveolar lavage fluid (BALF) from ARDS patients induces apoptosis, disrupting the alveolar-capillary barrier and increasing permeability[40-42], indicating that alveolar epithelial injury is in part associated with local upregulation of the Fas/FasL system and activation of the apoptotic cascade. Interestingly, beyond biochemical signaling, the mechanical forces present in ARDS also trigger apoptotic pathways. Mechanical ventilation, as the critical therapy for ARDS, contributes to aggravating lung damage (ventilator-induced lung injury (VILI)) with inappropriately high tidal volume. Piezo1, a mechanotransduction protein of type II pneumocytes, can be activated by increasing mechanical stretch of alveoli during ARDS, which is associated with the reduction of BCL2 and leads to increased apoptosis in ARDS[43]. Therefore, low tidal volume is recommended as a lung protective strategy for ARDS. In addition, DAPK1 is originally described as a proapoptotic Ca2+/calmodulin-regulated serine/threonine kinase involved in promoting alveolar epithelial cell apoptosis and VILI. Inhibition of DAPK1 appears to be protective against lung injury, suggesting that DAPK1 may be a potential therapeutic target for treating VILI[44]. These findings underscore the critical role of apoptosis in lung parenchymal cells during ARDS pathogenesis.
However, apoptosis is not immunologically silent. Under certain conditions, it can be a necessary tool of the immune system to fight against pathogens, though this may also aggravate inflammatory damage[45]. For example, death receptor-mediated apoptosis can activate the NF-κB pathway, promoting the expression of proinflammatory genes and enhancing immunogenic potential. Besides, when efferocytosis is delayed or impaired, apoptotic cells may undergo secondary necrosis, leading to the release of DAMPs (e.g., HMGB1, histone and nucleosomes)[46]. In ARDS, delayed apoptosis of neutrophils might have distinct functions in the pathogenesis. In sepsis-induced ARDS, delayed neutrophil apoptosis was associated with increased lung inflammation[47,48]. This prolonged neutrophil survival allows for sustained inflammatory activity and tissue damage. Notably, a novel mechanism has been identified whereby delayed neutrophil apoptosis aggravates neutrophil extracellular trap (NET) formation in ARDS[49]. While NETs were originally proposed as an innate defense mechanism for pathogen clearance, excessive NET formation can be detrimental. Indeed, elevated plasma NET levels are positively associated with both ARDS severity and mortality[50], suggesting that the protective function of NETs can become pathological when dysregulated. However, apoptosis can also limit immune activation. For instance, caspase3 proteolytically inactivates several critical proteins in the IFN signaling cascade, including cyclic GMP-AMP synthase, mitochondrial antiviral signaling protein, and interferon regulatory factor 3, thereby dampening type I interferon production triggered by mitochondrial DNA release during apoptotic cell death[51]. Caspase3-/- mice showed increased resistance to viral infection, indicating that apoptotic caspases maintain innate immunity against viral infection[51]. In addition, some specific anti-inflammatory metabolites such as AMP, GMP, creatine, spermidine, and glyceraldehyde 3-phosphate were found to be released from apoptotic lymphocytes and macrophages that preserve their plasma membrane integrity[52,53]. These specific metabolic pathways remain active in apoptosis-committed cells, and the metabolites are released through caspase-1- or caspase-3-dependent cleavage and activation of pannexin 1 channels[52,53]. These metabolites not only act as immunosuppressive DAMPs, but also promote cell proliferation and wound healing in lung graft rejection. Thus, apoptotic cells not only promote phagocytosis but also dampen inflammation and promote tissue repair in ARDS.
3. Necroptosis in ARDS
3.1 Molecular mechanism of necroptosis
Necroptosis, the first identified programmed form of necrosis exhibiting morphological features similar to necrosis[54], was initially described in 1996 when caspase-1 and caspase-8 inhibition triggered cell death in cowpox virus-infected pig kidney cells[55]. Necroptosis represents the first discovered mechanism that mediates necrosis. The activation of death receptors, including TNFR1, Fas, DR4/5, IFNAR, IFNAGR, and TLRs, by their cognate ligands initiates the necroptosis signaling cascade under apoptosis-deficient conditions[56]. Groundbreaking discoveries have since identified RIPK1, RIPK3, and MLKL as key mediators of necroptotic cell death[57-59] (Figure 2). Activation of RIPK1 in caspase-deficient cells triggers the recruitment of TRADD, FADD, and RIPK3 to achieve necrosome assembly[60,61]. During necroptosis, the interaction between activated RIPK1 and RIPK3 is mediated by their respective RIP homotypic interaction motif (RHIM) domain, which triggers the formation of a RHIM-mediated amyloid-like structure to promote phosphorylation of RIPK3. The phosphorylated RIPK3 further induces the phosphorylation and oligomerization of MLKL, leading to the exposure of the N-terminal four-helix bundle (4HB) domain[62-65]. Initial studies speculated that RIPK1/RIPK3/MLKL triggered cell death through mitochondrial destabilization, which was mediated by the phosphoglycerate PGAM5- and DRP1-dependent pathway[29,66]. However, the deficiency of PGAM5 or DRP1 could not defend the occurrence of TNF-induced necroptosis, resulting in the challenge of this theory[64,67]. Currently, two nonexclusive theories explain how MLKL compromises cell membrane integrity: (1) a platform in the membrane was constructed by MLKL to open the calcium or sodium ion channels, enabling ion influx, cell swelling and rupture[68,69]. (2) MLKL itself forms pores in the membrane via the interaction of charged amino acids in its 4HB domain with phosphatidylinositol phosphates in the plasma membrane[70,71]. However, a latest study found that SIGLEC12 was a mediator of plasma membrane rupture (PMR) during necroptosis[72,73]. The cleavage of SIGLEC12 during necroptosis to produce a 20-KDa fragment is required and sufficient to induce PMR. This finding could expand our understanding of how programmed necrosis is executed and offers a new target for inhibiting necroptosis[72,73].
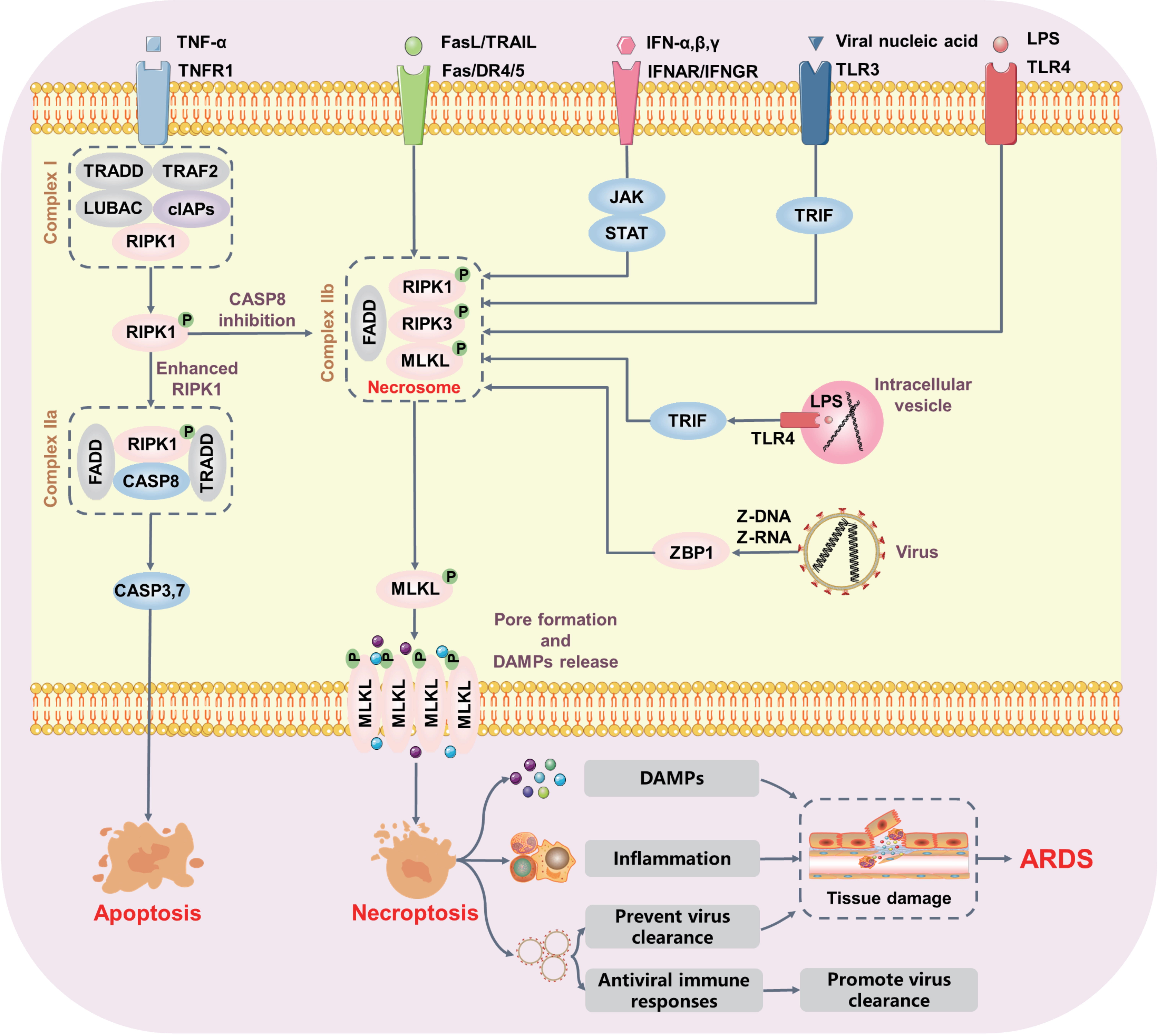
Figure 2. Molecular mechanisms of necroptosis in ARDS. Upon TNF activation, intracellular trimerized TNFR1 mediates the formation of transient complex I, which recruits multiple proteins including TRADD, LUBAC, TAB2, and cIAPs. In complex I, RIPK1 is rapidly polyubiquitylated, leading to the formation of two alternative cytosolic complexes. Complex IIa includes FADD, CASP8, and RIPK1 to promote apoptosis. When CASP8 activation is inhibited, RIPK1 binds to RIPK3 to form complex IIb. Polyubiquitylated RIPK3 in turn increases MLKL phosphorylation, resulting in pore formation and DAMP release. Activated necroptosis triggers excessive inflammation that aggravates lung tissue damage. Notably, activation of TLR3/4 or ZBP1 by pathogen-derived nucleic acids can also promote necroptosis in the absence of caspase function. ARDS: acute respiratory distress syndrome; TNF: tumor necrosis factor; TNFR1: TNF-receptor 1; TRADD: TNFR1-associated via death domain; LUBAC: linear ubiquitin chain assembly complex; TAB2: TGFβ-activated kinase 1-binding proteins 2; cIAPs: cellular inhibitor of apoptosis proteins; RIPK1: receptor-interacting protein kinase 1; FADD: Fas-associated protein with death domain; CASP: caspase; DAMP: damage-associated molecular pattern; TLR: toll-like receptor; ZBP1: Z-DNA binding protein 1.
It is now well established that necroptosis can be triggered by multiple stimuli, including activation of TNF family death domain receptors, Toll-like receptors (e.g., TLR3 and TLR4), and nucleic acid sensors (e.g., ZBP1, also known as DAI). Ligands such as TNF-α and FasL initiate the necroptosis pathway when caspase-8 activation is prevented by caspase inhibitors (e.g., Z-VAD-FMK) or by depletion of FADD[74,75]. TLR3 and TLR4 both utilize the RHIM-containing adaptor Toll/IL-1 receptor domain-containing adaptor inducing interferon-beta (TRIF) to recruit RIPK1 and RIPK3. Inhibiting RIPK1 prevents TLR3- or TLR4-mediated necroptosis in macrophages, indicating that the kinase activity of RIPK1 is necessary for TRIF-dependent necroptosis[76,77]. However, genetic studies in mice have suggested that TRIF can activate necroptosis in the absence of RIPK1[78,79]. Indeed, RIPK1 appears to suppress TRIF-induced necroptosis through its RHIM, as the inflammation and prenatal lethality in RIPK1 RHIM mutant mice are prevented by the loss of MLKL or combined loss of TRIF and ZBP1[80]. Consistently, primary human fibroblasts lacking RIPK1 are more resistant to lipopolysaccharide (LPS) or poly(I:C) induced cell death than control fibroblasts[81]. Similarly, ZBP1 is a sensor of cellular nucleic acids that can be activated through its binding to viral Z-DNA and Z-RNA, thereby driving necroptosis in response to viral infection[20,82]. Mechanistically, ZBP1 directly interacts with RIPK3 via its two RHIM domains to form a necrosome, thereby leading to MLKL oligomerization independent of RIPK1 and TNFR1[21]. However, the RHIM-dependent binding of ZBP1 to RIPK3 can be negatively regulated by the scaffolding function of RIPK1, which competes with ZBP1 for binding to RIPK3[21]. Activation of RIPK1 might relieve its RHIM-dependent suppression of TRIF or ZBP1 signaling, although this suppressive mechanism remains poorly understood. These findings suggest that activation of RIPK1 might relieve its RHIM-dependent suppression of TRIF or ZBP1 signaling, although this suppressive mechanism remains poorly understood.
