The good, the bad, and the iron: Ferroptosis and macrophages in ovarian cancer
Flavia Biamonte
1,#,*

,
Giuseppe Natali
2
,
Cristiana Galeano
1
,
Rosaria Lamanna
1
,
Lavinia Petriaggi
1
,
Francesco Saverio Costanzo
1
,
Emanuele Giorgio
1,#,*
,
Anna M. Battaglia
1
*Correspondence to:
Emanuele Giorgio, Department of Experimental and Clinical Medicine, Magna Graecia University of Catanzaro, Catanzaro 88100, Italy.
E-mail: emanuele.giorgio@unicz.it
Flavia Biamonte, Department of Experimental and Clinical Medicine, Magna Graecia University of Catanzaro, Catanzaro 88100, Italy. E-mail: flavia.biamonte@unicz.it
Flavia Biamonte, Department of Experimental and Clinical Medicine, Magna Graecia University of Catanzaro, Catanzaro 88100, Italy. E-mail: flavia.biamonte@unicz.it
Ferroptosis Oxid Stress. 2026;2:202605. 10.70401/fos.2026.0027
Received: February 16, 2026Accepted: May 08, 2026Published: May 11, 2026
Abstract
Ovarian cancer remains the most lethal gynaecological malignancy, due to late diagnosis, extensive peritoneal dissemination, and the common emergence of therapy resistance. While intrinsic genomic instability and DNA repair defects have long been considered the main biological events underlying ovarian cancer pathogenesis, it is now evident that disease progression is critically shaped by the tumor microenvironment (TME). Tumor-associated macrophages (TAMs) constitute the main immune population in both solid lesions and malignant ascites, orchestrating tumor growth, metastatic dissemination, immune evasion, and chemoresistance. In parallel, ferroptosis, an iron-dependent, lipid peroxidation-driven form of regulated cell death, has emerged as a therapeutic vulnerability in ovarian cancer, particularly in platinum- and Poly (ADP-ribose) polymerase (PARP) inhibitor-resistant disease. TAMs and ferroptosis engage in a bidirectional and context-dependent crosstalk: TAMs iron handling, redox activity, and polarization states modulate ferroptotic sensitivity of ovarian cancer cells, while ferroptotic stress reshapes TAMs phenotype, cytokine release, and immunosuppressive capacity. In this review, we reframe ovarian cancer as an immune-metabolic disease in which ferroptosis and TAM biology form a tightly coupled regulatory axis. We synthesize current mechanistic insights and propose that effective therapeutic ferroptosis induction requires concurrent modulation of TAMs plasticity.
Keywords
Ovarian cancer, ferroptosis, tumor-associated macrophages, iron metabolism, tumor microenvironment, immuno-metabolic crosstalk
1. Introduction
Epithelial ovarian cancer (EOC) accounts for a consistent fraction of gynaecological cancer mortality despite representing a minority of overall cancer incidence[1]. High-grade serous ovarian carcinoma (HGSOC), the dominant histotype (~70% of EOC), is defined by common TP53 mutations, chromosomal instability, and frequent defects in homologous recombination repair, most commonly involving breast cancer susceptibility gene 1/2 (BRCA1/2)[2]. These genomic liabilities have rationally shaped therapeutic strategies toward platinum-based chemotherapy and PARP inhibitors[3]. However, most patients relapse and ultimately develop therapy-refractory disease, highlighting the limits of interpreting EOC primarily through tumor cell-intrinsic genetics[4]. Ovarian cancer is increasingly recognized as an immune-metabolic disease, in which malignant cells coexist with a permissive microenvironment that enables dissemination and suppresses durable immune control[5]. The ovarian tumor microenvironment (TME) is enriched in immunosuppressive myeloid populations, dysregulated metabolic byproducts, and inflammatory mediators that collectively support growth while attenuating cytotoxic immunity[6,7]. In this scenario, tumor-associated macrophages (TAMs) dominate both solid lesions and malignant ascites (often 40-50% of immune cells), and a high TAM burden, particularly with M2-like features, is associated with advanced stage, platinum resistance, and lower survival[8]. In parallel, the emergence of ferroptosis, an iron-dependent, lipid peroxidation-driven form of regulated cell death, has uncovered a promising vulnerability in ovarian cancer. Ovarian cancer cells exhibit pronounced iron dependence, dysregulated lipid metabolism, and chronic oxidative stress, features that in principle lower the threshold for ferroptotic catastrophe[9,10]. Indeed, several preclinical studies support ferroptosis as a therapeutically actionable vulnerability. In platinum-resistant models, Tang et al. showed that olaparib synergized with arsenic trioxide to suppress tumor growth by jointly promoting apoptosis and ferroptosis, thereby overcoming resistance to conventional therapy[11]. In complementary intraperitoneal models, Jin et al. demonstrated that niraparib restricted metastatic dissemination through CD36-dependent lipid uptake and ferroptotic death, highlighting a mechanistic link between Poly (ADP-ribose) polymerase (PARP) inhibition, lipid remodeling, and ferroptosis[12]. Moreover, Miao et al. reported that ferroptosis suppressor protein 1 (FSP1) confers resistance to PARP inhibitors in BRCA-proficient ovarian cancer, indicating that endogenous anti-ferroptotic programs can blunt treatment efficacy[13]. These studies indicate that ferroptosis induction can directly suppress ovarian tumor growth and may resensitize resistant disease to platinum- and PARP inhibitor-based therapies by overcoming adaptive redox defenses. However, ferroptosis unfolds within an immune-metabolic niche that can attenuate, redirect, or even exploit this process to favor tumor adaptation and survival[14,15]. In this context, TAMs, through their privileged control of iron flux and redox signalling, can orchestrate whether ferroptotic stress becomes tumor-eradicating or merely adaptive[16]. Ferroptosis biology is even more complex when considering the distinctive features of ovarian cancer cell dissemination[17]. Ovarian cancer spreads predominantly through transcoelomic routes, forming malignant ascites and widespread peritoneal implants[18]. These spatially segregated niches impose divergent selective pressures: ascitic tumor cells often exist in macrophage-containing spheroids, where fluctuating oxygen and nutrient availability favour metabolic quiescence and heightened antioxidant buffering; omental metastases exploit adipocyte-derived fatty acids within lipid-rich, immune-tolerant niches structured around milky spots[19,20]. Consequently, ferroptotic vulnerability in ovarian cancer is unlikely to be uniform: ascitic spheroids may be primed for lipid peroxidation yet protected by macrophage-derived buffering cues, whereas omental metastases may be intrinsically ferroptosis-prone due to polyunsaturated fatty acids (PUFA) availability and iron flux but simultaneously protected by immunosuppressive stromal circuits[21]. These considerations suggest that ferroptosis and TAMs should be viewed as a single coupled regulatory system, and that ferroptosis-based interventions must be designed with the macrophage-rich, spatially heterogeneous ovarian TME explicitly in mind[22].
2. Molecular Determinants of Ferroptotic Vulnerability in Ovarian Cancer
Ferroptosis is defined by the iron-dependent accumulation of lipid peroxides within cellular membranes, culminating in catastrophic membrane failure and bioenergetic collapse[23]. Unlike apoptosis or necroptosis, ferroptosis is not executed by caspases or pore-forming machineries but reflects a metabolic inability to restrain self-propagating lipid radical chain reactions[24]. In ovarian cancer, multiple liabilities converge to prime this form of death: expansion of intracellular iron pools, enrichment of PUFA-containing phospholipids, and a critical dependence on antioxidant systems[25,26].
