SIRT3 at the crossroads of ferroptosis: Multidimensional regulation of the mitochondrial deacetylase Sirtuin 3 (SIRT3) on ferroptosis
Yixuan Chen
#
,
Qinyun Shi
#
,
Xiyu Wang
Xiangyun Wei
*
,
Rong Cai
*

*Correspondence to:
Xiangyun Wei, Department of Biochemistry & Molecular Cell Biology, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China.
E-mail: wxy18759692832@sjtu.edu.cn
Rong Cai, Department of Biochemistry & Molecular Cell Biology, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China. E-mail: rongcai@shsmu.edu.cn
Rong Cai, Department of Biochemistry & Molecular Cell Biology, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China. E-mail: rongcai@shsmu.edu.cn
Ferroptosis Oxid Stress. 2026;2:202603. 10.70401/fos.2026.0026
Received: January 27, 2026Accepted: April 16, 2026Published: April 20, 2026
Abstract
Ferroptosis, a regulated cell death modality, driven by iron-dependent lipid peroxidation, is intrinsically coupled with mitochondrial metabolic turbulence and redox dysregulation. While the mitochondrial sirtuin Sirtuin 3 (SIRT3) is canonically viewed as a master regulator of energy homeostasis, its defensive repertoire against ferroptosis extends far beyond the simplistic activation of antioxidant enzymes. In this review, we synthesize emerging evidence to construct an integrated “metabolic-structural” defense model orchestrated by SIRT3. We first delineate how SIRT3 functions as a metabolic rheostat, rewiring tricarboxylic acid (TCA) cycle flux via the deacetylation of isocitrate dehydrogenase 2 (IDH2) to sustain the nicotinamide adenine dinucleotide phosphate (NADPH)/glutathione (GSH) reservoir. Breaking away from the classical enzymatic paradigm, we highlight a novel non-enzymatic substrate regulatory mechanism where SIRT3 stabilizes the glutamate transporter SLC25A22 through specific deacetylation-ubiquitination crosstalk, thereby limiting ferroptotic susceptibility. Furthermore, we expand the SIRT3 signaling landscape by proposing a “SIRT3-nuclear factor erythroid 2-related factor 2 (Nrf2) deacetylation axis” that bridges mitochondrial stress signals to nuclear transcriptional defense, and by detailing its control over iron entry via the iron regulatory protein 1 (IRP1)-transferrin receptor 1 (TfR1) pathway. At the organelle level, we examine how SIRT3 remodels mitochondrial dynamics, upregulating optic atrophy-associated protein 1 (OPA1) while suppressing dynamin-related protein 1 (DRP1), to construct a “fusion network barrier” that dilutes ROS toxicity. We also posit a critical hypothesis: SIRT3 safeguards the integrity of mitochondria-associated endoplasmic reticulum membranes, preventing structural decoupling and calcium overload that triggers ferroptotic sensitization. Finally, we reconcile the context-dependent duality of SIRT3 in cancer and degenerative diseases, outlining future therapeutic strategies that leverage these multidimensional vulnerabilities.
Keywords
Sirtuin 3, ferroptosis, mitochondrial dynamics, SLC25A22, mitochondria-associated endoplasmic reticulum membranes, metabolic reprogramming
1. Introduction
Ferroptosis, a form of regulated cell death (RCD) distinct from apoptosis, is biochemically defined by the catastrophic accumulation of iron-dependent lipid peroxides and a breakdown in redox homeostasis. Unlike apoptotic cascades where mitochondrial reactive oxygen species (ROS) act primarily as signaling molecules, ferroptosis is driven by the failure of antioxidant defense systems to arrest the propagation of lipid peroxidation within cellular membranes[1]. While the absolute necessity of mitochondria in ferroptosis was historically a subject of debate, contemporary evidence has firmly established the organelle not merely as a metabolic powerhouse, but as the decisive “arbiter” of cell fate. The execution of ferroptosis hinges critically on mitochondria-mediated ROS generation, anaplerotic metabolism, and iron utilization[2]. Consequently, the maintenance of mitochondrial integrity constitutes a central checkpoint in determining cellular susceptibility to ferroptosis.
At the heart of this mitochondrial regulation lies Sirtuin 3 (SIRT3), the primary mitochondrial NAD+ dependent deacetylase. Structurally characterized by a conserved Rossmann fold and a zinc-binding motif, SIRT3 is robustly expressed in metabolically active tissues such as the heart, liver, and brain[3-5]. Classically, SIRT3 has been viewed as a master regulator of energy homeostasis, modulating the activity of metabolic enzymes to sustain ATP production. However, emerging research indicates that the defensive repertoire of SIRT3 extends far beyond this canonical view. It does not function merely as a passive antioxidant but acts as a dynamic “metabolic rheostat” and a “molecular switch” that governs the cellular threshold for ferroptosis through a sophisticated, multilayered defense strategy.
This review synthesizes recent advancements to propose an integrated “metabolic-structural” defense model orchestrated by SIRT3. We move beyond the traditional enzymatic paradigm, such as the regulation of isocitrate dehydrogenase 2 (IDH2) to sustain the nicotinamide adenine dinucleotide phosphate (NADPH)/glutathione (GSH) reservoir and superoxide dismutase 2 (SOD2) for ROS scavenging, to highlight novel, non-enzymatic substrate regulatory mechanisms. Specifically, we discuss how SIRT3 stabilizes the mitochondrial glutamate transporter mitochondrial solute carrier family 25 member 22 (SLC25A22) via specific deacetylation-ubiquitination crosstalk[6], and sustains the expression of the cystine/glutamate antiporter SLC7A11 through the activating transcription factor 4 (ATF4) signaling axis[7]. By coordinating these mechanisms, SIRT3 synergistically restores glutathione synthesis and reinforces the antioxidant barrier. Furthermore, we elucidate how SIRT3 orchestrates multidimensional mitochondrial quality control. This includes establishing an “anti-death barrier” by regulating mitochondrial dynamics (optic atrophy-associated protein 1/dynamin-related protein 1 (OPA1/DRP1) balance), preventing pathological mitophagy to restrict the labile iron pool (LIP), and stabilizing mitochondria-associated membranes (MAMs) to prevent calcium overload and subsequent ROS bursts.
Finally, we address the dualistic nature of SIRT3. While it functions as a guardian against ferroptosis in degenerative conditions, its “pro-survival” mechanisms can be hijacked in oncogenic contexts, acting as a double-edged sword in cancer progression. By dissecting these complex signaling axes, from the “SIRT3-ROS-iron regulatory protein 1 (IRP1)-transferrin receptor 1 (TfR1)” pathway to the stabilization of MAMs architecture, this review aims to provide a comprehensive framework for understanding SIRT3 as a central checkpoint in the ferroptotic landscape.
2. Overview of the Structure and Core Biological Functions of SIRT3
2.1 Structure
SIRT3, as an integrated “metabolic-structural” orchestrator in ferroptosis defense, requires a physical foundation that accommodates both organelle-level architecture and internal matrix biochemistry. This functional duality is inherently encoded in its processing-dependent isoform diversity and precise spatial partitioning.
Human SIRT3 is initially translated as a 44-kDa precursor protein containing an N-terminal targeting sequence[8]. Rather than serving merely as a transient intermediate, the retention of this full-length isoform, or its specific membrane-associated variants, anchors a distinct pool of SIRT3 to the mitochondrial outer membrane and peripheral compartments. This physical positioning dictates its “structural” capacity, enabling direct interaction with inter-organellar contact sites, particularly the MAMs. Conversely, importation into the matrix triggers the proteolytic cleavage of the N-terminus, yielding the 28-kDa mature enzyme encompassing the conserved catalytic core (residues 126-382)[3,8]. Stripped of its membrane-anchoring sequence, this truncated isoform localizes exclusively to the matrix to execute the “metabolic” orchestration.
The localization of this matrix-resident 28-kDa pool is subjected to strict spatiotemporal regulation rather than random diffusion. Proteomic analyses demonstrate that under basal physiological states, a fraction of mature SIRT3 is structurally sequestered through direct physical binding to the ATP synthase complex, specifically relying on the ATP5O subunit[4]. This protein-protein interaction (PPI) functions as a pH-sensitive reservoir. Acute mitochondrial stress induces sub-organellar pH fluctuations that disrupt this structural complex, triggering the rapid dissociation and mobilization of SIRT3[4]. This strictly regulated release mechanism guarantees that the deacetylase is promptly granted access to downstream targets, exactly at the onset of oxidative and metabolic crises.
Beyond spatial compartmentalization, the capacity of SIRT3 to regulate diverse targets, from soluble matrix enzymes to massive transmembrane transporters like SLC25A22[6], is defined by the biophysical architecture of its catalytic core. Primary crystallographic analyses of human SIRT3 reveal a highly dynamic active site cleft positioned between a large NAD+ binding Rossmann fold and a smaller zinc-binding module[8,9]. The transition of the cofactor binding loop upon NAD+ and substrate engagement exhibits extensive structural plasticity. This distinct conformational flexibility provides the steric tolerance required to bind substrates with drastically different physical sizes, structural folds, and hydrophobicity, establishing the definitive structural basis for its target promiscuity[9].
The diverse macroscopic phenomena governed by SIRT3 ultimately converge on a single biochemical absolute. The enzymatic removal of an acetyl group strips a neutral modification from a target lysine, restoring the inherent positive charge of the amino acid side chain. This localized electrostatic shift dictates fundamental physical changes: in metabolic enzymes, it reshapes the catalytic pocket to alter metabolic flux; in structural and transport proteins, it modifies intra- and inter-molecular electrostatic affinities to stabilize or disrupt physical tethering networks. This charge-dependent conformational reconfiguration constitutes the universal physical mechanism by which SIRT3 operates. Because this rapid structural-electrostatic coupling is directly proportional to neutralizing acute oxidative lipid damage[5], SIRT3 maintains robust baseline expression in tissues driven by high resting metabolic demand, including the heart, liver, brain, and kidneys[3,5], where it constantly scales the cellular energy threshold against ferroptotic vulnerability.
In this regard, a brief comparison with the other mitochondrial sirtuins further clarifies why SIRT3 occupies a privileged position in the ferroptosis landscape. Although SIRT3, Sirtuin 4 (SIRT4), and Sirtuin 5 (SIRT5) all reside within mitochondria and collectively buffer acyl stress, their biochemical specializations are not equivalent. SIRT3 is the dominant mitochondrial deacetylase and is uniquely positioned to couple protein deacetylation to ferroptosis-relevant processes at multiple hierarchical levels, ranging from matrix redox metabolism and antioxidant enzyme activation to iron handling, membrane architecture, and organelle quality control. By contrast, SIRT5 primarily functions as a desuccinylase, demalonylase, and deglutarylase, thereby governing a broader acidic acylation landscape whose relationship to ferroptosis appears largely indirect and metabolic rather than structurally integrative. SIRT4, in turn, remains mechanistically less defined; its reported activities, including adenosine diphosphate (ADP)-ribosyltransferase, lipoamidase/de-biotinylase, and the removal of branched-chain acyl adducts, suggest a role in nutrient sensing and amino acid flux, yet direct evidence placing SIRT4 at the core of ferroptotic execution remains comparatively limited. Thus, the distinction is not merely one of sub-mitochondrial localization or substrate class, but of systems-level reach: among the mitochondrial sirtuins, SIRT3 appears to be the only member for which a coherent ferroptosis-centered framework can currently be assembled, spanning enzymatic, structural, and spatiotemporal control layers[10].
2.2 Core biological functions
To comprehend the functional pleiotropy of SIRT3, one must first recognize the unique physicochemical reality of the mitochondrial matrix. Unlike the nuclear compartment, where protein acylation is tightly governed by the dynamic equilibrium of enzymatic “writers” and “erasers”, mitochondrial acylation is predominantly a spontaneous, non-enzymatic phenomenon. Driven by the high local concentrations of reactive acyl-CoA intermediates, lysine residues are continuously subjected to metabolism-driven chemical acylation[10]. In this harsh chemical landscape, SIRT3 does not function merely as a conventional signaling switch; rather, it operates as an indispensable NAD+ dependent quality control repair enzyme. Obligatorily coupled to the cleavage of NAD+ (yielding nicotinamide and 2′-O-acyl-ADP-ribose)[5], SIRT3 systematically strips these spontaneously acquired, charge-neutralizing, and sterically bulky acyl lesions from target proteins. Its versatile deacylase repertoire extends far beyond canonical deacetylation, encompassing the targeted removal of succinyl, crotonyl, and recently identified lactyl adducts generated under severe metabolic duress[10,11]. By restoring the native electrostatic potential and spatial profile of key lysine residues, this continuous physicochemical reversion forcibly reshapes catalytic pockets and macromolecular interaction interfaces. By translating the mitochondrial NAD+/NADH energy ratio into the relentless repair of structural and enzymatic lesions, SIRT3 orchestrates an adaptive continuum that bridges basal organelle housekeeping with extreme stress survival.
Under physiological baseline, this continuous deacylase-mediated repair mechanism synchronizes energy output with genomic and structural integrity. SIRT3 clears acyl lesions from key components of the oxidative phosphorylation (OXPHOS) machinery while simultaneously preserving the genetic blueprint of the organelle. Recent evidence establishes that SIRT3 acts as a critical fidelity protein that actively maintains mitochondrial DNA (mtDNA) replication[3] by specifically deacetylating polymerase gamma (Polγ) at highly conserved residues (e.g., K1039)[12]. This electrostatic restoration preserves Polγ’s native mtDNA binding capacity, preventing replication stalling, metabolic decline, and premature cellular senescence. Concurrently, SIRT3 sustains mitochondrial biogenesis and maintains membrane potential through the activation of the PGC-1α/TFAM transcriptional axis[13]. This basal optimization of mitochondrial mass and genetic stability provides the foundational structural resilience required to endure subsequent metabolic shocks.
