Non-neuronal ferroptosis in the central nervous system
Jack Winneberger
1,#
,
Lukas Raich
2,3,#
,
Tammo Potthast
2

,
Marcel S. Woo
2,4,5,6,*
*Correspondence to:
Marcel S. Woo, Translational Neurodegeneration Laboratory, Department of Neurology, University Medical Centre Hamburg Eppendorf, Hamburg 20246, Germany; Institute of Neuroimmunology and Multiple Sclerosis, University Medical Centre Hamburg Eppendorf, Hamburg 20246, Germany; Translational Neuroimaging Laboratory, McConnell Brain Imaging Centre (BIC), Montreal Neurological Institute, Montréal, QC H3A 2B4, Canada; Department of Neurology and Neurosurgery, Montreal Neurological Institute, McGill University, Montréal, QC H3A 2B4, Canada.
E-mail: m.woo@uke.de
Ferroptosis Oxid Stress. 2026;2:202609. 10.70401/fos.2026.0030
Received: March 04, 2026Accepted: June 05, 2026Published: June 05, 2026
Abstract
Ferroptosis, a lipid peroxidation-driven form of regulated cell death, has emerged as a central mechanism in neurological disease. While most studies have focused on neuronal vulnerability, non-neuronal cells, including oligodendrocytes, astrocytes, microglia, brain endothelial cells, and central nervous system (CNS) infiltrating T cells, play equally critical roles in shaping disease progression. These cell types regulate iron homeostasis, lipid metabolism, antioxidant defenses, and inflammatory signaling, thereby establishing the microenvironmental conditions that determine ferroptotic susceptibility within the CNS. Accumulating evidence demonstrates lipid peroxidation and ferroptosis-related signaling in demyelinating disorders, ischemic injury, small vessel disease, Alzheimer’s disease, Parkinson’s disease, and spinal cord injury. However, the contribution of non-neuronal cells to ferroptotic stress and execution remains comparatively underexplored. In this review, we synthesize emerging data highlighting cell type-specific dependencies on glutathione peroxidase 4 (GPX4), solute carrier family 7 member 11 (SLC7A11), ferroptosis suppressor protein 1 (FSP1), nuclear factor erythroid 2-related factor 2 (NRF2), peroxiredoxin (PRDX), thioredoxin (TRX), iron-handling proteins, and lipid remodeling pathways, and discuss how these regulatory networks differ across CNS-resident and CNS infiltrating T cells. We propose that ferroptosis in neurological disease is not solely a neuron-autonomous event, but a tissue-level process orchestrated by non-neuronal cells with distinct metabolic and immunological programs. Understanding these cell type-specific vulnerabilities and regulatory mechanisms will be essential for the development of targeted therapeutic strategies aimed at modulating ferroptotic stress in neuroinflammatory and neurodegenerative disorders.
Keywords
Neurodegeneration, neuroinflammation, glia cells, immune cells, endothelial cells
1. Introduction
Ferroptosis is a regulated form of cell death driven by iron-dependent lipid peroxidation and characterized by the accumulation of oxidized phospholipids within cellular membranes. Since its initial description[1], ferroptosis has been extensively studied across cancer biology, metabolic disorders, and neurodegeneration, and its molecular mechanisms have been comprehensively reviewed elsewhere[2-5]. While substantial progress has been made in defining the core biochemical framework of ferroptosis, its cell type-specific regulation and execution within the central nervous system (CNS) remain incompletely understood.
Neurological diseases represent one of the fastest growing global health challenges, with increasing prevalence driven by aging populations and improved survival from acute injury[6,7]. Particularly, neurodegenerative diseases pose a substantial burden due to the ageing society[8-10]. Understanding the underlying molecular processes is crucial to identify novel therapeutic strategies. The CNS is a highly specialized and tightly regulated organ system, characterized by complex cellular heterogeneity and finely tuned metabolic coupling between neurons, glia, endothelial cells, and infiltrating immune populations. These cells perform distinct yet interdependent functions, including myelination, barrier formation, immune surveillance, and metabolic support. Their unique lipid composition, iron handling, and antioxidant programs suggest that ferroptosis may manifest differently across CNS cell types compared to peripheral tissues. Increasing evidence implicates ferroptosis in a broad spectrum of neurological diseases, including Alzheimer’s disease (AD)[11-17], Parkinson’s disease (PD)[18-20], amyotrophic lateral sclerosis (ALS)[21,22], Huntington’s disease (HD)[23,24], multiple sclerosis (MS)[25-30], and stroke[31-34], where iron accumulation, glutathione depletion, mitochondrial dysfunction, and lipid peroxidation are recurrent pathological features[35]. Moreover, pharmacological inhibition of ferroptosis using iron chelators, radical trapping agents, or nuclear factor erythroid 2-related factor 2 (NRF2) activators ameliorates neuronal injury and functional deficits in multiple preclinical disease models[35-38], further supporting ferroptosis as a therapeutically relevant mechanism in neurodegeneration.