2.1 Iron metabolism and ferritinophagy
Intracellular iron availability is the “sine qua non” of ferroptosis[27]. In ovarian cancer, the canonical entry route is that of transferrin-bound Fe3+ uptake via transferrin receptor 1 (TFRC/TFR1), followed by endosomal acidification, ferrireduction (classically mediated by Six-Transmembrane Epithelial Antigen of the Prostate (STEAP), family metalloreductases), and export of Fe2+ into the cytosol through divalent metal transport systems such as divalent metal transporter 1 (DMT1) (SLC11A2), thereby feeding the labile iron pool (LIP), the kinetically accessible iron fraction that drives both metabolism and pro-oxidative reactions[22]. A recurring feature of HGSOC is an iron-addicted phenotype characterized by TFRC up-regulation together with reduced expression of the iron exporter ferroportin (SLC40A1/FPN), a configuration that expands the intracellular LIP[28,29]. Functionally, this iron addiction supports mitochondrial respiration, ribonucleotide reductase activity, and DNA synthesis, but it also imposes an intrinsic biochemical liability: excess LIP increases Fenton and Haber-Weiss chemistry, generating hydroxyl radicals and secondary reactive oxygen species (ROS) that preferentially attack PUFA-rich membrane phospholipids, thereby lowering the threshold for ferroptotic lipid peroxidation[27]. Because free iron is dangerous, cells buffer excess iron primarily into ferritin (FTH1/FTL). Ferroptotic competence, indeed, depends not only on iron uptake/export but also on the iron mobilization from storage, and here ferritinophagy becomes pivotal[30]. Ferritinophagy is a selective autophagic process mediated by the nuclear receptor coactivator 4 (NCOA4), which targets ferritin to lysosomes for degradation, thereby releasing Fe2+ back into the cytosol and expanding the LIP when cells are under metabolic stress[31]. This mechanism is highly relevant to ovarian cancer because therapy-resistant cells often rely on ferritin turnover and iron recycling to survive stress, creating a more accessible LIP that may be exploited to trigger ferroptosis when antioxidant defenses are inhibited[32,33]. In this regard, we have recently shown that differences in baseline LIP levels help explain heterogeneous ferroptosis responses in ovarian cancer: erastin triggered ferroptosis in cells with high LIP in a manner coupled to NCOA4-mediated ferritinophagy, whereas low-LIP cells were relatively resistant[34]. In agreement, Wang et al. showed that targeting the heme/iron axis via hemeoxygenase (HO-1) manipulation can enhance NCOA4-dependent ferritinophagy and restore both ferroptosis and cisplatin sensitivity[35]. Raphaël Rodriguez and colleagues further refine this concept by identifying lysosomes as a central redox-active iron hub in cancer cells, showing that pharmacologic activation of lysosomal iron preferentially eradicates persister cancer cells with an iron-rich, CD44high, drug-tolerant and metastatic phenotype. These findings suggest that ferritinophagy-derived iron and TFRC-mediated endolysosomal iron trafficking may converge within lysosomes to create a vulnerable pool of catalytic Fe2+ able to initiate ferroptotic damage when properly engaged, thereby linking iron storage turnover, stem-like phenotypes, and ferroptosis sensitivity[36].
Collectively, these findings support the hypothesis that, in ovarian cancer, ferroptosis susceptibility is governed less by static “iron abundance” and more by the dynamic flux between uptake, export, buffering, and mobilization from reservoirs[37].
2.2 Lipid peroxidation as the executioner
The execution phase of ferroptosis is driven by peroxidation of PUFAs esterified within membrane phospholipids. Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) orchestrate the incorporation of arachidonic acid into vulnerable phospholipid pools, generating substrates exquisitely sensitive to oxidation[38,39]. Then, lipid peroxidation can proceed enzymatically, for example via lipoxygenases, or through non-enzymatic iron-catalyzed radical propagation. Once initiated, oxidative damage spreads across membranes, collapsing ion gradients and organelle integrity[40]. Notably, ferroptosis lacks a single “point of no return”: it represents a systems-level failure of redox homeostasis in which lipid hydroperoxides overwhelm repair and detoxification capacity[41]. Ovarian cancer progression, particularly in ascites and omental metastases, depends on the continuous remodeling of membrane phospholipids, fatty-acid desaturation, and lipid trafficking. The abundance and identity of oxidizable substrates are actively shaped by the metastatic niche and by enzymes that control PUFA loading into membranes[42,43]. LPCAT3 has emerged as a clinically relevant determinant of arachidonic acid phospholipid remodeling, with experimental evidence linking LPCAT3-driven arachidonic acid metabolism to ferroptosis regulation as well as growth and metastatic behaviour[44]. This reinforces the concept that ferroptosis sensitivity in ovarian cancer is often governed upstream by phospholipid editing rather than by “ROS level” alone[45]. Dissemination niches actively supply fatty acids that can either promote or buffer ferroptotic execution. The omentum provides abundant lipids; adipocytes fuel metastatic outgrowth by transferring fatty acids to ovarian cancer cells[43]. This lipid abundance can, in principle, increase PUFA substrate availability and heighten susceptibility to lipid peroxidation; however, ovarian cancer frequently counters this threat by shifting toward desaturation programs that increase monounsaturated fatty acids (MUFAs), which are less peroxidation-prone and can dilute PUFA content in membranes[46]. In this regard, Tesfay et al. showed that stearoyl-CoA desaturase 1(SCD-1), the rate-limiting enzyme for MUFA synthesis, protects ovarian cancer cells from ferroptosis[47]. Diversely, ascites-derived ovarian cancer cells display enhanced fatty-acid desaturation activity (e.g., by the upregulation of including SCD-1/fatty acid desaturase 2 (FADS2)) in lipid-rich environments, associating desaturation with reduced oxidative stress, possibly raising the ferroptotic threshold in ascitic disease. These findings help explain why ferroptosis in ovarian cancer is often context-dependent: lipid-rich niches do not automatically sensitize tumors to ferroptosis because they simultaneously select for lipid desaturation and phospholipid remodeling states that resist peroxidation[25]. At the mechanistic level, lipid peroxidation in ovarian cancer can proceed via both enzymatic and non-enzymatic routes: iron-catalyzed radical propagation amplifies lipoxygenase-driven lipid oxidation and the balance between propagation and repair determines whether the cell crosses a lethality threshold[48]. This suggests that lipid peroxidation is not merely a terminal endpoint but can be pharmacologically engaged or avoided depending on how therapies reshape lipid uptake and membrane composition[49]. Recently, Jin et al. showed that niraparib can enhance fatty-acid uptake in the intraperitoneal setting and drive lipid peroxidation-associated ferroptotic death[12].
Taken together, these observations argue that in ovarian cancer, lipid peroxidation “execution” depends on whether metastatic environments (omentum vs. ascites) load membranes with oxidizable PUFA-PLs (e.g., via LPCAT3-linked arachidonic acid remodeling) or, conversely, drive protective MUFA/desaturation states (SCD1/FADS2) and glutathione peroxidase 4 (GPX4)-independent defences that prevent lipid hydroperoxides from reaching catastrophic levels[12,50].
2.3 Antioxidant defense systems
To survive in a chronically oxidizing environment, ovarian cancer cells rely on multi-layered antioxidant defences. The canonical ferroptosis checkpoint is the system Xc- - glutathione-GPX4 axis, in which cystine import sustains glutathione (GSH) synthesis and GPX4 detoxifies lipid hydroperoxides[51]. HGSOC frequently overexpresses GPX4, and this correlates with poor prognosis and platinum resistance, underscoring its role as a ferroptosis gatekeeper and a driver of therapy tolerance[52]. Inhibition of system Xc- or GPX4 selectively eliminates drug-tolerant persister cancer cells and restores chemosensitivity in preclinical models[53,54]. These dependencies arise within a highly stressful TME characterized by chronic oxidative pressure driven by hypoxia-reoxygenation cycles, inflammatory cytokines, and intense metabolic activity in both tumor and stromal cells[55]. Indeed, ascitic spheroids are exposed to unstable redox conditions, with fluctuating oxygen levels and limited access to nutrients and antioxidants[56]. To survive in this hostile environment, tumor cells rely on enhanced GSH synthesis and nuclear factor erythroid 2-related factor 2 (NRF2)-driven antioxidant defenses[57]. However, this protection is fragile, and even modest disruptions in cystine uptake, GPX4 activity, cofactor availability, or iron homeostasis can rapidly trigger lethal lipid peroxidation. As a result, ascitic spheroids often display relative ferroptosis resistance due to metabolic cooperation and strong antioxidant buffering. In contrast, omental metastases develop within lipid-rich, iron-enriched adipose niches that place tumor cells closer to a ferroptotic threshold, although this is partly counterbalanced by immunosuppressive stromal support[21,58]. These spatial asymmetries indicate that ferroptosis in ovarian cancer is governed not only by tumor-intrinsic genotype but by niche-specific immune-metabolic constraints, including TAMs-controlled iron flux and oxidative tone[59].
