The lysosomal iron rheostat: Orchestrating ferroptosis in cancer plasticity
Francesca Rizzollo
1,2,*

,
Patrizia Agostinis
1,2,*
*Correspondence to:
Francesca Rizzollo, Cell Death Research and Therapy Laboratory, Center for Cancer Biology, VIB, Leuven 3000, Belgium; Department of Cellular and Molecular Medicine, KU Leuven, Leuven 3000, Belgium.
E-mail: francesca.rizzollo@kuleuven.be
Patrizia Agostinis, Cell Death Research and Therapy Laboratory, Center for Cancer Biology, VIB, Leuven 3000, Belgium; Department of Cellular and Molecular Medicine, KU Leuven, Leuven 3000, Belgium. E-mail: patrizia.agostinis@kuleuven.be
Patrizia Agostinis, Cell Death Research and Therapy Laboratory, Center for Cancer Biology, VIB, Leuven 3000, Belgium; Department of Cellular and Molecular Medicine, KU Leuven, Leuven 3000, Belgium. E-mail: patrizia.agostinis@kuleuven.be
Ferroptosis Oxid Stress. 2026;2:202612. 10.70401/fos.2026.0029
Received: March 26, 2026Accepted: May 25, 2026Published: May 25, 2026
Abstract
Iron is indispensable for cellular metabolism yet potentially cytotoxic, making its intracellular handling a fundamental determinant of cell fate decisions. The endo-lysosomal system has recently emerged as a central iron rheostat that integrates transferrin uptake, ferritinophagy, and lysosomal iron export to control iron bioavailability for mitochondria and other iron-dependent pathways. Growing studies further show that lysosomal iron is not merely permissive for ferroptosis but can directly initiate lipid damage through localized iron activation, lysosomal lipid peroxidation, and lysosomal membrane permeabilization. At the same time, emerging studies on organelle contact sites reveal that ferroptosis arises from the failure of a coordinated multi-organellar communication system, in which lysosomes, the endoplasmic reticulum, and mitochondria exchange iron, lipids, and redox signals in an effort to metabolically adapt to stress. This perspective is particularly relevant to drug-tolerant persisters and mesenchymal cancer cell states, which rely on rewired lysosomal iron trafficking to sustain plasticity while becoming highly susceptible to ferroptosis. In this minireview, we discuss emerging insights into the spatial organization of iron metabolism and propose a model in which ferroptosis sensitivity depends on the intracellular routing, chemical reactivity, and release dynamics of iron, highlighting lysosomal iron handling as a key therapeutic vulnerability in minimal residual disease.
Keywords
Lysosomes, iron, ferroptosis, membrane contact sites, metabolism, drug-tolerant cancer cells
1. From Uptake to Fate: Iron’s Path Through the Endo-Lysosomal Network
Iron is an essential micronutrient required by all cell types, as it supports fundamental metabolic and biosynthetic processes, such as DNA synthesis and repair, oxygen handling, and mitochondrial energy metabolism[1]. These functions rely on iron’s ability to cycle between the ferrous (Fe2+) and ferric (Fe3+) states, its incorporation into prosthetic groups such as iron–sulfur (Fe–S) clusters and heme that act as cofactors for numerous proteins, and the tight regulation of cellular pathways governing iron uptake, export, degradation, and utilization[1]. At the same time, this redox flexibility makes iron potentially hazardous: when not properly buffered and compartmentalized, Fe2+ can react with hydrogen peroxide through Fenton chemistry, generating hydroxyl radicals that damage proteins, nucleic acids, and membrane lipids, thereby compromising cell viability[1,2].