3.2 Necroptosis in ARDS
Necroptosis is generally considered as a form of immunological cell death, due to its early membrane rupture and robust proinflammatory potential[83]. The immunogenicity of necroptosis is shaped not only by membrane disruption but also by RIPK1- and RIPK3-dependent transcriptional programs within dying cells[84]. RIPK1 promotes the transcription and translation of inducible DAMPs, such as TNF, IL-6, CXCL1, and type I IFNs, which are passively released upon membrane rupture and ultimately enhance immune cell recruitment and activation, thereby inducing inflammatory injury[84,85]. Currently, necroptosis has mainly been recognized as a downstream event of pattern recognition receptors in response to microbial infection. For example, in the context of Staphylococcus aureus (S. aureus) induced lung injury, necroptotic epithelial cell death is detrimental to the host[86]. However, RIPK3-mediated necroptosis in Yersinia infection is beneficial to the host[87,88]. Thus, cell death is not inherently “good” or “bad”, but rather serves as a double-edged sword whose biological consequence is dictated by the specific pathogen, infected tissue, and timing of execution. In addition, ZBP1, the innate sensor of nucleic acids, is considered the critical initiator of virus-induced necroptosis. Deletion of ZBP1 improves pulmonary tissue damage and lung inflammation during influenza A virus (IAV) infection; however, some studies report higher mortality in ZBP1 knockout mice due to increased viral titers and viral dissemination[19,20,89]. This divergence may be attributable to differences in host factors and the local microenvironment. This context dependency, influenced by host genetics, pathogen virulence, infection dose, and tissue microenvironment, suggests that necroptosis operates not as a simple on/off switch, but as a rheostat whose fine-tuning determines whether the host survives or succumbs to infection. Currently, some studies have focused on the critical role of RIPK3 in defending against virus infection. Mechanistically, RIPK3 can activate a parallel apoptosis pathway that is fully capable of restricting IAV in the absence of necroptosis. For example, MLKL knockout mice demonstrate a beneficial effect in IAV-induced ARDS, whereas dual deletion of MLKL/FADD or RIPK3 results in excessive lethality and high viral titers due to the absence of apoptosis[21]. However, recent studies have pointed out that necroptosis can also drive robust antiviral immune responses and promote effective viral clearance in mice where apoptosis is deficient, perhaps due to earlier deployment of necroptosis during the host anti-IAV response[90]. This discrepancy raises additional questions about how RIPK3 determines the choice between necroptosis and apoptosis at the single-cell level.
As for critically ill patients, MLKL and RIPK3 levels in peripheral blood increase over time and are predictive of poor survival. Interestingly, plasma RIPK3 levels are independently associated with ARDS severity[91-94]. Moreover, ARDS patients requiring ventilation display higher RIPK3 levels, which may contribute to the pathogenesis of VILI[95]. Thus, these findings emphasize that mechanical stress might activate RIPK3-mediated necroptosis. In line with these studies, plasma RIPK3 levels were positively correlated with the severity of COVID-19 in critically ill patients, who often require mechanical ventilation support[96]. Similarly, preclinical studies suggest that SARS-CoV-2 infection activates the ZBP1-RIPK3 pathway, thereby promoting local inflammation and lung injury[97,98]. Pharmacological inhibition of RIPK3 by GSK872 or genetic deletion of MLKL protected mice against SASRS-CoV-2 infection[99]. Collectively, while RIPK3 and MLKL levels are positively correlated with the severity of ARDS, the mentioned studies did not use analysis methods specific for phosphorylated RIPK3 and MLKL. Such tools[100] are now available and will be necessary to conclusively determine the role of necroptosis in ARDS.
4. Pyroptosis in ARDS
4.1 Molecular mechanism of pyroptosis
Pyroptosis is a type of cell death that is induced by activation of inflammasome sensors and subsequent formation of gasdermin pores in the plasma membrane[29]. Inflammasome sensors, such as the Nod-like receptor family, the DNA receptor Absent in Melanoma 2 and the Pyrin receptor, are activated by sensing specific DAMPs or PAMPs. When activated, inflammasome sensors form the oligomeric scaffold and recruit the adapter protein apoptosis-associated speck-like protein containing a CARD (ASC), thus seeding the formation of micron-sized polymeric structures known as inflammasomes (Figure 3). Within the canonical pathway[101,102], inflammasomes formation bridges ASC and procaspase-1, leading to dimerization and activation of procaspase-1 via autocleavage[102], which dimerizes as a proform and generates the active p33/p10 species. Active caspase-1 further mediates cleavage of the effector protein gasdermin D (GSDMD), generating an N-terminal fragment that is able to oligomerize and form pores in the plasma membrane, leading to pyroptotic cell death[103]. Activation of caspase-1 also cleaves pro-IL-1β and pro-IL-18 to produce mature IL-1β and IL-18, which are released through GSDMD pores[104,105]. Moreover, GSDMD pores promote membrane destabilization and cell lysis, eventually leading to the release of DAMPs such as HMGB1, allowing them to escape from cells through these pores. Interestingly, some escaping DAMPs further amplify pyroptotic signaling[106,107] or contribute to the pyroptosis in bystander cells[108]. As for the non-canonical pathway, caspase-11 in murine and caspase-4, and 5 has been discovered to promote pyroptosis in the absence of caspase-1 by stimulation with intracellular LPS and directly targeting GSDMD[109,110]. Remarkably, these caspases not only mediate pyroptosis but also act as act as intracellular LPS receptors[111]. However, the mechanisms by which caspases sense LPS remain elusive. Recent studies have revealed that guanylate-binding proteins (GBPs) are essential for the activation of caspase-11 and caspase-4, and 5[112,113]. GBP1 binds to LPS with high affinity, further allowing the formation of a multimolecular complex with GBP2 and GBP4, which finally promote the recruitment and activation of caspases. Interestingly, recent breakthroughs indicate that PMR in pyroptotic cells is triggered by ninjurin 1 (NINJ1)[72]. The NINJ1 deficient cells upon PAMP stimulation undergo pyroptosis in culture, but showed persistent ballooned morphology. Furthermore, eliminating NINJ1 does not prevent pyroptosis, but it does inhibit the release of large intracellular DAMPs (e.g., LDH and HMGB1). Mechanistically, NINJ1 acts downstream of GSDMD, appearing as an autoinhibited dimer in live cells but transitioning to an active oligomer through a conformational rearrangement of its TM1 domain[114]. This oligomerization further drives PMR, enabling the release of intracellular DAMPs. However, the mechanism of triggering oligomerization of NINJ1 still remains elusive. A plausible hypothesis is that NINJ1 senses alterations in membrane tension or lipid remodeling when dying cells become swollen[115]. Thus, it is necessary to elucidate the precise molecular events that govern NINJ1 activation.
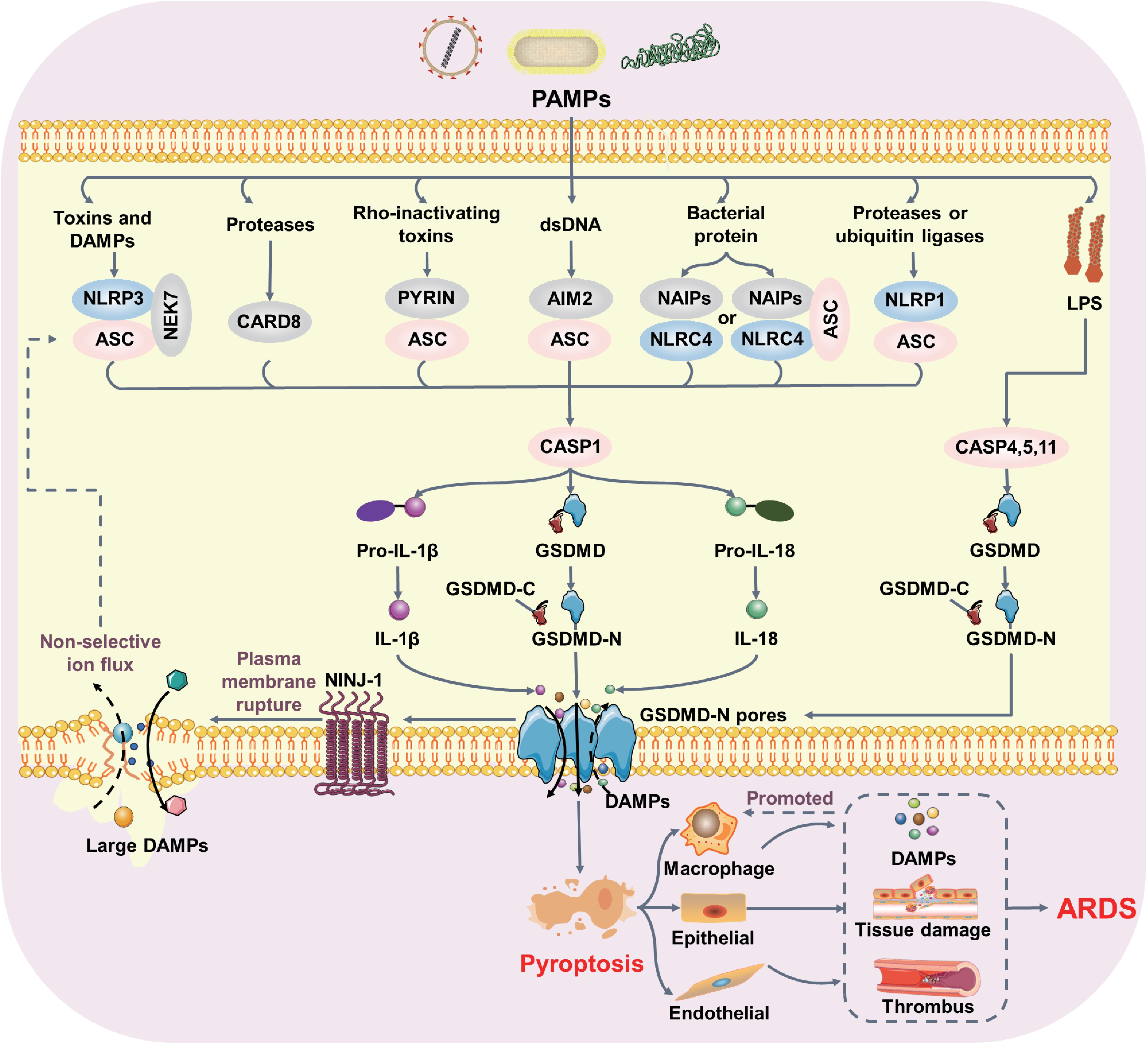
Figure 3. GSDMD-dependent pyroptosis in ARDS pathogenesis. Pathogenic bacterial and viral infections promote inflammasome formation, thereby activating CASP1, CASP4, and CASP5 in humans and CASP1 and CASP11 in mice. Activated caspases cleave GSDMD, releasing an N-terminal fragment that oligomerizes and forms pores in the plasma membrane. CASP1-mediated mature inflammatory cytokines (IL-1β and IL-18) are released through GSDMD pores. Moreover, GSDMD pores promote NINJ1-dependent cell rupture. Uncontrolled pyroptosis triggers excessive DAMP release, promotes thrombus formation, and ultimately aggravates lung injury. GSDMD: gasdermin D; ARDS: acute respiratory distress syndrome; CASP: caspase; NINJ1: ninjurin 1; DAMP: damage-associated molecular pattern.
4.2 Pyroptosis in ARDS
Pyroptosis-mediated tissue damage in ARDS likely results from an overactive, uncontrolled immune response and vascular dysfunction. Pyroptosis involves the release of cellular contents, which can exacerbate necroinflammation, leading to clinical deterioration and death in ARDS patients (Figure 3). This process occurs predominantly in professional phagocytic cells, including monocytes, macrophages, and dendritic cells, upon stimulation by pathogens such as bacteria or viruses, or even by pathogen-derived components such as LPS or viral genomes. Indeed, increased pyroptotic peripheral blood mononuclear cells in septic patients are strongly associated with clinical severity and 28-day mortality[116]. Analysis of BALF from ARDS patients showed that pyroptosis-related cytokines IL-1β and IL-18 were significantly elevated, whereas specific caspase-1 inhibition reduced DAMP production and alleviated lung inflammation while improving survival[117]. These findings suggest that inhibition of the inflammatory response and pyroptosis is crucial for improving patient survival in ARDS and other severe inflammatory conditions. However, future biomarker-based studies are needed to better understand the mechanisms underlying the therapeutic benefits and to identify patients who would most benefit from anti-pyroptotic interventions. Notably, as a critical mediator of PMR during pyroptosis, NINJ1 has emerged as an important player in ARDS pathogenesis. In IAV-infected mice, NINJ1 deletion prevents the release of DAMPs and IL-1β, and attenuates lung injury[118]. Interestingly, in the pyroptosis context, NINJ1 is not sufficient to fully trigger PMR by itself, and additional mechanical force is required for full PMR[119]. This finding suggests that mechanical ventilation may affect the plasma membrane integrity and exacerbate pyroptosis in ARDS.