To date, much of the focus on ferroptosis in the CNS has centered on neurons. Neuronal ferroptosis has been reviewed in depth elsewhere[36,38-40], demonstrating that neurons are highly dependent on selenium and glutathione peroxidase 4 (GPX4) for survival, and that neuronal GPX4 deletion robustly induces ferroptotic degeneration in vivo[32,41-43]. Conversely, deletion of acyl-CoA synthetase long-chain family member 4 (ACSL4)[44] is protective in models of inflammation-induced neurodegeneration[27], highlighting the importance of phospholipid remodeling in neuronal ferroptosis susceptibility. Under steady-state conditions, neurons express relatively low levels of NRF2 and solute carrier family 7 member 11 (SLC7A11)[45], rendering them particularly dependent on GPX4-mediated lipid peroxide detoxification. During inflammation, however, non-canonical signaling pathways can be activated that shift neuronal redox balance and promote glutathione-GPX4-dependent cell death programs[28,29]. Neuronal ferroptosis closely interacts with excitotoxicity, neuroinflammation, and mitochondrial metabolic dysfunction, positioning lipid peroxidation as a convergence point of multiple pathogenic pathways in neurodegenerative disease.
In contrast to neurons, the regulation and execution of ferroptosis in non-neuronal CNS cell types, including oligodendrocytes, astrocytes, microglia, brain endothelial cells, and CNS infiltrating T cells, have received less systematic attention. These cells possess distinct iron-handling mechanisms, lipid metabolic programs, and antioxidant networks that may either suppress or amplify ferroptotic stress within the neural microenvironment. Given their central roles in myelination, blood-brain barrier integrity, immune signaling, and metabolic support, ferroptosis in these populations may critically shape disease trajectories in neuroinflammatory and neurodegenerative disorders.
The aim of this review is to systematically examine current evidence for lipid peroxidation and ferroptosis across non-neuronal CNS and CNS-infiltrating immune cells (graphical visualizations are shown in Figure 1, Figure 2, Figure 3, Figure 4 and Figure 5). By comparing cell type-specific dependencies on key ferroptosis regulators and identifying emerging mechanistic themes and gaps, we seek to highlight how ferroptotic stress operates as a tissue-level process in neurological disease and to define priorities for future investigation.
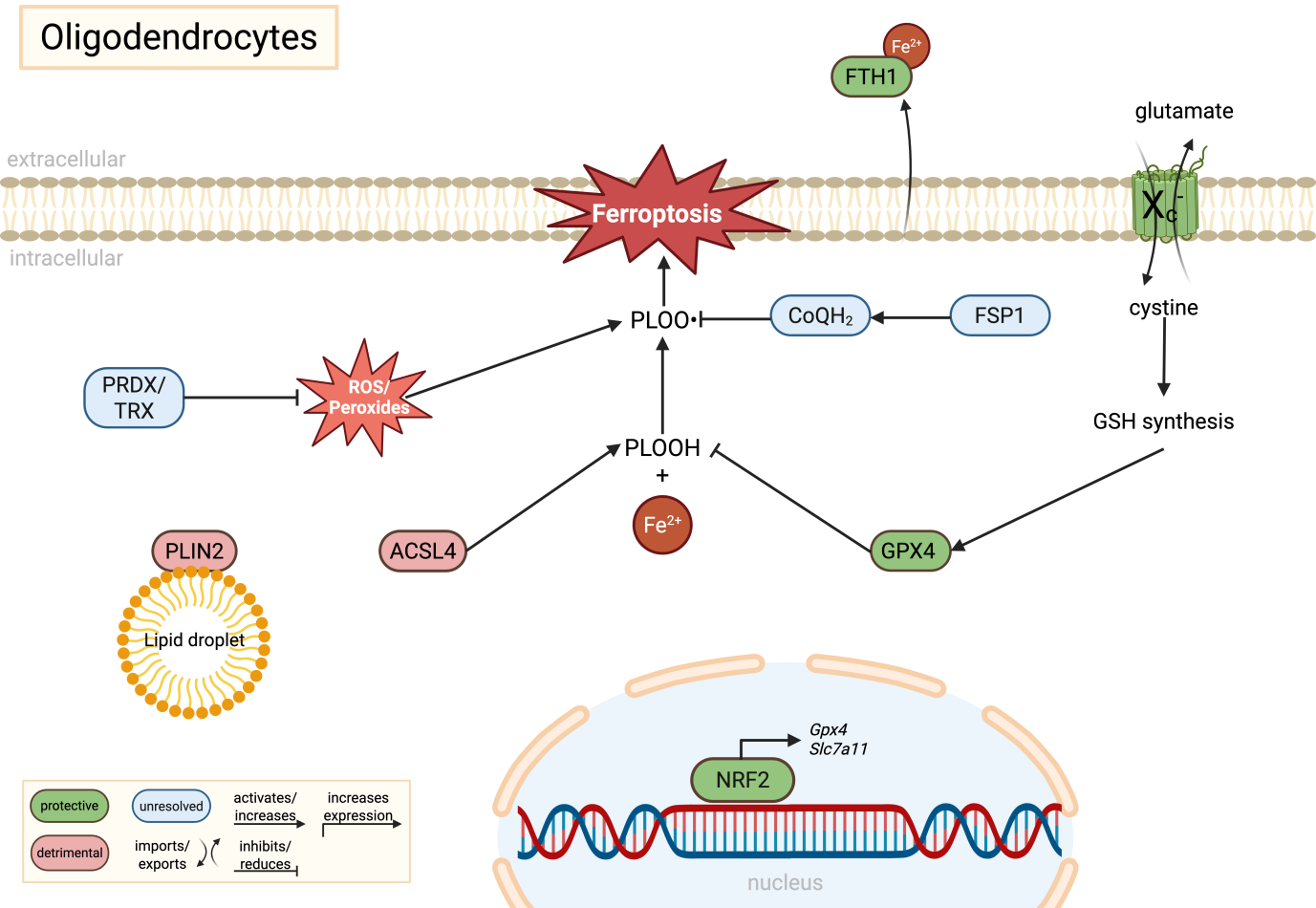{kind=link}
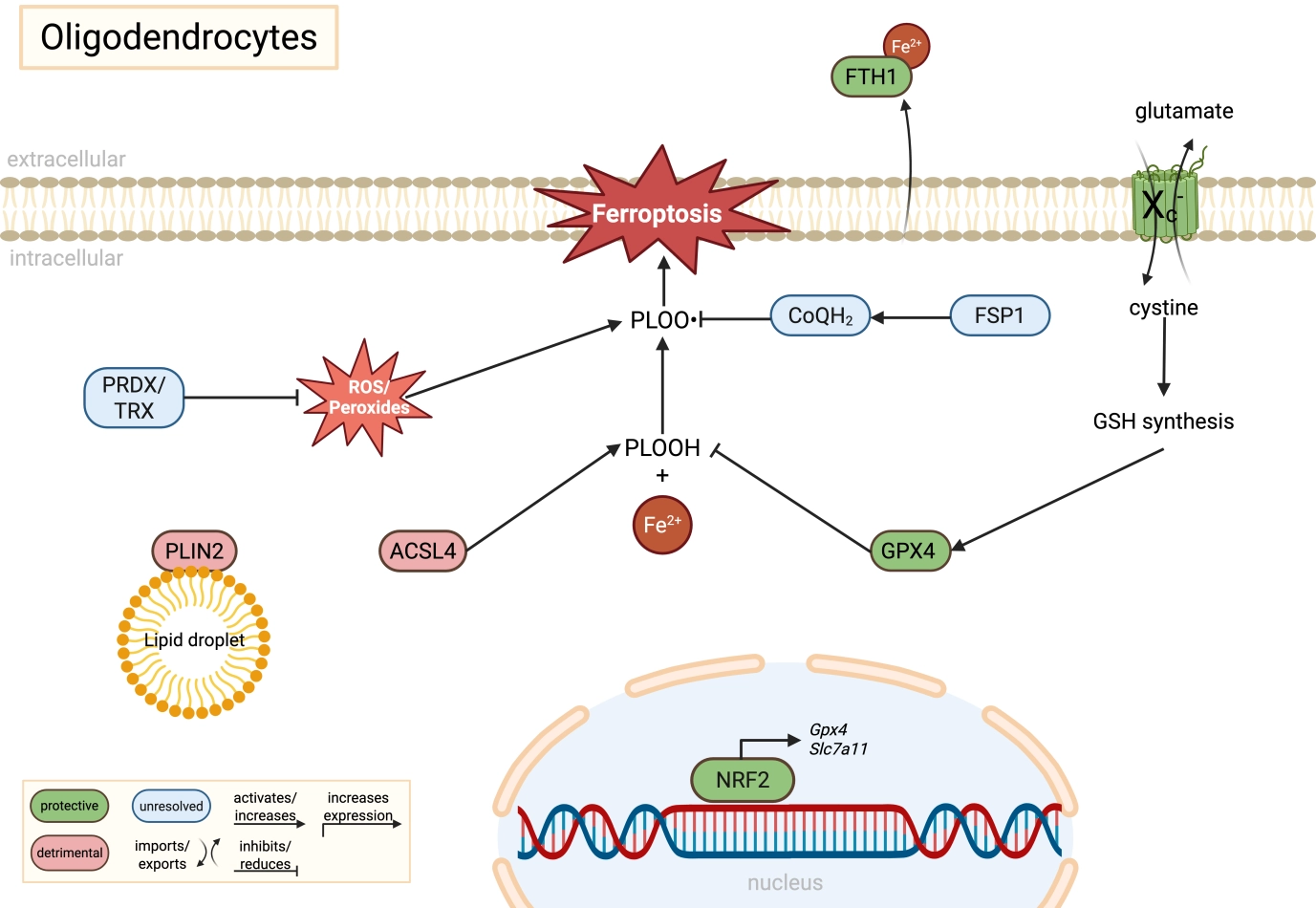
Figure 1. Molecular regulation of ferroptosis in oligodendrocytes. Oligodendrocytes are highly vulnerable to ferroptotic stress due to their elevated iron content and lipid-rich membranes required for myelin synthesis. GPX4-dependent detoxification of PLOOH is supported by cystine uptake through system Xc- (SLC7A11), glutathione synthesis, and potentially by alternative antioxidant pathways including FSP1/CoQH2 and PRDX/TRX systems, although their precise roles in oligodendrocytes remain insufficiently resolved. ACSL4-mediated phospholipid remodeling and PLIN2-associated lipid droplets expand the pool of peroxidation-prone lipids, thereby increasing ferroptotic susceptibility. In parallel, secretion of FTH1 may contribute to extracellular iron buffering and neuroprotection. Green indicates protective pathways, red indicates detrimental pathways, and blue indicates unresolved or context-dependent mechanisms. Created in BioRender. Winneberger, J. (2026) https://BioRender.com/arrl9t0. GPX4: glutathione peroxidase 4; SLC7A11: solute carrier family 7 member 11; PLOOH: phospholipid hydroperoxides; FSP1: ferroptosis suppressor protein 1; PRDX: peroxiredoxin; TRX: thioredoxin; ACSL4: acyl-CoA synthetase long-chain family member 4; PLIN2: perilipin-2; FTH1: ferritin heavy chain 1; NRF2: nuclear factor erythroid 2-related factor 2; GSH: glutathione.
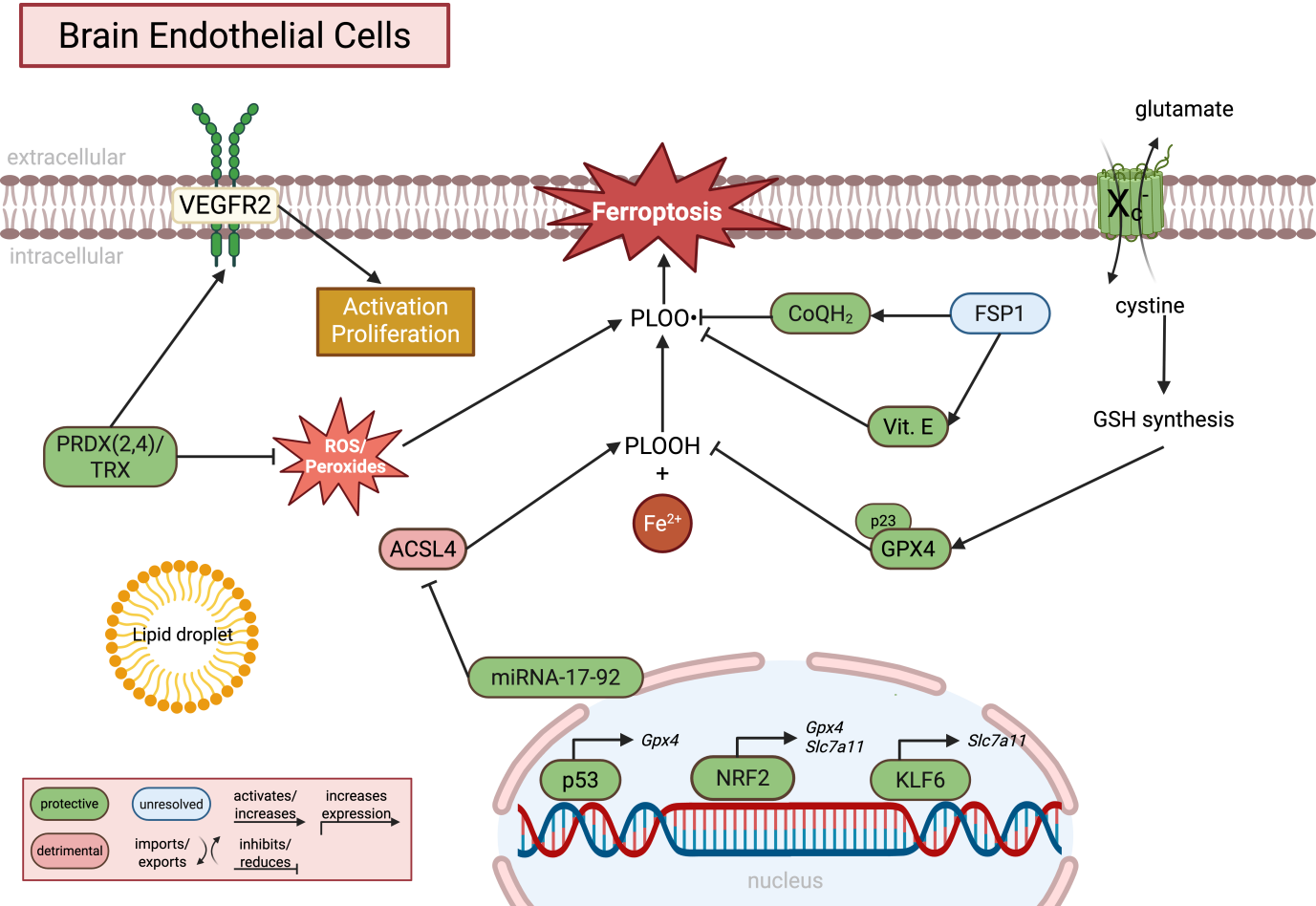{kind=link}
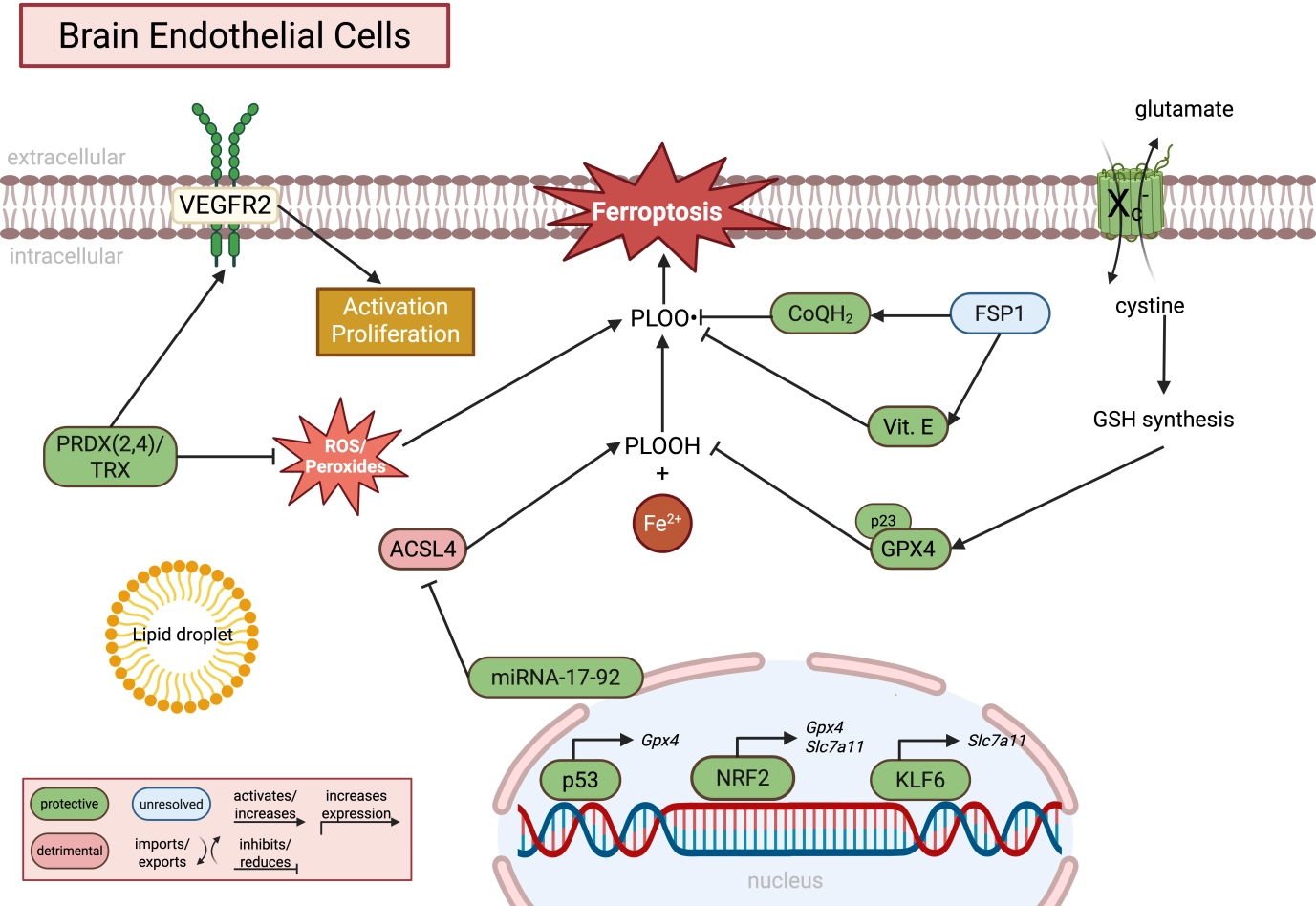
Figure 2. Molecular regulation of ferroptosis in brain endothelial cells. Brain endothelial cells rely on tightly regulated antioxidant and lipid metabolic pathways to maintain blood-brain barrier integrity and suppress lipid peroxidation. GPX4-dependent detoxification of PLOOH is supported by cystine uptake via system Xc- (SLC7A11), glutathione synthesis, and alternative antioxidant systems including FSP1/CoQH2, vitamin E, and the PRDX/TRX pathway. NRF2, KLF6, and p53 regulate the transcriptional expression of ferroptosis-associated genes, whereas miRNA-17-92 suppresses ACSL4 expression. In parallel, ROS/peroxide signaling contributes to endothelial activation, proliferation, and angiogenesis, highlighting that lipid peroxidation may exert both physiological signaling and pathological ferroptotic functions in BECs. Green indicates protective pathways, red indicates detrimental pathways, and blue indicates unresolved or context-dependent mechanisms. Created in BioRender. Winneberger, J. (2026) https://BioRender.com/swexm06. GPX4: glutathione peroxidase 4; PLOOH: phospholipid hydroperoxides; SLC7A11: solute carrier family 7 member 11; FSP1: ferroptosis suppressor protein 1; PRDX: peroxiredoxin; TRX: thioredoxin; NRF2: nuclear factor erythroid 2-related factor 2; KLF6: Krueppel-like factor 6; ACSL4: acyl-CoA synthetase long-chain family member 4; ROS: reactive oxygen species; BECs: brain endothelial cells.