When cells encounter acute oxidative or ischemic stress, this baseline lesion-repair apparatus rapidly specializes into a targeted defense against lipid peroxidation, as shown in Figure 1. The physiological intent of this metabolic rewiring is to systematically disarm the three indispensable drivers of ferroptosis: reactive oxygen species (ROS), oxidizable lipids, and catalytic iron. To neutralize oxidative triggers, SIRT3 couples central carbon flux with the organelle’s redox buffering capacity. By fine-tuning the kinetics of tricarboxylic acid (TCA) cycle enzymes (e.g., IDH2[14]), it continuously funnels metabolic intermediates toward the production of reducing equivalents (NADH and NADPH). This sustained NADPH pool serves as the absolute prerequisite for regenerating GSH, thereby supporting the direct SIRT3-mediated deacetylation and activation of primary antioxidant enzymes like manganese superoxide dismutase (MnSOD/SOD2)[15-17]. To restrict the “fuel” of ferroptosis, this SIRT3-sustained NADPH pool extends its protective reach to orchestrate endoplasmic reticulum (ER)-mediated lipid remodeling. Specifically, NADPH acts as a direct allosteric ligand for the ER-localized E3 ubiquitin ligase MARCHF6 via its unique C-terminal regulatory region[18]. This metabolic sensing hyperactivates MARCHF6, driving the rapid ubiquitin-proteasome degradation of core ferroptosis effectors, most notably acyl-CoA synthetase long-chain family member 4 (ACSL4) and p53[18]. Because ACSL4 dictates ferroptosis sensitivity by incorporating highly oxidizable polyunsaturated fatty acids into membrane phospholipids, its prompt clearance systematically starves the ferroptotic cascade of its essential lipid substrates. Crucially, this synergistic defense is completed by the regulation of intracellular iron handling. By modulating the IRP1 axis and stabilizing iron-sulfur clusters, SIRT3 actively restricts the accumulation of the intracellular LIP[19]. This precise sequestration of free iron deprives the mitochondrial matrix of the primary catalyst required for Fenton chemistry, effectively halting ferroptosis at its biochemical root.
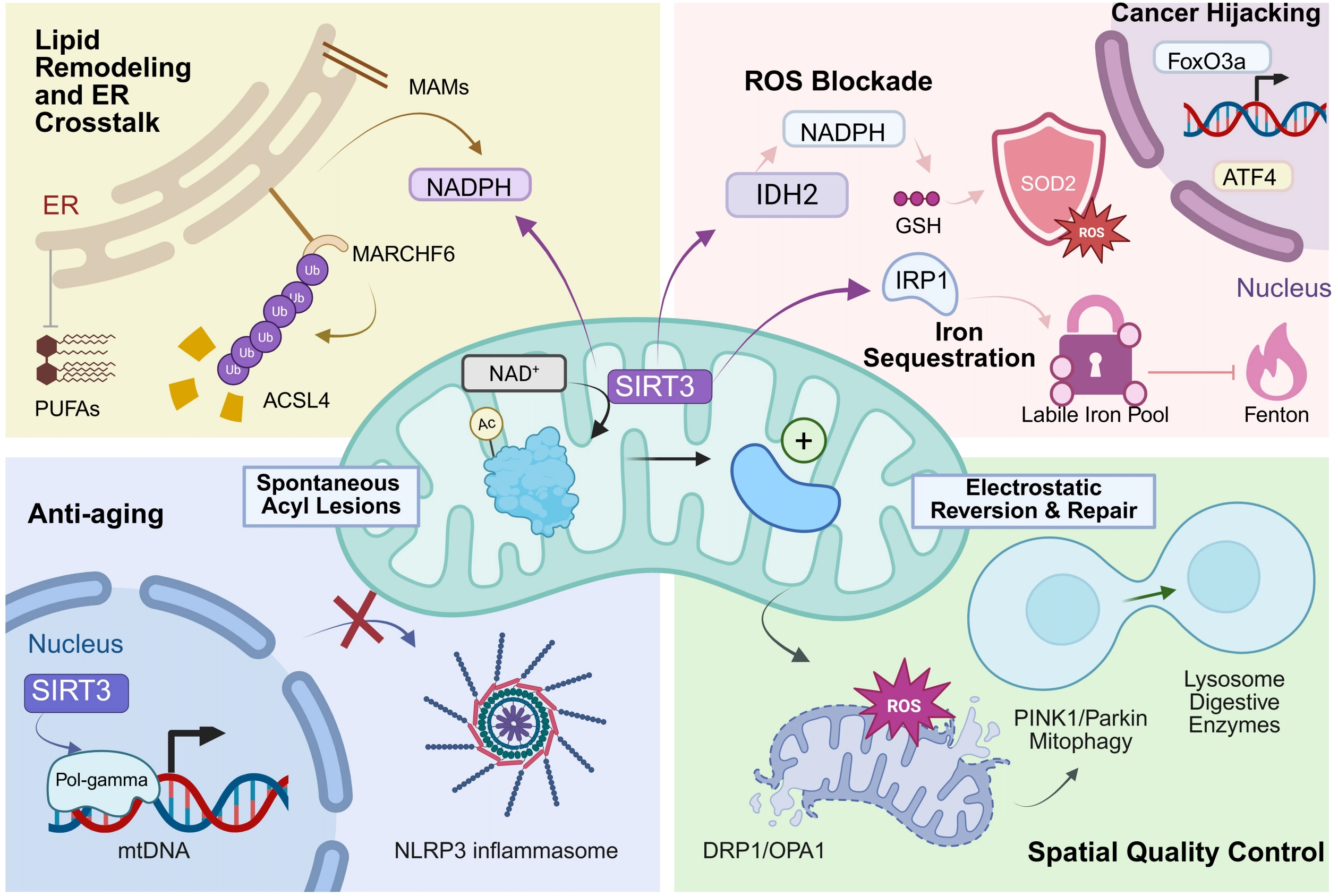
Figure 1. The multi-scale spatiotemporal defense mechanism of SIRT3 against ferroptosis and its role in cell fate determination. (Center) Core enzymatic logic: Residing primarily in the mitochondrion, SIRT3 utilizes NAD+ to remove spontaneous acyl lesions from target proteins. This deacetylation drives an electrostatic reversion that structurally and functionally repairs the compromised proteins; (Top Left) Lipid remodeling and ER crosstalk: At MAMs, SIRT3-driven NADPH production sustains the activity of the E3 ligase MARCHF6. This pathway selectively ubiquitinates and degrades ACSL4, thereby blocking the pathogenic integration of highly oxidizable PUFAs into cellular membranes; (Top Right) ROS/Iron blockade and Cancer hijacking: SIRT3 fuels the IDH2/SOD2 axis to neutralize ROS and restricts the Fenton reaction by sequestering the LIP via IRP1. Conversely, in oncogenic contexts, tumor cells actively hijack this defense network through FoxO3a and ATF4 transactivation to persistently evade ferroptosis and develop chemoresistance; (Bottom Right) Spatial quality control: When oxidative damage breaches the primary biochemical shields, SIRT3 manages structural integrity by modulating DRP1/OPA1-mediated mitochondrial dynamics. This physically isolates irrecoverably damaged, ROS-accumulating mitochondrial fragments, priming them for targeted clearance via PINK1/Parkin-dependent mitophagy and subsequent lysosomal digestion; (Bottom Left) Anti-aging and inflammatory suppression: In aging models, SIRT3 preserves mtDNA fidelity by stabilizing Pol-gamma. By strictly preventing the cytosolic leakage of mtDNA, SIRT3 effectively abrogates the aberrant assembly and activation of the NLRP3 inflammasome. Created in BioRender.com. SIRT3: Sirtuin 3; ER: endoplasmic reticulum; MAMs: mitochondria-associated membranes; ACSL4: acyl-CoA synthetase long-chain family member 4; PUFAs: polyunsaturated fatty acids; ROS: reactive oxygen species; IDH2: isocitrate dehydrogenase 2; SOD2: superoxide dismutase 2; LIP: labile iron pool; IRP1: iron regulatory protein 1; ATF4: activating transcription factor 4; DRP1: dynamin-related protein 1; OPA1: optic atrophy-associated protein 1; PINK1: PTEN-induced putative kinase 1; mtDNA: mitochondrial DNA.
Biochemical lesion repair alone is often insufficient to contain propagating lipid damage once membrane integrity is breached. To counter this, SIRT3 engages a broad spectrum of non-enzymatic substrates, including structural scaffolds, pore-forming complexes, and transmembrane carriers (such as the glutamate transporter SLC25A22[6]), to execute a rigorous spatial quality control network. This physical dimension dynamically adjusts mitochondrial substrate import and stabilizes the architecture of mitochondria-associated membranes[20], thereby restricting the pathological influx of calcium and peroxidized lipids from the endoplasmic reticulum. When localized oxidative damage exceeds repair thresholds, SIRT3 relies on non-enzymatic effectors to regulate mitochondrial dynamics (the OPA1/DRP1 fusion-fission machinery[21,22]) and initiate PTEN-induced putative kinase 1/Parkin-mediated mitophagy[23]. This coordinated structural response physically quarantines and degrades ROS-leaking, iron-overloaded mitochondrial fragments, effectively utilizing “spatial restriction” to prevent catastrophic chain reactions, as revealed in Figure 1.
The successful execution of this organelle-level quality control reverberates throughout the entire cellular ecosystem, fundamentally dictating long-term genetic adaptation and immune homeostasis, as presented in Figure 1. By rigorously containing mitochondrial ROS and preventing the leakage of damaged mtDNA into the cytosol, SIRT3 suppresses the aberrant activation of intracellular danger sensors. In the context of immune senescence, SIRT3 directly governs innate immune responses by promoting the ubiquitination and subsequent degradation of the NLRP3 inflammasome in macrophages, a mechanism that actively mitigates systemic aging phenotypes such as osteoporosis[24].
Expanding this immune-metabolic paradigm, the ferroptotic system is increasingly recognized as a central pathological driver beyond classical organ injuries, extending into complex endocrine-metabolic disorders such as polycystic ovary syndrome[25]. In such multisystem endocrinopathies, natural polyphenols operate as broad-spectrum sirtuin activators, exemplified by resveratrol, which has demonstrated a profound therapeutic capacity to blunt glutathione peroxidase 4 (GPX4)-dysregulated ferroptotic damage. This intersection strongly suggests that the SIRT3-coordinated antioxidant network functions at the critical nexus of reproductive endocrinology and systemic inflammatory repair, warranting broader exploration in metabolic syndromes.
Parallel to this immune-metabolic regulation, the acute enzymatic shifts governed by SIRT3 generate signaling intermediates that modulate core retrograde transcriptional networks, including the FoxO3a, p53, and ATF4 pathways[7,17,26]. This genetic feedback loop ensures that the acute chemical and physical defenses are continuously replenished by de novo protein synthesis.
The seamless integration of non-enzymatic lesion repair, specialized metabolic-redox coupling, and spatial quality control renders SIRT3 a profound determinant of cell fate in pathology, as visualized in Figure 1. In degenerative conditions, such as ischemia-reperfusion injury in acute kidney injury or chronic fibrosis, this multidimensional architecture acts as a vital guardian, continuously clearing metabolic lesions and lipid peroxides to suppress pathological cell death[24]. Conversely, this precise survival machinery represents a critical vulnerability in oncology[3]. Cancers characterized by severe metabolic constraints and high basal oxidative stress, such as lung adenocarcinoma (LUAD) and glioblastoma (GBM)[6,7], maliciously hijack the SIRT3 network. In these malignancies, the overexpression of SIRT3 hyperactivates the iron-lipid-redox metabolic triad and structural quality control systems to endure hostile microenvironments, effectively transforming a physiological protector into a potent driver of tumor chemoresistance and ferroptosis evasion.
3. Overview of Ferroptosis Function
Ferroptosis is an iron-dependent form of RCD that is morphologically and biochemically distinct from apoptosis, necrosis, and autophagy. Originally defined by Dixon et al. in 2012, it is characterized by intracellular iron overload and the lethal accumulation of lipid peroxides, culminating in membrane rupture and cell death[27,28]. The core mechanism of ferroptosis involves a redox imbalance, where GPX4 functions as a central regulator by reducing lipid hydroperoxides to maintain membrane stability[29]. Recently, ferroptosis suppressor protein 1 (FSP1) has been identified as a second critical molecular node that inhibits ferroptosis, functioning in parallel to GPX4[30]. Mounting evidence indicates that ferroptosis plays a pivotal role in diverse physiological and pathological processes, including tumor suppression, neurodegeneration, tissue injury, and immunoregulation, thereby positioning it as a promising therapeutic target for various diseases[1,31].
Pathologically, ferroptosis exerts a dual role depending on the disease context, as depicted in Figure 2. In the oncological landscape, ferroptosis operates fundamentally as a highly contextual “double-edged sword” rather than a unidirectional tumor-suppressive mechanism[32]. On the one hand, targeted pharmacological induction, such as the blockade of System Xc- or GPX4, effectively eradicates recalcitrant cancer cells, overcomes chemoresistance, and potentiates anti-tumor immunity via the release of immunostimulatory damage-associated molecular patterns. Conversely, depending on the specific cellular context and the tumor microenvironment (TME), ferroptosis can paradoxically exert pro-tumorigenic effects. For instance, the ferroptotic death of tumor-infiltrating immune cells or the paracrine release of specific oncogenic mediators (e.g., mutant kirsten rat sarcoma viral oncogene homolog) can drive macrophage polarization toward an immunosuppressive phenotype, thereby facilitating tumor immune evasion and progression[33,34]. In neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases, as well as in ischemic tissue injuries (e.g., myocardial infarction, hepatic ischemia-reperfusion, and acute kidney injury), aberrant ferroptosis acts as a pathogenic driver characterized by iron accumulation and mitochondrial dysfunction[35-37]. Consequently, inhibiting ferroptosis has emerged as a vital strategy for neuroprotection and organ preservation, underscoring the broad therapeutic potential of modulating iron metabolism and lipid peroxidation pathways across diverse clinical landscapes[31,38,39].