Pyroptosis in pulmonary parenchymal cells, such as epithelial and endothelial cells, is directly related to lung injury. Caspase-11-induced pyroptosis in pulmonary epithelial and endothelial cells leads to alveolar-capillary barrier damage in sepsis-induced ALI[15,120]. Moreover, caspase-11-dependent pyroptosis in endothelial cells drives vascular leakage and hypotension[15]. The influx of Ca2+ through GSDMD pores in the endothelium can promote lethal coagulation and clot formation in micro-vessels[121]. Intriguingly, GSDMD nanobodies enter cells through initial pores and then inhibit further pore formation[122]. Collectively, these studies indicate that GSDMD in the endothelium is druggable. Inhibiting GSDMD might provide a therapeutic benefit for ARDS.
Pyroptosis is regulated by an intrinsic repair mechanism that limits excessive inflammation. The initial GSDMD pores in the membrane were thought to be transient and removed by membrane repair mechanisms. The endosomal sorting complex required for transport III (ESCRT-III)-dependent membrane repair pathway sheds GSDMD-containing ectosomes to prevent cell lysis and DAMPs release, which attenuate inflammation and promote tissue repair[123]. However, the role of ESCRT-III-dependent membrane repair mechanisms in the pathophysiology of ARDS remains poorly understood and warrants further investigation.
5. Ferroptosis in ARDS
5.1 Molecular mechanism of Ferroptosis
Ferroptosis refers to the oxidative cell death characterized by iron-dependent accumulation of excessive lipid peroxides in the cell membrane, leading to PMR[124] (Figure 4). The initiation of ferroptosis involves three essential elements: inhibition of antioxidant systems, lipid peroxidation, and iron accumulation. Currently, the core mechanism of ferroptosis is the inhibition of system xc- (xCT)-GSH-GPX4 axis. Erastin and RSL3 are common small molecules that were originally applied to induce ferroptosis of RAS-mutant cancer cells by targeting system xCT and GPX4, respectively[124-126]. Genetic inactivation of GPX4 and overexpression of xCT protect cells from ferroptosis. Mechanistically, the antiporter xCT, encoded by SLC7A11 and SLC3A2, is specific for exporting glutamate in exchange for importing cystine[127]. Imported cystine is reduced to cysteine, which protects cells from oxidative damage by serving as a precursor for glutathione synthesis. GPX4, a selenoprotein with glutathione-dependent activity, plays a critical role in defending against lipid peroxidation[128]. Inactivation of GPX4 leads to the excessive production of reactive oxygen species (ROS), resulting in lethal accumulation of lipid peroxides and disruption of the plasma membrane, ultimately leading to organ damage, such as acute renal failure and embryonic death[129,130]. Interestingly, GPX4 is not the only antioxidant enzyme to eliminate phospholipid hydroperoxides; the existence of a GPX4-independent pathway can terminate ferroptosis. Among them, AIFM2 (also known as FSP1), a cytosolic NADH ubiquinone oxidoreductase, was identified as having the capability of reducing CoQ10 and inhibiting ferroptosis[131,132]. In addition, AIFM2 also contributes to membrane repair through the ESCRTIII-dependent pathway, enhancing its anti-ferroptotic effects[133].
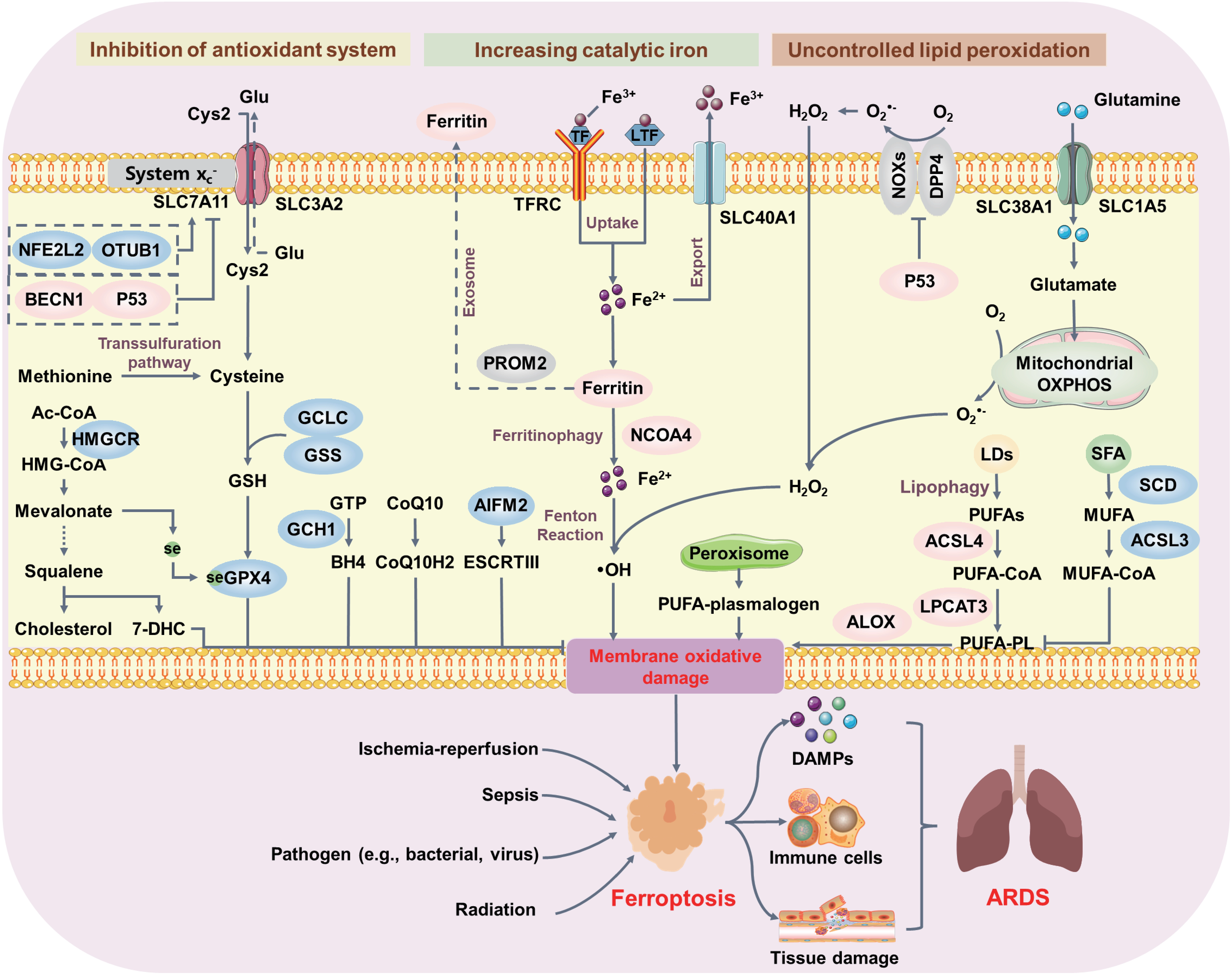
Figure 4. Core molecular mechanisms of ferroptosis in ARDS. Ferroptosis can occur through two major pathways: inhibiting the antioxidant SLC7A11-GSH-GPX4 system and increasing iron accumulation. Inhibition of system xc- decreases GSH production and subsequently suppresses GPX4 activity. Alternatively, CoQ10, BH4, and 7-DHC inhibit ferroptosis independently of GSH. The generation of PUFAs by ACSL4 and LPCAT3 or PUFA-plasmalogens by peroxisomes and subsequent activation of ALOX play pivotal roles in initiating lipid peroxidation. Additionally, NCOA4-mediated ferritinophagy degrades ferritin and increases intracellular catalytic iron, further activating the Fenton reaction to produce hydroxyl radicals involved in membrane oxidative stress. Lipid peroxidation-driven ferroptosis induces excessive tissue damage and uncontrolled inflammation, contributing to ARDS pathological processes. ARDS: acute respiratory distress syndrome; GSH: glutathione; GPX4: glutathione peroxidase 4; 7-DHC: 7-dehydrocholesterol; PUFAs: polyunsaturated fatty acids; ACSL4: acyl-CoA synthetase long-chain family member 4; LPCAT3: lysophosphatidylcholine acyltransferase 3; ALOX: arachidonate lipoxygenase; NCOA4: nuclear receptor coactivator 4.
Cell membranes, the primary target of oxidative damage in ferroptosis, can be influenced by metabolic pathways and membrane composition. In particular, increased polyunsaturated fatty acids (PUFAs) content in membranes enhances cellular sensitivity to ferroptotic inducers. To execute ferroptosis, susceptible PUFAs released from the plasma membrane undergo lipid peroxidation. This process is regulated by acyl-CoA synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), and lipoxygenases (LOXs)[134]. ACSL4 activates long-chain fatty acids by converting them into PUFA-CoA esters, which then enter at least two metabolic pathways[135-137]. In the first pathway, PUFA-CoA esters are incorporated into phosphatidylethanolamines by LPCAT3[136,137]. In the second pathway, sterol O-acyltransferase 1 catalyzes the formation of PUFA-CEs[138]. The products of both pathways serve as substrates for lipid peroxidation in a context-dependent manner. Unsurprisingly, loss of ACSL4 leads to the accumulation of unesterified PUFAs. Although these free PUFAs remain susceptible to peroxidation, they are insufficient to induce ferroptosis. Enhancing ACSL4 activity via protein kinase C-β-mediated phosphorylation of ACSL4 at Thr328 promotes the biosynthesis of PUFA-containing phospholipids during the ferroptotic process[139]. However, a recent study found that loss of ACSL4 severely tempers the ferroptotic cell death in many cell lines but does not totally block ferroptosis[140], suggesting the existence of compensatory mechanisms for the loss of ACSL4. PUFAs undergo further oxygenation catalyzed by LOXs, which introduce hydroperoxy groups (-OOH) into fatty acid chains, thereby initiating lipid peroxidation. Six isoforms (ALOX5, ALOX12, ALOX12B, ALOX15, ALOX15B, and ALOXE3) have been identified in humans, each with distinct substrate preferences and catalytic activities[141,142]. Among them, ALOX15 promotes phospholipid peroxidation and ferroptotic damage by peroxidizing PUFA-PE[143]. However, ALOX15 deficiency fails to block GPX4-loss-induced ferroptosis in acute renal failure models, revealing functional redundancy among ALOX isoforms. These observations underscore the importance of comprehensive ALOX expression profiling to determine which isoforms contribute to ferroptosis in specific biological contexts.
Increased iron uptake and reduced iron export via ferroportin promote ferroptotic cell death. Ferrous iron is transported into the cell via transferrin receptor protein. Intracellular iron is either bound to ferritin for storage or exported by ferroportin. Ferritin is composed of two subunits: a heavy chain (FTH1) that has ferroxidase activity to convert Fe2+ to Fe3+ for safe storage, and a light chain (FTL) without this activity[144]. During the ferroptotic process, the degradation of intracellular ferritin within lysosomes is mediated by the nuclear receptor coactivator 4 (NCOA4), leading to the release of iron[145]. The released iron then participates in the Fenton reaction, generating highly reactive lipid radicals that disrupt the plasma membrane[146].
Collectively, regardless of the enzymes that trigger lipid peroxidation, lipid peroxides initiate a chain reaction catalyzed by transition metals such as iron to generate lipid radicals. These radicals react with nearby lipids, amplifying lipid peroxidation in a self-propagating process, which eventually leads to the disruption of the plasma membrane[147]. How lipid peroxides compromise the integrity of the membrane remains elusive. Recent studies have demonstrated that NINJ1-dependent lysis is dispensable for the initial steps of ferroptosis[148,149]. Deficiency of NINJ1 protects macrophages and fibroblasts from PMR and inhibits the release of DAMPs. Therefore, by clarifying the mysteries of lipid peroxidation-driven ferroptosis, we may discover novel therapies for ferroptosis-related diseases.