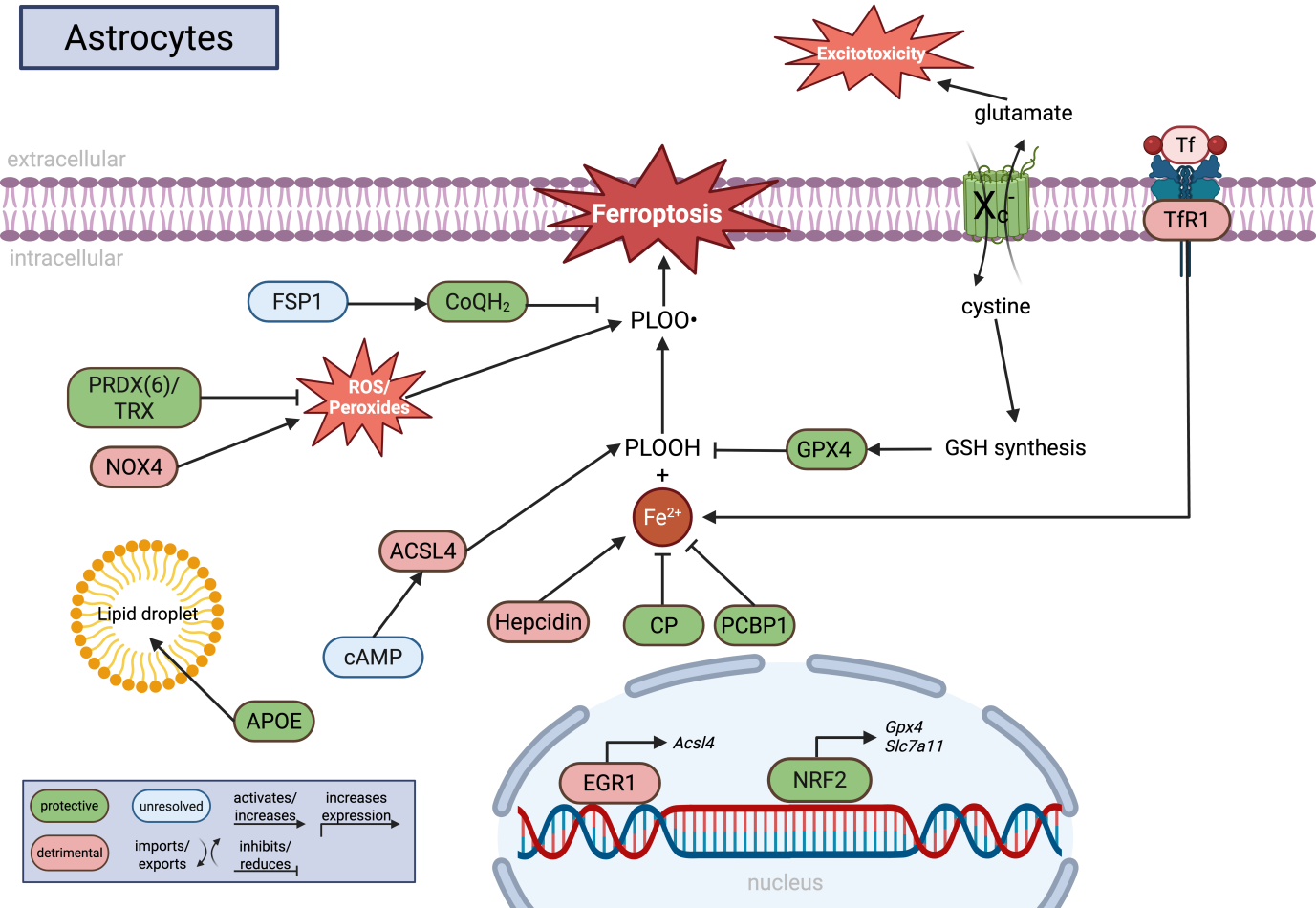{kind=link}
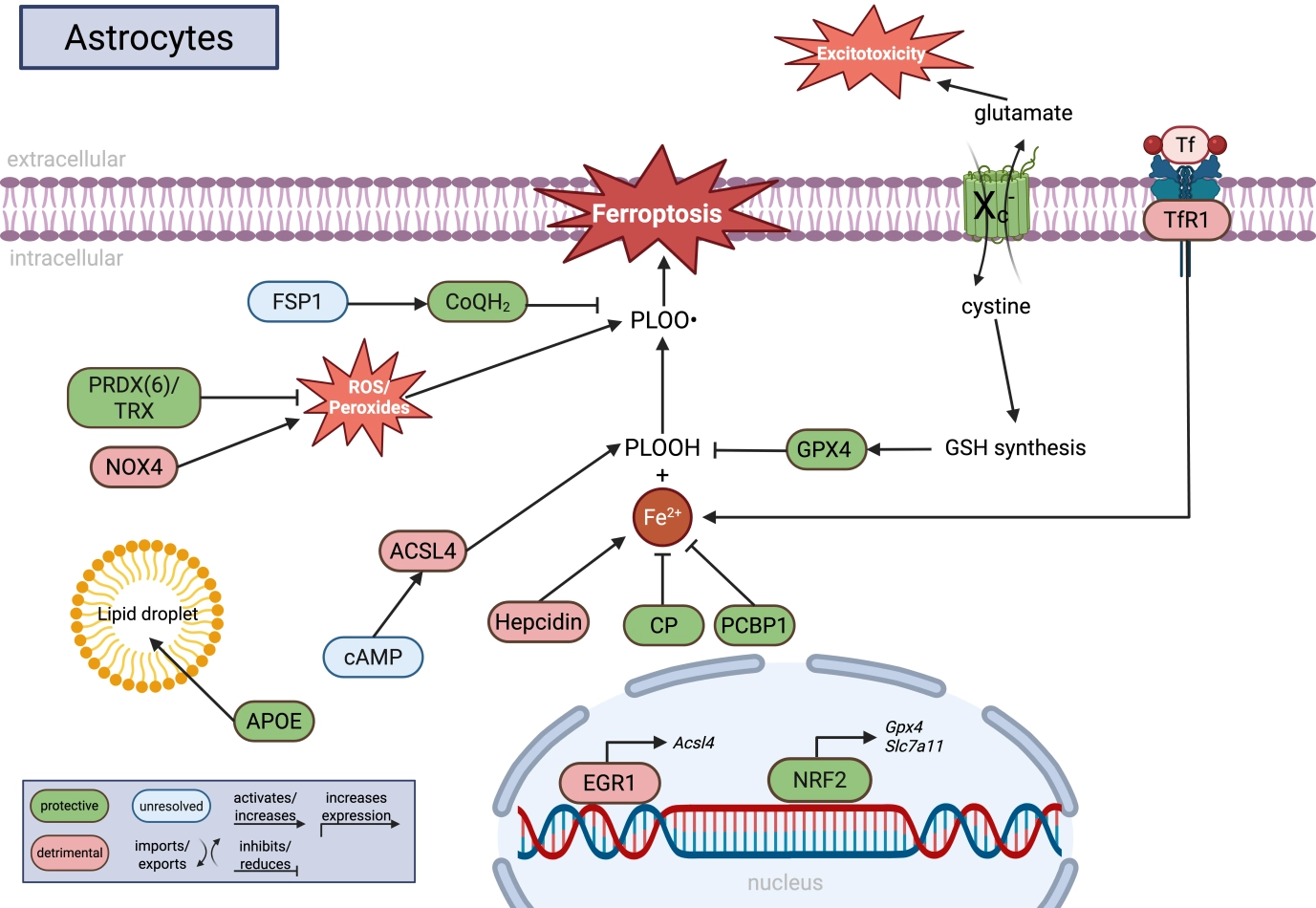
Figure 3. Molecular regulation of ferroptosis in astrocytes. Astrocytes coordinate iron metabolism, lipid homeostasis, antioxidant defense, and glutamate signaling to regulate ferroptotic stress within the CNS. GPX4-dependent detoxification of PLOOH is supported by cystine uptake through system Xc- (SLC7A11), glutathione synthesis, NRF2 signaling, and alternative antioxidant systems including FSP1/CoQH2 and PRDX6/TRX. Astrocytic iron handling is regulated by CP, PCBP1, hepcidin, and TfR1, whereas lipid remodeling pathways involving ACSL4, APOE, lipid droplets, and cAMP signaling influence the abundance of peroxidation-prone lipids. In parallel, NOX4-derived ROS production and glutamate release via system Xc- connect ferroptosis-related pathways to oxidative stress and excitotoxicity. Green indicates protective pathways, red indicates detrimental pathways, and blue indicates unresolved or context-dependent mechanisms. Created in BioRender. Winneberger, J. (2026) https://BioRender.com/80w9t9n. CNS: central nervous system; PLOOH: phospholipid hydroperoxides; SLC7A11: solute carrier family 7 member 11; FSP1: ferroptosis suppressor protein 1; PRDX6: peroxiredoxin 6; TRX: thioredoxin; NRF2: nuclear factor erythroid 2-related factor 2; CP: ceruloplasmin; PCBP1: poly(rC)-binding protein 1; TfR1: transferrin receptor 1; ACSL4: acyl-CoA synthetase long-chain family member 4; APOE: apolipoprotein E; cAMP: cyclic adenosine-monophosphate; NOX4: NADPH oxidase 4; ROS: reactive oxygen species; GPX4: glutathione peroxidase 4.