Beyond the canonical GSH/GPX4 system, ovarian cancer cells can exploit parallel lipid peroxide detoxification networks centered on the FSP1-dihydroorotate dehydrogenase (DHODH)-coenzyme Q10 (CoQ10)/vitamin K2 (VK2) axis, which has emerged as a major determinant of ferroptosis resistance. FSP1 (formerly AIFM2) functions at the plasma membrane as an NAD(P)H-dependent oxidoreductase that reduces ubiquinone (CoQ10) to ubiquinol, thereby trapping lipid radicals and preventing propagation of phospholipid peroxidation independently of GPX4[60]. This concept was first established by Bersuker et al., who identified the CoQ10-FSP1 pathway as a complementary ferroptosis defense system, and subsequently expanded by Doll et al., who demonstrated that FSP1 also reduces vitamin K to its hydroquinone form, revealing a second lipophilic antioxidant circuit able to suppress ferroptotic damage[60,61]. In parallel, DHODH provides a mitochondria-restricted ferroptosis defense by coupling pyrimidine synthesis to reduction of CoQ within the inner mitochondrial membrane, thus protecting mitochondrial phospholipids when GPX4 activity is compromised[62]. In ovarian cancer, these defense systems appear particularly relevant under therapeutic stress. FSP1 overexpression has been shown to confer resistance to PARP inhibition in BRCA-proficient ovarian cancer, indicating that ferroptosis defense may intersect with DNA damage tolerance pathways[13]. Additional studies reported that FSP1-driven ferroptosis resistance promotes tumor progression, dissemination, and chemoresistance, while ACSL1-dependent myristoylation of FSP1 enhances its membrane localization and supports peritoneal metastasis, highlighting the importance of post-translational control of this pathway[63,64]. These findings indicate that the FSP1-DHODH-CoQ10/VK2 network constitutes a multilayered antioxidant shield in ovarian cancer, buffering lipid peroxidation across distinct subcellular compartments and potentially explaining why some tumors remain refractory to GPX4-directed ferroptosis induction alone[65]. Therapeutically, co-targeting GPX4 together with FSP1 or DHODH may therefore represent a rational strategy to collapse metabolic redundancy and sensitize resistant ovarian tumors to ferroptotic cell death.
Finally, emerging evidence indicates that the guanosine triphosphate (GTP)-cyclohydrolase 1 (GCH1)-tetrahydrobiopterin (BH4) axis represents a distinct ferroptosis defense pathway with potential relevance in ovarian cancer. GCH1, the rate-limiting enzyme in de novo BH4 synthesis, was identified by Kraft et al. as a potent suppressor of ferroptosis through mechanisms that extend beyond classical redox signaling, including direct radical-trapping antioxidant activity and remodeling of membrane lipid composition toward species less permissive to peroxidation[66]. In this model, BH4 acts not only as a cofactor for aromatic amino acid hydroxylases and nitric oxide synthases, but also as a lipophilic antioxidant capable of intercepting phospholipid peroxyl radicals and preserving membrane integrity under oxidative stress. Complementary work by Soula et al. further integrated BH4 into a broader metabolic ferroptosis defense network, showing functional crosstalk between the GCH1/BH4 system and the FSP1-CoQ10 pathway, thereby suggesting that cancer cells may deploy multiple parallel radical-scavenging circuits to survive lipid peroxide stress[67]. In ovarian cancer, this pathway may be particularly important because highly proliferative and therapy-resistant cells experience chronic oxidative stress, elevated iron flux, and PUFA-rich membrane vulnerability, conditions that create strong selective pressure for non-canonical ferroptosis suppressors. Consistent with this concept, Hu et al. demonstrated that pharmacologic or genetic targeting of the GCH1/BH4 axis enhanced ferroptosis susceptibility in ovarian cancer models, supporting GCH1 as a therapeutically actionable metabolic node[66,67]. Mechanistically, activation of the GCH1/BH4 program may cooperate with GPX4, FSP1, and DHODH to buffer lipid peroxidation across cytosolic and membrane compartments, potentially contributing to resistance against platinum compounds, PARP inhibitors, or direct ferroptosis inducers (FINs). These findings suggest that the GTP-GCH1-BH4 axis constitutes an additional layer of antioxidant plasticity in ovarian cancer, and that dual or triple targeting of complementary ferroptosis defense systems may be required to effectively collapse redox adaptation in resistant tumors.
Collectively, ferroptosis vulnerability in ovarian cancer emerges from the dynamic intersection of iron availability, PUFA-enriched membrane architecture, and a multilayered antioxidant system centered not only on GPX4, but also on complementary FSP1-DHODH-CoQ10/VK2 and GCH1-BH4 defense systems. Thus, susceptibility to ferroptotic death is not dictated by any single molecular determinant, but by the balance between pro-oxidant pressure and the adaptive capacity of tumor cells to distribute redox protection across distinct subcellular compartments. This equilibrium is profoundly context dependent and shaped by disease niche, metabolic state, therapy exposure, and microenvironmental cues, which together generate marked spatial and temporal heterogeneity in ferroptosis sensitivity[68,69]. These concepts highlight why single-agent ferroptosis induction may be insufficient in many settings, whereas rational co-targeting of redundant antioxidant circuits could more effectively collapse redox resilience in resistant ovarian cancer populations. Furthermore, while ferroptosis induction has attracted major therapeutic interest, ferroptosis resistance should not be viewed as a passive absence of death signaling, but as an adaptive survival program selected during platinum exposure, PARP inhibition, hypoxia, and metastatic dissemination. This perspective has important translational implications, as inhibition of key suppressors may be more effective than direct ferroptosis induction alone, particularly in tumors that have evolved redundant antioxidant defenses. Table 1 and Figure 1 summarize the principal molecular determinants and therapeutic liabilities governing ferroptosis in ovarian cancer.