5.2 Ferroptosis in ARDS
Ferroptosis is an immunogenic cell death that leads to the release of multiple DAMPs, such as HMGB1 and ATP. These DAMPs not only damage the alveolar-capillary barrier but also trigger inflammasome activation and IL-1β release from immune cells, amplifying local and systemic inflammation. In ARDS patients, ferroptotic signatures in the BALF of ARDS patients are significantly associated with disease severity[150]. Alterations in serum markers of iron metabolism and oxidative stress, such as elevated malondialdehyde, catalytic iron, and lactoferrin, demonstrate that iron overload and lipid peroxidation are associated with severe COVID-19 cases and the development of inflammatory injury[151,152], suggesting the critical role of ferroptosis in virus infection. Recent studies have found that the decreased iron levels can impair the IAV replication. The IAV hemagglutinin induces ferritinophagy and facilitate lipid peroxidation by interacting with NCOA4, which further inhibits MAVS-mediated antiviral immunity. The inhibition of NCOA4 restricts iron release through ferritinophagy exhibiting the strong antiviral effects[153]. Besides, IAV infection disrupts xCT-GPX4 system, leading to lipid peroxidation and lung injury[154]. Notably, the GSK3β/KEAP1-NRF2 axis, a critical antioxidant pathway in cells, when activated, can inhibit ferroptosis by promoting the expression of GPX4 and SLC7A11, which ultimately ameliorates IAV-induced ARDS[155]. These findings emphasize that the virus triggers oxidative damage in lung tissues, potentially through hijacking the cellular antioxidant system. Besides, other pathogens, such as Mycobacterium tuberculosis (MTB), also induce ferroptosis in infected cells and mice. MTB triggers ferroptosis in macrophages by increasing lipid peroxide levels and reducing GSH or GPX4 expression, which can be blocked by iron chelation or the ferroptosis inhibitor Ferrostatin-1 (Fer-1)[156,157]. Inhibiting ferroptosis of MTB-infected lungs can significantly decrease bacteria loads and further improve the inflammation[156,157], which is inspiring us to find a novel strategy to defend MTB infection by focusing on ferroptosis. Notably, pathogens also commandeer host lipid metabolic enzymes to promote the production of lipid ROS and consequently trigger ferroptosis. P.aeruginosa expresses 15-lipoxygenase, which can generate 15-hydroperoxy-AA-PE by oxidizing host arachidonic acid-PE, thereby inducing ferroptosis in human bronchial epithelial cells[158]. This indicates that P. aeruginosa hijacks PUFAs in the plasma membrane to trigger ferroptosis and cause lung injury, underscoring the importance of lipid metabolism in pathogen-induced ferroptosis. Currently, promising new strategies are being developed for treating ARDS by targeting ferroptosis signaling. However, the complex interplay mechanisms between pathogen infection and ferroptosis remain elusive. Although pathogens that induce ARDS may disrupt iron metabolism and initiate lipid peroxidation pathways, the precise mechanisms vary across different pathogen species. In particular, the mechanisms by which pathogens hijack host metabolic pathways to trigger ferroptosis require further investigation.
6. Other Cell Death Patterns in ARDS
6.1 Autophagy in ARDS
Autophagy is a cellular degradation process that removes damaged or unnecessary cellular components to maintain cellular homeostasis, yet sustained or excessive autophagy can cause autophagy-dependent cell death mediated by autophagy-related proteins (ATGs), such as ATG4B, ATG5, and ATG7. In ARDS, autophagy is induced in response to diverse ARDS stimuli, including infection, oxidative stress, mechanical stretch, and trauma[159]. During acute pulmonary infection, autophagy activation may be a protective mechanism to promote the clearance of pathogens and ameliorate inflammatory damage. In polymicrobial sepsis-induced ARDS, genetic deletion of autophagy genes such as LC3B and BECN1 remarkably exaggerates the inflammation and organ damage, ultimately leading to death[160-162]. The underlying mechanism appears to be that lacking LC3 and BECN1 induces the activation of caspase-1 and increases the secretion of IL-1β[161], suggesting the potential connection between pyroptosis and autophagy. Similarly, in response to P.aeruginosa infection, ATG7 deficiency in mice impairs the pathogen clearance, enhances neutrophilic infiltration, increases IL-1β release, and results in the severe lung injury[163]. Recently, a novel mechanism was demonstrated whereby inhibition of mitophagy of alveolar epithelial cells during ARDS by silencing RUNX1 significantly aggravates inflammatory response, indicating that targeting RUNX1-mediated mitophagy represents a valuable therapeutic strategy for ARDS[164]. However, while autophagy plays a protective role in ARDS, the excessive accumulation of autophagosomes may trigger detrimental effects in certain contexts. For instance, inhibition of autophagy ameliorates the VILI[165,166]. Genetic deletion of LC3 abolishes mechanical ventilation-induced NLRP3 activation and subsequently reduces the release of DAMPs[166]. Moreover, inhibition of autophagy also prevents the activation of inflammatory pathways such as NF-κB and alleviates VILI[165]. More interestingly, even in sepsis-induced ARDS, LC3-mediated autophagy protects mice from lung injury in the early stage but plays a lethal role in the late stage of sepsis[167], indicating that the role of autophagy in lung injury is context- and stage-dependent. Besides, NCOA4-mediated ferritinophagy is a selective form of macroautophagy/autophagy that promotes the degradation of ferritin and release of DAMPs. Inhibition of ferritinophagy improves oxidative stress and decreases organ damage, suggesting the crosstalk mechanism between autophagy and ferroptosis has a critical role in ARDS pathogenesis[168]. These findings collectively emphasize that autophagy plays a dual role in ARDS pathogenesis, and its therapeutic modulation requires careful consideration of the disease stage, specific context, and the balance between protective and detrimental effects. Future therapeutic strategies should focus on fine-tuning autophagy activity rather than simply promoting or inhibiting it, potentially through stage-specific or cell-type-specific interventions.
6.2 NETosis in ARDS
NETosis is the process of NET generation, in which web-like DNA-protein structures are generated by various cells (e.g., neutrophils, other leukocytes, and epithelial cells) and released in response to infection and injury. Currently, the best-described pathways that lead to NET formation involve the production of ROS via NADPH oxidase (NOX). In response to stimuli, activated NOX induces ROS production, which further promotes the degradation of myeloperoxidase (MPO) and enables the translocation of neutrophil elastase (NE) from the cytosol into the nucleus. Subsequently, NE induces the degradation of histones, resulting in chromatin decondensation and DNA release. Moreover, ROS also activates peptidyl arginine deiminase 4 (PAD4), which promotes the citrullination of histones, further supporting chromatin decondensation. Additionally, NETosis can be activated by platelets, in which elevated Ca2+ directly activates PAD4, leading to chromatin decondensation[169,170]. However, although the exact mechanism whereby NETs are terminally released into the extracellular space remains unknown, the disassembly of the plasma membrane is thought to play a critical role in this process[171].
NETosis is a double-edged sword for the immune system. Elevated NETs can encapsulate, capture, and kill pathogens via antimicrobial proteins, such as cfDNA antimicrobial peptide-LL37, lactoferrin, MPO, and others. However, excessive release of NETs can trigger and amplify inflammatory responses, ultimately leading to organ damage and various diseases. Compared with healthy controls, BALF from ARDS patients contains higher levels of NETs, which can trigger damage to pulmonary epithelial cells[49,172]. In COVID-19, patients also have elevated amounts of cfNDA, MPO, and histones[173-175]. Moreover, elevated cfDNA is associated with the release of lactate dehydrogenase, while histones are positively correlated with platelet counts, supporting the important role of NETosis in COVID-19-related thrombosis[176,177]. Interestingly, hypoxic patients exhibit increased NETosis, indicating that the PaO2: FiO2 ratio correlates inversely with NETs levels. In particular, neutrophil activation and ROS production lead to the low oxygen availability in tissues, necessitating mechanical ventilation in ARDS patients[178]. However, mechanical ventilation can induce the formation of NETs, such that ARDS patients receiving mechanical ventilation have higher levels of cfDNA and MPO[175,179]. Thus, a more comprehensive analysis of the impact of oxygen levels and mechanical power on NETosis may provide a better evaluation of the role of neutrophils in ARDS pathogenesis. NETs components, particularly NE, also contribute to pathogen-induced lung injury. Genetic absence of NE impairs the generation of NETs in mice with P. aeruginosa and methicillin-resistant S. aureus infection[180,181]. Increased NE disrupts the integrity of the alveolar-capillary barrier, which promotes the degradation of cell adhesion molecules such as E-cadherin and VE-cadherin[182]. Collectively, these findings emphasize the damaging effect of NETs in pathogen-induced ARDS. However, activation of NETosis is also important in the context of sterile inflammation. In transfusion-induced ALI, platelet activation enhances the release of NETs in the lungs, which impairs the integrity of endothelial cells and promotes the accumulation of platelets, eventually resulting in thrombus formation and lung injury[183]. Notably, this mechanism also explains the formation of microvascular thrombosis in SARS-CoV-2 infection[184]. Activated platelets facilitate NETs release, and the released NETs further promote platelet adhesion, activation, and aggregation, leading to thrombosis, which generates a detrimental feedback loop. Interestingly, a recently discovered PAD4-independent NETosis pathway may involve other RCD patterns such as pyroptosis, necroptosis, and autophagy, indicating crosstalk between NETosis and other RCD patterns[185], which might collaboratively aggravate ARDS. PAD4 promotes the expression of NLRP3 and ASC, PAD4 promotes the expression of NLRP3 and ASC, further increasing the assembly of the inflammasome, and leading to inflammasome activation[186]. In addition, GSDMD in neutrophils is also involved in nuclear permeabilization and chromatin relaxation. GSDMD pores formation in the nuclear membrane enables the caspases-11 to access the chromatin, thereby mediating the degradation of histones and chromatin decondensation[187]. Furthermore, GSDMD pores in plasma membrane also enable the release of NETs[188]. Genetic deletion of GSDMD inhibits the production of NETs, which eventually attenuates the development of ARDS[189]. Lastly, delayed clearance of NETs also aggravates lung injury. The impaired ability of macrophages to remove NETs is associated with sustained inflammation in ARDS patients[172]. Increasing the levels of proinflammatory macrophages enhances the removal of NETs[190]. These findings highlight the importance of balanced NETs production and macrophage-mediated clearance in ARDS pathogenesis.
7. Crosstalk Among Cell Death Pathways in ARDS
Cell death pathways have originally been considered to function mutually exclusively with little or no overlap. However, accumulating evidence reveals that the biological relevance of different RCD patterns is complex, and various RCD pathways appear to operate synergistically to eliminate cells rather than exerting unique and isolated roles[22] (Figure 5). Classically, under certain conditions, apoptosis, pyroptosis and necroptosis are tightly connected and converge into PANoptosis, which is mediated by the PANoptosome. The critical role of caspase-8 as the mediator of PANoptosis was the earliest-discovered bridge between apoptosis and necroptosis. Mechanistically, FADD-mediated activation of caspase-8 triggers apoptosis, whereas inhibition of caspase-8 promotes necrosome formation and increases MLKL phosphorylation, thereby enhancing necroptosis[191]. Regarding pyroptosis, multiple studies have demonstrated that caspase-8 can function upstream of NLRP3, thereby promoting caspase-1 activation and IL-1β maturation[192]. During IAV infection, the pathogen activates ZBP1, which subsequently triggers NLRP3 activation and ultimately induces RIPK3/caspase-8-dependent cell death[19]. In line with this, RIPK3 has been reported to regulate caspase-8 activity and, consequently, NLRP3 inflammasome activation. In the absence of IAP proteins, stimuli such as LPS, TNF, or dsDNA can engage RIPK3 to promote caspase-8 activation, leading to apoptosis and NLRP3-dependent caspase-1 activation[193-195]. IAV-infected mice with RIPK3 absence inhibit the caspase-8-dependent apoptosis, which impairs the ability of virus clearance, eventually resulting in pulmonary edema and increased alveolar fibrin deposition[21]. However, in the presence of functional caspase-8, RIPK3 kinase activity is dispensable for NLRP3 activation and IL-1β release. Moreover, under these conditions, caspase-8 can directly cleave GSDMD, promoting membrane permeabilization and thereby facilitating NLRP3 activation and pyroptotic cell death as part of host defense against infection. However, the activation of NLRP3 and IL-1β can also be induced in a RIPK1/RIPK3 dependent and MLKL-independent manner during viral infection, indicating that RIPK1/RIPK3 can promote inflammasome activation through a necroptosis-independent signaling axis rather than via canonical MLKL-mediated necroptotic execution. Interestingly, in polymicrobial sepsis-induced multiorgan dysfunction, both RIPK3-dependent necroptosis and GSDMD-dependent pyroptosis pathways are activated and collaborate to amplify necroinflammation and DAMP release in macrophages and endothelial cells, ultimately aggravating lung injury[196]. Thus, given the tight crosstalk among apoptosis, necroptosis, and pyroptosis, the concept of PANoptosis was proposed to describe their functions in diseases. PANoptosis has been implicated in septic lung injury, which is associated with massive cytokine release. Inhibition of PANoptosis by simultaneously downregulating ZBP1, GSDMD, caspase-3/8, and MLKL can suppress cytokine storm and ultimately ameliorate lung injury[197]. Thus, these findings emphasize that the host employs multiple cell death pathways rather than a single mechanism to combat pathogen spread.