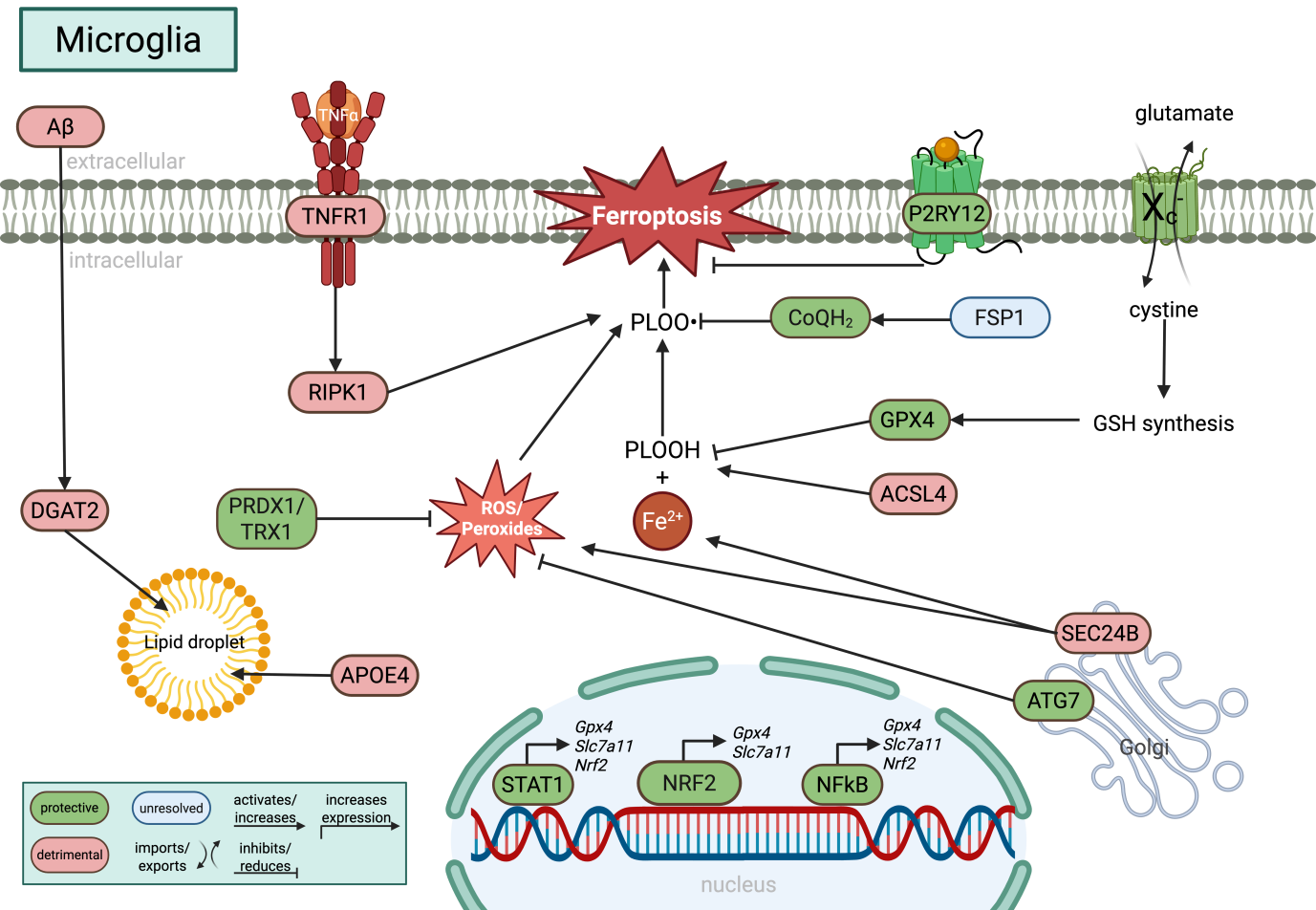{kind=link}
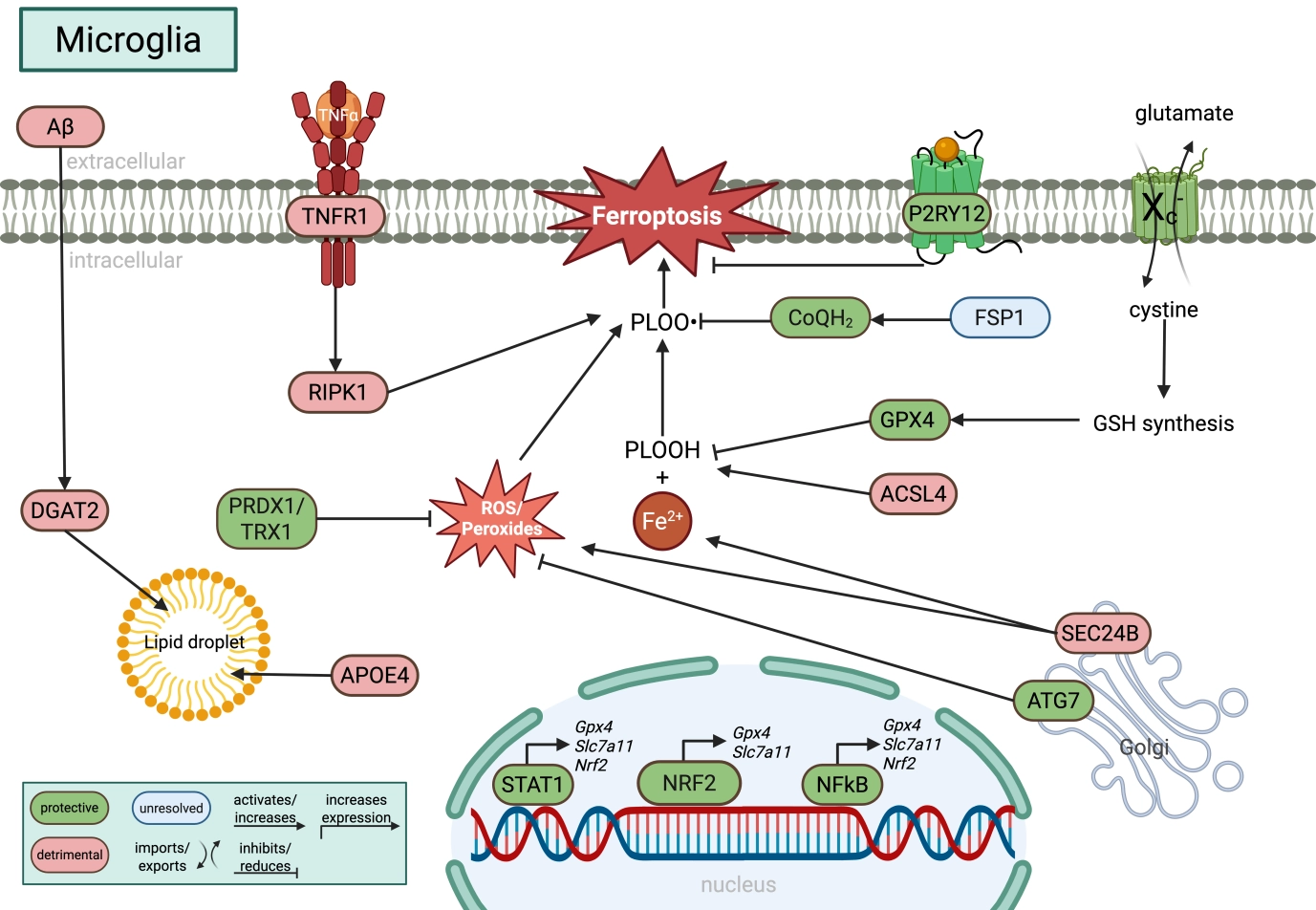
Figure 4. Molecular regulation of ferroptosis in microglia. Microglia integrate iron metabolism, inflammatory signaling, lipid remodeling, and antioxidant defense pathways that collectively determine ferroptotic sensitivity. GPX4-dependent detoxification of PLOOH is supported by cystine uptake via system Xc- (SLC7A11), glutathione synthesis, NRF2 signaling, and alternative antioxidant systems including FSP1/CoQH2 and PRDX1/TRX1. Inflammatory signaling pathways such as TNFα-RIPK1 promote lipid peroxidation, whereas STAT1-, NFκB-, and P2RY12-dependent signaling increase GPX4 and SLC7A11 expression. Lipid droplet accumulation, regulated by APOE4 and DGAT2, together with ACSL4-mediated phospholipid remodeling, expands the pool of peroxidation-prone lipids. In parallel, autophagy- and vesicle trafficking-associated proteins including ATG7 and SEC24B modulate ferroptotic stress, highlighting the close interplay between lipid metabolism, inflammation, and ferroptosis in microglia. Green indicates protective pathways, red indicates detrimental pathways, and blue indicates unresolved or context-dependent mechanisms. Created in BioRender. Winneberger, J. (2026) https://BioRender.com/2x46ir9. GPX4: glutathione peroxidase 4; PLOOH: phospholipid hydroperoxides; SLC7A11: solute carrier family 7 member 11; FSP1: ferroptosis suppressor protein 1; PRDX6: peroxiredoxin 6; TRX1: thioredoxin 1; TNFα: tumor-necrosis factor α; RIPK1: receptor-interacting serine/threonine-protein kinase 1; STAT1: signal transducer and activator of transcription 1; NFκB: nuclear factor kappa B; APOE4: apolipoprotein E 4; DGAT2: dacylglycerol O-acyltransferase 2; ACSL4: acyl-CoA synthetase long-chain family member 4; ATG7: autophagy related 7.
Figure 5. Molecular regulation of ferroptosis in CNS-infiltrating T-cells. Infiltrating T cells rely on tightly controlled antioxidant and metabolic pathways to balance activation, proliferation, and ferroptotic stress. GPX4-dependent detoxification of PLOOH is supported by cystine uptake through system Xc- (SLC7A11), glutathione synthesis, and alternative antioxidant pathways including FSP1/CoQH2 and PRDX/TRX systems. NRF2 signaling promotes GPX4 expression and redox homeostasis, whereas ACSL4-mediated phospholipid remodeling increases the pool of peroxidation-prone lipids. Interestingly, ferroptosis-related pathways are closely linked to T-cell activation and proliferation, suggesting that controlled lipid peroxidation may participate in metabolic signaling and immune effector functions beyond terminal ferroptotic cell death. Green indicates protective pathways, red indicates detrimental pathways, and blue indicates unresolved or context-dependent mechanisms. Created in BioRender. Winneberger, J. (2026) https://BioRender.com/59b299m. CNS: central nervous system; GPX4: glutathione peroxidase 4; PLOOH: phospholipid hydroperoxides; SLC7A11: solute carrier family 7 member 11; FSP1: ferroptosis suppressor protein 1; PRDX: peroxiredoxin; TRX: thioredoxin; NRF2: nuclear factor erythroid 2-related factor 2; ACSL4: acyl-CoA synthetase long-chain family member 4.
2. Oligodendrocyte Lineages
Among all CNS cell types, oligodendrocytes exhibit the highest iron content, which is required for myelin synthesis. In addition to myelination, oligodendrocytes provide essential trophic and metabolic support to neurons, underscoring their importance for neuronal integrity and circuit function. Oligodendrocyte pathology has classically been studied in demyelinating disorders such as multiple sclerosis (MS)[46] where oligodendrocytes accumulate the lipid peroxidation by-products malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE)[47,48]. However, emerging evidence indicates that oligodendrocyte dysfunction and death also contribute to acute ischemic injury, cerebral small vessel disease, and other primary neurodegenerative diseases, including AD[49-51] where lipid peroxidation in oligodendrocytes has not been systematically investigated.