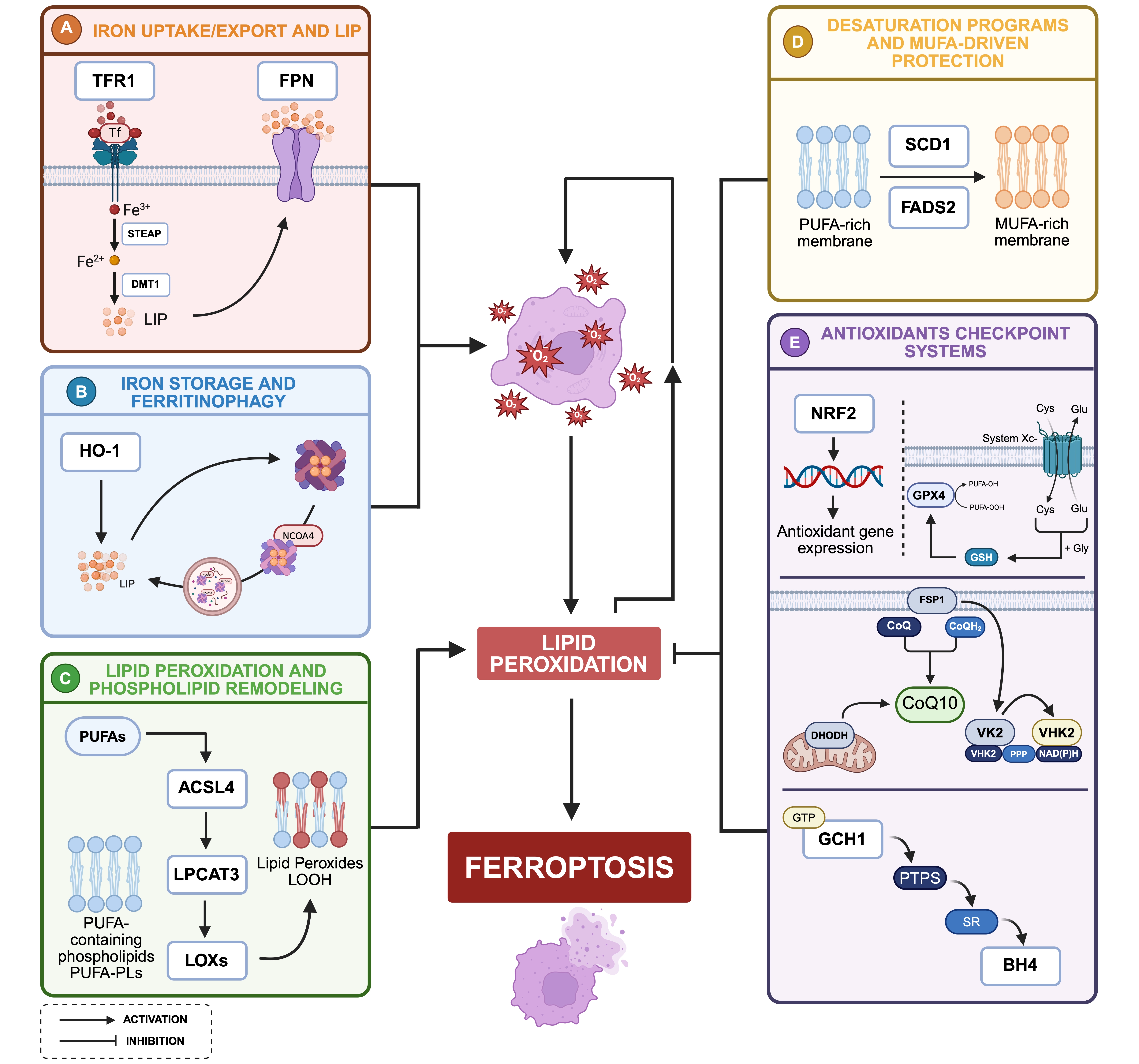
Figure 1. Molecular mechanisms governing ferroptosis in ovarian cancer. (A) Transferrin receptor mediated iron internalization, together with reduced ferroportin-dependent export, promotes intracellular iron retention and expansion of the LIP, thereby increasing redox active iron and ROS generation; (B) Intracellular iron is buffered within ferritin complexes and mobilized through NCOA4-mediated ferritinophagy, while HO-1 activity further contributes to iron release under stress conditions, dynamically amplifying ferroptotic susceptibility; (C) ACSL4 and LPCAT3 driven incorporation of PUFAs into membrane phospholipids generates oxidizable substrates that are targeted by enzymatic (e.g., lipoxygenases) and non-enzymatic iron-dependent reactions, leading to lipid hydroperoxide accumulation and membrane destabilization; (D) Lipid remodeling toward MUFAs through SCD1 and FADS2 reduces membrane susceptibility to peroxidation, conferring resistance particularly in lipid rich microenvironments; (E) The system Xc--glutathione-GPX4 axis, together with NRF2-dependent transcriptional programs, detoxifies lipid peroxides and sustains redox homeostasis. In parallel, the FSP1-CoQ10/VK2 system suppresses lipid peroxidation at the plasma membrane through regeneration of lipophilic radical-trapping antioxidants, independently of GPX4. Mitochondrial DHODH further supports ferroptosis resistance by reducing CoQ within the inner mitochondrial membrane. Finally, the GTP-GCH1–BH4 axis provides an additional defense mechanism, with BH4 acting as a radical-trapping antioxidant and modulator of membrane lipid composition. Ferroptosis sensitivity in ovarian cancer depends on the balance between iron availability, membrane lipid composition, and antioxidant defenses. Created in BioRender. Biamonte, F. (2026) https://BioRender.com/po5wm7g. LIP: labile iron pool; TFR1: transferrin receptor 1; FPN: ferroportin; STEAP: six-transmembrane epithelial antigen of the prostate; DMT1: divalent metal transporter 1; NCOA4: nuclear receptor coactivator 4; HO-1: heme oxygenase 1; PUFA: polyunsaturated fatty acids; MUFA: monounsaturated fatty acids; ACSL4: acyl-CoA synthetase long-chain family member 4; LPCAT3: lysophosphatidylcholine acyltransferase 3; LOXs: lipoxygenases; system Xc-: cystine/glutamate antiporter; GSH: glutathione; GPX4: glutathione peroxidase 4; NRF2: nuclear factor erythroid 2-related factor 2; ROS: reactive oxygen species; FSP1: ferroptosis suppressor protein 1; CoQ: coenzyme Q; VK2: vitamin K2; PPP: pentose phosphate pathway; NADPH: nicotinamide adenine dinucleotide phosphate; DHODH: dihydroorotate dehydrogenase; GCH1: GTP cyclohydrolase 1; PTPS: 6-pyruvoyl-tetrahydropterin synthase; SR: sepiapterin reductase; BH4: tetrahydrobiopterin.
Table 1. Key molecular determinants of ferroptosis vulnerability in ovarian cancer.
| Ferroptosis determinant | Key molecular components | Functional relevance in ovarian cancer | Ref. |
| Iron uptake/export and LIP | TFRC/TFR1, FPN/SLC40A1, STEAP, DMT1 | Increased intracellular Fe2+ promotes ROS generation, enhancing lipid peroxidation and ferroptosis susceptibility | [22,70] |
| Iron storage and ferritinophagy | Ferritin (FTH1/FTL), NCOA4, HO-1 | Ferritinophagy mediates ferritin degradation and iron release, expanding LIP and increasing ferroptosis sensitivity under therapy-induced stress | [31,71] |
| Lipid peroxidation and phospholipid remodeling | PUFA-PLs, ACSL4, LPCAT3, LOXs | Phospholipid remodeling increases oxidizable PUFA substrates and modulates ferroptotic vulnerability in ascites and omental metastases | [43,72] |
| Desaturation programs and MUFA-driven protection | SCD1, FADS2, MUFA | MUFA enrichment dilutes PUFA content and limits lipid peroxidation, attenuating ferroptosis in lipid-rich environments | [46,47] |
| Antioxidant checkpoint systems | System Xc- (SLC7A11), GSH, GPX4, NRF2, FSP1, CoQ10/VK2, DHODH, GCH1-BH4 | Prevents lipid hydroperoxide accumulation and supports redox buffering through complementary antioxidant systems, promoting platinum/PARPi tolerance and ferroptosis resistance | [52,53,62,64] |
LIP: labile iron pool; TFRC/TFR1: transferrin receptor 1; FPN/SLC40A1: ferroportin/solute carrier family 40 member 1; STEAP: six-transmembrane epithelial antigen of the prostate; DMT1: divalent metal transporter 1; FTH1, ferritin heavy chain 1; FTL: ferritin light chain; NCOA4: nuclear receptor coactivator 4; HO-1: heme oxygenase 1; PUFA: polyunsaturated fatty acids; PUFA-PLs: PUFA-containing phospholipids; ACSL4: acyl-CoA synthetase long-chain family member 4; LPCAT3: lysophosphatidylcholine acyltransferase 3; LOXs: lipoxygenases; SCD1: stearoyl-CoA desaturase 1; FADS2: fatty acid desaturase 2; MUFA: monounsaturated fatty acids; system Xc-: cystine/glutamate antiporter; SLC7A11: solute carrier family 7 member 11; GSH: glutathione; GPX4: glutathione peroxidase 4; NRF2: nuclear factor erythroid 2-related factor 2; ROS: reactive oxygen species; FSP1: ferroptosis suppressor protein 1; CoQ10: coenzyme Q10 (ubiquinone); VK2: vitamin K2 (menaquinone); DHODH: dihydroorotate dehydrogenase; GCH1: GTP cyclohydrolase 1; BH4: tetrahydrobiopterin.
3. TAMs in Ovarian Cancer: Origin, Dominance, and Plasticity
3.1 Ontogeny and spatial organization
TAMs represent the predominant immune population in ovarian cancer, accounting for up to 40-50% of leukocytes in both solid lesions and malignant ascites[52]. TAMs derive from embryonically seeded tissue-resident macrophages and bone marrow-derived monocytes recruited from circulation. Single-cell transcriptomic and lineage-tracing analyses suggest that resident macrophages mainly contribute to TAM pools within omental metastases and milky spots, whereas monocyte-derived macrophages dominate in ascites[20,73]. These populations are functionally distinct: resident macrophages exhibit enhanced lipid-handling capacity and iron-retention programs, while monocyte-derived TAMs display greater inflammatory responsiveness and cytokine plasticity[74].