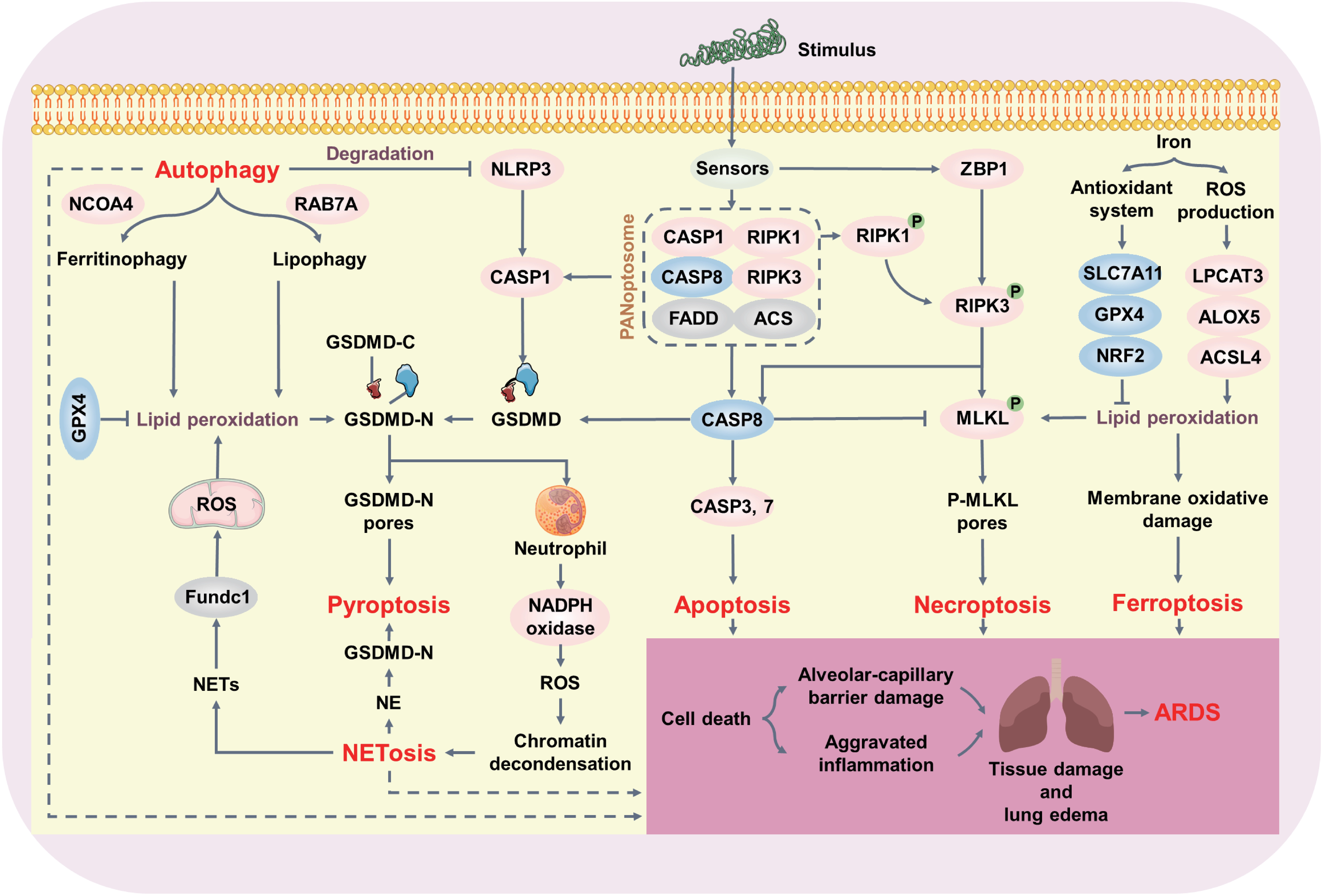
Figure 5. Crosstalk among cell death pathways in ARDS. Diverse initiator and effector molecules involved in various RCD types are interchangeably used to execute cell death. For example, activated CASP8 not only induces apoptosis but also promotes GSDMD cleavage to trigger pyroptotic cell death. Additionally, procaspase-8 assembly with RIPK1 and FADD can activate necroptosis, indicating connectivity between apoptosis and necroptosis. Lipid peroxidation is an emerging mechanism linking various forms of cell death. Excessive lipid peroxidation, the prominent feature of ferroptosis, can activate executioners of pyroptosis (e.g., GSDMD) and necroptosis (e.g., MLKL). Autophagy is also involved in lipid peroxidation through ferritinophagy and lipophagy while promoting NLRP3 degradation, suggesting that autophagy is an important regulatory mechanism for pyroptosis and ferroptosis. NETosis occurs downstream of other RCD patterns and can be triggered by activated GSDMD. Released NE and NETs, in turn, induce necroptosis and ferroptosis, respectively. Ultimately, these cell death patterns coordinate and cooperate to promote ARDS development. ARDS: acute respiratory distress syndrome; RCD: regulated cell death; CASP: caspase; GSDMD: gasdermin D; RIPK1: receptor-interacting protein kinase 1; FADD: Fas-associated protein with death domain; MLKL: mixed lineage kinase domain-like; NLRP3: NOD-like receptor family, pyrin domain-containing protein 3; NE: neutrophil elastase; NETs: neutrophil extracellular traps.
Oxidative stress is another pivotal mechanism in ARDS pathogenesis that plays a crucial role in crosstalk among various cell death patterns. In particular, GPX4, as a peroxidation inhibiting protein on plasma, protects cells from oxidative damage. For instance, GPX4 knockout followed by uncontrolled lipid peroxidation can notably drive GSDMD-mediated pyroptosis in lethal polymicrobial sepsis, indicating pyroptosis initiation through ferroptotic processes[28]. Impaired GPX4 function in macrophages promotes the activation of caspase-11-mediated noncanonical inflammasome, leading to generation of activated N-terminal of GSDMD. Furthermore, GPX4 also inhibits the expression of caspase-1-dependent NLRP3 inflammasome, indicating that GPX4 has a broad role in regulating pyroptosis. In contrast, treatment with lipid peroxides (e.g., 4-hydroxynonenal (4-HNE)) at physiological concentrations or increased endogenous 4-HNE by inhibiting GPX4 suppresses NLRP3 activation and further reduces lung inflammation and injury[198]. These paradoxical findings emphasize that lipid peroxidation products exert concentration-dependent, cell type-specific, and temporally dynamic effects on inflammasome regulation, warranting further investigation into the precise thresholds and molecular targets governing the transition from anti-inflammatory to pro-pyroptotic signaling. In addition, oxidative stress has been considered an essential downstream effector of the RIPK1-RIPK3 pathway. TNF-α dramatically triggers ferritin degradation and increases reactive iron to participate in the Fenton reaction, thereby resulting in lipid ROS production and ultimately ferroptosis. Interestingly, RIPK3 deletion does not inhibit lipid peroxidation driven by GPX4 deficiency, indicating that lipid peroxidation-mediated ferroptosis is an upstream effector of the necrosome independent of TNF-α stimulation[27,199]. In a transgenic animal with anemia, deletion of GPX4 aggravates lipid peroxidation that triggers RIPK3-dependent necroptosis in erythrocytes. However, additional work should elucidate whether lipid peroxidation products affect the posttranslational modification of proteins in necroptotic pathway. As for autophagy, NCOA4-mediated ferritinophagy is essential for cellular metabolic stress, whereas disrupting iron balance and contributes to pathological process. Inhibiting the expression of NCOA4 and FTH1 in macrophages remarkably improves the sepsis-induced ARDS[168,200]. RAB7A-mediated lipophagy is another autophagic process that selectively degrades lipid droplets to generate ROS, which may exacerbate organ damage[201]. However, the role of lipophagy-induced peroxidation in ARDS pathogenesis still lacks the direct evidence.
As mentioned above, NETosis may occur downstream of other cell death modalities, including pyroptosis, necroptosis, and ferroptosis. Activated GSDMD in neutrophils is required for NET extrusion, which can subsequently trigger pulmonary inflammation[189]. Interestingly, components of secreted NETs (e.g., NE) can also act upstream of caspases to produce GSDMD fragments, thereby indicating a mutually reinforcing feedback loop between NETosis and pyroptosis[202]. This bidirectional interaction may further compromise host defense and exacerbate inflammatory damage in sepsis-induced ALI. Moreover, the interplay between pyroptosis and NETosis creates a vicious cycle in ARDS pathogenesis. Pyroptosis-derived IL-1β and IL-18 not only amplify inflammatory responses but also prime neutrophils for enhanced NET formation, while NETs themselves serve as a scaffold for inflammasome assembly and activation in adjacent immune cells. This cascade effect contributes to the overwhelming inflammatory storm characteristic of severe ARDS[188,203]. Similarly, such a reciprocal relationship is also observed between ferroptosis and NETosis. In LPS-induced sepsis, lipid peroxidation directly damages granule membranes and increases lysosomal membrane permeability, resulting in NET release[188]. Elevated NET formation conversely promotes mitochondrial ROS production and lipid peroxidation, thereby leading to endothelial cell ferroptosis[204]. This ferroptosis-NETosis axis is particularly detrimental in ARDS, as endothelial ferroptosis disrupts the alveolar-capillary barrier, leading to increased vascular permeability and pulmonary edema, hallmarks of ARDS[146]. Furthermore, ferroptotic endothelial cells release DAMPs that further activate neutrophils and perpetuate NET formation, establishing another pathological feedback loop[205]. Recently, ferroptosis has been considered a type of autophagy-dependent cell death due to degradation of anti-ferroptotic proteins such as ferritin, GPX4, and SLC40A1/ferroportin-1, as well as lipid droplets[206]. Inhibiting ferroptosis by reducing autophagy has proven beneficial for improving ALI[207]. However, the role of autophagy in ARDS is context-dependent and exhibits a dual nature. While excessive autophagy may promote ferroptosis by degrading protective proteins, basal autophagy is essential for maintaining cellular homeostasis and limiting inflammation[208]. In alveolar epithelial cells, autophagy-mediated clearance of damaged mitochondria (mitophagy) prevents excessive ROS accumulation and subsequent ferroptosis, thereby preserving epithelial barrier integrity[209]. Regarding pyroptosis, autophagy appears to block pyroptosis by eliminating DAMPs, PAMPs, and pyroptotic proteins (e.g., NLRP3 and GSDMD). Inhibition of autophagy through ATG7 deficiency dampens autophagosome formation in alveolar macrophages and promotes inflammasome activation, thereby triggering pyroptosis and resulting in pulmonary neutrophil infiltration and severe lung injury[163]. Mechanistically, selective autophagy (xenophagy and mitophagy) targets inflammasome components for lysosomal degradation, effectively serving as a negative feedback mechanism to prevent excessive pyroptosis[210]. Disruption of this regulatory mechanism, as observed in ARDS patients with autophagy gene polymorphisms, is associated with heightened inflammasome activity and worse clinical outcomes[161].
The temporal dynamics of cell death pathway activation further complicate ARDS pathogenesis. During the early exudative phase, pyroptosis and NETosis predominate, driving acute inflammation and barrier disruption. As ARDS progresses to the proliferative phase, ferroptosis and necroptosis become more prominent, contributing to fibroproliferative responses and impaired tissue repair. This sequential activation pattern suggests that therapeutic strategies targeting cell death pathways may need to be tailored to specific disease stages. Collectively, these findings highlight that multiple cell death pathways do not operate in isolation but rather coordinate and interact through complex molecular networks to regulate inflammatory responses and immune defense, which are crucial for ARDS pathogenesis. Understanding these intricate interactions is essential for developing more effective therapeutic strategies that can simultaneously target multiple pathological mechanisms.
8. Conclusions and Future Perspectives
RCD occurs through various subroutines that cause cells to undergo lysis in different ways, leading to distinct morphological changes and immune consequences. In ARDS, although cell death is beneficial for pathogen elimination, excessive cell death can cause systemic inflammation and pathology. Consequently, ARDS patients must carefully balance cell death activation to prevent uncontrolled inflammation while simultaneously promoting pathogen clearance to limit infectious disease progression. This delicate equilibrium is particularly challenging because the threshold between protective and pathological cell death varies dynamically depending on disease stage, pathogen load, host immune status, and underlying comorbidities. Clinical observations reveal that patients with hyperinflammatory ARDS phenotypes exhibit elevated levels of cell death markers (e.g., circulating cell-free DNA, HMGB1, and histones) compared to those with hypoinflammatory phenotypes, suggesting that personalized modulation of cell death pathways may be necessary[211,212]. Accumulating evidence suggests that multiple cell death pathways collectively participate in ARDS pathogenesis through their distinct mechanisms. Recent single-cell transcriptomic analyses of ARDS lung tissue have revealed substantial heterogeneity in cell death pathway activation across different cell populations. While alveolar macrophages predominantly exhibit pyroptotic signatures, alveolar epithelial type II cells show enrichment of ferroptosis-related genes, and endothelial cells display markers of both apoptosis and necroptosis[213,214]. This cell-type-specific engagement of RCD pathways underscores the complexity of therapeutic targeting and suggests that comprehensive, multi-pathway approaches may be more effective than single-target strategies. Importantly, establishing a standardized panel of biomarkers and functional assays, including genetic and pharmacological approaches, to identify whether RCD is occurring in a single or mixed form is required. Current diagnostic challenges include the lack of specific biomarkers that can discriminate between different RCD modalities in clinical samples. While GSDMD-N terminal fragments indicate pyroptosis and phosphorylated MLKL suggests necroptosis, these markers often coexist in ARDS patients, complicating interpretation[215]. The distinct lethal subroutines that operate either in parallel, in a hierarchical fashion, or through dynamic transitions require further exploration. Particularly, longitudinal monitoring of RCD biomarkers in BALF and plasma may enable real-time assessment of disease trajectory and therapeutic response, facilitating adaptive treatment strategies. Currently, the emerging connectivity of RCD pathways and its physiological implications in ARDS are gradually gaining attention. Systems biology approaches integrating proteomics, metabolomics, and transcriptomics data have begun to reveal unexpected molecular hubs that integrate multiple cell death signals. Disruption of these regulatory nodes in ARDS may lead to dysregulated, uncontrolled cell death. Understanding these integration points could reveal novel therapeutic opportunities for simultaneously modulating multiple pathways. Additional research is needed to better understand the contexts in which RCD pathways are coordinated, to elucidate the functional roles of this crosstalk in ARDS, and to determine why these interactions vary under different conditions. Finally, regardless of the underlying biology and complex mechanisms governing cell death, targeting cell death pathways holds enormous promise. The development of novel therapeutic agents that selectively inhibit or activate RCD pathways has great potential for preventing overwhelming inflammation and facilitating pathogen elimination. Several promising therapeutic strategies are currently under investigation. Necrostatin-1 and its derivatives, which inhibit RIPK1-mediated necroptosis, have shown efficacy in preclinical ARDS models and are being evaluated in early-phase clinical trials[57]. NLRP3 inflammasome inhibitors such as MCC950 effectively reduce pyroptosis-driven inflammation and are progressing toward clinical development[216]. Ferroptosis inhibitors including ferrostatin-1 and liproxstatin-1 demonstrate protective effects against VILI[146]. Importantly, combination therapies targeting multiple cell death pathways simultaneously may offer superior efficacy compared to monotherapies. For example, dual inhibition of necroptosis and pyroptosis synergistically reduces lung injury severity in experimental models[217]. Beyond small-molecule inhibitors, emerging modalities such as siRNA-based therapeutics, antibody-drug conjugates targeting cell death executioners, and engineered extracellular vesicles for targeted delivery of anti-cell death cargo represent innovative approaches with potential for clinical translation[218].