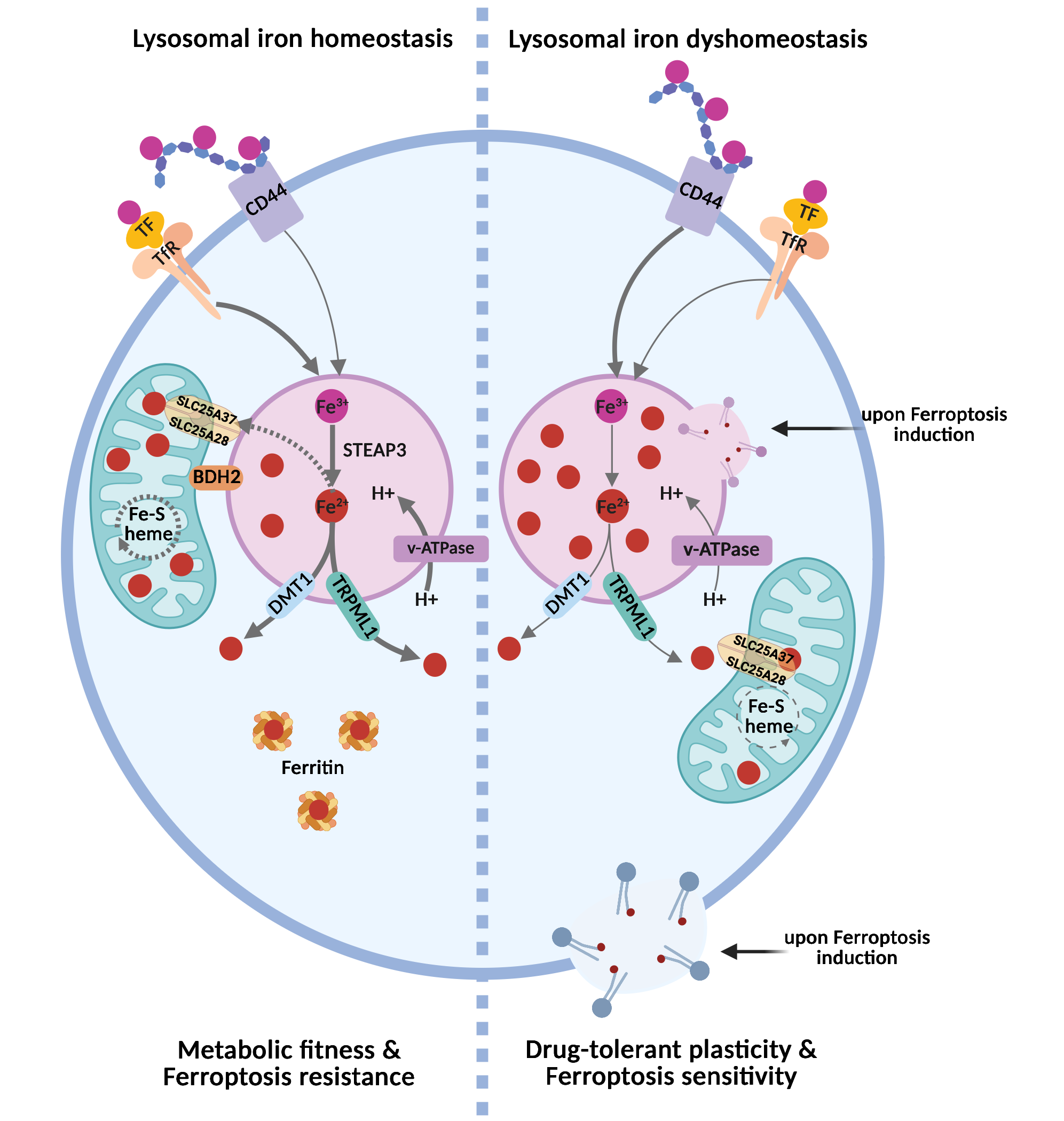
Therefore, maintaining iron homeostasis requires the coordinated control of iron uptake, storage, recycling, and intracellular distribution. The endo-lysosomal system is at the center of this network, serving not only as a degradative compartment but also as a key hub for iron uptake, mobilization, and temporary storage[1,3] (Figure 1).