Oligodendrocytes and their precursor cells robustly express GPX4, highlighting a strong dependence on lipid peroxide detoxification. GPX4 expression is consistently reduced in oligodendrocytes and oligodendrocyte precursor cells (OPCs) in mouse models of demyelination[47,52,53] and has also been reported to be altered in hemorrhagic stroke[54,55], chronic cerebral hypoperfusion[56] and neurodegenerative disease contexts[57]. In addition, the system Xc- is downregulated in oligodendrocytes in MS, as suggested by epigenetic and transcriptional studies[58]. By contrast, the expression and functional relevance of alternative ferroptosis-modulating pathways, including peroxiredoxins, ferroptosis suppressor protein 1 (FSP1), or related antioxidant systems, have not been systematically investigated in oligodendrocyte lineage cells.
Functional studies in vitro support a critical role for ferroptosis in oligodendrocytes. Pharmacological inhibition of GPX4 or SLC7A11 as well as iron overload induces lipid peroxidation and oligodendrocyte death in vitro, which are blocked by radical trapping agents (RTA) such as Lip-1 and Fer-1[55,59-62]. Interestingly, susceptibility to erastin-induced ferroptosis decreases during maturation in an oligodendrocyte cell line[59], suggesting developmental stage-dependent differences in glutathione metabolism and redox buffering capacity during oligodendrocyte maturation. Additionally, hemin-induced ferroptosis correlates with GPX4 downregulation[54], providing further evidence for the central role of GPX4 in maintaining redox homeostasis. Interestingly, perilipin-2 (PLIN2), a key regulator of lipid droplet formation, is upregulated in oligodendrocytes in a mouse model of ischemic stroke[55] and its deletion limits oligodendrocyte death in contrast to gastric cancer cells where PLIN2 deletion increases ferroptosis[63], suggesting that lipid droplets are a relevant source of lipid peroxidation in oligodendrocytes. Along these lines, inhibitors of the fatty acid binding proteins 5 and 7 reduce lipid peroxidation[48] in preclinical MS models, highlighting that the oxidized lipid pool and sources are highly cell type, tissue and context specific (Figure 1).
While systemic treatments with RTAs ameliorate lipid peroxidation in mouse models of demyelination and cerebral ischemia[53,55,56], it is unclear whether this is a direct effect on oligodendrocytes or an indirect effect. Therefore, cell type-specific genetic deletion in vivo studies should be prioritized. Although selenium is essential for myelin production and oligodendrocyte development[64,65], it is unclear how selenium is transported to oligodendrocytes, how intracellular selenium metabolism is regulated, and whether selenium availability becomes rate-limiting for GPX4 activity in disease states. Moreover, the contribution of GPX4-independent ferroptosis-suppressing pathways, such as the FSP1-CoQ10[66,67] axis, peroxiredoxin (PRDX) or thioredoxin (TRX) systems[68,69], remains largely unexplored in oligodendrocytes. Beyond cell-autonomous mechanisms, oligodendrocytes may also modulate ferroptotic stress in neighboring cells. Notably, oligodendrocytes can secrete ferritin heavy chain 1 (FTH1), which exerts neuroprotective effects[70], pointing toward ferritin-mediated iron buffering as an additional layer of ferroptosis regulation. Whether such mechanisms are altered in demyelinating, ischemic, or neurodegenerative diseases remains unknown.
3. Brain Endothelial Cells
Brain endothelial cells (BECs) are central regulators of the blood brain barrier (BBB) and CNS homeostasis, controlling the exchange of nutrients, metabolites, and immune signals between the circulation and the brain. Disruption of BBB integrity is a shared pathological feature across virtually all neuroinflammatory and neurodegenerative diseases, including MS, stroke, AD, and related disorders. In vivo evidence from preclinical models and histopathological human postmortem studies of ischemic stroke, traumatic brain injury, and AD demonstrates lipid peroxidation and oxidative stress in vascular cells[71-74]. The unique lipid composition with enrichment of polyunsaturated fatty acids (PUFAs) in specific membrane domains and exceptionally low membrane turnover and transcytosis of brain endothelial cells[75,76], while essential for BBB integrity, may predispose these cells to lipid peroxidation when antioxidant defenses or iron homeostasis are compromised.
This concept is supported by in vivo evidence demonstrating that endothelial cell-specific deletion of GPX4 alone does not elicit an overt phenotype but leads to pronounced BBB dysfunction and thrombus formation only when combined with vitamin E deficiency[77], similarly to hematopoietic stem cells and hepatocellular cells[78,79]. In addition, BECs exhibit high expression of the antioxidant transcription factor NRF2, which plays a central role in maintaining redox homeostasis at the BBB. Notably, NRF2 deletion exacerbates oxidative stress and BBB dysfunction in mouse models of stroke and cerebral hemorrhage, correlating with reduced expression of GPX4 and SLC7A11[80-86]. However, direct evidence linking these changes to lipid peroxidation-driven cell death remains limited. Congruently, pharmacological activation of NRF2 ameliorates BBB disruption in preclinical brain injury models[80,81], underscoring the importance of NRF2-regulated antioxidant programs in preserving endothelial barrier integrity. In addition to the Xc-/GPX4-axis, periredoxins crucially maintain the functionality of endothelial cells. Deletion of peroxiredoxin 4 (PRDX4) in BECs increases the vulnerability to cerebral ischemia/reperfusion injury in vivo[87], and PRDX2 is an essential antioxidant enzyme during steady-state to prevent the oxidation and inactivation of the vascular endothelial growth factor receptor 2 (VEGFR2)[87]. Vascular endothelial growth factor (VEGF)-signalling is essential for angiogenesis, and can be therapeutically increased by thioredoxin mimetic peptides[87]. Furthermore, endothelial deletion of the thioredoxin reductase 2 (TXNRD2) increases vascular stiffness and leads to vascular hypertrophy in mice[88], underlining the close interaction between the redox balance and endothelial functionality. Systemic treatment with RTAs like ferrostatin-1 or liproxstatin-1[71,72,88-90] counteracts BBB dysfunction in preclinical brain injury and infectious models; however, cell type specific models are required for further mechanistic studies. In addition, SLC7A11 is downregulated in BECs in mouse models of stroke, MS, traumatic brain injury, and epilepsy[91]. However, further studies are warranted to answer whether the endothelial SLC7A11 downregulation is functionally and phenotypically relevant in these mouse models.
Ferroptosis in brain endothelial cells has thus far been investigated predominantly in vitro. Beyond direct inhibition of GPX4 or SLC7A11, physiologically relevant stressors such as hypoxia, lipopolysaccharide, and altered shear stress induce lipid peroxidation and ferroptotic cell death in endothelial cells, concomitant with reduced expression of GPX4 and SLC7A11[72,92,93] while increasing the expression of TRX and PRDX proteins[94,95]. In cell culture models, co-enzyme Q10 depletion also increases the susceptibility to ferroptosis[96,97]. However, its endogenous synthesis and regulation in BECs remains unexplored. Interestingly, overexpression of the transcription factor Krueppel-like factor 6 (KLF6) increases SLC7A11 expression and co-enzyme Q10 synthesis[93] while p53 increases GPX4 expression[98], thereby, acting synergistically with NRF2. Notably, because hypoxia and shear stress are known to activate NRF2 signaling in endothelial cells[99,100], these findings suggest that additional regulatory mechanisms in addition to NRF2-mediated adaptive responses regulate the endothelial vulnerability to ferroptosis under pathological conditions. In addition to transcriptional regulation, the GPX4-stability is modulated by direct binding to p23[92] that reduces GPX4-degradation, which is mechanistically unresolved. It is likely that GPX4-stability and degradation are highly cell type specific. Interestingly, the miRNA-17-92 protects endothelial cells from ferroptosis by targeting ACSL4[101], indicating that the key players of ferroptosis are regulated on several levels in endothelial cells (Figure 2).
A major limitation of current ferroptosis research in brain endothelial cells is the paucity of in vivo studies. Given that endothelial GPX4 deletion induces overt endothelial dysfunction only in the context of concurrent vitamin E deficiency[77], identifying the compensatory antioxidant mechanisms that protect brain endothelial cells under physiological conditions represents a critical priority. Furthermore, it is essential to distinguish between endothelial dysfunction at the BBB and overt endothelial cell death. Ferroptotic cell death represents the terminal stage of uncontrolled lipid peroxidation, whereas sublethal lipid peroxidation is likely to exert important regulatory and signaling functions that modulate BBB permeability, transport, and inflammatory responses. Accordingly, future studies should dissect how lipid peroxidation and the modulation of key ferroptosis-related proteins and enzymes influence BBB function independently of cell death, and how endothelial cell death itself reshapes the surrounding microenvironment, including immune cell infiltration and neuroinflammation.
4. Astrocytes
Astrocytes occupy a central position in the regulation of ferroptosis in the CNS. In contrast to neurons and oligodendrocytes, astrocytes are generally more resistant to ferroptotic cell death and function as metabolic and redox regulators that shape ferroptosis susceptibility in neighboring cells, particularly neurons[102-104]. This role is rooted in their specialized capacity to control iron homeostasis, lipid metabolism, cystine-glutamate exchange, and NRF2-dependent antioxidant programs, positioning astrocytes as key gatekeepers of ferroptotic stress across the neurovascular unit.
Astrocytes play a pivotal role in cerebral iron handling and act as buffers that limit iron-induced oxidative stress. Astrocytic expression of ceruloplasmin (CP) facilitates iron export and its ferroxidase activity maintains iron in a redox-safe state, thereby preventing iron accumulation and secondary lipid peroxidation. Loss or dysfunction of astrocytic CP results in iron overload, increased oxidative stress, age-dependent neurodegeneration, and impaired myelination in vivo[105-109] underscoring the importance of astrocyte-mediated iron regulation for long-term brain homeostasis. In a stroke mouse model, transferrin receptor 1 dependent iron-overload also induces death in astrocytes themselves[110]. Along these lines, poly(rC)-binding protein 1 (PCBP1)-dependent iron stabilization reduces lipid peroxidation in astrocytes[111] while iron retention via injury-induced hepcidin expression increases ferroptosis in astrocytes in vivo[112]. Perivascular astrocytes appear to exert antioxidative functions in regions exposed to fluctuating iron levels, suggesting spatial specialization of astrocytic iron buffering.
Astrocytes are highly specialized for lipid uptake, storage, and redistribution, enabling them to buffer excess fatty acids and protect neurons from lipid-induced toxicity[113]. They efficiently take up PUFAs and channel them into lipid droplets, thereby limiting the incorporation of peroxidation-prone lipids into neuronal membranes. Lipid droplet metabolism is profoundly regulated by the apolipoprotein E (APOE)[114]. Genetic APOE deletion or the APOE4 genotype, which is the greatest genetic risk factor for AD, promote lipid accumulation while impairing lipid export and turnover, thereby increasing the availability and residence time of oxidizable lipids[115,116]. This shift renders astrocytes, and indirectly neurons, more vulnerable to lipid peroxidation and ferroptosis. Intriguingly, astrocytic lipid peroxidation is detectable in AD mouse models and postmortem AD tissues, further supporting the need to study ferroptotic cell death in astrocytes. This is additionally regulated by NADPH oxidase 4 (NOX4)-dependent reactive oxygen species (ROS) production that sensitizes astrocytes to lipid peroxidation and ferroptotic cell death in AD mouse models[117,118]. Enzymes regulating fatty acid activation and phospholipid remodeling, including ACSL4, further shape this balance[119,120]. In vivo, early growth response 1 (Egr1) dependent ACSL4 expression drives ferroptosis in astrocytes in a mouse model of autoimmune CNS inflammation[121]. Cyclic adenosine-monophosphate (cAMP), a key 2nd messenger of astrocytes that shapes neuroglia coupling, induces ACSL4 expression in cultured astrocytes[122]. This might point towards a physiological role of activity-dependent lipid peroxidation for neuroglia coupling.