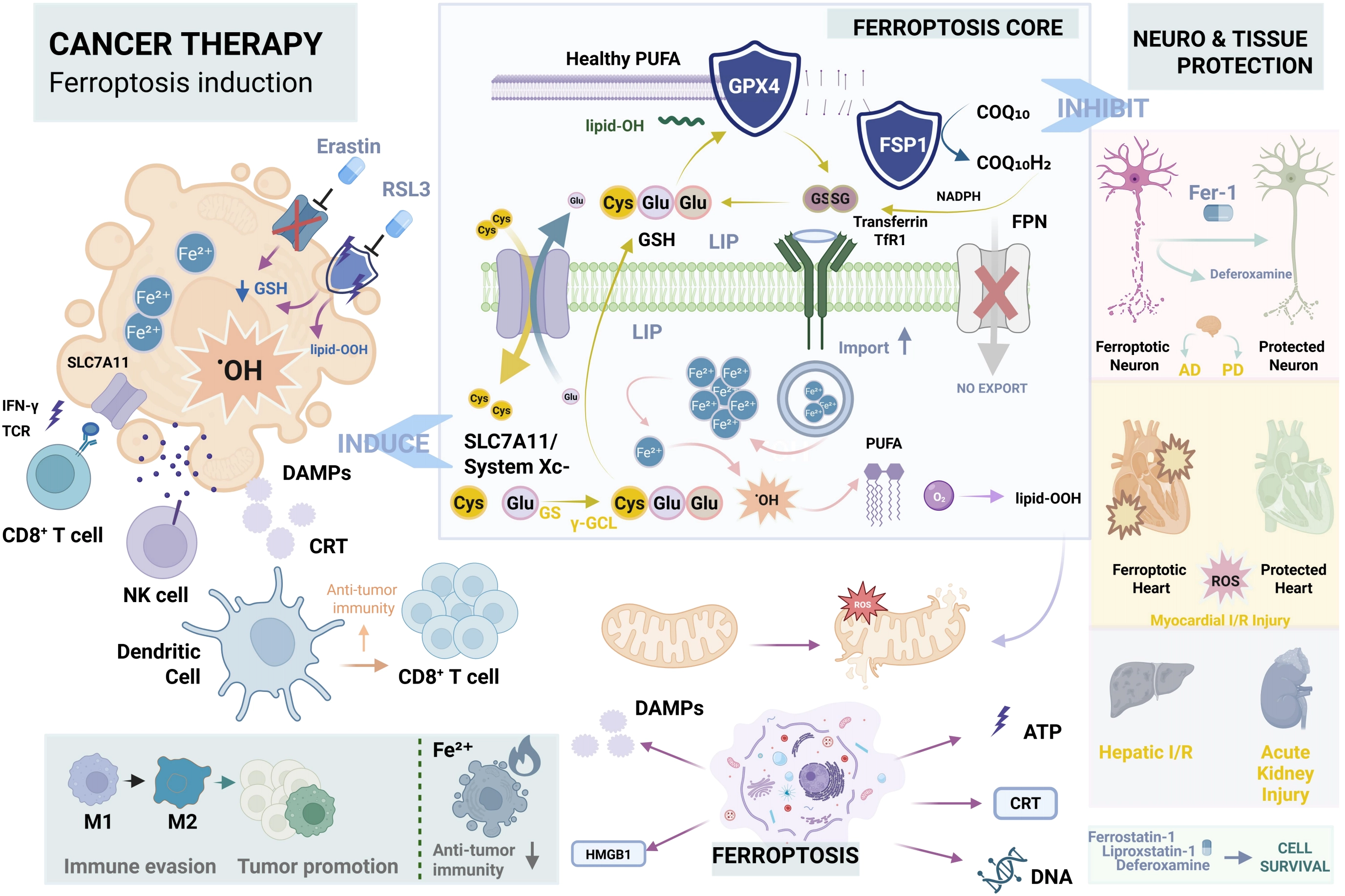
Figure 2. The core mechanism of ferroptosis and its bidirectional therapeutic strategies. (Center) Ferroptosis Core: Iron overload drives the Fenton reaction, converting PUFAs into lethal lipid peroxides (lipid-OOH). This pathogenic process is heavily restricted by the GSH/GPX4 and FSP1 defense axes; (Left) Cancer Therapy (Induction): Targeted inducers (e.g., Erastin, RSL3) trigger cancer cell death and potentiate anti-tumor immunity via the release of DAMPs. However, ferroptosis operates as a “double-edged sword” within the tumor microenvironment, potentially facilitating tumor immune evasion via M2 macrophage polarization; (Right) Tissue Protection (Inhibition): Conversely, aberrant ferroptosis drives pathogenesis in neurodegenerative diseases (AD, PD) and ischemia-reperfusion (I/R) injuries. Pharmacological inhibitors (e.g., Fer-1, Deferoxamine) block lipid peroxidation to confer robust neuroprotection and organ preservation. Created in BioRender.com. PUFAs: polyunsaturated fatty acids; GSH: glutathione; GPX4: glutathione peroxidase 4; FSP1: ferroptosis suppressor protein 1; AD: Alzheimer’s diseases; PD: Parkinson’s diseases; I/R: ischemia-reperfusion; DAMPs: damage-associated molecular patterns.
Collectively, the significance of ferroptosis extends beyond its role as a cell death mechanism to encompass disease pathology and therapeutic innovation. Figure 2 exemplifies that preclinical studies have demonstrated the promise of ferroptosis inducers (e.g., erastin) and inhibitors (e.g., ferrostatin-1) for cancer therapy and tissue protection, respectively[1,31].
4. SIRT3-Regulated Ferroptosis Network: Enzymatic and Non-Enzymatic Substrates
4.1 Regulation of metabolic and antioxidant enzymes
A hallmark feature of ferroptosis is lipid peroxidation resulting from the accumulation of ROS. This stands in contrast to apoptosis, where mitochondrial ROS primarily function as signaling molecules or direct damaging agents to trigger apoptotic cascades. Consequently, ROS imbalance likely serves as a critical “molecular switch” determining cell fate towards ferroptosis or alternative death modalities[1]. Given that metabolic and antioxidant enzymes play pivotal defensive roles in modulating redox imbalance, we prioritize their discussion here.
In delineating the regulation of the ferroptotic antioxidant system by SIRT3, it is imperative to recognize that SIRT3 does not function as an isolated or unidirectional checkpoint. For example, a bidirectional, reciprocal regulatory mechanism exists between SIRT3 and PGC-1α. Specifically, PGC-1α, acting as a master transcriptional coactivator for mitochondrial biogenesis, significantly upregulates SIRT3 transcription. Conversely, SIRT3 activates PGC-1α through deacetylation. This mutual reinforcement mechanism maintains mitochondrial homeostasis and potentiates the activity of antioxidant enzymes, such as SOD2, thereby synergistically mitigating ROS accumulation[5,40]. Furthermore, this regulatory loop frequently functions in concert with SIRT1. Upon sensing an elevated NAD+/NADH ratio, SIRT1 deacetylates PGC-1α, which subsequently promotes SIRT3 activation. This establishes the canonical PGC-1α/SIRT1/SIRT3 antioxidant axis, collectively preserving cellular redox equilibrium[13,41].
Further research indicates that the AMP-activated protein kinase (AMPK)/SIRT3/PGC-1α axis constitutes a master regulator orchestrating cellular energy metabolism and ferroptotic fate determination. AMPK, functioning as a cellular energy stress sensor, not only serves as an upstream activator of PGC-1α but also exhibits significant interdependence with SIRT3, as shown in Figure 3: AMPK activation relies on SIRT3 expression, while SIRT3 deficiency leads to compromised AMPK phosphorylation. This positive feedback loop is critical for maintaining mitochondrial function[13,40]. Based on these interactions, SIRT3-dependent metabolic reprogramming offers a novel mechanistic explanation for cellular evasion of ferroptosis. Recent studies have demonstrated that under metabolic stress, activation of the AMPK signaling pathway inhibits the synthesis of polyunsaturated fatty acid (PUFA) by phosphorylating key enzymes such as acetyl-CoA carboxylase, thereby reducing lipid peroxidation substrates[42]. Given the crucial role of SIRT3 in sustaining AMPK activity, it is plausible to postulate that SIRT3 acts through this AMPK-mediated metabolic checkpoint to limit PUFA synthesis, thereby abrogating the initiation of ferroptosis at its source.
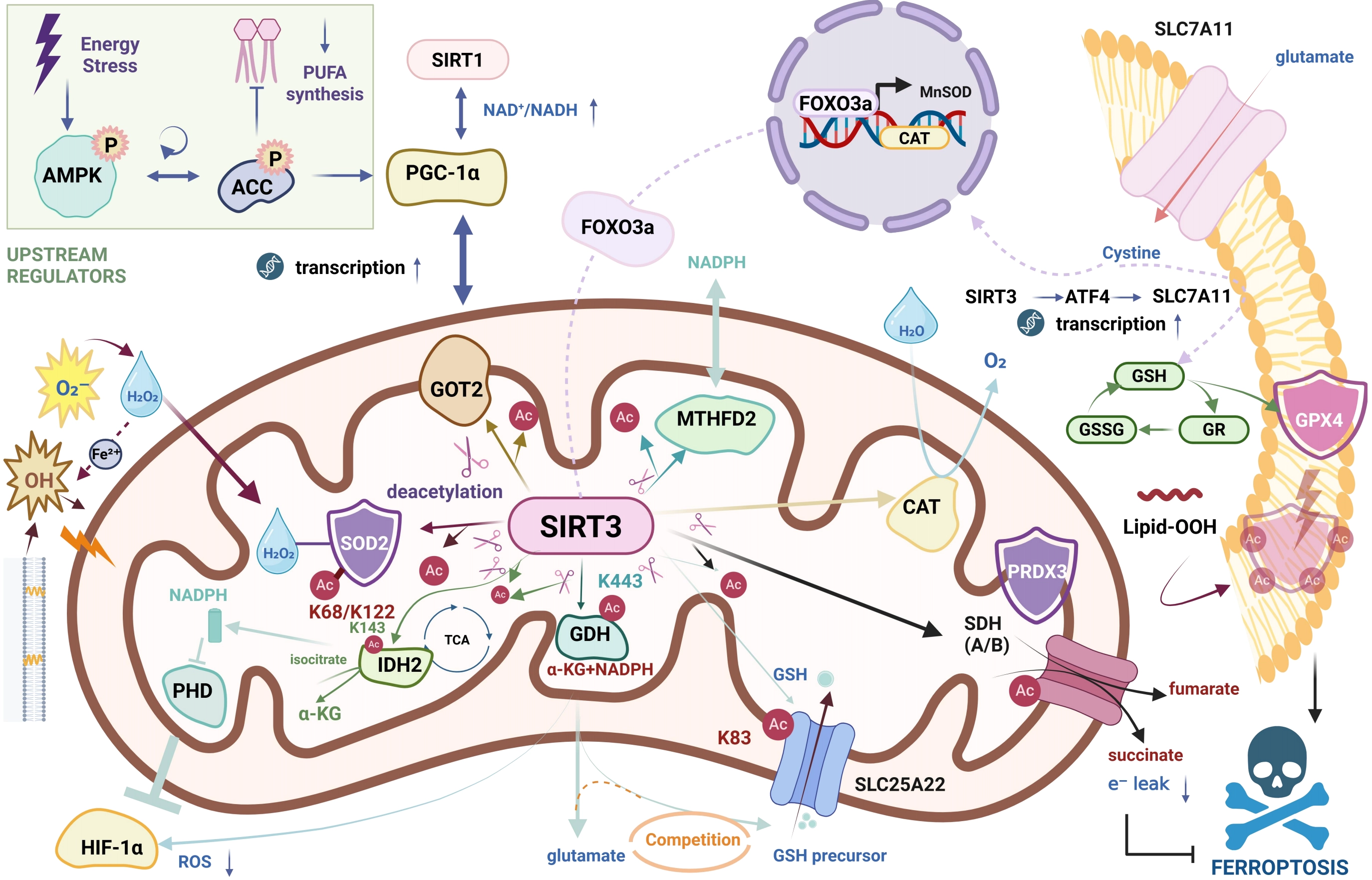
Figure 3. Mechanistic overview of SIRT3-mediated enzymatic regulation in ferroptosis inhibition. SIRT3 acts as a mitochondrial metabolic checkpoint regulated by the AMPK/PGC-1α signaling axis. It suppresses lipid peroxidation and ferroptosis through three primary mechanisms: (1) ROS Scavenging: Deacetylation and activation of MnSOD, PRDX3, and CAT (via FOXO3a) to eliminate ROS; (2) GPX4 Regulation: Direct deacetylation of GPX4 and stabilization of SLC25A22 to support GSH biosynthesis; and (3) NADPH Regeneration: Activation of metabolic enzymes IDH2, GDH, and MTHFD2 to produce NADPH, essential for redox balance. Additionally, SIRT3 regulates SDH to prevent electron leakage. Created in BioRender.com. SIRT3: Sirtuin 3; AMPK: AMP-activated protein kinase; ROS: reactive oxygen species; MnSOD: manganese superoxide dismutase; PRDX3: peroxiredoxin 3; CAT: catalase; GPX4: glutathione peroxidase 4; GSH: glutathione; NADPH: nicotinamide adenine dinucleotide phosphate; IDH2: isocitrate dehydrogenase 2; GDH: glutamate dehydrogenase; MTHFD2: mitochondrial methylenetetrahydrofolate dehydrogenase/cyclohydrolase; SDH: succinate dehydrogenase.
4.1.1 MnSOD
SIRT3 enhances the enzymatic activity of MnSOD by deacetylating specific lysine residues, notably K68 and K122, thereby facilitating the dismutation of superoxide anions (O2-) into hydrogen peroxide (H2O2)[15,43], displayed in Figure 3. It is critical to underscore the backbone function of MnSOD within the ferroptosis antioxidant defense system. Specifically, in the presence of mitochondrial SOD, O2 is converted to H2O2, which subsequently diffuses from the mitochondria into the cytosol. Under conditions of iron overload favorable for the Fenton reaction, H2O2 can generate highly reactive hydroxyl radicals; however, catalase is responsible for detoxifying H2O2 into water and oxygen[1].
In liver cancer models, under cadmium-induced oxidative stress, SIRT3 activates the autophagy pathway (ULK1-LC3 axis) via the SOD2-mROS signaling axis to mitigate cytotoxicity and cell death[16]. In the context of neurodegenerative diseases, SIRT3 plays a pivotal role in protecting neurons from oxidative injury. For instance, in Parkinson’s disease models, SIRT3 modulates oxidative stress by deacetylating MnSOD at the lysine 68 residue, while its deficiency compromises mitochondrial function in dopaminergic neurons[43,44]. Furthermore, SIRT3 restores mitochondrial fusion-fission homeostasis by deacetylating MnSOD, thereby reducing oxidative damage and alleviating pathology in Alzheimer’s disease[44]. Regarding cardiovascular pathology, studies on cardiac hypertrophy indicate that pressure overload suppresses the expression of antioxidant enzymes; conversely, SIRT3 overexpression restores the enzymatic activity of catalase and MnSOD, thereby antagonizing ROS production and preventing pathological remodeling[17]. Cross-disease studies reveal that the regulation of ROS homeostasis via MnSOD activity represents a conserved mechanism governed by SIRT3.
To elucidate the upstream regulation of SOD and the interplay between antioxidant enzymes, we briefly describe the regulatory network. Mechanistically, SIRT3 promotes the translocation of FoxO3a from the mitochondria to the nucleus, rather than merely affecting cytosolic localization. This nuclear accumulation initiates the transcription of downstream antioxidant genes, including MnSOD and catalase (CAT), thereby triggering the associated antioxidant defense system[17,43,45]. For example, this SIRT3-FOXO3a axis has been demonstrated to protect cells from oxidative stress-induced injury by upregulating the antioxidant capacity[17,45].