Thus, understanding the mechanisms of various cell death processes in ARDS at the molecular level may help identify therapeutic targets for lung injury and improve patient outcomes. Future research directions should prioritize: (1) developing high-resolution spatial mapping technologies to visualize cell death pathway activation in situ within intact lung tissue; (2) establishing standardized protocols for RCD biomarker measurement to enable cross-study comparisons and meta-analyses; (3) conducting mechanistic studies to identify druggable regulatory nodes that control cell death pathway selection; (4) performing preclinical studies in large animal models that better recapitulate human ARDS pathophysiology; and (5) designing adaptive clinical trials that incorporate real-time biomarker monitoring to optimize treatment strategies. By integrating these multidisciplinary approaches, we can transform our understanding of cell death in ARDS from descriptive observations to actionable therapeutic insights, ultimately improving outcomes for this devastating syndrome.
Authors contribution
Huang Y: Conceptualization, funding acquisition.
Zheng Y: Conceptualization, funding acquisition, writing-original draft, visualization.
Zhong N, Sang L: Writing-review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This study was funded by National Key Research and Development Program of China (Grant No. 2022YFC2504402), National Natural Science Foundation of China (Grant No. 82270081), Major Project of Guangzhou National Laboratory (Grant No. GZNL2023A03004), and the Clinical and Epidemiological Research Project of State Key Laboratory of Respiratory Disease (Grant No. SKLRD-L-202503).
Copyright
© The Author(s) 2026.
References
-
1. Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute respiratory distress syndrome: The berlin definition. JAMA. 2012;307(23):2526-2533.[DOI]
-
2. Matthay MA, Arabi Y, Arroliga AC, Bernard G, Bersten AD, Brochard LJ, et al. A new global definition of acute respiratory distress syndrome. Am J Respir Crit Care Med. 2024;209(1):37-47.[DOI]
-
3. Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American-European consensus conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149(3):818-824.[DOI]
-
4. Raghavendran K, Napolitano LM. Definition of ALI/ARDS. Crit Care Clin. 2011;27(3):429-437.[DOI]
-
5. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5:18.[DOI]
-
6. Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. Lancet. 2021;398(10300):622-637.[DOI]
-
7. Bos LDJ, Ware LB. Acute respiratory distress syndrome: Causes, pathophysiology, and phenotypes. Lancet. 2022;400(10358):1145-1156.[DOI]
-
8. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8):788.[DOI]
-
9. Zoulikha M, Xiao Q, Boafo GF, Sallam MA, Chen Z, He W. Pulmonary delivery of siRNA against acute lung injury/acute respiratory distress syndrome. Acta Pharm Sin B. 2022;12(2):600-620.[DOI]
-
10. Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med. 1982;3(1):35-56.[DOI]
-
11. Newton K, Strasser A, Kayagaki N, Dixit VM. Cell death. Cell. 2024;187(2):235-256.[DOI]
-
12. Libby P, Lüscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. 2020;41(32):3038-3044.[DOI]
-
13. Zelic M, Roderick JE, O’Donnell JA, Lehman J, Lim SE, Janardhan HP, et al. RIP kinase 1-dependent endothelial necroptosis underlies systemic inflammatory response syndrome. J Clin Invest. 2018;128(5):2064-2075.[DOI]
-
14. Zhuang W, Zhou J, Zhong L, Lv J, Zhong X, Liu G, et al. CXCR1 drives the pathogenesis of EAE and ARDS via boosting dendritic cells-dependent inflammation. Cell Death Dis. 2023;14(9):608.[DOI]
-
15. Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. 2017;127(11):4124-4135.[DOI]
-
16. Li B, Liu J, He W, Zhou Y, Zhao M, Xia C, et al. Inhibition of macrophage inflammasome assembly and pyroptosis with GC-1 ameliorates acute lung injury. Theranostics. 2025;15(6):2360-2374.[DOI]
-
17. Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: An auto-amplification loop causes organ failure. Nat Rev Immunol. 2014;14(11):759-767.[DOI]
-
18. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13(12):862-874.[DOI]
-
19. Kuriakose T, Man SM, Malireddi RKS, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1(2):eaag2045.[DOI]
-
20. Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, et al. DAI senses influenza a virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe. 2016;20(5):674-681.[DOI]
-
21. Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH, Ingram JP, et al. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza a virus. Cell Host Microbe. 2016;20(1):13-24.[DOI]
-
22. Bedoui S, Herold MJ, Strasser A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol. 2020;21(11):678-695.[DOI]
-
23. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99-103.[DOI]
-
24. Tsuchiya K, Nakajima S, Hosojima S, Nguyen DT, Hattori T, Le TM, et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat Commun. 2019;10:2091.[DOI]
-
25. Zhang JY, Zhou B, Sun RY, Ai YL, Cheng K, Li FN, et al. The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res. 2021;31(9):980-997.[DOI]
-
26. Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem Biol. 2017;24(4):507-514.[DOI]
-
27. Canli Ö, Alankuş YB, Grootjans S, Vegi N, Hültner L, Hoppe PS, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127(1):139-148.[DOI]
-
28. Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. 2018;24(1):97-108.[DOI]
-
29. Yuan J, Ofengeim D. A guide to cell death pathways. Nat Rev Mol Cell Biol. 2024;25(5):379-395.[DOI]
-
30. Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell. 1996;86(1):147-157.[DOI]
-
31. Green DR. The mitochondrial pathway of apoptosis part II: The BCL-2 protein family. Cold Spring Harb Perspect Biol. 2022;14(6):a041046.[DOI]
-
32. Lakhani SA, Masud A, Kuida K, Porter JGA, Booth CJ, Mehal WZ, et al. Caspases 3 and 7: Key mediators of mitochondrial events of apoptosis. Science. 2006;311(5762):847-851.[DOI]
-
33. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2):181-190.[DOI]
-
34. Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, et al. Compartmentalization of TNF receptor 1 signaling internalized TNF receptosomes as death signaling vesicles. Immunity. 2004;21(3):415-428.[DOI]
-
35. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491-501.[DOI]
-
36. Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94(4):481-490.[DOI]
-
37. Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14(16):2060-2071.[DOI]
-
38. Bachofen M, Weibel ER. Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am Rev Respir Dis. 1977;116(4):589-615.[DOI]
-
39. Fujita M, Kuwano K, Kunitake R, Hagimoto N, Miyazaki H, Kaneko Y, et al. Endothelial cell apoptosis in lipopolysaccharide-induced lung injury in mice. Int Arch Allergy Immunol. 1998;117(3):202-208.[DOI]
-
40. Matute-Bello G, Liles WC, Steinberg KP, Kiener PA, Mongovin S, Chi EY, et al. Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J Immunol. 1999;163(4):2217-2225.[DOI]
-
41. Albertine KH, Soulier MF, Wang Z, Ishizaka A, Hashimoto S, Zimmerman GA, et al. Fas and Fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am J Pathol. 2002;161(5):1783-1796.[DOI]
-
42. Glavan BJ, Holden TD, Goss CH, Black RA, Neff MJ, Nathens AB, et al. Genetic variation in the FAS Gene and associations with acute lung injury. Am J Respir Crit Care Med. 2011;183(3):356-363.[DOI]
-
43. Liang GP, Xu J, Cao LL, Zeng YH, Chen BX, Yang J, et al. Piezo1 induced apoptosis of type II pneumocytes during ARDS. Respir Res. 2019;20:118.[DOI]
-
44. Wang Y, Yang Y, Chen L, Xiong W, Song L, Li B, et al. Death-associated protein kinase 1 mediates ventilator-induced lung injury in mice by promoting alveolar epithelial cell apoptosis. Anesthesiology. 2020;133(4):905-918.[DOI]
-
45. Chen R, Zou J, Liu J, Kang R, Tang D. DAMPs in the immunogenicity of cell death. Mol Cell. 2025;85(20):3874-3889.[DOI]
-
46. Tanzer MC, Frauenstein A, Stafford CA, Phulphagar K, Mann M, Meissner F. Quantitative and dynamic catalogs of proteins released during apoptotic and necroptotic cell death. Cell Rep. 2020;30(4):1260-1270.[DOI]
-
47. Liu G, Park YJ, Tsuruta Y, Lorne E, Abraham E. p53 attenuates lipopolysaccharide-induced NF-κB activation and acute lung injury. J Immunol. 2009;182(8):5063-5071.[DOI]
-
48. Wang JF, Wang YP, Xie J, Zhao ZZ, Gupta S, Guo Y, et al. Upregulated PD-L1 delays human neutrophil apoptosis and promotes lung injury in an experimental mouse model of sepsis. Blood. 2021;138(9):806-810.[DOI]
-
49. Song C, Li H, Mao Z, Peng L, Liu B, Lin F, et al. Delayed neutrophil apoptosis may enhance NET formation in ARDS. Respir Res. 2022;23:155.[DOI]
-
50. Lefrançais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight. 2018;3(3):e98178.[DOI]
-
51. Ning X, Wang Y, Jing M, Sha M, Lv M, Gao P, et al. Apoptotic caspases suppress type I interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol Cell. 2019;74(1):19-31.[DOI]
-
52. Medina CB, Mehrotra P, Arandjelovic S, Perry JSA, Guo Y, Morioka S, et al. Metabolites released from apoptotic cells act as tissue messengers. Nature. 2020;580(7801):130-135.[DOI]
-
53. Rao H, Ding Q, Liu A, Qiu H, Luo J. The synergistic role of P2rx7 and Panx1 in regulating alveolar macrophage pyroptosis and exosome-mediated ferroptosis of alveolar epithelial cells in lipopolysaccharide-induced acute respiratory distress syndrome. FASEB J. 2025;39(16):e70904.[DOI]
-
54. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311-320.[DOI]
-
55. Ray CA, Pickup DJ. The mode of death of pig kidney cells infected with cowpox virus is governed by the expression of the crmA Gene. Virology. 1996;217(1):384-391.[DOI]
-
56. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106-1121.[DOI]
-
57. Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313-321.[DOI]
-
58. Cho Y, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112-1123.[DOI]
-
59. Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23(8):994-1006.[DOI]
-
60. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150(2):339-350.[DOI]
-
61. Wu X, Ma Y, Zhao K, Zhang J, Sun Y, Li Y, et al. The structure of a minimum amyloid fibril core formed by necroptosis-mediating RHIM of human RIPK3. Proc Natl Acad Sci U S A. 2021;118(14):e2022933118.[DOI]
-
62. Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: Mechanisms and relevance to disease. Annu Rev Pathol Mech Dis. 2017;12:103-130.[DOI]
-
63. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213-227.[DOI]
-
64. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39(3):443-453.[DOI]
-
65. Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A. 2014;111(42):15072-15077.[DOI]
-
66. Xie Y, Zhu S, Zhong M, Yang M, Sun X, Liu J, et al. Inhibition of aurora kinase a induces necroptosis in pancreatic carcinoma. Gastroenterology. 2017;153(5):1429-1443.[DOI]
-
67. Remijsen Q, Goossens V, Grootjans S, Van den Haute C, Vanlangenakker N, Dondelinger Y, et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 2014;5(1):e1004.[DOI]
-
68. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16(1):55-65.[DOI]
-
69. Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24(1):105-121.[DOI]
-
70. Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7(4):971-981.[DOI]
-
71. Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, et al. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol Cell. 2016;61(4):589-601.[DOI]
-
72. Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, Li Q, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. 2021;591(7848):131-136.[DOI]
-
73. Noh H, Hashem Z, Boms E, Najafov A. SIGLEC12 mediates plasma membrane rupture during necroptotic cell death. Nature. 2026;649(8096):460-466.[DOI]
-
74. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1(6):489-495.[DOI]