Astrocytes are the predominant cell type expressing the cystine-glutamate antiporter system Xc-[45], making them central regulators of cystine uptake and glutathione synthesis in the brain. Through this pathway, astrocytes maintain their own redox balance and supply cysteine-derived metabolites to neighboring cells, thereby suppressing ferroptosis in more vulnerable populations. At the same time, system Xc--mediated glutamate release introduces a pathological trade-off[123], as excessive glutamate export under inflammatory conditions may exacerbate excitotoxic signaling and indirectly promote neuronal injury[28,29,124,125]. Thus, astrocytic system Xc- activity represents a finely tuned balance between ferroptosis protection and excitotoxic risk. Intriguingly, SLC7A11 has been recently identified to be located in lysosomes where it mediates slow H+ efflux, and its deletion induces overacidification and subsequent reduced degradation of proteins[126]. This links glutathione metabolism directly with protein aggregation, a hallmark of neurodegenerative diseases. The expression of SLC7A11 is mostly controlled by NRF2 in astrocytes[127,128]. Astrocytes have the most robust NRF2-dependent antioxidant responses among CNS cell types, enabling strong basal and inducible defenses against oxidative stress. Impairment of NRF2 signaling in astrocytes promotes oxidative stress and has been linked to astrocytic ferroptosis in an AD mouse model[129], whereas NRF2 activation preserves astrocyte viability and enhances neuroprotection[130,131]. Importantly, astrocytic NRF2 activity not only protects astrocytes themselves but also indirectly shields neurons and oligodendrocytes from ferroptotic injury by maintaining tissue-level redox balance. Interestingly, inflammatory cytokines and lipopolysaccharide (LPS) induce SLC7A11 expression in cultured astrocytes[132], rendering them more susceptible to erastin induced ferroptosis. Since this correlates with decreased GPX4 expression after LPS stimulation[133], this suggests SLC7A11 as a major anti-oxidative system under inflammatory conditions and possibly yet undiscovered glutathione (GSH)-dependent anti-oxidative pathways. Astrocytic GPX4 is furthermore transcriptionally downregulated in an in vivo model of chronic stress, which correlates with lipid peroxidation and can be counteracted with the iron-chelator deferiprone[111]. Interestingly, in an acute in vivo brain injury model, GPX4 is upregulated in astrocytes while it is downregulated in neurons[134], further supporting that astrocytes are essential for the anti-oxidative defense system of neurons. However, in vivo studies with astrocyte-specific GPX4 deletion are required to causally link GPX4 to lipid peroxidation and ferroptosis in astrocytes.
In addition to the Xc-/GPX4-axis, co-enzyme Q10 protects astrocytes against oxidative damage and death in vitro[135-137]. However, its regulation in vivo and the role of FSP1 in astrocytes remain unexplored. Astrocytes also highly express peroxiredoxin 6 (PRDX6) and its deletion promotes while its overexpression inhibits the development of AD pathologies in preclinical in vivo models[138,139]. Although in vitro studies suggest that thioredoxins maintain the expression of perioredoxins[140,141], further in vivo studies with astrocyte-specific deletions are required to delineate Xc-/GPX4-independent anti-ferroptotic defense in astrocytes (Figure 3).
Although astrocytes are critical regulators of iron and lipid metabolism in the brain, there is only limited evidence for direct lipid peroxidation and ferroptotic cell death in vivo. Histological studies provide evidence for astrocytic lipid peroxidation in AD[117,118] and potentially ferroptosis-sensitive astrocyte populations have been identified by single-cell sequencing studies[133,142]. However, these need to be interpreted with caution since a lot of ferroptosis key players are regulated by their enzymatic activity, protein degradation, or cellular localization rather than their transcriptional expression. Astrocytic dysfunction, atrophy, and eventual death are key features of neurodegenerative diseases and it is likely that ferroptosis plays an important role. It is critical to understand astrocyte pathologies since they also modulate neuronal ferroptosis by lipid detoxification and secretion of soluble factors like lactoferrin or other unknown factors. Cell-type specific deletion in vivo studies are required to address which enzymes regulate ferroptosis in astrocytes.
5. Microglia
Microglia are the resident myeloid cells of the CNS with a pivotal position at the intersection of iron metabolism, lipid handling, and inflammatory signaling. Under physiological conditions, microglia contribute to tissue homeostasis by clearing debris, recycling iron, and coordinating repair processes. These same functions render microglia particularly prone to accumulating labile iron and oxidizable lipids in disease states, thereby lowering the threshold for lipid peroxidation. Iron-accumulating microglia have been increasingly recognized as drivers of neurodegenerative and neuroinflammatory diseases[143,144]. As a result, microglia are increasingly recognized not only as responders to ferroptosis in the parenchyma, but as cells that themselves experience ferroptotic stress and actively shape ferroptosis susceptibility in surrounding neural cells.
Microglial vulnerability to ferroptosis reflects a balance between iron availability, lipid composition, and antioxidant capacity. Microglia rely on the SLC7A11-GSH-GPX4 axis[145-150] to suppress phospholipid peroxidation, and disruption of this pathway repeatedly emerges as a common feature of microglial ferroptosis across experimental systems. In addition, some studies suggest that co-enzyme Q10 protects microglia from oxidative injury[151,152]. However, these rely on systemic or indirect co-enzyme Q10 modulation and therefore need to be interpreted carefully. In parallel, disease-associated changes in the overload of labile iron[153-156] and lipid remodeling enzymes, including those that promote incorporation of PUFAs into membrane phospholipids, expand the pool of peroxidation-prone substrates. A prominent manifestation of altered microglial lipid handling is the accumulation of intracellular lipid droplets. Lipid droplet-accumulating microglia represent a dysfunctional and proinflammatory state that becomes increasingly prevalent with aging and neurodegeneration. While lipid droplets may initially buffer excess fatty acids and limit membrane lipid peroxidation, chronic stress appears to convert them into reservoirs of oxidized lipids, thereby amplifying ferroptotic stress and rendering them dysfunctional[153,157-160]. Notably, the homozygous APOE4 genotype is associated with an accumulation of lipid droplets[161] and amyloid-β stimulates Diacylglycerol-O-Acyltransferase 2 mediated lipid droplet synthesis[162] that strongly inhibits the phagocytic capacity in AD, positioning microglia lipid metabolism as central players in AD pathology.
Crucially, microglia do not need to undergo terminal ferroptotic death to exert pathogenic effects. Increasing evidence supports the existence of sublethal ferroptotic stress states in microglia[163] and ACSL4-dependent lipid remodelling[164] impairs cellular function without immediate cell loss. Such states are associated with altered cytokine and chemokine release, reduced phagocytic efficiency, and metabolic rewiring, collectively reshaping the tissue environment in a manner that sensitizes neurons and oligodendrocytes to secondary injury[165]. This distinction between ferroptotic stress and ferroptotic death is particularly relevant for chronic neurodegenerative diseases, where microglia persist over long time scales and may oscillate between adaptive and maladaptive phenotypes.
In addition to impaired phagocytic clearance in AD, ferroptotic signalling in microglia has been implicated in maintaining a pro-inflammatory microenvironment in white matter abnormalities during aging[166], Parkinson’s disease[147,167], cerebral ischemia-reperfusion injury[168,169] and spinal cord injury[170,171]. While tumor-necrosis factor α (TNFα) and its downstream signaling receptor-interacting serine/threonine-protein kinase 1 (RIPK1) promote ferroptosis in microglia[172,173], interferon-induced signal transducer and activator of transcription 1 (STAT1), NRF2 or nuclear factor kappa B (NFκB) signalling increases the resilience to ferroptosis by inducing GPX4 and SLC7A11 expression[150,174]. These are also transcriptionally regulated by lactate[167] and adenosine-nucleotides via the purinergic receptor P2Y12[175], further underlining that ferroptotic sensitivity is associated with microglial injury responses. Additionally, PRDX1 protects microglia from oxidative cell death[176,177] while restricting oxidative tissue damage in in vivo stroke models[178]. Similarly, thioredoxin-1 (TRX1) regulates microglia polarization into an anti-inflammatory phenotype[179,180]. However, these redox systems have been mostly investigated in vitro or by indirect system modulation in vivo. Targeted approaches are required to further study the interplay between microglia activation states and the PRDX/TRX-axis. Interestingly, α-synuclein mutations that impair autophagy, the vesicle trafficking protein SEC24B, and autophagy related 7 (ATG7) are important regulators of microglial ferroptosis[147,163,181], suggesting an important interplay between autophagy and lipid peroxidation in microglia (Figure 4).
Despite substantial progress, most evidence for microglial ferroptosis still derives from in vitro or acute injury models, and in vivo studies that clearly distinguish ferroptotic signaling from terminal cell death remain limited. This distinction is particularly important given that ferroptotic stress may coexist or interact with other inflammatory death pathways, such as pyroptosis in AD, that decisively drives protein aggregation[182], and that the integration of ferroptotic signaling with broader immune responses is still poorly understood.
6. CNS Infiltrating T Cells
In addition to classical inflammatory brain disorders, increasing evidence supports a role for CNS-infiltrating peripheral T cells in neurodegenerative diseases[183-185]. Ischemic and inflammatory microenvironments expose these cells to oxidized lipids, iron dysregulation, and oxidative stress, suggesting that ferroptosis-related mechanisms may influence their survival and effector function within the CNS. While most studies have focused on how ferroptotic death in tumor cells shapes immune responses[186,187], emerging data indicate that ferroptosis pathways directly regulate immune cell biology. Cytotoxic T cells critically depend on GPX4-mediated lipid peroxide detoxification, and disruption of this antioxidant axis impairs their survival and function[188-191]. Conversely, ferroptosis-suppressing mechanisms such as FSP1 are upregulated in activated regulatory T cells and its deletion increases T-cell mediated cytotoxicity against tumors[192]. Interestingly, proliferative T cells strongly upregulate the expression of the PRDX/TRX-axis. While PRDX2-deletion increases proliferation, activation and death of effector T cells[193,194], TRX1 overexpression promotes a central memory phenotype and decreases the effector functions of T cells[195,196]. This suggests that compartmentalized regulation of oxidation differentially affects T cell proliferation, activation and phenotype conversion into memory phenotypes. Ferroptosis inhibition increases the cytotoxicity and durability of engineered T cells[197] while systemic RTA treatment or genetic modulation of lipid remodeling enzymes like ACSL4 inhibit T cell activation and CNS infiltration in experimental autoimmune and infectious models[27,198]. These findings point towards highly immune cell type specific ferroptosis mechanisms.
The oxidative stress-responsive transcription factor NRF2 represents an additional metabolic checkpoint in T cells. NRF2 supports antioxidant gene expression and oxidative phosphorylation, thereby enhancing ferroptosis resistance and maintaining cellular redox balance[199-201]. Inhibition of NRF2 in cytotoxic T cells has been shown to increase their proliferation and effector function, suggesting that antioxidant support can limit maximal immune activation and that NRF2 is not the dominant anti-oxidative factor in T cells[202-204]. Together, these findings indicate that ferroptosis-related pathways and NRF2 signaling not only regulate immune cell survival but also shape inflammatory potency. How these mechanisms operate within CNS-infiltrating immune cells, and whether ferroptosis primarily limits immune persistence or modulates inflammatory amplification in neurodegenerative disease, remains to be clarified (Figure 5).