4.1.2 IDH2
IDH2 is a pivotal mitochondrial metabolic enzyme responsible for catalyzing the oxidative decarboxylation of isocitrate to generate α-ketoglutarate (α-KG) while concomitantly producing NADPH[43]. As a crucial intracellular reducing equivalent, NADPH plays a central role in maintaining the reduced state of GSH and defending against ferroptosis. Evidence indicates that the downregulation of IDH2 leads to a depletion of the mitochondrial NADPH pool, thereby significantly sensitizing cancer cells to erastin-induced ferroptosis[46], as visualized in Figure 3.
As the primary mitochondrial deacetylase, SIRT3 exerts significant regulatory control over antioxidant defense and metabolic reprogramming by modifying IDH2. Specifically, SIRT3 has been confirmed to deacetylate IDH2 at the lys143 residue. This modification promotes the dissociation of the IDH2 dimer, resulting in decreased ROS levels and the hindrance of cancer progression in both in vitro and in vivo models[5]. Furthermore, SIRT3-mediated activation of IDH2 augments the production of α-KG. Since α-KG functions as a co-substrate for prolyl hydroxylases (PHDs), this upregulation promotes PHD activity and the subsequent degradation of hypoxia-inducible factor 1α, thereby mitigating hypoxia-induced ROS accumulation[5,26].
In specific tumor microenvironments, SIRT3-mediated IDH2 activation is also intimately linked to metabolic adaptation. For instance, diffuse large B-cell lymphoma cells exhibit elevated SIRT3 expression, which, through the deacetylation and activation of IDH2 and glutamate dehydrogenase (GDH), facilitates the efficient entry of amino acids, such as glutamine, into the TCA cycle, shown in Figure 3. This enables cancer cells to generate substantial metabolic intermediates (e.g., acetyl-CoA) to support cell growth and proliferation, while simultaneously suppressing autophagy and its associated catabolism of macromolecules[26]. In summary, by activating IDH2 through deacetylation, SIRT3 not only maintains mitochondrial NADPH levels to resist ferroptosis but also supports tumor cell survival by modulating ROS levels and metabolic flux.
4.1.3 GPX4
Traditional paradigms of SIRT3 research have predominantly focused on the direct deacetylation of metabolic enzymes (e.g., IDH2, SOD2), following a linear logic of “deacetylation-conformational change-enzymatic activation.” However, recent evidence regarding the regulation of the core ferroptosis enzyme, GPX4, suggests the necessity of adopting a higher-dimensional integrative perspective: “substrate accessibility dictates enzyme function”[6,7].
GPX4 serves as the cornerstone of the cellular antioxidant defense system, responsible for reducing toxic lipid hydroperoxides to non-toxic alcohols, thereby arresting the onset of ferroptosis[29]. As a critical mitochondrial NAD+-dependent deacetylase, SIRT3 engages in extensive molecular crosstalk with GPX4. The central mechanism involves the maintenance of GPX4 enzymatic activity and protein stability via deacetylation. Research indicates that SIRT3 specifically removes acetyl groups from mitochondrial proteins to uphold mitochondrial homeostasis[47]. Conversely, in the pathological progression of ferroptosis, the downregulation of SIRT3 is frequently accompanied by an aberrant elevation in GPX4 acetylation levels. For instance, in models of cadmium-induced renal cytotoxicity, SIRT3 deficiency has been shown to directly promote mitochondrial GPX4 acetylation, leading to a loss of antioxidant function and the triggering of severe ferroptosis[47].
Recent investigations into neurodegenerative disorders have further corroborated this direct regulatory mechanism. In mouse models of perioperative neurocognitive disorder, anesthesia and surgical stress induced a significant downregulation of SIRT3 in hippocampal neurons, resulting in a surge of mitochondrial GPX4 acetylation. Activation of SIRT3 via the small-molecule agonist Honokiol (HKL), or genetic overexpression of SIRT3, significantly attenuated mitochondrial GPX4 acetylation, thereby restoring its enzymatic activity and inhibiting neuronal ferroptosis. Notably, this neuroprotective effect is strictly GPX4-dependent, as gene silencing of GPX4 or treatment with the inhibitor Erastin completely abolished the cognitive improvements conferred by HKL[47]. These findings confirm that SIRT3 functions not merely as a general regulator of oxidative stress but as a specific modulator directly governing the functional state of GPX4 via deacetylation.
Beyond direct modification, SIRT3 reinforces GPX4 function indirectly by orchestrating the upstream metabolic environment and associated signaling axes. SIRT3 optimizes mitochondrial metabolism by regulating mitochondrial ROS homeostasis, thereby alleviating the oxidative pressure placed on the GPX4 system[47]. Furthermore, SIRT3 may synergize with the p53 pathway (Sirt3/p53 axis) to further promote GPX4 deacetylation and stability[47,48]. Our research revealed that SIRT3 deacetylates and stabilizes the mitochondrial glutamate transporter SLC25A22; this ensures a continuous mitochondrial supply of glutamate, a critical precursor for GSH synthesis. This upregulation of intracellular GSH provides the GPX4 enzymatic system with sufficient reducing substrates to eliminate lipid peroxides[6]. Simultaneously, we also discovered that SIRT3 maintains high expression levels of SLC7A11 via the transcription factor ATF4, guaranteeing cystine uptake and subsequent cysteine generation, which constitutes the rate-limiting step in GSH synthesis. Notably, Figure 3 shows that deficiency in SIRT3 leads to insufficient cystine intake and the exhaustion of the GSH pool, rendering GPX4 “functionally inactive” due to cofactor deprivation, thus precipitating severe ferroptosis[7]. This mechanism unveils a novel dimension of SIRT3-mediated enzymatic regulation: the maintenance of antioxidant catalytic efficiency through the optimization of upstream substrate transport and synthesis.
4.1.4 Other enzymes
In addition to the aforementioned core antioxidant and metabolic enzymes, SIRT3 constructs a multidimensional ferroptosis defense network by deacetylating a series of key enzymes located at metabolic crossroads. These enzymes not only act as a “second line of defense” downstream of SOD2 to scavenge hydrogen peroxide (H2O2), but also influence the supply of GSH synthesis substrates and reducing power (NADPH) through the regulation of the TCA cycle and glutamine metabolic flux.
4.2 The secondary line of defense in H2O2 scavenging: CAT and PRDX
Although SOD2 efficiently scavenges superoxide anions, its metabolic product, H2O2, if not degraded in a timely manner, can directly trigger lipid peroxidation via the Fenton reaction. In Figure 3, it is evident that SIRT3 plays a critical relay role in this process. Studies indicate that SIRT3 enhances the H2O2 decomposing capacity of CAT via deacetylation, thereby mitigating ROS toxicity; conversely, the inhibition of CAT activity has been confirmed to lead to H2O2 accumulation, indirectly promoting lipid peroxidation. Furthermore, SIRT3 deacetylates FOXO3a, promoting its nuclear translocation and the subsequent transcriptional upregulation of CAT, thus establishing a dual safeguard mechanism of “deacetylation-mediated activation and transcriptional upregulation”[17,43,45].
Simultaneously, while peroxisomes contribute to ferroptosis primarily by mediating lipid synthesis rather than ROS production[46], peroxiredoxin 3 (PRDX3), a mitochondria-specific peroxidase, serves as a direct substrate of SIRT3. During ferroptosis, the accumulation of mitochondrial lipid peroxides is a pivotal event. Given that PRDX3 functions as the primary mitochondrial H2O2 scavenger, SIRT3 may indirectly influence the levels of mitochondrial lipid peroxides and consequently regulate the onset of ferroptosis by maintaining PRDX3 activity[49].
4.3 GDH: Regulation of glutamine metabolism
Glutamate dehydrogenase (GDH/GLUD1) serves as a pivotal hub in glutamine metabolism, and the impact of its deacetylation by SIRT3 (e.g., at the K443 residue) on ferroptosis regulation is highly complex[43].
On one hand, SIRT3-mediated activation of GDH enhances the conversion of glutamate to α-KG. Figure 3 presents that this process not only facilitates the efficient entry of amino acids into the TCA cycle to generate metabolic intermediates (such as acetyl-CoA) but also couples with the production of NADPH, thereby providing the reducing power necessary for GSH regeneration[26,43].
However, it is noteworthy that the hyperactivation of GDH may exert a “double-edged sword” effect. Although GDH promotes anaplerosis, glutamate itself is one of the three essential precursors for GSH synthesis. Consequently, SIRT3-enhanced GDH activity accelerates the consumption of glutamate into the TCA cycle, potentially creating substrate competition with the GSH synthetic pathway under specific metabolic stresses. Furthermore, active glutaminolysis is intrinsically recognized as a driving factor for ferroptosis[46]. Thus, the regulation of GDH by SIRT3 embodies a delicate equilibrium between maintaining energy production and orchestrating metabolic adaptations to resist ferroptosis.
4.4 Homeostasis of the mitochondrial electron transport chain: Succinate dehydrogenase (SDH)
Electron leakage from the mitochondrial electron transport chain (ETC) constitutes a primary source of ROS burst during the initiation of ferroptosis. SIRT3 plays a critical role in mitigating this process by deacetylating the A (SDHA) and B (SDHB) subunits of succinate dehydrogenase (SDH/Complex II), thereby enhancing electron transport efficiency and averting succinate accumulation[43], as summarized in Figure 3. Research indicates that the inhibition of SIRT3 results in SDH inactivation via hyperacetylation and consequent succinate accumulation. This metabolic shift exacerbates inflammation and metabolic dysregulation through epigenetic mechanisms[50]. By optimizing the functionality of SDH and other ETC components (e.g., Complexes I and IV), SIRT3 sustains mitochondrial membrane potential, thereby reducing ROS leakage at the source and mitigating the downstream risk of lipid peroxidation[43].
4.5 Auxiliary regulation of NADPH generation: Heterogeneity of mitochondrial methylenetetrahydrofolate dehydrogenase/cyclohydrolase (MTHFD2) and glutamate oxaloacetate transaminase 2 (GOT2)
Beyond IDH2, SIRT3 exerts influence over the NADPH pool through the modulation of other metabolic enzymes; however, this regulation displays distinct substrate specificity. SIRT3-mediated deacetylation and activation of MTHFD2 promotes NADPH production, thereby mitigating oxidative stress[51], as shown in Figure 3. Conversely, studies indicate that SIRT3-mediated deacetylation of glutamate oxaloacetate transaminase 2 (GOT2) paradoxically inhibits NADPH generation, precipitating cell death in specific tumor contexts under oxidative stress[52]. This dichotomy suggests that SIRT3-mediated regulation of ferroptosis is not a monolithic linear suppression but rather a precision-tuned network regulation contingent upon cell type and metabolic context.
5. SIRT3-Regulated Non-Enzymatic Substrates in Iron Metabolism and Lipid Peroxidation
5.1 SLC25A22
SLC25A22, an inner mitochondrial membrane glutamate transporter, serves as a key non-enzymatic substrate of SIRT3 in ferroptosis regulation. To fully address its role, it is necessary to distinguish how deacetylation affects the protein’s inherent transport activity versus its regulatory state. Our findings in LUAD models indicate that SIRT3-mediated deacetylation does not alter the transport kinetics or substrate affinity of SLC25A22. Rather, it specifically regulates protein turnover. By directly interacting with and deacetylating SLC25A22 at lysine 83 (K83), SIRT3 suppresses the transporter’s ubiquitination, preventing its degradation through the ubiquitin-proteasome system. Consequently, the SIRT3-SLC25A22 axis functions primarily to stabilize the protein; by extending the half-life of SLC25A22, SIRT3 ensures continuous mitochondrial glutamate import during RSL3-induced ferroptotic stress[6].
To understand how this stabilized pool of SLC25A22 actively resists ferroptosis, recent independent studies provide important context regarding its broader tumor-suppressive role. In pancreatic ductal adenocarcinoma models, SLC25A22 has been shown to block lipid peroxidation and cell death through iron-independent mechanisms[53]. The sustained mitochondrial glutamate influx provided by SLC25A22 feeds two distinct anti-ferroptotic pathways. First, it supplies the necessary precursors for GSH synthesis. Second, it activates the AMPK pathway. AMPK activation then upregulates stearoyl-CoA desaturase, which shifts lipid metabolism toward the synthesis of monounsaturated fatty acids (MUFAs)[53]. Since MUFAs replace easily oxidized PUFAs in cell membranes, this shift deprives the ferroptotic process of its primary lipid targets. Ultimately, a two-tiered defense mechanism emerges: SIRT3 first dictates the regulatory status of SLC25A22 by maintaining its stability via post-translational modification; this stable structural foundation then allows SLC25A22 to execute its transport function, rewiring cellular lipid metabolism to resist ferroptosis.
5.2 TfR1: The TfR1 axis and ferritinophagy
The expansion of the intracellular LIP serves as the absolute prerequisite for driving the Fenton reaction and initiating lipid peroxidation during ferroptosis[31]. Consequently, understanding SIRT3-mediated ferroptosis defense requires looking beyond its classical enzymatic antioxidant targets. We must examine how SIRT3 dictates the regulatory status of key non-enzymatic structural proteins to restrict the accumulation of “iron fuel” at its source. While SIRT3 does not directly alter the intrinsic iron-binding pockets or transport kinetics of these membrane carriers, it exerts profound indirect control over their expression and structural conformation by modulating the mitochondrial redox microenvironment.
A primary example of this regulatory control is the IRP1 and TfR1 axis. IRP1 functions as a highly sensitive metabolic switch. Under normal, iron-replete conditions, it exhibits cytosolic aconitase activity (its basal functional state). However, oxidative stress forces IRP1 to undergo a conformational rearrangement into an RNA-binding protein (its active regulatory state). Research demonstrates that SIRT3 serves as the upstream gatekeeper of this switch[54]. When SIRT3 is deficient, the resulting surge in mitochondrial ROS shifts IRP1 into its RNA-binding conformation. This activated IRP1 binds specifically to iron-responsive elements (IREs) on TfR1 messenger RNA (mRNA), stabilizing the transcript and causing massive TfR1 overexpression. Rather than directly interacting with the transporter, SIRT3 deficiency causes a loss of regulatory control over the IRP1 switch, triggering an unchecked influx of extracellular iron that directly expands the cytosolic LIP[54].