{kind=link}
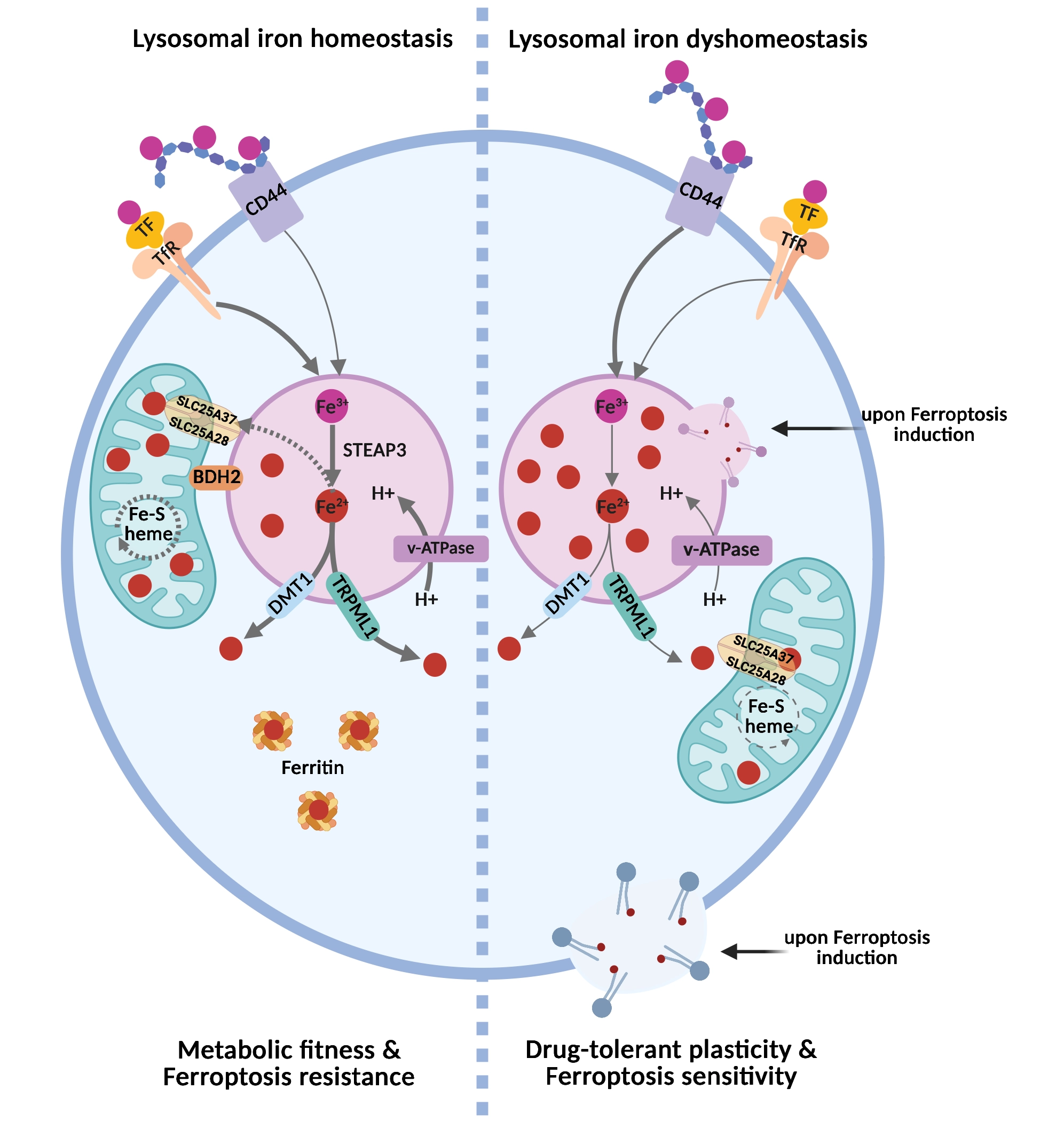
Figure 1. The lysosomal iron rheostat controls inter-organelle iron routing and ferroptosis sensitivity. In the homeostatic state (left), intracellular iron is derived mainly from the uptake of holo-transferrin via receptor-mediated endocytosis following binding to the Trf. In acidic lysosomes, Fe3+ is reduced to Fe2+ by STEAP3 and redistributed through DMT1/TRPML1-mediated export into the cytosol, where it is stored in ferritin nanocages. In these cells, BDH2 at the interface between mitochondria and lysosomes favors lysosome-to-mitochondria iron transfer via the mitochondrial inner membrane transporters SLC25A28 (mitoferrin-2) and SLC25A37 (mitoferrin-1). This preserves mitochondrial iron utilization, Fe–S/heme synthesis, and oxidative metabolism, resulting in ferroptosis resistance. In the dyshomeostatic state (right), characteristic of the drug-tolerant cancer cell state, higher expression of CD44 mediates the endocytosis of iron-bound hyaluronates, which, together with defective inter-organelle iron routing, causes lysosomal iron accumulation, reduced mitochondrial iron availability, and metabolic stress. Under these conditions, lysosomal iron becomes a redox-active pool that can promote lysosomal lipid peroxidation, lysosomal membrane permeabilization, and propagation of oxidative damage to other membranes, thereby sensitizing plastic, drug-tolerant cancer cells to ferroptosis. Created in BioRender. Agostinis, P. (2026) https://BioRender.com/ob8cgax. Trf: transferrin receptor; Fe3+: ferric iron; Fe2+: ferrous iron; STEAP3: six-transmembrane epithelial antigen of prostate 3; DMT1: divalent metal transporter 1; TRPML1: transient receptor potential mucolipin 1; BDH2: 3-hydroxybutyrate dehydrogenase 2.