-
75. Woznicki JA, Saini N, Flood P, Rajaram S, Lee CM, Stamou P, et al. TNF-α synergises with IFN-γ to induce caspase-8-JAK1/2-STAT1-dependent death of intestinal epithelial cells. Cell Death Dis. 2021;12(10):864.[DOI]
-
76. He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108(50):20054-20059.[DOI]
-
77. Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, et al. Cutting edge: RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol. 2014;193(4):1539-1543.[DOI]
-
78. Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157(5):1189-1202.[DOI]
-
79. Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature. 2016;540(7631):129-133.[DOI]
-
80. Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature. 2016;540(7631):124-128.[DOI]
-
81. Cuchet-Lourenço D, Eletto D, Wu C, Plagnol V, Papapietro O, Curtis J, et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science. 2018;361(6404):810-813.[DOI]
-
82. Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature. 2020;580(7803):391-395.[DOI]
-
83. Meier P, Legrand AJ, Adam D, Silke J. Immunogenic cell death in cancer: Targeting necroptosis to induce antitumour immunity. Nat Rev Cancer. 2024;24(5):299-315.[DOI]
-
84. Snyder AG, Hubbard NW, Messmer MN, Kofman SB, Hagan CE, Orozco SL, et al. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol. 2019;4(36):eaaw2004.[DOI]
-
85. Chen KS, Manoury-Battais S, Kanaya N, Vogiatzi I, Borges P, Kruize SJ, et al. An inducible RIPK3-driven necroptotic system enhances cancer cell-based immunotherapy and ensures safety. J Clin Invest. 2025;135(2):e181143.[DOI]
-
86. Greenlee-Wacker MC, Rigby KM, Kobayashi SD, Porter AR, DeLeo FR, Nauseef WM. Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J Immunol. 2014;192(10):4709-4717.[DOI]
-
87. Philip NH, Dillon CP, Snyder AG, Fitzgerald P, Wynosky-Dolfi MA, Zwack EE, et al. Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-κB and MAPK signaling. Proc Natl Acad Sci U S A. 2014;111(20):7385-7390.[DOI]
-
88. Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci U S A. 2014;111(20):7391-7396.[DOI]
-
89. Gautam A, Boyd DF, Nikhar S, Zhang T, Siokas I, Van de Velde LA, et al. Necroptosis blockade prevents lung injury in severe influenza. Nature. 2024;628(8009):835-843.[DOI]
-
90. Shubina M, Tummers B, Boyd DF, Zhang T, Yin C, Gautam A, et al. Necroptosis restricts influenza A virus as a stand-alone cell death mechanism. J Exp Med. 2020;217(11):e20191259.[DOI]
-
91. Shashaty MGS, Reilly JP, Faust HE, Forker CM, Ittner CAG, Zhang PX, et al. Plasma receptor interacting protein kinase-3 levels are associated with acute respiratory distress syndrome in sepsis and trauma: A cohort study. Crit Care. 2019;23:235.[DOI]
-
92. Nakamura H, Kinjo T, Arakaki W, Miyagi K, Tateyama M, Fujita J. Serum levels of receptor-interacting protein kinase-3 in patients with COVID-19. Crit Care. 2020;24:484.[DOI]
-
93. Ma KC, Schenck EJ, Siempos II, Cloonan SM, Finkelzstein EJ, Pabon MA, et al. Circulating RIPK3 levels are associated with mortality and organ failure during critical illness. JCI Insight. 2018;3(13):e99692.[DOI]
-
94. Vucur M, Roderburg C, Kaiser L, Schneider AT, Roy S, Loosen SH, et al. Elevated serum levels of mixed lineage kinase domain-like protein predict survival of patients during intensive care unit treatment. Dis Markers. 2018;2018:1983421.[DOI]
-
95. Siempos II, Ma KC, Imamura M, Baron RM, Fredenburgh LE, Huh JW, et al. RIPK3 mediates pathogenesis of experimental ventilator-induced lung injury. JCI Insight. 2018;3(9):e97102.[DOI]
-
96. Ruskowski K, Neb H, Talbot SR, Choorapoikayil S, Adam EH, von Knethen A, et al. Persistently elevated plasma concentrations of RIPK3, MLKL, HMGB1, and RIPK1 in patients with COVID-19 in the intensive care unit. Am J Respir Cell Mol Biol. 2022;67(3):405-408.[DOI]
-
97. Li S, Zhang Y, Guan Z, Ye M, Li H, You M, et al. SARS-CoV-2 Z-RNA activates the ZBP1-RIPK3 pathway to promote virus-induced inflammatory responses. Cell Res. 2023;33(3):201-214.[DOI]
-
98. Komiya Y, Kamiya M, Oba S, Kawata D, Iwai H, Shintaku H, et al. Necroptosis in alveolar epithelial cells drives lung inflammation and injury caused by SARS-CoV-2 infection. Biochim Biophys Acta Mol Basis Dis. 2024;1870(8):167472.[DOI]
-
99. Han Y, Zhu J, Yang L, Nilsson-Payant BE, Hurtado R, Lacko LA, et al. SARS-CoV-2 infection induces ferroptosis of sinoatrial node pacemaker cells. Circ Res. 2022;130(7):963-977.[DOI]
-
100. Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016;23(1):76-88.[DOI]
-
101. Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol. 2016;26(13):R568-R572.[DOI]
-
102. Broz P, Dixit VM. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407-420.[DOI]
-
103. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660-665.[DOI]
-
104. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1βprocessing in monocytes. Nature. 1992;356(6372):768-774.[DOI]
-
105. Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, et al. Activation of interferon-γ inducing factor mediated by interleukin-1β converting enzyme. Science. 1997;275(5297):206-209.[DOI]
-
106. Cui Y, Yang Y, Tao W, Peng W, Luo D, Zhao N, et al. Neutrophil extracellular traps induce alveolar macrophage pyroptosis by regulating NLRP3 deubiquitination, aggravating the development of septic lung injury. J Inflamm Res. 2023;16:861-877.[DOI]
-
107. Liu W, Ren Y, Wang T, Wang M, Xu Y, Zhang J, et al. Blocking CIRP protects against acute pancreatitis by improving mitochondrial function and suppressing pyroptosis in acinar cells. Cell Death Discov. 2024;10:156.[DOI]
-
108. Wright SS, Kumari P, Fraile-Ágreda V, Wang C, Shivcharan S, Kappelhoff S, et al. Transplantation of gasdermin pores by extracellular vesicles propagates pyroptosis to bystander cells. Cell. 2025;188(2):280-291.[DOI]
-
109. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117-121.[DOI]
-
110. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: Mechanisms and diseases. Sig Transduct Target Ther. 2021;6:128.[DOI]
-
111. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187-192.[DOI]
-
112. Santos JC, Dick MS, Lagrange B, Degrandi D, Pfeffer K, Yamamoto M, et al. LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. EMBO J. 2018;37(6):e98089.[DOI]
-
113. Santos JC, Boucher D, Schneider LK, Demarco B, Dilucca M, Shkarina K, et al. Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat Commun. 2020;11:3276.[DOI]
-
114. Pourmal S, Truong ME, Johnson MC, Yang Y, Zhou L, Alegre K, et al. Autoinhibition of dimeric NINJ1 prevents plasma membrane rupture. Nature. 2025;637(8045):446-452.[DOI]
-
115. Dondelinger Y, Priem D, Huyghe J, Delanghe T, Vandenabeele P, Bertrand MJM. NINJ1 is activated by cell swelling to regulate plasma membrane permeabilization during regulated necrosis. Cell Death Dis. 2023;14(11):755.[DOI]
-
116. Wang Y, Liu Y, Liu Q, Zheng Q, Dong X, Liu X, et al. Caspase-1-dependent pyroptosis of peripheral blood mononuclear cells is associated with the severity and mortality of septic patients. BioMed Res Int. 2020;2020:9152140.[DOI]
-
117. Peukert K, Fox M, Schulz S, Feuerborn C, Frede S, Putensen C, et al. Inhibition of caspase-1 with tetracycline ameliorates acute lung injury. Am J Respir Crit Care Med. 2021;204(1):53-63.[DOI]
-
118. Xu Y, Zheng Y, Liu Y, Wei C, Ren J, Zuo W, et al. Ninjurin-1 mediates cell lysis and detrimental inflammation of PANoptosis during influenza A virus infection. Sig Transduct Target Ther. 2025;10:307.[DOI]
-
119. Zhu Y, Xiao F, Wang Y, Wang Y, Li J, Zhong D, et al. NINJ1 regulates plasma membrane fragility under mechanical strain. Nature. 2025;644(8078):1088-1096.[DOI]
-
120. Wang J, Sahoo M, Lantier L, Warawa J, Cordero H, Deobald K, et al. Caspase-11-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLoS Pathog. 2018;14(5):e1007105.[DOI]
-
121. Zhang H, Zeng L, Xie M, Liu J, Zhou B, Wu R, et al. TMEM173 drives lethal coagulation in sepsis. Cell Host Microbe. 2020;27(4):556-570.[DOI]
-
122. Schiffelers LDJ, Tesfamariam YM, Jenster LM, Diehl S, Binder SC, Normann S, et al. Antagonistic nanobodies implicate mechanism of GSDMD pore formation and potential therapeutic application. Nat Commun. 2024;15:8266.[DOI]
-
123. Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. 2018;362(6417):956-960.[DOI]
-
124. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-1072.[DOI]
-
125. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285-296.[DOI]
-
126. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15(3):234-245.[DOI]
-
127. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12(8):599-620.[DOI]
-
128. Hadian K, Stockwell BR. The therapeutic potential of targeting regulated non-apoptotic cell death. Nat Rev Drug Discov. 2023;22(9):723-742.[DOI]
-
129. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180-1191.[DOI]
-
130. Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34(4):496-502.[DOI]
-
131. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688-692.[DOI]
-
132. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693-698.[DOI]
-
133. Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022;608(7924):778-783.[DOI]
-
134. Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. 2022;82(12):2215-2227.[DOI]
-
135. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10(7):1604-1609.[DOI]
-
136. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91-98.[DOI]
-
137. Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81-90.[DOI]
-
138. Lin Z, Liu J, Long F, Kang R, Kroemer G, Tang D, et al. The lipid flippase SLC47A1 blocks metabolic vulnerability to ferroptosis. Nat Commun. 2022;13:7965.[DOI]
-
139. Zhang HL, Hu BX, Li ZL, Du T, Shan JL, Ye ZP, et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol. 2022;24(1):88-98.[DOI]
-
140. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585(7826):603-608.[DOI]
-
141. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966-E4975.[DOI]
-
142. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21(5):579-591.[DOI]
-
143. Dar HH, Mikulska-Ruminska K, Tyurina YY, Luci DK, Yasgar A, Samovich SN, et al. Discovering selective antiferroptotic inhibitors of the 15LOX/PEBP1 complex noninterfering with biosynthesis of lipid mediators. Proc Natl Acad Sci U S A. 2023;120(25):e2218896120.[DOI]
-
144. Lawson DM, Treffry A, Artymiuk PJ, Harrison PM, Yewdall SJ, Luzzago A, et al. Identification of the ferroxidase centre in ferritin. FEBS Lett. 1989;254:207-210.[DOI]
-
145. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105-109.[DOI]
-
146. Yoshida M, Minagawa S, Araya J, Sakamoto T, Hara H, Tsubouchi K, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10:3145.[DOI]
-
147. Riegman M, Sagie L, Galed C, Levin T, Steinberg N, Dixon SJ, et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol. 2020;22(9):1042-1048.[DOI]
-
148. Hirata Y, Cai R, Volchuk A, Steinberg BE, Saito Y, Matsuzawa A, et al. Lipid peroxidation increases membrane tension, Piezo1 gating, and cation permeability to execute ferroptosis. Curr Biol. 2023;33(7):1282-1294.[DOI]
-
149. Ramos S, Hartenian E, Santos JC, Walch P, Broz P. NINJ1 induces plasma membrane rupture and release of damage-associated molecular pattern molecules during ferroptosis. EMBO J. 2024;43(7):1164-1186.[DOI]
-
150. Ma A, Feng Z, Li Y, Wu Q, Xiong H, Dong M, et al. Ferroptosis-related signature and immune infiltration characterization in acute lung injury/acute respiratory distress syndrome. Respir Res. 2023;24:154.[DOI]
-
151. Van Coillie S, Van San E, Goetschalckx I, Wiernicki B, Mukhopadhyay B, Tonnus W, et al. Targeting ferroptosis protects against experimental (multi)organ dysfunction and death. Nat Commun. 2022;13:1046.[DOI]
-
152. Peleman C, Van Coillie S, Ligthart S, Choi SM, De Waele J, Depuydt P, et al. Ferroptosis and pyroptosis signatures in critical COVID-19 patients. Cell Death Differ. 2023;30(9):2066-2077.[DOI]