Furthermore, the cellular LIP is governed not only by external iron uptake but also by internal iron mobilization, predominantly through ferritinophagy. Ferritinophagy is an autophagic process mediated by nuclear receptor coactivator 4 (NCOA4), which targets the principal iron-storage protein, ferritin (FTH1), for lysosomal degradation and subsequent release of free iron[31]. Emerging evidence indicates a functional intersection between SIRT3 biology and this internal iron release mechanism. Because SIRT3 is essential for maintaining autophagic flux and mitochondrial quality control, its depletion often leads to dysregulated, maladaptive autophagy under stress. In this context, the loss of SIRT3 exacerbates NCOA4-mediated ferritinophagy, indiscriminately dumping sequestered iron from ferritin cages into the cytoplasm. This internal iron hemorrhage, combined with the TfR1-mediated external iron influx, creates a catastrophic expansion of the LIP that sensitizes cells to ferroptosis[31].
The biological significance of this dual iron-regulatory failure (TfR1 hyperactivation and unregulated ferritinophagy) has been validated across multiple disease models. In pancreatic cancer, low SIRT3 expression tightly correlates with elevated TfR1 levels and fatal intracellular iron accumulation, highlighting the tumor-suppressive role of the SIRT3-iron axis[54]. Similarly, in GBM, inhibiting SIRT3 triggers a surge in ROS and a localized accumulation of ferrous iron (Fe2+) within mitochondria. Although this oxidative shift concurrently suppresses the SLC7A11-ATF4 anti-ferroptotic pathway, the massive accumulation of mitochondrial lipid peroxides driven by the expanded LIP remains the primary executioner of GBM cell death[7]. Additionally, in neurodegenerative conditions like Friedreich’s ataxia, SIRT3 impairment leads to the hyperacetylation of mitochondrial proteins and aberrant IRP1 activation, ultimately causing pathological mitochondrial iron deposition.
In summary, SIRT3 establishes a multi-tiered molecular barrier against iron overload. By scavenging ROS to keep the IRP1-TfR1 uptake switch in an “off” regulatory state, and by potentially stabilizing ferritin pools against excessive NCOA4-mediated autophagic degradation, SIRT3 tightly constrains the cellular LIP, effectively halting ferroptosis before the Fenton reaction can begin.
5.3 p53
Beyond its canonical role in cell cycle arrest and apoptosis, p53 has recently been established as a pivotal non-enzymatic regulator of ferroptosis. However, the dualistic nature of p53, acting as either a “pro-survival” or “pro-death” agent in ferroptosis, is strictly contingent upon its post-translational modification status[48]. This review posits that SIRT3 serves as a decisive metabolic checkpoint by determining the functional orientation of p53 via the “SIRT3-p53 acetylation axis”.
First, the acetylation status of p53 is a critical determinant for its ferroptotic induction. Evidence suggests that acetylated p53 transcriptionally represses key anti-ferroptotic proteins, such as SLC7A11, thereby sensitizing cells to ferroptosis. Importantly, Figure 4 shows that the mitochondrial deacetylase SIRT3 possesses the capacity to reverse this process, acting as a molecular brake on p53-mediated ferroptosis[44,48].
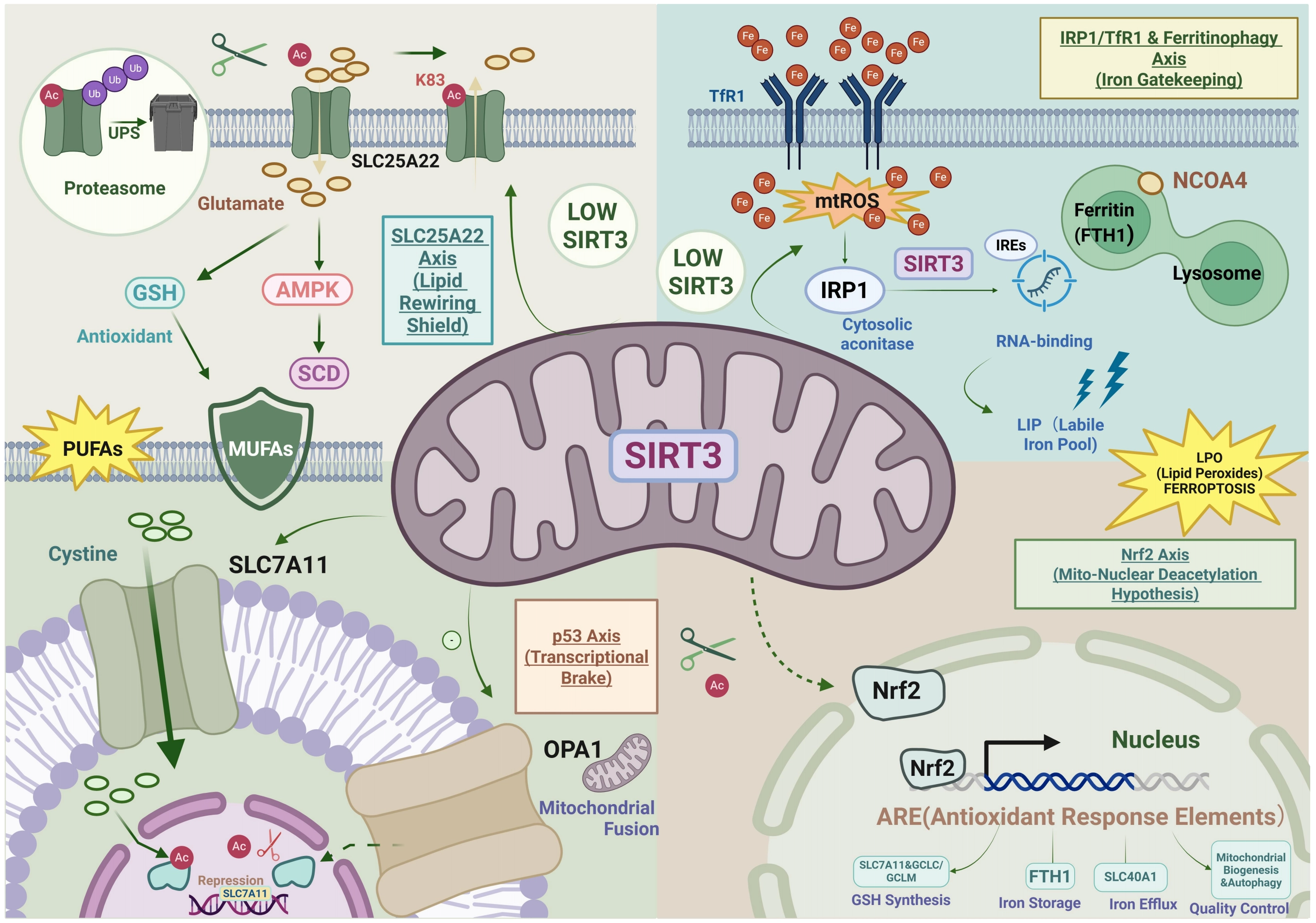
Figure 4. SIRT3 governs ferroptosis defense through the regulation of non-enzymatic substrates. SIRT3 functions as a central checkpoint against ferroptosis via four distinct axes: SIRT3 deacetylates SLC25A22 at Lys83 (K83), preventing its ubiquitination and proteasomal degradation to stabilize mitochondrial glutamate transport. By suppressing mitochondrial ROS, SIRT3 maintains IRP1 in its cytosolic aconitase form, preventing TfR1 mRNA stabilization and restricting the LIP. SIRT3 deacetylates p53, relieving its transcriptional repression of the cystine transporter SLC7A11 to restore GSH synthesis and GPX4 activity. A hypothetical axis proposes that SIRT3 may deacetylate Nrf2, facilitating its nuclear translocation and the transcription of antioxidant genes (FTH1, SLC40A1) via ARE. Created in BioRender.com. SIRT3: Sirtuin 3; ROS: reactive oxygen species; IRP1: iron regulatory protein 1; TfR1: transferrin receptor 1; mRNA: messenger RNA; LIP: labile iron pool; GSH: glutathione; GPX4: glutathione peroxidase 4; Nrf2: nuclear factor erythroid 2-related factor 2; FTH1: ferritin; ARE: Antioxidant Response Elements.
Second, SIRT3-mediated deacetylation of p53 constitutes a pivotal “molecular brake” on the ferroptotic cascade, particularly under conditions of metabolic stress. While acetylated p53 is known to suppress the expression of the cystine/glutamate antiporter SLC7A11 and activate glutaminase 2 to promote ferroptosis[48], SIRT3 counters this process by maintaining p53 in a deacetylated, inactive state. Evidence from cardiac models demonstrates that SIRT3 deficiency leads to hyperacetylation of p53, which directly facilitates ferroptosis and drives myocardial fibrosis[44]. Therefore, we postulate that SIRT3 functions as a pro-survival guardian: by removing acetyl groups from p53, SIRT3 prevents the p53-mediated transcriptional repression of SLC7A11, thereby preserving intracellular GSH levels and redox homeostasis, as elucidated in Figure 4. Furthermore, beyond transcriptional regulation, SIRT3 may synergize with this axis by stabilizing mitochondrial dynamics; SIRT3 has been shown to enhance the expression and acetylation of OPA1, promoting mitochondrial fusion and attenuating fibrosis[44]. Consequently, the “SIRT3-p53 axis” likely serves as a dual-layer checkpoint, simultaneously blocking the nuclear transcriptional initiation of ferroptosis and maintaining mitochondrial integrity to resist oxidative insults.
5.4 Nrf2: A potential deacetylation target of SIRT3 along the mito-nuclear axis
Although Nrf2 is widely recognized as the “master regulator” of cellular defense against ferroptosis, whether it serves as a direct non-enzymatic substrate subject to epigenetic regulation by the mitochondrial deacetylase SIRT3 remains a significant scientific hiatus. Nrf2 plays a decisive transcriptional role in the anti-ferroptosis network. Upon recognizing Antioxidant Response Elements, it drives three critical defense modules, as summarized in Figure 4: (i) upregulating the cystine transporter SLC7A11 and glutamate-cysteine ligase (GCLC/GCLM) to secure the substrate supply for GPX4-mediated lipid peroxide scavenging[35,55]; (ii) modulating the transcription of FTH1 and Ferroportin (SLC40A1) to prevent the Fenton reaction triggered by LIP overload[35]; and (iii) maintaining mitochondrial biogenesis and autophagic flux, functions that spatially and functionally overlap with SIRT3 biology[35].
However, current mechanistic understandings of Nrf2 are predominantly confined to the Keap1-p62 ubiquitin-proteasome degradation pathway[55], while its acetylation status is frequently overlooked, as shown in Figure 4. A precedent for SIRT3-mediated transcriptional regulation has been established: SIRT3 deacetylates the transcription factor p53 at Lysine 320 (K320), thereby inhibiting its pro-ferroptotic activity and protecting pulmonary epithelial cells from PM2.5-induced injury[56]. Based on this, it is logical to hypothesize that SIRT3 may modulate Nrf2 via a similar deacetylation mechanism.
Notably, while targeted knockdown of SIRT3 in glioblastoma models did not alter the total protein abundance of Nrf2[7], our observation does not preclude the possibility that SIRT3 regulates Nrf2 at the level of transcriptional activity. SIRT3 may deacetylate critical lysine residues on Nrf2 to facilitate its nuclear translocation or enhance its DNA-binding affinity, rather than affecting its stability. Consequently, establishing the existence of a “SIRT3-Nrf2 deacetylation axis” would fill a critical gap connecting mitochondrial metabolic signaling (SIRT3) with Nrf2, offering a novel theoretical perspective on how cells integrate metabolic stress with adaptive defense responses, as suggested by Figure 4, with question marks indicating the need for prospective validation.
6. The Mitochondrial Centrality of SIRT3 in Ferroptosis
In the majority of mammalian cells, mitochondria function not merely as metabolic powerhouses but as decisive arbiters of cell fate. While the absolute necessity of mitochondria in ferroptosis has historically been a subject of debate, the execution of lipid peroxidation and ferroptosis hinges critically on mitochondria-mediated reactive ROS generation and metabolic reprogramming[2]. As the primary mitochondrial NAD+ dependent deacetylase, SIRT3 functions far beyond the role of a mere antioxidant; instead, it orchestrates a multilayered defense strategy against ferroptosis through multidimensional mitochondrial quality control mechanisms.
Ferroptosis fundamentally represents a catastrophic failure of cellular lipid quality control (LQC), leading to the autocatalytic propagation of toxic lipid radicals[57]. As the primary intracellular hubs of iron, reactive oxygen species, and polyunsaturated fatty acids, mitochondria are highly vulnerable to such lipid electrophile-induced membrane rupture. Consequently, neutralizing ferroptosis requires more than localized enzymatic antioxidants; it demands a robust physical defense architecture. In this context, SIRT3 transcends its canonical role as a metabolic deacetylase to orchestrate a hierarchical, spatiotemporal LQC continuum. This structural defense strictly operates across three logical dimensions: preemptive isolation, damage quarantine, and terminal clearance. First, at the inter-organelle boundary, SIRT3 stabilizes MAMs to physically restrict the pathological influx of pro-ferroptotic lipids and calcium from the endoplasmic reticulum. Second, should localized oxidation breach this barrier, SIRT3 initiates topological remodeling via mitochondrial dynamics, asymmetrically driving fission to segregate peroxidized, iron-leaking segments from the healthy network. Finally, this structural quarantine seamlessly dictates the terminal execution phase, wherein SIRT3 primes these irreparably damaged fragments for targeted mitophagy and lysosomal degradation. Together, MAMs stabilization, dynamic remodeling, and mitophagy function not as isolated events, but as an integrated spatial barrier that prevents localized lipid damage from triggering a global ferroptotic collapse.
6.1 The role of SIRT3 in interorganellar communication: MAMs
MAMs are not merely signaling microdomains; they are highly dynamic, physical scaffolds maintained by specific tethering complexes. The primary structural bridge facilitating ER-to-mitochondria Ca2+ transfer is the tripartite complex comprising the ER-resident inositol 1,4,5-trisphosphate receptor (IP3R), the cytosolic linker GRP75, and the mitochondrial voltage-dependent anion channel 1 (VDAC1)[20], as illustrated in Figure 5. The structural integrity and optimal physical distance of MAMs are paramount for the precise spatiotemporal control of ER-to-mitochondria Ca2+ transfer.