Canonically, the uptake of extracellular iron is mediated by the transferrin (Tf)–transferrin receptor (TfR) axis. In this pathway, ferric iron (Fe3+) binds transferrin, which engages TfR1 or TfR2 at the plasma membrane and is subsequently internalized via endocytosis. Within the acidified compartments of the late endosomes and lysosomes, Fe3+ is reduced to Fe2+ by the ferrireductase six-transmembrane epithelial antigen of prostate 3 (STEAP3), which requires luminal acidity for activity. The resulting Fe2+ is then mobilized to the cytosol through SLC11A2 (divalent metal transporter 1, DMT1) or via the lysosomal cation channel Mucolipin 1 (MCOLIN1, also known as transient receptor potential mucolipin 1 or TRPML1)[1]. Once released into the cytosol, Fe2+ enters the labile iron pool (LIP). The LIP represents a metabolically accessible iron fraction, and because of its redox activity and potential cytotoxicity, it is tightly regulated. Cytosolic iron is transiently buffered by reduced glutathione (GSH) via thiol-mediated chelation. This chelation is also required for the function of iron chaperones such as Poly(rC)-Binding Protein 1 (PCBP1), which delivers iron to ferritin for sequestration or to iron-dependent enzymes[4-6]. To prevent iron-induced toxicity, excess cytosolic iron is either exported via ferroportin (FPN/SLC40A1) or stored within ferritin nanocages composed of ferritin heavy (FTH) and light (FTL) chains[1].
Ferritin-bound iron can be remobilized through lysosomal transport pathways leading to ferritin degradation regulated by the nuclear receptor coactivator 4 (NCOA4). Under conditions of iron depletion, NCOA4 binds ferritin and directs it to lysosomes for degradation via both LC3-dependent and LC3-independent pathways[7,8] involving tax1-binding protein 1 (TAX1BP1)[9]. In both pathways, NCOA4 drives ferritin phase separation, leading to the formation of ferritin–NCOA4 condensates, which facilitate TAX1BP1-dependent lysosomal degradation of ferritin[10,11]. Additionally, lysosomes receive iron also from the degradation of mitochondria via mitophagy[1].
A key determinant of this cycle is lysosomal pH. Vacuolar-type ATPase (v-ATPase)-driven acidification maintains iron solubility, supports iron release from endocytic cargo, and enables its efficient reutilization after ferritin degradation[1,12]. Conversely, loss of lysosomal acidification traps iron within endolysosomal compartments and creates a state of functional iron deficiency despite unchanged total cellular iron levels[12-14]. As a result, cells develop defective mitochondrial respiration, altered Fe–S-dependent metabolism, activation of pseudo-hypoxic/hypoxia inducible factor (HIF) signaling, impaired proliferation, and, in some settings, inflammatory signaling and non-apoptotic cell death[12-14].
Different pathological stress signals, including but not limited to oxidative stress, inflammation, and aging-related conditions or diseases, can alter homeostatic control of lysosomal iron and the labile iron pool[15]. However, determining how these stress conditions influence the levels and distribution of redox-active iron within the lysosomes and other compartments remains technically challenging. Current approaches, including the use of fluorescent iron sensors, lysosome-targeted probes, biochemical methodologies, and even the highly sensitive inductively coupled plasma mass spectrometry (ICP-MS), are all subject to limitations. Consequently, this area remains in need of further methodological development.
Despite these methodological limitations, growing studies indicate that lysosomes do not simply mediate iron entry into the cell, but they also determine where iron is retained, in which chemical state it is maintained, and which downstream pathways it can support. This is why endo-lysosomal iron handling has consequences that extend beyond nutrient supply to the control of cell fate. In cancer models, altered endosomal iron routing rewires mitochondrial metabolism and changes invasive and metastatic behavior[16], whereas selective activation of lysosomal iron can directly trigger ferroptosis in iron-rich, plastic, and drug-tolerant cancer cell states[17].
2. When Lysosomal Iron Turns Lethal: Fuelling Ferroptosis
Ferroptosis is a regulated form of cell death driven by iron-dependent phospholipid peroxidation and the ensuing loss of membrane integrity[2,18]. Although iron has long been viewed as a permissive factor in this process, accumulating evidence indicates that its subcellular localization is equally, if not more, important[17,19-22]. In this regard, lysosomes are emerging as major determinants of ferroptotic susceptibility because they can concentrate high levels of redox-active iron, thereby creating a local pool of catalytic iron that can drive membrane lipid oxidation[17] (Figure 1).