-
153. Ouyang A, Chen T, Feng Y, Zou J, Tu S, Jiang M, et al. The hemagglutinin of influenza a virus induces ferroptosis to facilitate viral replication. Adv Sci. 2024;11(39):2404365.[DOI]
-
154. Zheng Y, Zhang Y, Chen Y, Deng X, Liu B, Xu Q, et al. Indoleamine 2,3-dioxygenase 1 drives epithelial cells ferroptosis in influenza-induced acute lung injury. Redox Biol. 2025;81:103572.[DOI]
-
155. Liu C, Wu X, Bing X, Qi W, Zhu F, Guo N, et al. H1N1 influenza virus infection through NRF2-KEAP1-GCLC pathway induces ferroptosis in nasal mucosal epithelial cells. Free Radic Biol Med. 2023;204:226-242.[DOI]
-
156. Amaral EP, Costa DL, Namasivayam S, Riteau N, Kamenyeva O, Mittereder L, et al. A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J Exp Med. 2019;216(3):556-570.[DOI]
-
157. Amaral EP, Foreman TW, Namasivayam S, Hilligan KL, Kauffman KD, Barbosa Bomfim CC, et al. GPX4 regulates cellular necrosis and host resistance in Mycobacterium tuberculosis infection. J Exp Med. 2022;219(11):e20220504.[DOI]
-
158. Dar HH, Tyurina YY, Mikulska-Ruminska K, Shrivastava I, Ting HC, Tyurin VA, et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J Clin Invest. 2018;128(10):4639-4653.[DOI]
-
159. Ornatowski W, Lu Q, Yegambaram M, Garcia AE, Zemskov EA, Maltepe E, et al. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 2020;36:101679.[DOI]
-
160. Carchman EH, Rao J, Loughran PA, Rosengart MR, Zuckerbraun BS. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology. 2011;53(6):2053-2062.[DOI]
-
161. Nakahira K, Haspel JA, Rathinam VAK, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222-230.[DOI]
-
162. Zhao H, Chen H, Meng X, Yang G, Hu Y, Xie K, et al. Autophagy activation improves lung injury and inflammation in sepsis. Inflammation. 2019;42(2):426-439.[DOI]
-
163. Pu Q, Li Y, Lan L, Deng X, Liang H, Ma F, et al. Atg7 deficiency intensifies inflammasome activation and pyroptosis in Pseudomonas sepsis. J Immunol. 2017;198(8):3205-3213.[DOI]
-
164. Tang X, Zhong L, Tian X, Zou Y, Hu S, Liu J, et al. RUNX1 promotes mitophagy and alleviates pulmonary inflammation during acute lung injury. Sig Transduct Target Ther. 2023;8:288.[DOI]
-
165. López-Alonso I, Aguirre A, González-López A, Fernández ÁF, Amado-Rodríguez L, Astudillo A, et al. Impairment of autophagy decreases ventilator-induced lung injury by blockade of the NF-κB pathway. Am J Physiol Lung Cell Mol Physiol. 2013;304(12):L844-L852.[DOI]
-
166. Zhang Y, Liu G, Dull RO, Schwartz DE, Hu G. Autophagy in pulmonary macrophages mediates lung inflammatory injury via NLRP3 inflammasome activation during mechanical ventilation. Am J Physiol Lung Cell Mol Physiol. 2014;307(2):L173-L185.[DOI]
-
167. Lo S, Yuan SF, Hsu C, Cheng YJ, Chang YF, Hsueh HW, et al. Lc3 over-expression improves survival and attenuates lung injury through increasing autophagosomal clearance in septic mice. Ann Surg. 2013;257(2):352-363.[DOI]
-
168. Shao N, Yu H, Li X, Han M, Chen C, Zhu J, et al. Ferritinophagy and organ injury. Autophagy. 2026.[DOI]
-
169. Huang J, Hong W, Wan M, Zheng L. Molecular mechanisms and therapeutic target of NETosis in diseases. MedComm. 2022;3(3):e162.[DOI]
-
170. Poli V, Zanoni I. Neutrophil intrinsic and extrinsic regulation of NETosis in health and disease. Trends Microbiol. 2023;31(3):280-293.[DOI]
-
171. Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014;8(3):883-896.[DOI]
-
172. Grégoire M, Uhel F, Lesouhaitier M, Gacouin A, Guirriec M, Mourcin F, et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur Respir J. 2018;52(2):1702590.[DOI]
-
173. Ng H, Havervall S, Rosell A, Aguilera K, Parv K, von Meijenfeldt FA, et al. Circulating markers of neutrophil extracellular traps are of prognostic value in patients with COVID-19. Arterioscler Thromb Vasc Biol. 2021;41(2):988-994.[DOI]
-
174. Ouwendijk WJD, Raadsen MP, van Kampen JJA, Verdijk RM, von der Thusen JH, Guo L, et al. High levels of neutrophil extracellular traps persist in the lower respiratory tract of critically ill patients with coronavirus disease 2019. J Infect Dis. 2021;223(9):1512-1521.[DOI]
-
175. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5(11):e138999.[DOI]
-
176. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136(10):1169-1179.[DOI]
-
177. Burkard P, Schonhart C, Vögtle T, Köhler D, Tang L, Johnson D, et al. A key role for platelet GPVI in neutrophil recruitment, migration, and NETosis in the early stages of acute lung injury. Blood. 2023;142(17):1463-1477.[DOI]
-
178. Lodge KM, Vassallo A, Liu B, Long M, Tong Z, Newby PR, et al. Hypoxia increases the potential for neutrophil-mediated endothelial damage in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2022;205(8):903-916.[DOI]
-
179. Yildiz C, Palaniyar N, Otulakowski G, Khan MA, Post M, Kuebler WM, et al. Mechanical ventilation induces neutrophil extracellular trap formation. Anesthesiology. 2015;122(4):864-875.[DOI]
-
180. Kolaczkowska E, Jenne CN, Surewaard BGJ, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673.[DOI]
-
181. Thanabalasuriar A, Scott BNV, Peiseler M, Willson ME, Zeng Z, Warrener P, et al. Neutrophil extracellular traps confine pseudomonas aeruginosa ocular biofilms and restrict brain invasion. Cell Host Microbe. 2019;25(4):526-536.[DOI]
-
182. Zhou X, Jin J, Lv T, Song Y. A narrative review: The role of NETs in acute respiratory distress syndrome/acute lung injury. Int J Mol Sci. 2024;25(3):1464.[DOI]
-
183. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122(7):2661-2671.[DOI]
-
184. Nicolai L, Leunig A, Brambs S, Kaiser R, Weinberger T, Weigand M, et al. Immunothrombotic dysregulation in COVID-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation. 2020;142(12):1176-1189.[DOI]
-
185. Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, et al. Neutrophil extracellular trap formation is associated with IL-1β and autophagy-related signaling in gout. PLoS One. 2011;6(12):e29318.[DOI]
-
186. Münzer P, Negro R, Fukui S, di Meglio L, Aymonnier K, Chu L, et al. NLRP3 inflammasome assembly in neutrophils is supported by PAD4 and promotes NETosis under sterile conditions. Front Immunol. 2021;12:683803.[DOI]
-
187. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol. 2018;3(26):eaar6676.[DOI]
-
188. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3(26):eaar6689.[DOI]
-
189. Xie J, Zhu CL, Wan XJ, Zhao ZZ, Meng Y, Li P, et al. GSDMD-mediated NETosis promotes the development of acute respiratory distress syndrome. Eur J Immunol. 2023;53:2250011.[DOI]
-
190. Haider P, Kral-Pointner JB, Mayer J, Richter M, Kaun C, Brostjan C, et al. Neutrophil extracellular trap degradation by differently polarized macrophage subsets. Arterioscler Thromb Vasc Biol. 2020;40(9):2265-2278.[DOI]
-
191. Pandeya A, Kanneganti TD. Therapeutic potential of PANoptosis: Innate sensors, inflammasomes, and RIPKs in PANoptosomes. Trends Mol Med. 2024;30(1):74-88.[DOI]
-
192. Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363-367.[DOI]
-
193. Yabal M, Müller N, Adler H, Knies N, Groß CJ, Damgaard RB, et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 2014;7(6):1796-1808.[DOI]
-
194. Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282.[DOI]
-
195. Lawlor KE, Feltham R, Yabal M, Conos SA, Chen KW, Ziehe S, et al. XIAP loss triggers RIPK3- and caspase-8-driven IL-1β activation and cell death as a consequence of TLR-MyD88-induced cIAP1-TRAF2 degradation. Cell Rep. 2017;20(3):668-682.[DOI]
-
196. Chen H, Li Y, Wu J, Li G, Tao X, Lai K, et al. RIPK3 collaborates with GSDMD to drive tissue injury in lethal polymicrobial sepsis. Cell Death Differ. 2020;27(9):2568-2585.[DOI]
-
197. Cui Y, Wang X, Lin F, Li W, Zhao Y, Zhu F, et al. miR-29a-3p improves acute lung injury by reducing alveolar epithelial cell PANoptosis. Aging Dis. 2022;13(3):899.[DOI]
-
198. Hsu CG, Chávez CL, Zhang C, Sowden M, Yan C, Berk BC. The lipid peroxidation product 4-hydroxynonenal inhibits NLRP3 inflammasome activation and macrophage pyroptosis. Cell Death Differ. 2022;29(9):1790-1803.[DOI]
-
199. Maniam P, Essilfie AT, Kalimutho M, Ling D, Frazer DM, Phipps S, et al. Increased susceptibility of cystic fibrosis airway epithelial cells to ferroptosis. Biol Res. 2021;54:38.[DOI]
-
200. Xu W, Wu Y, Wang S, Hu S, Wang Y, Zhou W, et al. Melatonin alleviates septic ARDS by inhibiting NCOA4-mediated ferritinophagy in alveolar macrophages. Cell Death Discov. 2024;10:253.[DOI]
-
201. Shin DW. Lipophagy: Molecular mechanisms and implications in metabolic disorders. Mol Cells. 2020;43(8):686-693.[DOI]
-
202. Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep. 2018;22(11):2924-2936.[DOI]
-
203. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349(6245):316-320.[DOI]
-
204. Chu C, Wang X, Yang C, Chen F, Shi L, Xu W, et al. Neutrophil extracellular traps drive intestinal microvascular endothelial ferroptosis by impairing Fundc1-dependent mitophagy. Redox Biol. 2023;67:102906.[DOI]
-
205. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: The role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18(5):280-296.[DOI]
-
206. Xie Y, Kang R, Klionsky DJ, Tang D. GPX4 in cell death, autophagy, and disease. Autophagy. 2023;19(10):2621-2638.[DOI]
-
207. Li L, Wu D, Deng S, Li J, Zhang F, Zou Y, et al. NVP-AUY922 alleviates radiation-induced lung injury via inhibition of autophagy-dependent ferroptosis. Cell Death Discov. 2022;8:86.[DOI]
-
208. Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124(9):3987-4003.[DOI]
-
209. Bueno M, Lai YC, Romero Y, Brands J, Croix CMS, Kamga C, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. 2015;125(2):521-538.[DOI]
-
210. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255-263.[DOI]
-
211. Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA. Subphenotypes in acute respiratory distress syndrome: Latent class analysis of data from two randomised controlled trials. Lancet Respir Med. 2014;2(8):611-620.[DOI]
-
212. Sinha P, Delucchi KL, Thompson BT, McAuley DF, Matthay MA, Calfee CS, et al. Latent class analysis of ARDS subphenotypes: A secondary analysis of the statins for acutely injured lungs from sepsis (SAILS) study. Intensive Care Med. 2018;44(11):1859-1869.[DOI]
-
213. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med. 2019;199(12):1517-1536.[DOI]
-
214. Melms JC, Biermann J, Huang H, Wang Y, Nair A, Tagore S, et al. A molecular single-cell lung atlas of lethal COVID-19. Nature. 2021;595(7865):114-119.[DOI]
-
215. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10:1689.[DOI]
-
216. Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248-255.[DOI]
-
217. Zheng M, Karki R, Vogel P, Kanneganti TD. Caspase-6 is a key regulator of innate immunity, inflammasome activation, and host defense. Cell. 2020;181(3):674-687.[DOI]
-
218. Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. 2019;31(51):1904197.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Zheng Y, Zhong N, Sang L, Huang Y. The role of multiple modes of cell death in the pathogenesis of acute respiratory distress syndrome. Ferroptosis Oxid Stress. 2026;2:202602. https://doi.org/10.70401/fos.2026.0024
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Apoptosis in ARDS
- 3. Necroptosis in ARDS
- 4. Pyroptosis in ARDS
- 5. Ferroptosis in ARDS
- 6. Other Cell Death Patterns in ARDS
- 7. Crosstalk Among Cell Death Pathways in ARDS
- 8. Conclusions and Future Perspectives
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Zheng Y, Zhong N, Sang L, Huang Y. The role of multiple modes of cell death in the pathogenesis of acute respiratory distress syndrome. Ferroptosis Oxid Stress. 2026;2:202602. https://doi.org/10.70401/fos.2026.0024
copy
Share Link
copy