3.2 TAM plasticity in ovarian cancer shapes the TME
Macrophage diversity is commonly described using the M1/M2 polarization model. Classically activated M1-like macrophages, typically induced by interferon-gamma (IFN-γ) in combination with microbial/Toll-like receptor (TLR) signals, produce inflammatory cytokines and reactive oxygen/nitrogen species and support antigen presentation and cytotoxic immunity. Alternatively activated M2-like macrophages, instead, driven by IL-4/IL-13 and reinforced by IL-10-rich “resolving” signals, promote immune regulation, tissue repair, angiogenesis, and extracellular matrix remodelling. Although the field increasingly recognizes that the M1/M2 dichotomy is an oversimplification, since macrophages in vivo rarely exist as pure M1 or M2 states, it remains clinically useful to distinguish tumor-restraining from tumor-promoting functional programs[75,76]. Indeed, single-cell and integrative immune profiling of ovarian cancer TME show that TAMs populate a continuum of activation states combining inflammatory, metabolic, angiogenic, and immunosuppressive features[7,77].
However, the M2-like states remain predominant across both solid lesions and ascites. Ovarian cancer TAMs, indeed, directly enable metastatic fitness by supplying trophic signals (e.g., epidermal growth factor, EGF-driven programs), promoting angiogenesis and invasion through vascular-endothelial growth factor (VEGF) and matrix-remodelling enzymes, and, notably, stabilizing multicellular spheroids during transcoelomic dissemination. In a mechanistic study, macrophages localized at the core of ovarian cancer spheroids secreted EGF and promoted reciprocal upregulation of αMβ2 integrin on macrophages and intercellular adhesion molecule 1 (ICAM-1) on tumor cells, strengthening heterotypic adhesion and supporting early metastatic spread[78]. TAMs also impose an immunosuppressive barrier via IL-10/TGF-β and immune checkpoints; notably, B7-H4-expressing TAMs have been identified as a distinct suppressive subpopulation in ovarian carcinoma and ascites, contributing to impaired T-cell function[79]. M2-type TAMs phenotypes are not fixed; they can change depending on signals from the TME. Factors released by the tumor and ascites fluid, such as IL-6, leukemia inhibitory factor (LIF), and Colony-Stimulating Factor 1, together with metabolic changes, push macrophages into an immune-suppressive, tumor-supporting state. Standard cancer treatments can also reprogram these macrophages, making them more involved in tissue repair and suppression of anti-tumor immunity[8,80].
Collectively, the ovarian cancer literature supports a model in which TAM plasticity is continuously instructed by spatial niche, checkpoint-mediated immune suppression, and therapy-induced reprogramming, making TAMs both a major determinant of clinical outcome and a necessary co-target when designing therapeutic strategies[81]. Figure 2 outlines the spatially distinct TAM programs in ascites vs omental metastases.
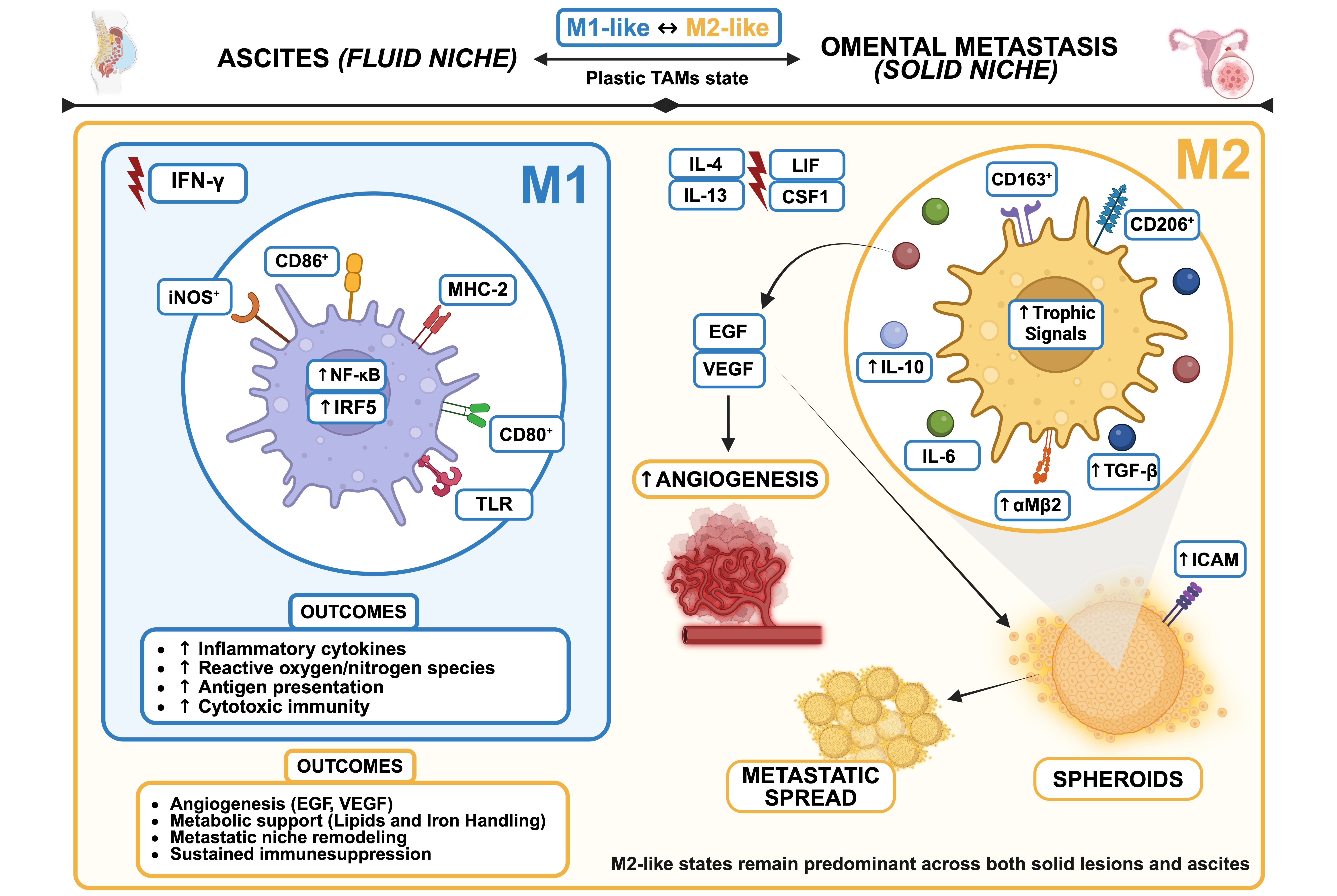
Figure 2. TAMs plasticity and niche-specific reprogramming in EOC. Spatial context governs macrophage function and metastatic support. Macrophage diversity in the ovarian cancer TME is classically framed within the M1-M2 polarization model, although this represents a functional continuum rather than discrete states. M1-like macrophages, induced by IFN-γ and TLR signaling, exhibit pro-inflammatory and cytotoxic functions, supporting antigen presentation and immune activation. In contrast, M2-like macrophages, driven by IL-4/IL-13 and reinforced by tumor-derived signals, promote immune regulation, tissue remodeling, and angiogenesis. In ovarian cancer, TAMs predominantly adopt M2-like programs across both ascites and solid lesions. These cells actively sustain metastatic fitness by promoting angiogenesis and invasion, providing trophic support through factors such as EGF and VEGF, and stabilizing multicellular spheroids during transcoelomic dissemination. Macrophages within spheroid cores secrete EGF and enhance reciprocal αMβ2 integrin and ICAM-1 expression, strengthening tumor-macrophage adhesion and facilitating early metastatic spread. TAMs further contribute to immune evasion through IL-10, TGF-β, and immune checkpoint pathways. Importantly, TAM phenotypes remain highly plastic and are continuously shaped by cytokines (including IL-6, LIF, and CSF1), metabolic cues, and therapeutic interventions, which collectively reinforce immunosuppressive and tumor-supportive states. Overall, spatial context and microenvironmental signals dynamically instruct TAM function, making these cells key drivers of disease progression and attractive therapeutic targets. Created in BioRender. Biamonte, F. (2026) https://BioRender.com/6akoz17. TAMs: tumor-associated macrophages; CD206: cluster of differentiation 206; TGF-β: transforming growth factor beta; CSF-1: colony-stimulating factor 1; LIF: leukemia inhibitory factor; B7-H4: B7 homolog 4 (VTCN1); αMβ2: integrin alpha M beta 2; ICAM: intercellular adhesion molecule; EGF: epidermal growth factor; CD163: cluster of differentiation 163; IL-10: interleukin-10; VEGF: vascular endothelial growth factor; ICAM-1: intercellular adhesion molecule 1.