7. Physiological Role of Ferroptosis
While the majority of the literature (including this review) focuses on ferroptosis in diseases, it is unclear whether there is a physiological role for ferroptosis in the brain. While controlled oxidation of lipids is a well-established signaling cascade in neurons and non-neuronal cells (e.g. by phospholipases or lipoxygenases[205-208]), it is less clear whether uncontrolled lipid peroxidation participates in homeostatic signaling. Since the generation of reactive oxygen species, particularly from mitochondria, represents an important second messenger coupling cellular respiration to distinct activation and proliferation states, it is likely that lipid peroxidation also participates in these signaling processes. Along these lines, most in vivo evidence currently derives from BECs, where oxidative stress can be induced by physiological stimuli, like shear stress[93,94], and the PRDX/TRX system plays a central role in regulating angiogenesis and vascular morphology under homeostatic conditions[88,209]. However, it remains important to distinguish whether these phenotypes are directly mediated by modulation of lipid peroxidation or reflect additional ferroptosis-independent functions of the PRDX/TRX system.
Ferroptosis-related pathways appear to play important roles during brain development, although current evidence suggests that this primarily reflects the need to maintain redox homeostasis during periods of high metabolic activity rather than physiological ferroptotic cell death itself. Complete deletion of GPX4[42] or the glutamate-cysteine ligase[210] is embryonically lethal, underscoring the essential role of lipid peroxide detoxification for cellular survival and developmental integrity. In both neuronal and non-neuronal lineages, expression of ferroptosis-associated regulators changes dynamically during differentiation, proliferation, and migration[211,212], likely reflecting increased oxidative stress and metabolic demand during these processes. Along these lines, retinoic acid signaling has been implicated in the induction of anti-ferroptotic programs in differentiating neurons[213], and because retinoic acid is also a central regulator of glial and vascular development, similar mechanisms may operate across multiple CNS cell types. Along these lines, the epigenetic regulator G9A suppresses the expression of ferroptosis-associated genes under neuroinflammatory conditions[30]. Since G9A plays an important role for brain development and is associated with neurodevelopmental disorders[214,215], similar regulatory mechanisms may likewise operate in glia cell differentiation. However, despite these developmental links to antioxidant and lipid metabolic pathways, there is currently no convincing evidence that ferroptotic cell death itself acts as a physiological mechanism controlling cell proliferation, migration, or developmental patterning in the brain.
Ferroptosis is likely to be a relevant mechanism to prevent uncontrolled proliferation and the rise of primary brain tumors, including neuroblastoma and astrocytic tumors. Increasing evidence suggests that brain tumor cells actively suppress ferroptosis sensitivity through metabolic adaptations, including lipid remodeling programs that reduce the abundance of peroxidation-prone phospholipids and thereby limit lipid peroxide accumulation[216-218]. In line, pharmacological or genetic enhancement of ferroptosis sensitivity can improve the efficacy of existing therapeutic approaches[219-223], supporting ferroptosis induction as a promising strategy for brain tumor treatment. These findings further raise the possibility that ferroptosis may represent an endogenous tumor-suppressive mechanism within the CNS. At the same time, the interaction between tumor cells and surrounding non-neuronal CNS and infiltrating immune populations likely shapes ferroptotic sensitivity and therapeutic response, highlighting an important area for future investigation.
8. Conclusion
Accumulating evidence supports the presence of lipid peroxidation across multiple inflammatory, degenerative, ischemic and traumatic neurological diseases. However, much of the current literature relies on altered expression of canonical ferroptosis regulators as indirect evidence, rather than on direct demonstration of lipid peroxidation-driven cell death. Changes in GPX4, SLC7A11, NRF2, ACSL4, PRDX or TRX expression provide important mechanistic clues but do not, by themselves, establish ferroptosis as the dominant execution mechanism in vivo. Future studies must therefore distinguish transcriptional or metabolic adaptation from bona fide ferroptotic cell death.
Across cell types, distinct regulatory dependencies emerge. Oligodendrocytes appear highly dependent on GPX4-mediated lipid peroxide detoxification, consistent with their iron-rich and lipid-dense physiology, while the functional relevance of SLC7A11 and alternative antioxidant systems remains incompletely defined. Brain endothelial cells exhibit strong NRF2 activity and require GPX4 and the PRDX/TRX system particularly under conditions of antioxidant compromise, yet direct in vivo evidence for endothelial ferroptotic death is limited. Astrocytes display robust NRF2- and SLC7A11-driven antioxidant programs and act as regulators of ferroptotic stress within the neurovascular unit, whereas direct GPX4-dependent ferroptosis in vivo remains insufficiently demonstrated. Microglia show pronounced sensitivity to iron accumulation and lipid remodeling via ACSL4, with emerging evidence supporting GPX4-dependent ferroptotic stress states that may amplify neuroinflammation. In infiltrating immune cells, GPX4 is critical for survival of cytotoxic T cells, while NRF2 and the PRDX/TRX-system function as a metabolic checkpoint that shapes effector states rather than serving solely as ferroptosis suppressors.
Notably, several ferroptosis-modulating systems, including vitamin E-dependent lipid scavenging[224,225], the FSP1-CoQ10 axis[66,67], and PRDX/TRX[68,69] pathways, remain poorly investigated in vivo across non-neuronal CNS cell types. Whether these systems provide redundancy, compensation, or cell type-specific protection is largely unknown.
A critical conceptual distinction must also be maintained between lipid peroxidation-induced signaling and terminal ferroptotic cell death. Sublethal lipid peroxidation can profoundly alter cell function, inflammatory output, and intercellular communication without necessarily leading to membrane rupture or execution of ferroptosis. Particularly in chronic neurodegenerative diseases, such ferroptotic stress states may be more relevant than overt cell death. Importantly, as demonstrated in neurons, ferroptosis and lipid peroxidation should be investigated not only under pathological conditions but also in steady-state physiology[41-43], where tightly regulated lipid oxidation may contribute to metabolic signaling, cell-cell communication, and tissue homeostasis. Along these lines, particular attention should be drawn to the localization of lipid peroxidation. In neurons, the GPX4-localization determines its anti-ferroptotic activity[41]. Particularly, the differential effects of the Xc-/GPX4 and PRDX/TRX-axes on different CNS-resident cell types underline the importance of compartment-specific lipid peroxidation and oxidative challenges for cellular homeostasis and signalling.
Mechanistically, progress will require systematic in vitro screening approaches in purified cell populations, combined with rigorous in vivo genetic modulation using cell type-specific deletion or overexpression of ferroptosis regulators. Such strategies are essential to identify novel, cell-specific ferroptosis regulators and to disentangle adaptive redox responses from true ferroptotic execution. A deeper understanding of how ferroptotic stress is differentially regulated across CNS cell types may enable the development of precision biomarkers[226] and therapies that selectively modulate lipid peroxidation in defined cellular compartments, reshaping neuroinflammatory and neurodegenerative disease trajectories.
Acknowledgements
The figures in the manuscript were generated using biorender. During the preparation of this work, the authors used generative AI (ChatGPT, OpenAI, GPT-5.5) solely to improve the language, grammar, and readability of the text. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the final output.
Authors contribution
Winneberger J: Visualization, investigation, writing-original draft.
Raich L: Investigation, writing-original draft.
Potthast T: Investigation.
Woo MS: Investigation, supervision, funding acquisition, writing-original draft.
All authors agree to the final version of the manuscript.
Conflicts of interest
The authors disclose no competing interests.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This research is supported by the German Research Foundation (WO 2835/1-1 to MSW) and Corona Foundation (S0199/10110/2025 to MSW).
Copyright
© The Author(s) 2026.
References
-
2. Mishima E, Nakamura T, Doll S, Proneth B, Fedorova M, Pratt DA, et al. Recommendations for robust and reproducible research on ferroptosis. Nat Rev Mol Cell Biol. 2025;26(8):615-630.[DOI]
-
3. Jiang X, Stockwell BR, Conrad M. Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266-282.[DOI]
-
4. Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25(6):424-442.[DOI]
-
6. Steinmetz JD, Seeher KM, Schiess N, Nichols E, Cao B, Servili C, et al. Global, regional, and national burden of disorders affecting the nervous system, 1990–2021: A systematic analysis for the Global Burden of Disease Study 2021. Lancet Neurol. 2024;23(4):344-381.[DOI]
-
7. Naghavi M, Ong KL, Aali A, Ababneh HS, Abate YH, Abbafati C, et al. Global burden of 288 causes of death and life expectancy decomposition in 204 countries and territories and 811 subnational locations, 1990–2021: A systematic analysis for the Global Burden of Disease Study 2021. Lancet. 2024;403(10440):2100-2132.[DOI]
-
8. Nichols E, Vos T. The estimation of the global prevalence of dementia from 1990-2019 and forecasted prevalence through 2050: An analysis for the Global Burden of Disease (GBD) study 2019. Alzheimers Dement. 2021;17(S10):e051496.[DOI]
-
9. Chen S, Cao Z, Nandi A, Counts N, Jiao L, Prettner K, et al. The global macroeconomic burden of Alzheimer’s disease and other dementias: Estimates and projections for 152 countries or territories. Lancet Glob Heal. 2024;12(9):e1534-e1543.[DOI]
-
10. Livingston G, Huntley J, Liu KY, Costafreda SG, Selbæk G, Alladi S, et al. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. Lancet. 2024;404(10452):572-628.[DOI]
-
13. Ayton S, Fazlollahi A, Bourgeat P, Raniga P, Ng A, Lim YY, et al. Cerebral quantitative susceptibility mapping predicts amyloid-β-related cognitive decline. Brain. 2017;140(8):2112-2119.[DOI]
-
14. Bao WD, Pang P, Zhou XT, Hu F, Xiong W, Chen K, et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021;28(5):1548-1562.[DOI]
-
24. Muller M, Leavitt BR. Iron dysregulation in Huntington’s disease. J Neurochem. 2014;130(3):328-350.[DOI]
-
28. Woo MS, Mayer C, Binkle-Ladisch L, Sonner JK, Rosenkranz SC, Shaposhnykov A, et al. STING orchestrates the neuronal inflammatory stress response in multiple sclerosis. Cell. 2024;187(15):4043-4060.e30.[DOI]
-
29. Woo MS, Brand J, Bal LC, Moritz M, Walkenhorst M, Vieira V, et al. The immunoproteasome disturbs neuronal metabolism and drives neurodegeneration in multiple sclerosis. Cell. 2026;189(10):3164-3174.[DOI]
-
31. Karuppagounder SS, Alin L, Chen Y, Brand D, Bourassa MW, Dietrich K, et al. N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Neurol. 2018;84(6):854-872.[DOI]
-
36. Kalisvaart ACJ, Ratan RR. Cracking the neuronal ferroptosis code: In vitro insights into mechanisms and treatment of stroke. Ferroptosis Oxid Stress. 2026;2(2):202518.[DOI]
-
38. Lei P, Walker T, Ayton S. Neuroferroptosis in health and diseases. Nat Rev Neurosci. 2025;26(8):497-511.[DOI]
-
39. Nguyen TPM, Alves F, Lane DJR, Bush AI, Ayton S. Triggering ferroptosis in neurodegenerative diseases. Trends Neurosci. 2025;48(10):750-765.[DOI]
-
42. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172(3):409-422.e21.[DOI]
-
43. Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8(3):237-248.[DOI]
-
46. Falcão AM, van Bruggen D, Marques S, Meijer M, Jäkel S, Agirre E, et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med. 2018;24(12):1837-1844.[DOI]
-
50. Ma Q, Tian JL, Lou Y, Guo R, Ma XR, Wu JB, et al. Oligodendrocytes drive neuroinflammation and neurodegeneration in Parkinson’s disease via the prosaposin-GPR37-IL-6 axis. Cell Rep. 2025;44(2):115266.[DOI]
-
54. Shen D, Wu W, Liu J, Lan T, Xiao Z, Gai K, et al. Ferroptosis in oligodendrocyte progenitor cells mediates white matter injury after hemorrhagic stroke. Cell Death Dis. 2022;13:259.[DOI]
-
56. Liu Q, Liu J, Li S, Xu J, He P, Li C, et al. Lcn2-induced oligodendrocyte ferroptosis contributes to white matter damage in chronic cerebral hypoperfusion. Glia. 2025;73(11):2305-2321.[DOI]
-
61. Li Y, Wang B, Yang J, Liu R, Xie J, Wang J. Iron overload causes ferroptosis but not apoptosis in MO3.13 oligodendrocytes. Neurochem Res. 2023;48(3):830-838.[DOI]
-
64. Ma C, Wurlitzer K, Nunes LGA, Hoffmann PR, Pitts MW. Iron and selenium: At the crossroads of development and death in oligodendrocytes. Arch Biochem Biophys. 2025;771:110509.[DOI]
-
65. Gu J, Royland JE, Wiggins RC, Konat GW. Selenium is required for normal upregulation of myelin genes in differentiating oligodendrocytes. J Neurosci Res. 1997;47(6):626-635.[DOI]
-
67. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693-698.[DOI]
-
68. Ito J, Nakamura T, Toyama T, Chen D, Berndt C, Poschmann G, et al. PRDX6 dictates ferroptosis sensitivity by directing cellular selenium utilization. Mol Cell. 2024;84(23):4629-4644.e9.[DOI]
-
69. Chen Z, Inague A, Kaushal K, Fazeli G, Schilling D, Xavier da Silva TN, et al. PRDX6 contributes to selenocysteine metabolism and ferroptosis resistance. Mol Cell. 2024;84(23):4645-4659.e9.[DOI]
-
71. Fang J, Yuan Q, Du Z, Fei M, Zhang Q, Yang L, et al. Ferroptosis in brain microvascular endothelial cells mediates blood-brain barrier disruption after traumatic brain injury. Biochem Biophys Res Commun. 2022;619:34-41.[DOI]
-
75. Bénistant C, Dehouck MP, Fruchart JC, Cecchelli R, Lagarde M. Fatty acid composition of brain capillary endothelial cells: Effect of the coculture with astrocytes. J Lipid Res. 1995;36(11):2311-2319.[DOI]
-
78. Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016;9:22-31.[DOI]
-
79. Hu Q, Zhang Y, Lou H, Ou Z, Liu J, Duan W, et al. GPX4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis. 2021;12:706.[DOI]
-
82. Warpsinski G, Smith MJ, Srivastava S, Keeley TP, Siow RCM, Fraser PA, et al. Nrf2-regulated redox signaling in brain endothelial cells adapted to physiological oxygen levels: Consequences for sulforaphane mediated protection against hypoxia-reoxygenation. Redox Biol. 2020;37:101708.