Recent studies now provide direct evidence for a causal role of lysosomal iron in ferroptotic execution[17]. Cañeque et al. showed that chemical activation of lysosomal iron is sufficient to trigger oxidation of membrane lipids and ferroptosis, whereas iron inactivation is protective[17].
Another study showed that following lysosomal lipid peroxidation, lysosomal membrane permeabilization drives lysosomal iron leakage into the cytosol, which then propagates lipid peroxidation via radical-chain reaction to membranes of proximal organelles, precipitating ferroptosis[22]. Together, these findings redefine lysosomal iron from a merely permissive factor to a key effector of ferroptotic damage.
Importantly, the ferroptotic potential of lysosomal iron depends not only on how much iron is stored, but also on how it is mobilized, retained, or redirected. In senescent and treatment-resistant cells, lysosomal alkalinization causes retention of Fe2+ and lipid radicals within lysosomes and suppresses cystine-deprivation-induced lipid peroxidation and ferroptosis, whereas restoration of lysosomal acidity re-sensitizes cells to ferroptotic death[23]. Likewise, disrupting endosomal acidification or transferrin endocytosis suppresses cysteine-deprivation-induced ferroptosis by lowering the rise in cellular labile iron, highlighting that endo-lysosomal iron supply can be essential for ferroptosis in a stimulus-specific manner[24]. In melanoma, defective 3-hydroxybutyrate dehydrogenase 2 (BDH2)-dependent lysosome-to-mitochondria Fe2+ transfer causes lysosomal Fe2+ sequestration and primes mesenchymal cell states for ferroptosis, reinforcing the idea that lysosomal iron routing, rather than iron abundance alone, can determine ferroptotic vulnerability[21].
Taken together, current evidence supports an emerging and context-dependent model in which lysosomes are not passive iron depots but active regulators of ferroptotic cell fate. By controlling iron concentration, chemical reactivity, intracellular release, and inter-organelle transfer, lysosomes can determine whether iron supports metabolic adaptation or is converted into a lethal catalyst of lipid peroxidation.
3. Iron Channeling Across Organelle Borders
The classical view that metabolites and ions move between organelles mainly through the bulk cytosol has been revised by the recognition that membrane contact sites (MCSs) are privileged platforms for inter-organelle communication[25-27]. Through these nanometer-scale interfaces, organelles exchange lipids, ions, metabolites, and redox signals without membrane fusion, allowing spatially restricted control of metabolism and stress responses. Within this network, the endoplasmic reticulum (ER) occupies a central position because it establishes contacts with mitochondria, lysosomes, lipid droplets, and peroxisomes, thereby integrating lipid metabolism, organelle dynamics, and stress signaling[25-27].
From a ferroptosis perspective, the organelle contact-site framework may help reconcile seemingly different views on where the process begins. The ER has long been considered a major ferroptosis-relevant membrane system[2,28]. Recent super-resolution live imaging now indicates that phospholipid peroxidation is first enriched at ER–mitochondria contact sites (EMCSs), which, as they expand, act as conduits through which ferroptotic-prone polyunsaturated fatty acids (PUFAs) propagate into mitochondrial membranes[29]. Consistent with this model, genetic perturbation of key EMCS components, including protein kinase R-like endoplasmic reticulum kinase (PERK) and mitofusin 2 (MFN2)[29], as well as pharmacological inhibition of Sigma1 receptor[30] confer protection from ferroptosis across different cellular systems. In parallel, recent studies position lysosomes within the same organellar network as key compartments where redox-active iron can initiate or amplify lipid damage[17,22]. These observations are therefore better viewed as complementary rather than contradictory, suggesting a multi-organellar model in which lysosomes provide catalytic iron, the ER provides oxidation-prone phospholipid substrates, and mitochondria amplify the ensuing metabolic and redox stress.
Consistent with this coordinated view, lysosomes and mitochondria form direct membrane contact sites and can also engage with the ER in tripartite junctions[31,32]. Earlier studies proposed that endosomal compartments could deliver iron directly to mitochondria through transient “kiss-and-run” interactions[33-35], but the molecular basis of iron transfer at mitochondria–lysosome contacts remained unresolved. In triple-negative breast cancer, DMT1-dependent endosome–mitochondria interactions were subsequently shown to sustain mitochondrial iron translocation and metastatic outgrowth, highlighting how organelle-restricted iron routing can shape cancer cell fitness[16].