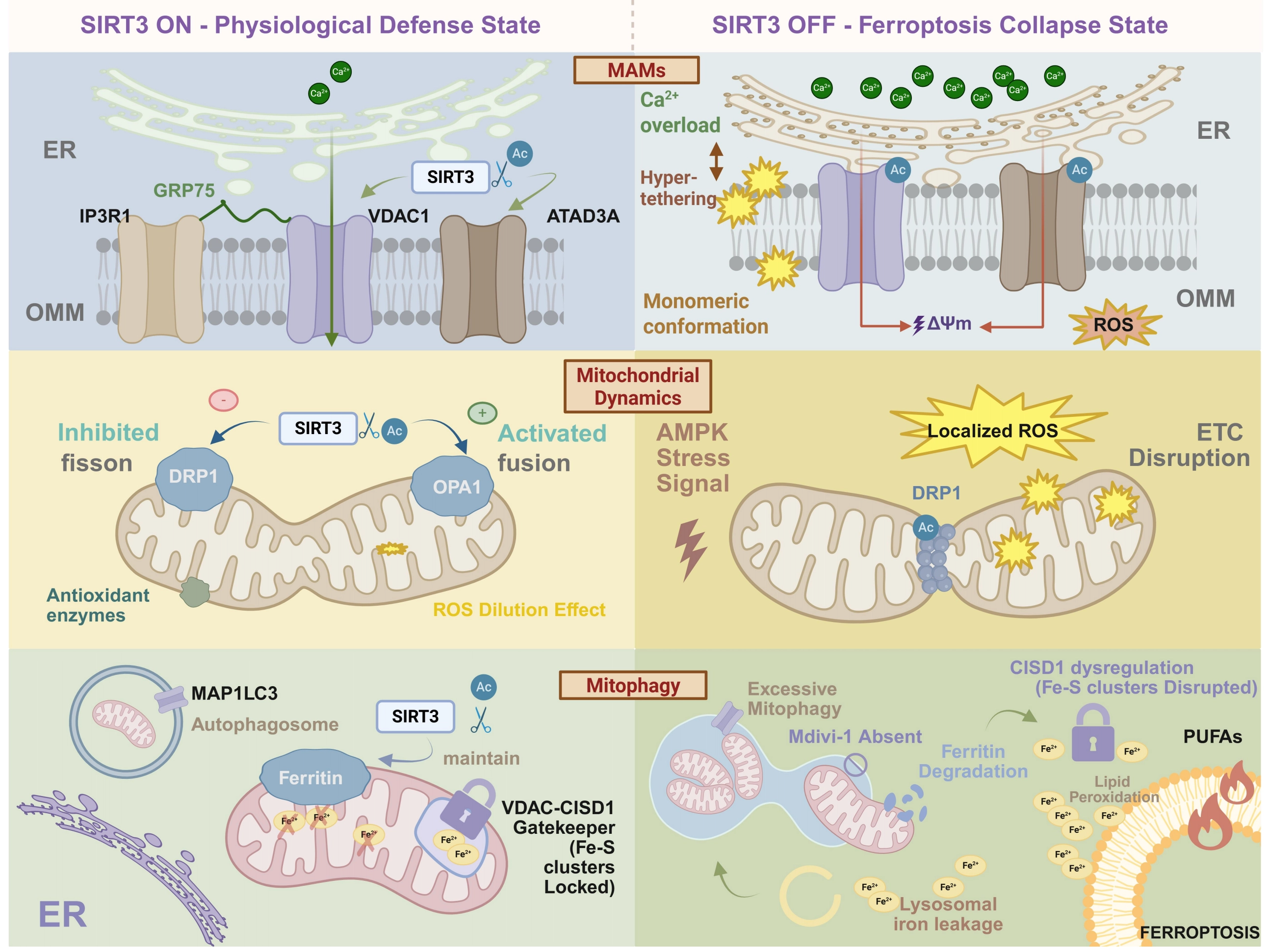
Figure 5. The spatiotemporal architecture of SIRT3-mediated mitochondrial quality control against ferroptosis. The schematic contrasts the SIRT3-driven physiological defense state (Left) with the ferroptotic collapse state triggered by SIRT3 depletion (Right) across three mitochondrial dimensions. (Top) MAMs Stabilization: SIRT3 deacetylates VDAC1 and ATAD3A to maintain a healthy ER-mitochondria interorganellar cleft. SIRT3 loss traps ATAD3A in a pathogenic monomeric conformation, driving VDAC1-GRP75-IP3R1 “hyper-tethering.” This structural collapse funnels excessive ER-derived Ca2+ into mitochondria, dissipating membrane potential (ΔΨm) and inciting a localized mtROS burst. (Middle) Topological Remodeling: SIRT3 dictates mitochondrial dynamics by inhibiting DRP1-mediated fission and activating OPA1-driven fusion. The resulting interconnected network buffers oxidative damage via a “ROS dilution effect”. Conversely, SIRT3 deficiency permits stress-induced excessive fragmentation, transforming isolated mitochondria into platforms for ETC disruption and localized ROS accumulation. (Bottom) Terminal Mitophagy Checkpoint: SIRT3 prevents autophagy-mediated iron hemorrhage by stabilizing mitochondrial ferritin and maintaining the VDAC-CISD1 “gatekeeper” to securely lock Fe-S clusters. Unchecked, excessive mitophagy degrades ferritin cages and dismantles Fe-S clusters, unleashing a catastrophic expansion of free iron (lysosomal iron leakage). This iron fuels the terminal peroxidation of PUFAs, executing ferroptosis. Created in BioRender.com. SIRT3: Sirtuin 3; MAMs: mitochondria-associated membranes; VDAC1: voltage-dependent anion channel 1; ER: endoplasmic reticulum; OPA1: optic atrophy-associated protein 1; ROS: reactive oxygen species; PUFAs: polyunsaturated fatty acids.
Emerging evidence demonstrates that SIRT3 orchestrates the interorganellar gap distance by strictly dictating the electrostatic state and quaternary structure of key tethering proteins. At the outer mitochondrial membrane (OMM), SIRT3 directly targets and deacetylates VDAC1. Under physiological conditions, this hypoacetylated state physically restricts VDAC1’s binding affinity to the GRP75-IP3R machinery, maintaining a healthy interorganellar cleft. Conversely, SIRT3 depletion traps VDAC1 in a hyperacetylated state. This subtle enzymatic shift removes local positive charges, inducing a conformational alteration that dramatically amplifies VDAC1’s affinity for GRP75, physically pulling the ER and mitochondria into a state of pathological “hyper-tethering”[58].
Furthermore, this physical remodeling is synergistically governed by the mitochondrial structural protein ATAD3A. Mechanistically, SIRT3 specifically deacetylates ATAD3A at the lysine 134 (K134) residue, a targeted modification that strictly drives ATAD3A self-oligomerization. The absence of SIRT3-dependent deacetylation forces ATAD3A into a pathogenic monomeric conformation. Crucially, these aberrant monomers act as a structural glue that heavily interacts with and stabilizes the IP3R1-GRP75-VDAC1 supercomplex, thereby further shrinking the ER-mitochondria distance and rigidifying the MAM architecture[59].
Figure 5 shows that such mechanically rigidified and excessively coupled MAM architecture abolishes the spatiotemporal buffering of Ca2+. The resulting unrestrained Ca2+ leakage from the ER provokes an acute mitochondrial calcium overload. This ionic collapse instantly dismantles the electrochemical gradient (ΔΨm) and triggers a massive, localized burst of mitochondrial reactive oxygen species (mitochondrial ROS; mtROS)[60].
Collectively, this review posits a structurally refined hypothesis: SIRT3 physically guards the interorganellar boundary by maintaining tethering proteins in an electrostatic and oligomeric state that prevents pathological membrane fusion. When SIRT3 is lost, the resulting MAM “hyper-coupling” orchestrates a lethal architectural collapse, funneling an excess of ER-derived Ca2+ directly into the mitochondria. This structural failure establishes the precise peroxidative microenvironment necessary to execute the terminal lipid peroxidation cascade of ferroptosis.
6.2 Mitochondrial dynamics
SIRT3 remodels mitochondrial dynamics to establish an anti-death barrier. The physical morphology of mitochondria, governed by the dynamic equilibrium between fusion and fission, directly dictates cellular sensitivity to ferroptosis[61]. Under normal physiological conditions (the “mitochondrial life cycle”), mitochondria segregate damaged components via fission and exchange contents through selective fusion, a mechanism critical for maintaining metabolic adaptability[62]. However, under pathological stress, such as energy deprivation or oxidative injury, this equilibrium frequently undergoes a fatal deviation. Toyama et al. demonstrated that AMPK-mediated stress responses rapidly induce mitochondrial fission[62]. In the context of ferroptosis, this DRP1-mediated excessive fission transitions from an adaptive response to a “lethal prelude,” accompanied by a burst of ROS and loss of membrane potential[61]. This fragmented mitochondrial morphology not only destabilizes the electron transport chain but also precipitates the acute accumulation of local oxidative damage, rendering cells highly susceptible to the cascade of ferroptosis.
At this critical juncture, SIRT3 constructs a pivotal “anti-death barrier” by reshaping mitochondrial dynamics. SIRT3 exerts a dual regulatory effect via its deacetylase activity: Figure 5 depicts that it downregulates DRP1 to inhibit pathological fragmentation[21] while simultaneously upregulating OPA1 to promote mitochondrial networking[22]. In Parkinson’s disease models, acetylation levels of DRP1 at Lysine 711 (K711) are significantly elevated; SIRT3-mediated deacetylation of this site inhibits DRP1 activity, thereby mitigating mitochondrial defects[21]. This mechanism offers significant translational insights for ferroptosis research. Concurrently, the direct deacetylation of OPA1 by SIRT3 functions as a molecular switch driving morphological remodeling. This not only significantly enhances cristae density and the complexity of the fusion network but also establishes the structural foundation for cellular resistance against metabolic stress by optimizing bioenergetics[22].
This morphological “fusion” carries profound biochemical significance, it is not merely the connectivity of membrane structures but a strategic defense mechanism. A highly interconnected mitochondrial network effectively mitigates local oxidative damage through the “dilution effect” and buffers ROS impact by sharing antioxidant enzyme systems, as shown in Figure 5. Furthermore, fusion mechanisms are postulated to physically restrict the propagation of lipid peroxidation products across the membrane system[61]. Thus, the essence of SIRT3 function lies in preventing mitochondria from transforming into a “death signal platform” that propagates oxidative stress waves. By maintaining reticular homeostasis, SIRT3 erects a robust defense line against ferroptosis across both physical and metabolic dimensions.
6.3 Mitophagy
Within the framework of mitochondrial centrality, the interplay between mitophagy and ferroptosis constitutes a pivotal checkpoint in determining cell fate. Accumulating evidence substantiates that mitophagy acts as a critical node in the ferroptosis regulatory network[23,37]. While the conventional view posits that mitophagy confers protection by eliminating damaged mitochondria, under specific pathological stress conditions, its excessive activation paradoxically functions as a driver of ferroptosis[19]. For instance, in the context of CISD3 (CDGSH iron sulfur domain 1, a mitochondria-localized protein) deficiency, the activation of mitophagy has been shown to alleviate lipid peroxidation and ferroptosis by clearing dysfunctional mitochondria[37]. However, this protective role is not absolute. Studies indicate that while inhibition of mitochondrial Complex I triggers mitophagy via mtROS, the ultimate cell death, including necroptosis and ferroptosis, is fundamentally a consequence of “mitophagy-dependent ROS generation”, implying that the autophagic process itself exacerbates the accumulation of lethal ROS[23,37].
Mitochondria serve not only as the primary site of ROS production but also as the hub for iron-sulfur (Fe-S) cluster synthesis and storage. Research highlights that excessive mitophagy precipitates the massive release of iron from Fe-S clusters, thereby expanding the intracellular LIP. This, in turn, triggers lysosomal iron leakage and initiates ferroptosis[19,63], as highlighted in Figure 5. Concurrently, enhanced mitochondrial fragmentation and mitophagy liberate sequestered iron reserves, providing the catalytic fuel for lipid peroxidation and sensitizing cells to ferroptosis[63,64]. These findings underscore that the magnitude of mitophagy directly dictates whether mitochondria function as executioners or inhibitors of ferroptosis.
In this context, SIRT3 emerges as a master regulator of mitochondrial quality control, a role that appears universally conserved across different osteoporosis models. Wang et al. recently reported in a type 2 diabetic osteoporosis model that mitochondrial ferritin deficiency promotes ferroptosis precisely by mediating aberrant mitophagy in osteoblasts[37]. This lends further credence to the hypothesis that SIRT3 may curb autophagy-mediated iron leakage by maintaining the stability or expression of mitochondrial ferritin. Furthermore, at the level of molecular interaction, the crosstalk between the outer mitochondrial membrane protein VDAC and CISD1 is regarded as a key “gatekeeper” of mitochondrial iron homeostasis, with its dysfunction directly impacting iron-dependent cell death trajectories[23]. Thus, the mechanism of SIRT3 extends beyond merely suppressing autophagic flux at a macroscopic level; it involves the precise regulation of these key protein stabilities, thereby severing the vicious cycle of “excessive mitophagy–ferritin degradation–CISD1 dysregulation–ferroptosis”.
The regulation of mitophagy by SIRT3 adds a novel dimension to our understanding of ferroptosis initiation and offers potential therapeutic targets. Experimental results using the mitophagy inhibitor Mdivi-1 align highly with the protective effects of SIRT3: both achieve ferroptosis blockade by restoring mitochondrial homeostasis and inhibiting autophagic flux[63,65]. This further corroborates that, in specific pathological contexts, the execution of ferroptosis is strictly dependent on unchecked mitophagy. Moreover, complex crosstalk and feedback loops exist between ferroptosis and autophagy; ferroptosis induction itself is accompanied by increased MAP1LC3 conversion and autophagosome formation. Some studies have even proposed novel selective autophagy modes, such as “clockophagy,” which facilitate ferroptosis through oxidative damage[66].
Therefore, SIRT3 is not merely a simple inhibitor. Instead, it modulates the sensitivity of mitochondria to stress signals. By preventing the autophagic flux from shifting toward a “pro-death” mode, specifically, by preventing autophagy from evolving into a source of excess ROS and LIP release, SIRT3 establishes a novel mechanistic link between mitochondrial quality control and ferroptosis suppression.
7. The Dual Oncogenic Role of SIRT3 and Its Context-Dependent Crosstalk with Ferroptosis
As the primary mitochondrial deacetylase, the function of SIRT3 in tumor biology has long been a subject of debate. The traditional binary paradigm, which rigidly defines SIRT3 as either a tumor suppressor or an oncogene[5], is increasingly insufficient to capture its complex biological behavior. Emerging evidence underscores that the role of SIRT3 is highly “context-dependent”, whether SIRT3 suppresses tumorigenesis or drives therapy resistance is strictly dictated by distinct cellular conditions, specifically, the stage of malignant progression, the intrinsic metabolic wiring of the tumor, and the oxidative stress threshold within the TME[5,67].