4. Ferroptosis and TAMs: A Bidirectional Crosstalk in Ovarian Cancer
Macrophages are the principal “iron recyclers” of peripheral tissues and therefore possess a uniquely flexible iron-handling machinery. This includes TFRC1-mediated uptake, ferritin storage, FPN/SLC40A1 export, and hepcidin-driven FPN internalization, that enables them to either retain or donate iron depending on inflammatory context[82,83]. Across different contexts, a conserved immune-metabolic pattern has emerged: inflammatory/M1-like macrophages preferentially retain iron and are characterized by elevated ferritin and reduced FPN-mediated export reinforced by cytokine-driven hepcidin, whereas alternatively activated/M2-like macrophages adopt an “iron-release” phenotype, with higher FPN and related programs that increase extracellular iron availability[84,85]. From a ferroptosis standpoint, this dichotomy has direct consequences: iron-retaining macrophages can buffer the TME against iron-driven lipid peroxidation, whereas iron-donating TAMs can increase local redox-active iron and lower the ferroptotic threshold in neighbouring cells[86]. Importantly, TAM iron-related phenotypes are not fixed but, rather, continuously shaped by interactions between the tumor and surrounding stromal cells. Tumor-educated macrophages can acquire iron-release programs via secretion of high-affinity carriers such as lipocalin-2 (LCN2), which delivers iron to tumor cells and elevates intracellular iron content[87]. In principle, this is highly relevant to ferroptosis, because increasing intratumor iron can sensitize membranes to peroxidation; however, in practice the outcome depends on whether iron delivery is coupled to collapse of antioxidant defences (e.g., GPX4-dependent buffering) or instead fuels adaptive redox remodelling[1,88]. The ovarian cancer setting adds a further layer of complexity because iron handling is tightly linked with inflammation and systemic iron-restriction programs[89]. The ratio of M1 to M2 macrophages in the peritoneal cavity is linked to higher levels of IL-6, hepcidin, ferritin, and oxidative stress, and to lower levels of free iron. This means ascites is not simply iron-rich or iron-poor. Instead, iron is constantly redistributed between free iron, protein-bound iron, and iron stored inside cells[90,91]. Mechanistically, IL-6 increases hepcidin, which causes macrophages and stromal cells to trap iron, leading to overall iron restriction in the body, a common feature of cancer-related inflammation and anemia. However, in specific local areas depending on TAM subtype and activity, redox-active iron can still accumulate. As a result, macrophages may create a paradox: limiting iron systemically while allowing localized iron availability near the tumor, where it can support tumor progression[92,93]. Omental metastases are enriched in resident-like TAMs with high HO-1 expression. These macrophages are immunosuppressive and associated with poor prognosis[94]. Because HO-1 releases iron from heme, they are closely involved in controlling iron metabolism and oxidative balance, two key factors that govern sensitivity to ferroptosis[95]. This creates a further paradox: releasing iron could promote ferroptosis, but at the same time these macrophages also support immune suppression and help tumors adapt metabolically[96]. Accordingly, increased iron flux does not automatically translate into therapeutically productive ferroptosis in vivo. In fact, TAMs can protect tumor cells from ferroptosis by promoting iron export or redistribution inside cancer cells, especially under hypoxic conditions[22]. Together, these observations support the idea that macrophages do not merely modulate ferroptosis, but define the iron topology of the ovarian tumor ecosystem, shaping where redox-active iron accumulates, where it is buffered, and whether lipid peroxidation can cross the lethality threshold[1,97]. A major unresolved challenge is that most studies still infer macrophage iron states indirectly from bulk markers or M1/M2 labels. Moving forward, spatially resolved mapping of TAM subsets that donate or retain iron in ascites and omental lesions, and understanding how these dynamics rewire ferroptosis sensitivity during therapy, will be essential before ferroptosis can be rationally exploited in ovarian cancer[77,98].
In ovarian cancer, ROS are not just harmful waste products of stress. They act as signaling molecules that connect macrophage activity, tumor adaptation to oxidative stress, and sensitivity to ferroptosis[1]. Macrophages produce ROS through mitochondrial metabolism and especially through NOX2 enzymes, which generate controlled bursts of superoxide that influence the oxidative environment of ascites and peritoneal tumors[99]. Macrophage-derived ROS intensify oxidative pressure on adjacent tumor cells, increasing lipid peroxide burden, particularly in regions already primed by high LIP and PUFA-rich membranes. However, in real tumors their effects are more complex, because ROS act as second messengers that reshape gene expression in both tumor cells and TAMs[100]. In macrophages, ROS intersect with signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappa B (NF-κB) signalling to modulate polarization and cytokine output, reinforcing IL-10/TGF-β-dominant programs and sustaining immune suppression under chronic oxidative conditions[101,102]. In tumor cells, ROS drive the activation of NRF2, an antioxidant adaptive response that reinforces iron homeostasis control and confers robust resistance to ferroptosis[103]. Finally, recent studies highlight that macrophages can protect tumor cells from ferroptosis through non-cell-autonomous mechanisms. They can reduce lipid peroxidation caused by GPX4 inhibitors and rescue cancer cells through direct cell contact and by releasing extracellular vesicles[104]. Hence, in TAM-rich tumors, such as ovarian cancer, ferroptotic stress is therefore counteracted not only by tumor-intrinsic defense systems, such as NRF2, GPX4, and FSP1, but also by macrophage-mediated “rescue circuits” that reduce the oxidative damage of tumor cell membranes.
Overall, ROS in ovarian cancer should be viewed as a two-side force: they can promote ferroptosis, but they can simultaneously activate adaptive pathways and macrophage buffering mechanisms that neutralize ferroptosis and reinforce an immunosuppressive niche. The dual role of TAMs in shaping ferroptosis and tumor adaptation is summarized in Table 2.
Table 2. Overview of TAM-mediated processes shaping ferroptosis and adaptive tumor programs in ovarian cancer.
| TAM-driven mechanism | How it can promote ferroptosis | How it can promote tumor adaptation | Ref. |
| M1-like iron retention vs M2-like iron release programs | FPN-driven iron export increases local redox-active iron availability and enhances lipid peroxidation | Iron sequestration limits ferroptotic stress, whereas iron availability may support tumor growth and immunosuppression | [85,86] |
| LCN2-dependent iron handling | LCN2-mediated iron delivery promotes tumor iron accumulation and oxidative stress | Enhanced iron availability can support redox adaptation and therapy tolerance unless antioxidant defences fail | [87] |
| Inflammatory iron restriction in ascites | Discrete niches may still accumulate redox-active iron, sustaining ferroptotic stress | IL-6-driven hepcidin signaling sustains chronic inflammation and promotes iron sequestration by macrophages | [91,105] |
| Heme catabolism axis in omental metastases | HO-1-mediated heme degradation releases iron, enhancing redox stress | Iron mobilization may reinforce immune tolerance and metabolic adaptation | [95,96] |
| ROS signaling and macrophage rescue circuits | TAM-derived ROS enhance oxidative damage and lipid peroxidation in iron-rich tumor cells | Chronic ROS activates NRF2/STAT3 programs promoting immune suppression and stress tolerance | [95,100] |
TAM: tumor-associated macrophage; FPN: ferroportin; LCN2: lipocalin-2; IL-6: interleukin-6; HO-1: heme oxygenase 1; ROS: reactive oxygen species; NRF2: nuclear factor erythroid 2-related factor 2; STAT3: signal transducer and activator of transcription 3.