Recently, BDH2 was shown to act as a central component of lysosome-to-mitochondria iron trafficking in melanoma[21]. BDH2, previously identified as the mammalian functional analogue of bacterial EntA, generates the siderophore-like metabolite 2,5-dihydroxybenzoic acid (2,5-DHBA), which facilitates mitochondrial iron delivery[21,36]. In melanoma cells, BDH2 localizes at mitochondria–lysosome contacts (MLCs), where BDH2-dependent iron transfer sustains mitochondrial bioenergetics, which in turn preserves lysosomal acidification and MLC formation[21]. Loss of BDH2 disrupts this feed-forward circuit, causing defective MLC tethering, lysosomal iron sequestration, and heightened ferroptosis sensitivity in mesenchymal-like melanoma cells[21]. Of note, a recent study shows that the close apposition of mitochondria and lysosomes, is required to sustain lysosomal pH[37]. These findings suggest that, through MLCs, mitochondria and lysosomes engage in a metabolic symbiosis in which their physical proximity enables iron-dependent ATP production, while ATP-driven v-ATPase activity preserves lysosomal pH.
Together, these findings argue that ferroptosis is unlikely to arise from one organelle acting in isolation; rather, it reflects failure of an interdependent iron, lipid, and redox network that spans lysosomes, mitochondria, and the ER.
4. Living on the Edge: Iron Dependency in Drug-Tolerant Persister Cancer Cells
The multi-organellar view of ferroptosis outlined above becomes particularly relevant to drug-tolerant persister states, where iron trafficking is rewired to sustain survival yet simultaneously creates an exploitable ferroptosis liability.
Growing evidence indicates that cancer stem-like or mesenchymal populations frequently display elevated iron demand together with altered lysosomal iron handling, creating a state in which iron supports adaptation but also generates a liability to ferroptosis[17,20,21,38].
Under therapeutic pressure, a subset of cancer cells enters a reversible, non-mutational state characterized by metabolic quiescence and adaptive stress responses that enable transient survival. First described by Sharma et al., such phenotypic plasticity creates a latent reservoir of drug-tolerant persister cells (DTP), marked as the minimal residual disease (MDR) population with epithelial-to-mesenchymal plasticity, which can later re-expand, fostering the emergence of stable and genetically encoded resistance mechanisms that drive relapse[39]. Consistently, therapeutic strategies aimed at eliminating DTP before their mutagenic conversion could markedly enhance treatment durability and delay the onset of clonal resistance.
The link between this state and ferroptosis was established by the landmark 2017 studies by Viswanathan et al. and Hangauer et al., which showed that therapy-resistant mesenchymal states and drug-tolerant persisters depend on the GSH– glutathione peroxidase 4 (GPX4) axis to survive oxidative stress and become selectively vulnerable to ferroptosis when this defence is disabled[40,41]. Subsequent work further showed that, in some cellular contexts, FSP1 provides a parallel ferroptosis-suppressive pathway that can compensate for GPX4 loss[42,43].
However, this dependence is intimately linked to altered iron handling, as iron availability crucially dictates the ferroptotic lipid peroxidation potential. Accordingly, DTPs often exhibit enhanced iron uptake and homeostasis, a double-edged adaptation that sustains metabolic plasticity but places them near a ferroptotic threshold. In this regard, Rodriguez and colleagues showed that stem-like mesenchymal breast cancer cells utilize CD44-hyaluronan-iron complexes to internalize iron through the endo-lysosomal route and fuel their epigenetic plasticity[19]. Importantly, selective manipulation of lysosomal iron reactivity was later shown to be sufficient to trigger ferroptosis in CD44high mesenchymal-like cancer cells, positioning lysosomal iron handling as a causal regulatory node rather than a secondary amplifier of oxidative stress[17]. These observations support the idea that the heightened iron uptake of persister-like cells is a double-edged adaptation: it sustains plasticity and survival under stress yet keeps these cells near a lethal oxidative threshold.
Consistent with this view, during targeted therapy with mitogen-activated protein kinase (MAPK) inhibitors, the emerging minimal residual disease (MRD) population enriched in mesenchymal-like melanoma cells displays a concomitant downregulation of BDH2 and RAB7, which, by weakening MLCs, results in aberrant lysosomal iron compartmentalization[21]. This ‘iron mis-trafficking phenotype’ sensitizes the mesenchymal-like, drug-tolerant melanoma cells to ferroptosis inducers, and determines their ferroptotic susceptibility both in vitro and in vivo, during hematogeneous metastatic colonization[21], reinforcing the notion that lysosomal dysfunction and altered inter-organelle communication are vital regulators of ferroptosis sensitivity.