As depicted in Figure 6, during the earliest stages of tumorigenesis, SIRT3 predominantly functions as a classical tumor suppressor by acting as a strict gatekeeper of mitochondrial integrity[3]. Under basal physiological conditions, normal cells rely on SIRT3 to scavenge steady-state ROS. When SIRT3 is lost or epigenetically silenced, cells experience a chronic, moderate elevation in mitochondrial superoxide[3]. Crucially, in this specific cellular context, such as in pre-malignant epithelial tissues, this ROS accumulation does not reach the lethal threshold required to trigger massive lipid peroxidation and ferroptosis. Instead, it acts as a subtle “mutagenic” signaling molecule, inflicting mitochondrial DNA damage and precipitating genomic instability. Furthermore, SIRT3 deficiency irreversibly impairs mitochondrial complexes I and III, forcing the cell into a compensatory glycolytic shift (the Warburg effect)[3]. Therefore, in early-stage contexts, the physiological presence of SIRT3 thwarts malignant transformation by neutralizing mutagenic ROS and preventing pre-cancerous metabolic reprogramming.
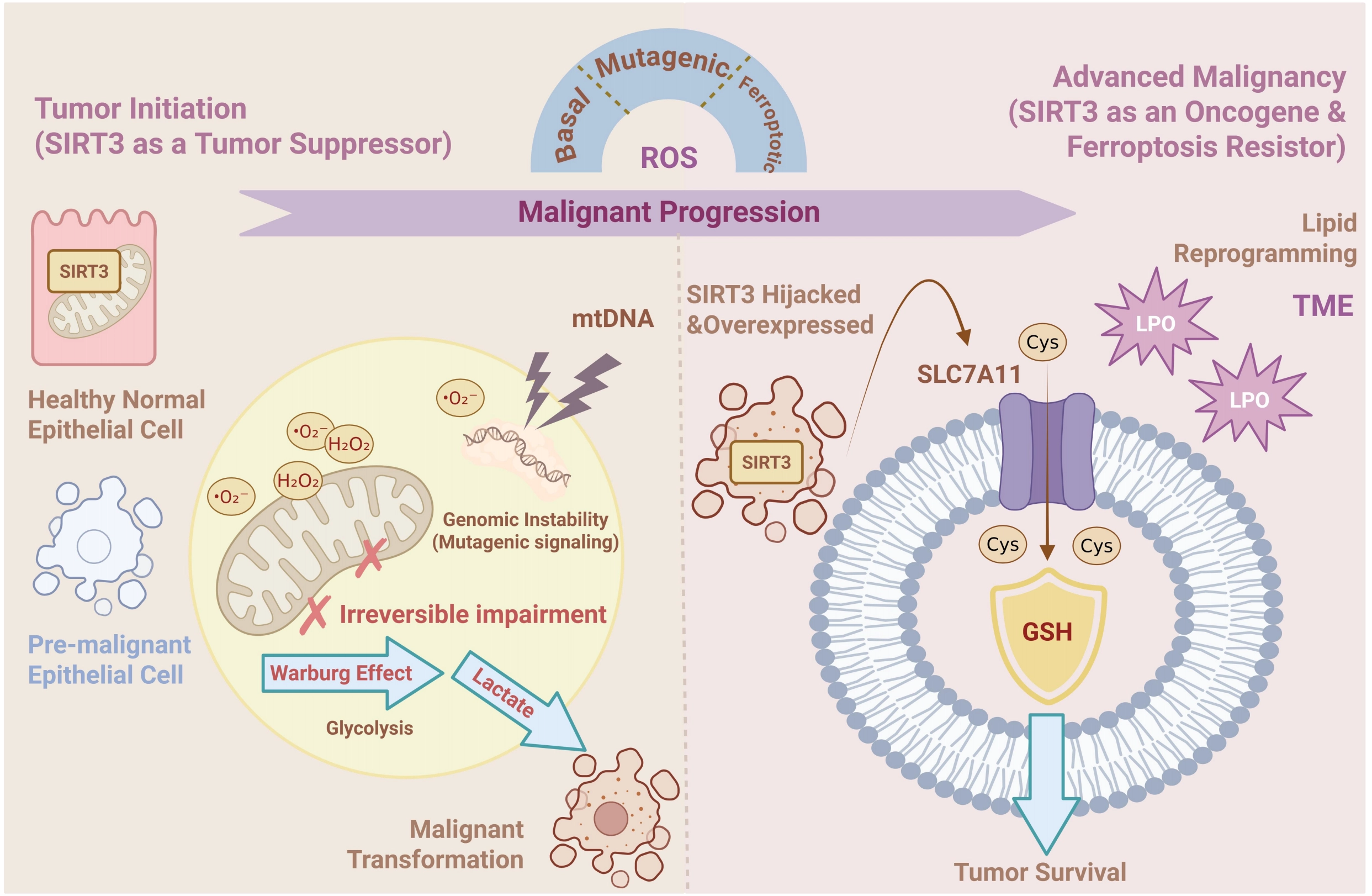
Figure 6. The context-dependent dual role of SIRT3 in ferroptosis and tumorigenesis. During tumor initiation, SIRT3 functions as a tumor suppressor; its deficiency precipitates mitochondrial ROS accumulation and genomic instability. This induces mitochondrial dysfunction (Complex I/III) and a glycolytic shift (Warburg effect), creating a mutagenic environment that drives tumorigenesis. In established tumors, SIRT3 shifts to an oncogenic role to confer ferroptosis resistance. Overexpressed SIRT3 upregulates the cystine transporter SLC7A11 via the ATF4 transcription factor, enhancing GSH synthesis and GPX4 activity. This signaling axis efficiently scavenges lethal lipid peroxides, thereby blocking the ferroptosis pathway and ensuring tumor survival under metabolic stress. Created in BioRender.com. SIRT3: Sirtuin 3; ROS: reactive oxygen species; GSH: glutathione; GPX4: glutathione peroxidase 4; ATF4: activating transcription factor 4.
Conversely, in established and advanced malignancies, the cellular context drastically changes. Advanced tumors face immense oxidative and metabolic stress generated by rapid proliferation, a hypoxic TME, and the administration of chemoradiotherapy. In these highly stressed conditions, cancer cells actively hijack SIRT3, shifting its role to a potent driver of ferroptosis resistance[67]. By exquisitely coordinating antioxidant defenses, overexpressed SIRT3 maintains ROS levels below the “critical lethal threshold”, allowing ROS to drive pro-tumorigenic signaling while preventing the execution of lipid peroxidation-induced death[5]. This oncogenic adaptation is particularly pronounced in highly aggressive and metabolically flexible cancers, such as GBM. In GBM models, robust SIRT3 expression is heavily relied upon for tumor survival under therapy-induced stress. Mechanistically, SIRT3 stabilizes the expression of the cystine transporter SLC7A11, which directly bolsters GSH biosynthesis and hyperactivates the GPX4 defense system[7]. Furthermore, as recent immuno-metabolic models suggest, therapy-resistant cancer cells frequently undergo lipid reprogramming to evade ferroptosis[67]. In this hostile landscape, SIRT3 acts as an indispensable metabolic buffer, empowering cancer cells to tolerate toxic lipid peroxides and actively driving acquired resistance against ferroptosis-inducing therapies.
In summary, the role of SIRT3 in ferroptosis operates on a dynamic, stage-specific continuum. We propose an integrative model: during tumor initiation, SIRT3 deficiency promotes carcinogenesis by permitting ROS-mediated genomic damage (loss of tumor-suppressive function); whereas during tumor progression and therapeutic intervention, SIRT3 overexpression shields cancer cells from lethal metabolic stress and drives chemoresistance (gain of oncogenic function). Consequently, therapeutic strategies must be precisely tailored to the specific cellular context, leveraging SIRT3 activation for chemoprevention in early precancerous stages, while deploying targeted SIRT3 inhibitors to dismantle the ferroptosis-resistance machinery in advanced, refractory tumors.
8. Clinical Applications and Future Perspectives
The pivotal role of SIRT3 in orchestrating mitochondrial homeostasis and oxidative stress responses positions it as a premier target for therapeutic intervention in ferroptosis-associated pathologies. Current preclinical and clinical developments underscore a distinct “bidirectional therapeutic strategy”: in oncology, the objective is to induce ferroptosis in cancer cells by inhibiting SIRT3[68], whereas in cardiovascular and neurodegenerative diseases, the aim is to protect tissue function by activating SIRT3 to suppress ferroptosis[69].
In the context of oncology, particularly in refractory triple-negative breast cancer (TNBC), malignant cells frequently exploit SIRT3 to bolster antioxidant defenses and evade cell death[2], as shown in Figure 7. Consequently, the development of SIRT3 inhibitors to disrupt redox homeostasis and trigger ferroptosis represents a promising therapeutic avenue. Recent findings identify the natural compound Eriocalyxin B (Eri B) as a novel SIRT3 inhibitor with potent anti-tumor activity[2]. Mechanistically, Eri B directly targets STAT3 to repress SIRT3 transcription, thereby dismantling the SIRT3-SREBP1-PPARα axis governing lipid metabolism. Concurrently, it inhibits the Nrf2-GPX4 signaling pathway, precipitating the accumulation of lethal lipid peroxides and inducing ferroptosis in TNBC models both in vitro and in vivo[2]. This discovery not only establishes Eri B as a ferroptosis inducer but also provides robust preclinical evidence for targeting SIRT3 in TNBC treatment. Furthermore, to mitigate the non-specific toxicity of small molecules, the integration of antibody-drug conjugates (ADCs) and nanodelivery systems will be critical for enhancing the clinical potential of SIRT3 inhibitors[5].
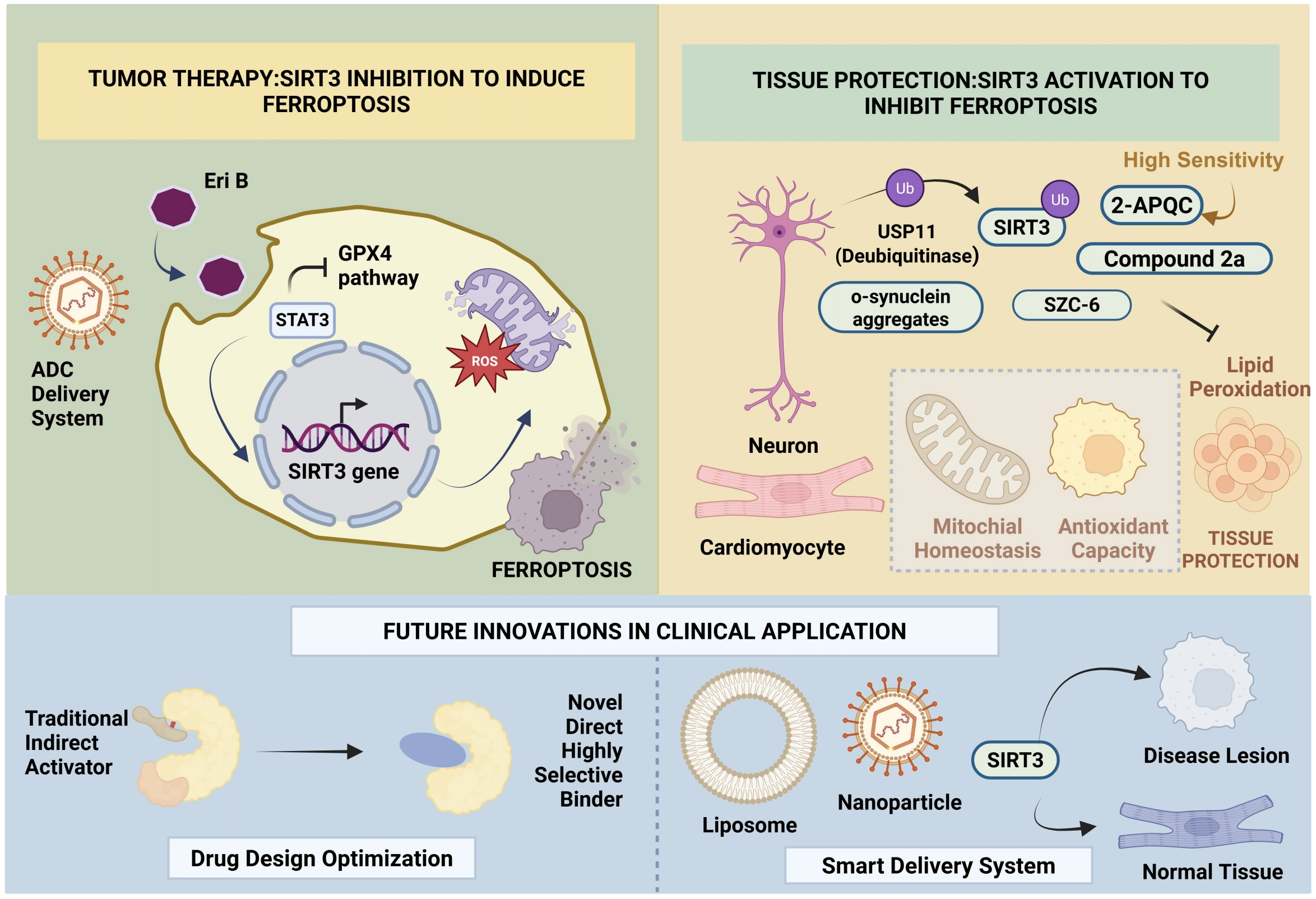
Figure 7. Bidirectional therapeutic strategies and future clinical innovations targeting SIRT3-mediated ferroptosis. The therapeutic approach bifurcates based on disease pathology. In oncology, SIRT3 inhibition (e.g., via Eriocalyxin B) disrupts the STAT3-SIRT3 axis and suppresses the Nrf2-GPX4 pathway, precipitating lethal lipid peroxidation to kill cancer cells. Conversely, in non-neoplastic diseases, SIRT3 activation—achieved via USP11 stabilization or novel specific agonists (e.g., 2-APQC, SZC-6, Compound 2a)—restores mitochondrial homeostasis and antioxidant capacity to block pathological ferroptosis. Future translation relies on two pillars: (1) Drug Design Optimization, shifting from non-selective indirect modulators to high-affinity direct binders (e.g., 2-APQC), and (2) Intelligent Delivery Systems (Liposomes, ADCs, Nanoparticles) to ensure precise lesion targeting and minimize off-target toxicity. Created in BioRender.com. SIRT3: Sirtuin 3; Nrf2: nuclear factor erythroid 2-related factor 2; GPX4: glutathione peroxidase 4; ADCs: antibody-drug conjugates.