4.1 Ferroptotic stress reprograms TAMs phenotype
Ferroptosis is not only a terminal event for the dying cell; it is also a microenvironmental signal that is sensed and decoded by macrophages[27,106]. Mechanistically, ferroptosis generates and releases a spectrum of oxidized phospholipids, lipid hydroperoxides, and reactive aldehydes (e.g., 4-Hydroxynonenal (4-HNE), malondialdehyde (MDA)) that can engage pattern-recognition and redox-sensitive pathways in innate immune cells[107,108]. In parallel, ferroptotic cancer cells release danger-associated molecular patterns (DAMPs) such as high mobility group box 1 (HMGB1), in an autophagy-dependent manner, which can signal through receptors including receptor for advanced glycation end-products (RAGE) and TLRs to shape macrophage recruitment and activation[109]. Importantly, the macrophage response to ferroptosis is graded rather than binary and depends on the intensity, timing, and spatial distribution of ferroptotic damage. Conceptually, ferroptosis can produce either (i) an immunostimulatory “danger” context or (ii) an immunoregulatory/conditioning context, and the same biochemical mediators can support either outcome, depending on kinetics and dose[110]. In ovarian cancer, partial ferroptotic stress may be more common than complete ferroptotic elimination, especially in spatially buffered niches such as ascitic spheroids and omental implants. Under these conditions, oxidized lipid signals may preferentially drive macrophage programs that favor tissue remodeling and immune suppression rather than antitumor clearance. This is consistent with broader immunology work showing that DAMP release during ferroptosis does not automatically guarantee immunogenicity: “early” ferroptotic states can be immunogenic, whereas lethal or sublethal ferroptotic stress can be immunosuppressive, in part because oxidized lipids can impair antigen-presenting function and reshape cytokine outputs[111]. Lipid peroxidation byproducts, such as 4-HNE, can further modulate macrophage signalling pathways including NF-κB, NRF2, and mitogen-activated protein kinase (MAPK) altering cytokine production and inflammasome activity, again emphasizing that ferroptosis-derived chemistry is inherently immunologically “active”[112]. Notably, ovarian cancer provides direct proof-of-principle that sublethal ferroptotic stress can reprogram macrophages into pro-tumoral states. In a well-defined model, low-dose erastin exposure promoted the invasion and migration of ferroptosis-resistant ovarian cancer cells by inducing STAT3-dependent M2 polarization of macrophages, with a central role for the interleukin-8 (IL-8)/C-X-C motif chemokine ligand 8 (CXCL8) axis. This finding is conceptually important because it demonstrates a non-intuitive mechanism of harm (Figure 3): ferroptosis induction can act as a selective pressure that (i) spares resistant tumor cells and (ii) simultaneously conditions the macrophage compartment to become more pro-metastatic[113].
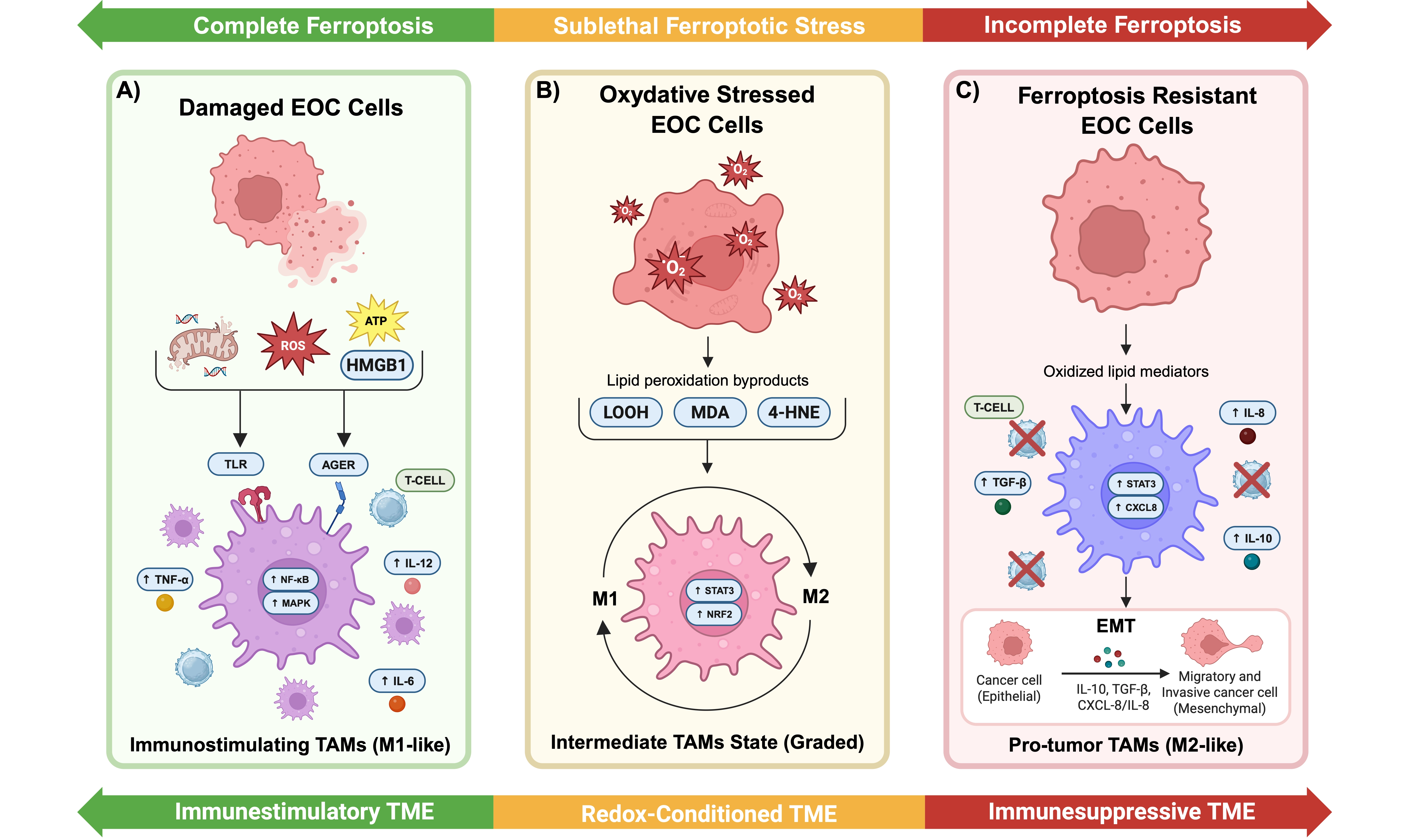
Figure 3. Ferroptosis intensity dictates macrophage polarization and tumor fate. The intensity and persistence of ferroptotic stress shape macrophage function along a functional continuum rather than through a binary outcome. (A) Complete ferroptosis is characterized by extensive lipid peroxidation, membrane disruption, mitochondrial damage, and robust release of DAMPs such as HMGB1. This acute ferroptotic burst promotes immunostimulatory TAM activation (M1-like phenotype), supporting T cell-mediated antitumor immunity; (B) Sublethal ferroptotic stress, commonly observed in ascites and solid tumor implants, allows tumor cell survival while generating persistent redox mediators, including oxidized phospholipids, 4-HNE, and MDA. These signals condition TAMs through redox-sensitive pathways such as STAT3 and NRF2, inducing a graded and plastic functional state rather than full polarization; (C) Chronic ferroptosis incompleteness drives stable TAM reprogramming toward an M2-like, protumoral phenotype. These macrophages secrete CXCL8/IL-8 and other pro-tumorigenic mediators, promoting EMT, migration, invasion, and establishment of an immunosuppressive niche characterized by marginalized or dysfunctional T cells. Collectively, ferroptosis emerges as an active microenvironmental regulator that dynamically rewires TAM biology and influences metastatic progression. Created in BioRender. Biamonte, F. (2026) https://BioRender.com/7tjjm6w. EOC: epithelial ovarian cancer; ROS: reactive oxygen species; ATP: adenosine triphosphate; HMGB1: high mobility group box 1; TLR: Toll-like receptor; AGER: advanced glycosylation end-product specific receptor; TAMs: tumor-associated macrophages; TNF-α: tumor necrosis factor alpha; NF-κB: nuclear factor kappa B; MAPK: mitogen-activated protein kinase; T-cell: T lymphocyte; IL-12: interleukin-12; IL-6: interleukin-6; LOOH: lipid hydroperoxides; MDA: malondialdehyde; 4-HNE: 4-hydroxynonenal; STAT3: signal transducer and activator of transcription 3; NRF2: nuclear factor erythroid 2-related factor 2; CXCL8: C-X-C motif chemokine ligand 8; IL-8: interleukin-8; TGF-β: transforming growth factor beta; IL-10: interleukin-10; EMT: epithelial-to-mesenchymal transition; TME: tumor microenvironment; DAMPs: danger-associated molecular patterns; RAGE: receptor for advanced glycation end-products.