Together with evidence that altered endosome–mitochondria iron routing can reshape mitochondrial metabolism and metastatic outgrowth in mesenchymal breast cancer cells[16], these findings argue that persister-cell fitness depends not simply on iron abundance, but on how iron is compartmentalized and exchanged between organelles[16,17,21]. In this sense, therapy-induced plasticity creates a distinct “lysosomal vulnerability,” in which acidic iron-rich compartments can become local sites of reactive oxygen species (ROS) generation and lipid damage when iron trafficking is perturbed.
This insight has already inspired some lysosome-targeted ferroptosis strategies. Salinomycin and its more potent derivative ironomycin selectively sequester iron in lysosomes of breast cancer stem-like cells, leading to lysosomal ROS production, membrane permeabilization, and cell death consistent with ferroptosis[44]. More recently, Cañeque et al. introduced fentomycin-1, a small molecule that activates lysosomal Fe3+ to oxidatively degrade phospholipids and selectively kill iron-rich CD44high sarcoma and pancreatic ductal adenocarcinoma (PDAC) cells that drive metastasis and drug tolerance[17]. Furthermore, while the ferroptosis inhibitor liproxstatin-1 protects by inactivating iron in lysosomes, fentomycin-1 kills DTPs in vitro and curbs intranodal breast cancer metastasis in vivo[17].
Together, these studies support the idea that organelle-targeted ferroptosis induction may create a therapeutic window for eradicating residual drug-tolerant populations before they convert into stable resistant clones.
5. Concluding Remarks and Perspectives
Over the past 15-20 years, the view of iron metabolism in cancer has evolved from viewing iron primarily as a resource supporting tumors to recognizing iron as a cancer addiction, and more recently, to considering compartmentalized iron handling as a potential liability of drug-tolerant cancer cell states. Following the identification of key molecular components of ferroptosis in 2012, a decisive turn came in 2017 when high-mesenchymal, and drug-tolerant persister states across multiple cancer types were shown to be selectively dependent on GPX4 and acutely vulnerable to lipid peroxidation[40,41]. This observation established ferroptosis as a core vulnerability of non-genetic drug-tolerant or mesenchymal states, a concept that has since expanded to different cancer models and cancer cell states that rely on lipid peroxide detoxification to survive. Within this framework, lysosomes emerged as the center of ferroptosis-relevant iron biology. Rather than serving only as terminal degradative compartments or transient iron stores, lysosomes integrate transferrin uptake, ferritinophagy, v-ATPase-dependent acidification, and iron export to determine whether iron is routed to other cellular compartments, such as mitochondria, or retained in a redox-active state[1,12,21]. This evolution from correlative association to causal function is supported by recent evidence that activation of lysosomal iron can trigger ferroptosis in vulnerable cancers[17]. In parallel, mitochondria–lysosome iron transfer was shown to tune mesenchymal-state vulnerability, reinforcing the organelle-level control of ferroptotic sensitivity. It has also emerged that ferroptosis resistance can also be organelle-based ferritin/exosomal iron export[45], lysosomal exocytosis[46], and niche-driven switching from GPX4 to ferroptosis suppressor protein 1 (FSP1) dependence[47].
At the same time, some studies argue against a strictly lysosome-centric model of ferroptosis initiation[28,48]. Endoplasmic reticulum–mitochondria contact sites can act as proximal hotspots of phospholipid peroxidation, occurring within minutes of ferroptosis induction[29], and emerging work on endosome–mitochondria and lysosome–mitochondria iron transfer indicates that ferroptosis sensitivity is shaped less by total iron abundance than by how iron is packaged, trafficked, and exchanged between organelles[16,21]. Within this framework, ferroptosis can be viewed not as the failure of a single organelle, but as a breakdown of a coordinated inter-organelle metabolic network that normally preserves lipid and iron homeostasis. When key defense mechanisms fail, this collapse drives the toxic accumulation of membrane-damaging lipid peroxides and the progressive decline of mitochondria bioenergetics and function, ultimately culminating in cell death.
This perspective is especially relevant to plastic cancer cell states. Drug-tolerant persister and mesenchymal cells increase their dependence on lysosomal iron trafficking to sustain epigenetic plasticity and stress adaptation, yet this same dependence exposes them to a therapeutic vulnerability[19,38,40]. Such rewiring likely extends beyond mitochondria-lysosome contacts to broader alterations in organelle organization and signaling, including dysfunctional lipid exchange between the ER and mitochondria, as well as the involvement of other organelles, such as lipid droplets, which collectively shape stress resilience and metabolic reprogramming.