Conversely, in cardiovascular diseases, neurodegenerative disorders, and intervertebral disc degeneration (IVDD), SIRT3 downregulation is often a precipitating factor for mitochondrial dysfunction and pathological ferroptosis, as displayed in Figure 7. Thus, restoring SIRT3 activity or stoichiometry is a vital therapeutic strategy. The deubiquitinase USP11 has been identified as a direct stabilizer of SIRT3. USP11 prevents SIRT3 degradation via deubiquitination, thereby fortifying cellular resilience against oxidative stress, inhibiting ferroptosis, and ameliorating pain behaviors associated with IVDD[70].
Given the limitations of traditional SIRT3 activators (e.g., honokiol, resveratrol), such as poor selectivity and indirect mechanisms often reliant on polyphenol antioxidant structures or the AMPK/Nrf2 pathway[5], structure-based screening technologies have catalyzed the discovery of highly specific small molecules. For instance, 2-APQC, a novel structure-specific SIRT3 activator, has been proven to specifically modulate mitochondrial homeostasis and alleviate cardiac hypertrophy and fibrosis; it is currently undergoing preclinical evaluation[44,71].
In the realms of neurology and cardiology, significant breakthroughs have been achieved. In Parkinson’s disease (PD) models, the novel spiro-grafted macrocyclic sulfonamide, Compound 2a, protects dopaminergic neurons from ferroptosis and clears α-synuclein aggregates by activating SIRT3[44]. Moreover, the SIRT3-targeting agent SZC-6 has advanced to clinical trials for the treatment of cardiac hypertrophy, marking a substantial step toward the clinical application of SIRT3 activators in ferroptosis-related diseases[44].
Despite the immense therapeutic potential of SIRT3 modulation, clinical translation remains impeded by challenges related to off-target effects and bioavailability. Traditional small-molecule agonists (e.g., STA-5326) often operate via indirect pathways, limiting their translational efficacy[71]. This review posits that future clinical innovation will converge on two dimensions, as outlined in Figure 7:
1. Optimized Drug Design: The development of compounds like 2-APQC that eschew general antioxidant properties in favor of direct, high-affinity binding to the SIRT3 catalytic pocket[44,71].
2. Intelligent Delivery Systems: The utilization of tumor-targeted delivery platforms, such as liposomes, nanoparticles, and ADCs. These systems will precisely deliver SIRT3 modulators to lesion sites, resolving issues of rapid clearance while minimizing specific “off-target” toxicity to normal tissues, thereby paving the way for the clinical implementation of SIRT3-targeted therapeutics[5].
Clinical translation remains at an early stage, and a central unmet need is the development of truly isoform-specific SIRT3 modulators, both activators and inhibitors, that can distinguish SIRT3 from other sirtuin family members and, more importantly, from indirect antioxidant or NAD+ modulating effects.
Although structure-guided compounds such as 2-APQC have provided proof-of-concept that direct pharmacological activation of SIRT3 is feasible, it remains unclear whether currently available agents possess sufficient selectivity, mitochondrial target engagement, pharmacokinetic robustness, and tissue exposure to support durable clinical benefit[71].
In parallel, the field lacks standardized pharmacodynamic biomarkers that can verify on-target SIRT3 modulation in vivo, such as substrate-specific acetylation signatures, mitochondrial acetylome readouts, or ferroptosis-linked metabolic indices that are deployable in preclinical and early-phase clinical studies.
Overcoming this analytical bottleneck requires a fundamental shift in how ferroptosis and acylation metrics are quantified. Advancing the translational frontier is strictly contingent upon the rigorous development, validation, and transparent sharing of novel experimental methodologies[72]. Currently, assessing SIRT3 activity mainly relies on lab-specific biochemical assays that lack cross-trial reproducibility. To delve into the SIRT3 modulation, the field must establish more robust technological frameworks, ranging from optimized tissue quantification protocols for specific acyl-adducts to non-invasive metabolic imaging of lipid flux[72]. Without formalizing these methodological standards, substantiating hypotheses in vivo, target engagement will remain intractable, making it nearly impossible to distinguish true SIRT3 regulation from off-target antioxidant noise in clinical settings.
Another actionable gap lies in patient stratification: because the biological consequence of SIRT3 modulation is profoundly context-dependent, future trials will require biomarkers capable of identifying SIRT3-addicted, ferroptosis-resistant tumors on the one hand, and SIRT3-deficient, ferroptosis-vulnerable degenerative tissues on the other[28,29].
This challenge is compounded by the dual nature of ferroptosis itself, whose therapeutic efficacy may be constrained by tumor microenvironmental heterogeneity, lineage-specific defense circuits, and the risk of injuring normal tissues that also rely on SIRT3 to preserve mitochondrial integrity[27,29].
Accordingly, the next phase of translational research should move beyond general claims of “activation” or “inhibition” and instead prioritize four concrete objectives: (i) rational design of direct, isoform-biased SIRT3 ligands; (ii) establishment of target-engagement and response biomarkers; (iii) development of mitochondria- and tissue-selective delivery platforms; and (iv) definition of disease-specific therapeutic windows that account for the divergent roles of SIRT3 in cancer versus non-neoplastic injury[28,31].
Only when these mechanistic, pharmacological, and clinical-stratification gaps are addressed can SIRT3-directed ferroptosis therapy progress from an attractive conceptual framework to a credible precision-medicine strategy.
Acknowledgements
GEMINI3 was used solely for language refinement, including improvements in grammar, clarity, and readability, and had no role in the development of scientific ideas or conclusions. The authors take full responsibility for the integrity, originality, and accuracy of the work.
Authors contribution
Cai R: Conceptualization, supervision, writing-review & editing.
Chen Y, Shi Q: Writing-original draft, writing-review & editing.
Wei X, Wang X: Writing-review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Funding
This work was supported by the National Natural Science Foundation of China (Grant No.82173352)
Copyright
© The Author(s) 2026.
References
-
2. Chen X, He J, Ge G, Ma S, Chen Z, Zhang L, et al. Eriocalyxin B induces ferroptosis through SIRT3 inhibition in triple-negative breast cancer. Phytomedicine. 2025;148:157257.[DOI]
-
5. Zhang J, Ye J, Zhu S, Han B, Liu B. Context-dependent role of SIRT3 in cancer. Trends Pharmacol Sci. 2024;45(2):173-190.[DOI]
-
6. Wei X, Wang T, Xing Z, Shi Q, Gu J, Fan Q, et al. Sirtuin 3 protects lung adenocarcinoma from ferroptosis by deacetylating and stabilizing mitochondrial glutamate transporter solute carrier family 25 member A22. Antioxidants. 2025;14(4):403.[DOI]
-
9. Moniot S, Weyand M, Steegborn C. Structures, substrates, and regulators of mammalian sirtuins–opportunities and challenges for drug development. Front Pharmacol. 2012;3:16.[DOI]
-
11. Jin J, Bai L, Wang D, Ding W, Cao Z, Yan P, et al. SIRT3-dependent delactylation of cyclin E2 prevents hepatocellular carcinoma growth. EMBO Rep. 2023;24(5):e56052.[DOI]
-
13. Wu M, Zhang C, Xie M, Zhen Y, Lai B, Liu J, et al. Compartmentally scavenging hepatic oxidants through AMPK/SIRT3-PGC1α axis improves mitochondrial biogenesis and glucose catabolism. Free Radic Biol Med. 2021;168:117-128.[DOI]
-
17. Qin X, Cai P, Liu C, Chen K, Jiang X, Chen W, et al. Cardioprotective effect of ultrasound‐targeted destruction of Sirt3‐loaded cationic microbubbles in a large animal model of pathological cardiac hypertrophy. Acta Biomater. 2023;164:604-625.[DOI]
-
18. Nguyen KT, Mun SH, Yang J, Lee J, Seok OH, Kim E, et al. The MARCHF6 E3 ubiquitin ligase acts as an NADPH sensor for the regulation of ferroptosis. Nat Cell Biol. 2022;24(8):1239-1251.[DOI]
-
19. Rademaker G, Boumahd Y, Peiffer R, Anania S, Wissocq T, Liégeois M, et al. Myoferlin targeting triggers mitophagy and primes ferroptosis in pancreatic cancer cells. Redox Biol. 2022;53:102324.[DOI]
-
22. Wang R, Xu H, Tan B, Yi Q, Sun Y, Xiang H, et al. SIRT3 promotes metabolic maturation of human iPSC-derived cardiomyocytes via OPA1-controlled mitochondrial dynamics. Free Radic Biol Med. 2023;195:270-282.[DOI]
-
23. Su L, Zhang J, Gomez H, Kellum JA, Peng Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy. 2023;19(2):401-414.[DOI]
-
24. Wu Y, Ling S, Mao J, Wang H, Wang B, Che Z, et al. Sirt3 genetically engineered apoptotic bodies alleviate skeletal aging by limiting aggravated NLRP3 inflammasome activation of senescent macrophages. Adv Sci. 2026.[DOI]
-
25. Cai J, Wan F, Yang J, Wang Y, Wang J. Resveratrol alleviates polycystic ovary syndrome symptoms by reducing inflammation, oxidative stress, and ferroptosis features in a mouse model: A prospective study. Clin Exp Obstet Gynecol. 2025;52(11):41496.[DOI]
-
27. Ubellacker JM, Dixon SJ. Prospects for ferroptosis therapies in cancer. Nat Cancer. 2025;6(8):1326-1336.[DOI]
-
29. Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25(6):424-442.[DOI]
-
30. Wu K, Vaughan AJ, Bossowski JP, Hao Y, Ziogou A, Kim SM, et al. Targeting FSP1 triggers ferroptosis in lung cancer. Nature. 2026;649(8096):487-495.[DOI]
-
31. Sun S, Shen J, Jiang J, Wang F, Min J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Sig Transduct Target Ther. 2023;8:372.[DOI]
-
32. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: The role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18(5):280-296.[DOI]
-
33. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212(4):555-568.[DOI]
-
34. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16(11):2069-2083.[DOI]
-
41. Rakshe PS, Dutta BJ, Chib S, Maurya N, Singh S. Unveiling the interplay of AMPK/SIRT1/PGC-1α axis in brain health: Promising targets against aging and NDDs. Ageing Res Rev. 2024;96:102255.[DOI]
-
43. Trinh D, Al Halabi L, Brar H, Kametani M, Nash JE. The role of SIRT3 in homeostasis and cellular health. Front Cell Neurosci. 2024;18:1434459.[DOI]
-
44. Cheng Y, Zhao A, Li Y, Li C, Miao X, Yang W, et al. Roles of SIRT3 in cardiovascular and neurodegenerative diseases. Ageing Res Rev. 2025;104:102654.[DOI]
-
45. Ning Y, Dou X, Wang Z, Shi K, Wang Z, Ding C, et al. SIRT3: A potential therapeutic target for liver fibrosis. Pharmacol Ther. 2024;257:108639.[DOI]
-
48. Liu Y, Su Z, Tavana O, Gu W. Understanding the complexity of p53 in a new era of tumor suppression. Cancer Cell. 2024;42(6):946-967.[DOI]
-
53. Liu Y, Wang Y, Lin Z, Kang R, Tang D, Liu J. SLC25A22 as a key mitochondrial transporter against ferroptosis by producing glutathione and monounsaturated fatty acids. Antioxid Redox Signal. 2023;39(1-3):166-185.[DOI]
-
54. Jeong SM, Lee J, Finley LS, Schmidt PJ, Fleming MD, Haigis MC. SIRT3 regulates cellular iron metabolism and cancer growth by repressing iron regulatory protein 1. Oncogene. 2015;34(16):2115-2124.[DOI]
-
56. Li N, Xiong R, Li G, Wang B, Geng Q. PM2.5 contributed to pulmonary epithelial senescence and ferroptosis by regulating USP3-SIRT3-P53 axis. Free Radic Biol Med. 2023;205:291-304.[DOI]
-
60. Rodríguez LR, Calap-Quintana P, Lapeña-Luzón T, Pallardó FV, Schneuwly S, Navarro JA, et al. Oxidative stress modulates rearrangement of endoplasmic reticulum-mitochondria contacts and calcium dysregulation in a Friedreich’s ataxia model. Redox Biol. 2020;37:101762.[DOI]
-
62. Tábara LC, Segawa M, Prudent J. Molecular mechanisms of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2025;26(2):123-146.[DOI]
-
68. Shen G, Liu J, Wang Y, Deng Z, Deng F. Ferroptosis in cancer and inflammatory diseases: Mechanisms and therapeutic implications. MedComm. 2025;6(9):e70349.[DOI]
-
69. Cao Y, Wang Y, Zhao N, Yuan Z, Zhang L, Jin P. Unraveling the roles of mitochondrial sirtuins in aging-related diseases: From mechanistic insights to therapeutic strategies. Metabolism. 2025;172:156356.[DOI]
-
70. Zhu J, Sun R, Sun K, Yan C, Jiang J, Kong F, et al. The deubiquitinase USP11 ameliorates intervertebral disc degeneration by regulating oxidative stress-induced ferroptosis via deubiquitinating and stabilizing Sirt3. Redox Biol. 2023;62:102707.[DOI]
-
72. Adams JC. Physiological Reports begins a new manuscript category, the Methods article. Physiol Rep. 2023;11(17):e15808.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Chen Y, Shi Q, Wang X, Wei X, Cai R. SIRT3 at the crossroads of ferroptosis: Multidimensional regulation of the mitochondrial deacetylase Sirtuin 3 (SIRT3) on ferroptosis. Ferroptosis Oxid Stress. 2026;2:202603. https://doi.org/10.70401/fos.2026.0026
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Overview of the Structure and Core Biological Functions of SIRT3
- 3. Overview of Ferroptosis Function
- 4. SIRT3-Regulated Ferroptosis Network: Enzymatic and Non-Enzymatic Substrates
- 5. SIRT3-Regulated Non-Enzymatic Substrates in Iron Metabolism and Lipid Peroxidation
- 6. The Mitochondrial Centrality of SIRT3 in Ferroptosis
- 7. The Dual Oncogenic Role of SIRT3 and Its Context-Dependent Crosstalk with Ferroptosis
- 8. Clinical Applications and Future Perspectives
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Chen Y, Shi Q, Wang X, Wei X, Cai R. SIRT3 at the crossroads of ferroptosis: Multidimensional regulation of the mitochondrial deacetylase Sirtuin 3 (SIRT3) on ferroptosis. Ferroptosis Oxid Stress. 2026;2:202603. https://doi.org/10.70401/fos.2026.0026
copy
Share Link
copy