Taken together, these data establish a clinically relevant warning: in a TAMs-rich tumor such as ovarian cancer, ferroptosis-based interventions must be evaluated not only by tumor-cell killing but by the macrophage phenotype they induce[114,115]. Ferroptosis induction is most likely to be therapeutically productive when it is sufficiently strong and spatially pervasive to (a) avoid generating a prolonged “conditioning” phase dominated by oxidized lipid mediators[111] and (b) prevent STAT3/CXCL8-driven M2 polarization that can amplify invasion, immune suppression, and metastatic competence. In practical terms, this suggests using combination therapies that pair ferroptosis induction with TAM-modulating strategies (e.g., STAT3 inhibition, CXCL8/CXCR2 blockade, or macrophage repolarization approaches) to limit adaptive resistance driven by the TME[116]. Another important aspect to consider is that standard and emerging treatment modalities for ovarian cancer are unlikely to act solely on tumor cells, but also remodel the ferroptosis-TAM regulatory axis. Platinum compounds increase intracellular ROS, mitochondrial dysfunction, and iron-dependent lipid damage, potentially lowering the ferroptotic threshold; however, repeated exposure can also select NRF2/GPX4-high resistant clones while promoting recruitment of immunosuppressive TAM populations involved in tissue repair and relapse[8,35,51,52]. PARP inhibitors may similarly enhance ferroptotic priming through replication stress, mitochondrial dysfunction, and altered lipid metabolism, yet they can simultaneously induce adaptive antioxidant programs and macrophage-derived rescue signals that buffer lethal peroxidation[11,13,47]. Anti-angiogenic therapies, by exacerbating hypoxia, metabolic stress, and vascular remodeling, may further influence macrophage polarization and iron handling, thereby shifting whether oxidative pressure culminates in ferroptosis or in redox adaptation[7,55]. Likewise, taxanes and other cytotoxic agents can trigger immunogenic stress responses but also increase CSF1-, IL6-, or STAT3-dependent TAM reprogramming after treatment[70,80]. These observations suggest that the clinical efficacy of several ovarian cancer therapies may partly depend on how they drive the ferroptosis-TAM circuit. Rational combinations integrating chemotherapy, PARP inhibition, FINs, and TAM-targeting strategies may therefore be required to convert treatment-induced oxidative stress into durable therapeutic benefits.
5. Conclusions
Ferroptosis and TAMs are not independent actors in ovarian cancer. They constitute a tightly coupled immune-metabolic axis governed by iron flux, lipid peroxidation, and redox signalling. TAMs shape the biochemical environment that determines whether ferroptotic stress is controlled or progresses to lethal membrane damage. In turn, signals released during ferroptosis can reprogram macrophage phenotype, cytokine production, and the immune environment. This reciprocity alters how ferroptosis should be conceptualized, not as a cell-autonomous death program, but as a microenvironmental event with consequences for the tumor ecosystem. In ovarian cancer, defined by spatial heterogeneity, macrophage dominance, and intrinsic redox fragility, ferroptosis represents both an opportunity and a risk. When engaged decisively and within an immune context that favours antitumor activation, ferroptosis can dismantle therapy resistance and expose otherwise inaccessible vulnerabilities. When partial, spatially confined, or uncoupled from immune modulation, the same process may instead reinforce immunosuppression, select for fitter resistant clones, and accelerate metastatic evolution. The therapeutic implication is profound: ferroptosis induction must be integrated with strategies that constrain TAM plasticity, prevent macrophage reprogramming, and restore immune competence. Several unresolved questions define the next phase of the field and will determine whether ferroptosis can be translated into durable clinical benefit: i) Which TAM subsets are mechanistically responsible for buffering vs amplifying ferroptotic stress in vivo? ii) How does ferroptotic vulnerability differ between ascitic spheroids and solid metastatic niches, and can spatial redox and iron mapping guide therapeutic targeting? iii) Can molecular biomarkers stratify patients prospectively for ferroptosis-based strategies? iv) Which molecular switches determine whether ferroptosis becomes immunogenic or merely conditioning? Addressing these questions will require integrative approaches combining single-cell and spatial omics, functional redox imaging, and models that faithfully recapitulate the ascitic and metastatic microenvironments of advanced ovarian cancer.
Acknowledgments
We thank past and present members of the Biochemistry and Cellular Biology Laboratory for their contributions to this work. All Figures were created using Biorender.com. ChatGPT-based language editing tools were used to improve grammar of the manuscript. All authors carefully reviewed and revised the edited text and take full responsibility for the final content, accuracy, and integrity of the manuscript.
Authors contribution
Biamonte F, Giorgio E, Battaglia AM: Conceptualization, visualization, writing-original draft, writing-review & editing.
Natali G, Galeano C, Lamanna R, Petriaggi L, Costanzo FS: Conceptualization, writing-review & editing.
All authors reviewed and approved the final manuscript prior to submission.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Funding
This work is supported by grants from the Unione Europea – Next Generation Eu, Missione 4 Componente 1 (CUP F53D23003840006) and PRIN – Progetti di Ricerca di Interesse Nazionale – Bando 2022 (Prot. 2022R85H27).
Copyright
© The Author(s) 2026.
References
-
14. Liu J, Dong R, Yuan B, Xie Y, Feng Z, Zhou S, et al. Immune cells dying from ferroptosis: Mechanisms and therapeutic opportunities. Cell Death Dis. 2025;16(1):878.[DOI]
-
22. Cui K, Wang K, Huang Z. Ferroptosis and the tumor microenvironment. J Exp Clin Cancer Res. 2024;43(1):315.[DOI]
-
28. Torti SV, Torti FM. Iron and cancer: More ore to be mined. Nat Rev Cancer. 2013;13(5):342-355.[DOI]
-
36. Cañeque T, Baron L, Müller S, Carmona A, Colombeau L, Versini A, et al. Activation of lysosomal iron triggers ferroptosis in cancer. Nature. 2025;642(8067):492-500.[DOI]
-
42. Vasseur S, Guillaumond F. Lipids in cancer: A global view of the contribution of lipid pathways to metastatic formation and treatment resistance. Oncogenesis. 2022;11(1):46.[DOI]
-
53. Cerra C, Tancock MRC, Thio N, Koo A, Wong A, J Cowley K, et al. Exploiting dysregulated iron homeostasis to eradicate persistent high-grade serous ovarian cancer. Cell Death Discov. 2025;11(1):423.[DOI]
-
60. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688-692.[DOI]
-
62. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586-590.[DOI]
-
64. Zhang Q, Li N, Deng L, Jiang X, Zhang Y, Lee LTO, et al. ACSL1-induced ferroptosis and platinum resistance in ovarian cancer by increasing FSP1 N-myristylation and stability. Cell Death Discov. 2023;9(1):83.[DOI]
-
68. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551(7679):247-250.[DOI]
-
90. Madeddu C, Gramignano G, Kotsonis P, Coghe F, Atzeni V, Scartozzi M, et al. Microenvironmental M1 tumor-associated macrophage polarization influences cancer-related anemia in advanced ovarian cancer: Key role of interleukin-6. Haematologica. 2018;103(9):e388-e391.[DOI]
-
93. Duan X, He K, Li J, Cheng M, Song H, Liu J, et al. Tumor associated macrophages deliver iron to tumor cells via Lcn2. Int J Physiol Pathophysiol Pharmacol. 2018;10(2):105-114.
-
116. Ren M, Chen LL, Jiang LY, Yu HH, Ji HZ. The CXCL8-CXCR2 axis promotes M2 macrophage polarization in ovarian cancer via RASGRP4-mediated mTOR-STAT3 signaling. Apoptosis. 2025;30(7):1839-1851.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Biamonte F, Natali G, Galeano C, Lamanna R, Petriaggi L, Costanzo FS, et al. The good, the bad, and the iron: Ferroptosis and macrophages in ovarian cancer. Ferroptosis Oxid Stress. 2026;2:202605. https://doi.org/10.70401/fos.2026.0027
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Molecular Determinants of Ferroptotic Vulnerability in Ovarian Cancer
- 3. TAMs in Ovarian Cancer: Origin, Dominance, and Plasticity
- 4. Ferroptosis and TAMs: A Bidirectional Crosstalk in Ovarian Cancer
- 5. Conclusions
- Acknowledgments
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Funding
- References
- Copyright
Science Exploration Style
Biamonte F, Natali G, Galeano C, Lamanna R, Petriaggi L, Costanzo FS, et al. The good, the bad, and the iron: Ferroptosis and macrophages in ovarian cancer. Ferroptosis Oxid Stress. 2026;2:202605. https://doi.org/10.70401/fos.2026.0027
copy
Share Link
copy