Consequently, several outstanding questions remain. Among these, how is the labile iron pool generated and partitioned across resistant cell states in vivo? Which defense axis, e.g., GPX4, FSP1, or others, dominates and in which contexts? Does targeting lysosomal iron trafficking and metabolism provide an effective strategy to eliminate persister-prone clones, and in which tissues or contexts? Can we identify biomarkers that render tumors most likely to respond to lysosomal iron-directed ferroptosis therapies?
Addressing these gaps will require defining how this dynamic network is altered in drug-tolerant persisters and how these adaptations can be selectively exploited to overcome non-genetic resistance.
Acknowledgements
The authors recognize and appreciate the significant contributions of many researchers whose work has advanced this field but were not included here due to space limitations. Automated language-editing software (Grammarly) was solely used to improve readability. The authors take full responsibility for the integrity, originality, and accuracy of the work.
Authors contribution
Rizzollo F, Agostinis P: Conceptualization, methodology, writing-original draft, writing-review & editing.
Conflicts of interest
Patrizia Agostinis is an Editorial Board Member of Ferroptosis and Oxidative Stress. The other author declares no conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by grants from the FWO (G0A3320N), the Stichting tegen Kanker (F/2022/2037), the KU Leuven C14/21/095 InterAction consortium, the EOS DECODE consortium N 30837538, the EOS MetaNiche consortium N 40007532, and the iBOF/21/053 ATLANTIS network and a Doctoral fellowship from the FWO (11L7622N).
Copyright
© The Author(s) 2026.
References
-
1. Rizzollo F, More S, Vangheluwe P, Agostinis P. The lysosome as a master regulator of iron metabolism. Trends Biochem Sci. 2021;46(12):960-975.[DOI]
-
3. Kurz T, Eaton JW, Brunk UT. The role of lysosomes in iron metabolism and recycling. Int J Biochem Cell Biol. 2011;43(12):1686-1697.[DOI]
-
17. Cañeque T, Baron L, Müller S, Carmona A, Colombeau L, Versini A, et al. Activation of lysosomal iron triggers ferroptosis in cancer. Nature. 2025;642(8067):492-500.[DOI]
-
24. Liu X, Zhao Z, Bian Z, Benthani FA, Hu Y, Liang D, et al. Endocytosis is essential for cysteine-deprivation-induced ferroptosis. Mol Cell. 2025;85(17):3333-3342.e4.[DOI]
-
26. Voeltz GK, Sawyer EM, Hajnóczky G, Prinz WA. Making the connection: How membrane contact sites have changed our view of organelle biology. Cell. 2024;187(2):257-270.[DOI]
-
29. Sassano ML, Tyurina YY, Diokmetzidou A, Vervoort E, Tyurin VA, More S, et al. Endoplasmic reticulum–mitochondria contacts are prime hotspots of phospholipid peroxidation driving ferroptosis. Nat Cell Biol. 2025;27(6):902-917.[DOI]
-
30. Zhang Z, Zhou H, Gu W, Wei Y, Mou S, Wang Y, et al. CGI1746 targets σ1R to modulate ferroptosis through mitochondria-associated membranes. Nat Chem Biol. 2024;20(6):699-709.[DOI]
-
34. Hamdi A, Roshan TM, Kahawita TM, Mason AB, Sheftel AD, Ponka P. Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism. Biochim Biophys Acta BBA Mol Cell Res. 2016;1863(12):2859-2867.[DOI]
-
37. Tian Z, Chen R, Fang G, Qiu K, Wu W, Shao X, et al. Mitochondria acidify lysosomes through membrane contacts. Cell Rep. 2026;45(3):117112.[DOI]
-
40. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551(7679):247-250.[DOI]
-
41. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453-457.[DOI]
-
43. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688-692.[DOI]
-
48. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73(2):354-363.e3.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Rizzollo F, Agostinis P. The lysosomal iron rheostat: Orchestrating ferroptosis in cancer plasticity. Ferroptosis Oxid Stress. 2026;2:202612. https://doi.org/10.70401/fos.2026.0029
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. From Uptake to Fate: Iron’s Path Through the Endo-Lysosomal Network
- 2. When Lysosomal Iron Turns Lethal: Fuelling Ferroptosis
- 3. Iron Channeling Across Organelle Borders
- 4. Living on the Edge: Iron Dependency in Drug-Tolerant Persister Cancer Cells
- 5. Concluding Remarks and Perspectives
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Rizzollo F, Agostinis P. The lysosomal iron rheostat: Orchestrating ferroptosis in cancer plasticity. Ferroptosis Oxid Stress. 2026;2:202612. https://doi.org/10.70401/fos.2026.0029
copy
Share Link
copy