-
85. Zou H, Leah T, Huang Z, He X, Mameli E, Caporali A, et al. Endothelial cell Nrf2 controls neuroinflammation following a systemic insult. iScience. 2025;28(6):112630.[DOI]
-
86. Cazalla E, Cuadrado A, García-Yagüe ÁJ. Role of the transcription factor NRF2 in maintaining the integrity of the Blood-Brain Barrier. Fluids Barriers CNS. 2024;21(1):93.[DOI]
-
93. Cui J, Fan Z, Ding S, Zhang J, Shen H, Zaidi SA, et al. Abnormal shear stress induces ferroptosis in endothelial cells via KLF6 downregulation. eLife. 2025;14:RP109140.[DOI]
-
94. Mowbray AL, Kang DH, Rhee SG, Kang SW, Jo H. Laminar shear stress up-regulates peroxiredoxins (PRX) in endothelial cells. J Biol Chem. 2008;283(3):1622-1627.[DOI]
-
100. McSweeney SR, Warabi E, Siow RCM. Nrf2 as an endothelial mechanosensitive transcription factor: Going with the flow. Hypertension. 2016;67(1):20-29.[DOI]
-
101. Xiao FJ, Zhang D, Wu Y, Jia QH, Zhang L, Li YX, et al. miRNA-17-92 protects endothelial cells from erastin-induced ferroptosis through targeting the A20-ACSL4 axis. Biochem Biophys Res Commun. 2019;515(3):448-454.[DOI]
-
102. Fan YG, Ge RL, Ren H, Jia RJ, Wu TY, Lei XF, et al. Astrocyte-derived lactoferrin inhibits neuronal ferroptosis by reducing iron content and GPX4 degradation in APP/PS1 transgenic mice. Pharmacol Res. 2024;209:107404.[DOI]
-
110. Guo Y, Wang Y, Ni Y, Bo B, He J, Zhu Y, et al. Iron overload mediates the differential cell fate of astrocytes from neurons and its regulatory mechanisms in ischemic stroke. Adv Sci. 2026;13(4):e07384.[DOI]
-
111. Zhang J, Zhao B, Jia M, Zhao Y, Lu Y, Xi W, et al. Astrocytic PCBP1 suppresses ferroptosis to restore glutamatergic homeostasis and mitigate stress-induced depression in male mice. Adv Sci. 2026;13(10):e13438.[DOI]
-
112. Davaanyam D, Lee H, Seol SI, Oh SA, Kim SW, Lee JK. HMGB1 induces hepcidin upregulation in astrocytes and causes an acute iron surge and subsequent ferroptosis in the postischemic brain. Exp Mol Med. 2023;55(11):2402-2416.[DOI]
-
114. Windham IA, Powers AE, Ragusa JV, Wallace ED, Zanellati MC, Williams VH, et al. APOE traffics to astrocyte lipid droplets and modulates triglyceride saturation and droplet size. J Cell Biol. 2024;223(4):e202305003.[DOI]
-
117. Park MW, Cha HW, Kim J, Kim JH, Yang H, Yoon S, et al. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. 2021;41:101947.[DOI]
-
118. Maimaiti Y, Su T, Zhang Z, Ma L, Zhang Y, Xu H. NOX4-mediated astrocyte ferroptosis in Alzheimer’s disease. Cell Biosci. 2024;14(1):88.[DOI]
-
121. Wen H, Zi Y, Liu Z, Bai Y, Lin J, Wang H, et al. ACSL4-mediated astrocyte ferroptosis augments neuroinflammation and exacerbates NMOSD pathology. Cell Death Differ. 2026;1-17.[DOI]
-
126. Zhou N, Chen J, Hu M, Wen N, Cai W, Li P, et al. SLC7A11 is an unconventional H+ transporter in lysosomes. Cell. 2025;188(13):3441-3458.e25.[DOI]
-
128. Lacher SE, Krznarich J, Levings DC, Pathak SS, Pufall M, Yang YM, et al. The glucocorticoid receptor inhibits NRF2-mediated expression of SLC7A11. Free Radic Biol Med. 2025;241:53-63.[DOI]
-
129. Tang Z, Chen Z, Guo M, Peng Y, Xiao Y, Guan Z, et al. NRF2 deficiency promotes ferroptosis of astrocytes mediated by oxidative stress in Alzheimer’s disease. Mol Neurobiol. 2024;61(10):7517-7533.[DOI]
-
134. Savaskan NE, Borchert A, Bräuer AU, Kuhn H. Role for glutathione peroxidase-4 in brain development and neuronal apoptosis: Specific induction of enzyme expression in reactive astrocytes following brain injury. Free Radic Biol Med. 2007;43(2):191-201.[DOI]
-
138. Pankiewicz JE, Lizińczyk AM, Franco LA, Diaz JR, Gootman A, Sadowski MJ. Role of peroxiredoxin 6 in the development of tau pathology. Alzheimers Dement. 2023;19(S13):e072575.[DOI]
-
139. Pankiewicz JE, Diaz JR, Martá-Ariza M, Lizińczyk AM, Franco LA, Sadowski MJ. Peroxiredoxin 6 mediates protective function of astrocytes in Aβ proteostasis. Mol Neurodegener. 2020;15(1):50.[DOI]
-
140. Islam MI, Sultana S, Padmanabhan N, Rashid MU, Siddiqui TJ, Coombs KM, et al. Thioredoxin-1 protein interactions in neuronal survival and neurodegeneration. Biochim Biophys Acta BBA Mol Basis Dis. 2025;1871(1):167548.[DOI]
-
145. Bussiere R, Tulsian N, Wieder C, McConnaughie D, Tynan E, Lowe A, et al. Modelling ferroptosis in a human microglial line by sequential exposure to iron and GPX4 inhibition. bioRxiv [Preprint]. 2026.[DOI]
-
146. Li HL, Ohmiya H, Sakamoto S, Yugami M, Oki A, Furusawa M, et al. Microglial dynamics and ferroptosis induction in humaniPSC-derived neuron–astrocyte–microglia tri-cultures. FEBS Open Bio. 2026;2211-5463.70182.[DOI]
-
150. Sun W, Li H, Shen Y, Xiao H. Resveratrol attenuates rotenone-induced inflammation and oxidative stress via STAT1 and Nrf2/Keap1/SLC7A11 pathway in a microglia cell line. Pathol Res Pract. 2021;225:153576.[DOI]
-
152. Bhardwaj M, Kumar A. Neuroprotective mechanism of Coenzyme Q10 (CoQ10) against PTZ induced kindling and associated cognitive dysfunction: Possible role of microglia inhibition. Pharmacol Rep. 2016;68(6):1301-1311.[DOI]
-
153. Rocha KCE, Xiang Q, Qian C, Wang L, Yuan W, Beldona V, et al. Labile iron overload reprograms microglia and neurons for lipid droplet synthesis in the aging brain. bioRxiv [Preprint]. 2025.[DOI]
-
156. McIntosh A, Mela V, Harty C, Minogue AM, Costello DA, Kerskens C, et al. Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol. 2019;29(5):606-621.[DOI]
-
157. Marschallinger J, Iram T, Zardeneta M, Lee SE, Lehallier B, Haney MS, et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci. 2020;23(2):194-208.[DOI]
-
158. Li Y, Munoz-Mayorga D, Nie Y, Kang N, Tao Y, Lagerwall J, et al. Microglial lipid droplet accumulation in tauopathy brain is regulated by neuronal AMPK. Cell Metab. 2024;36(6):1351-1370.e8.[DOI]
-
159. Sung S, Kim HJ, Cha SJ, Nahm M, Kim SH, Kwon MS. Microglial lipid droplets as therapeutic targets in age-related neurodegenerative diseases. npj Aging. 2026;12:2.[DOI]
-
160. Wu X, Miller JA, Lee BTK, Wang Y, Ruedl C. Reducing microglial lipid load enhances β amyloid phagocytosis in an Alzheimer’s disease mouse model. Sci Adv. 2025;11(6):eadq6038.[DOI]
-
161. Haney MS, Pálovics R, Munson CN, Long C, Johansson PK, Yip O, et al. APOE4/4 is linked to damaging lipid droplets in Alzheimer’s disease microglia. Nature. 2024;628(8006):154-161.[DOI]
-
162. Prakash P, Manchanda P, Paouri E, Bisht K, Sharma K, Rajpoot J, et al. Amyloid-β induces lipid droplet-mediated microglial dysfunction via the enzyme DGAT2 in Alzheimer’s disease. Immunity. 2025;58(6):1536-1552.e8.[DOI]
-
167. Qin Q, Wang D, Qu Y, Li J, An K, Mao Z, et al. Enhanced glycolysis-derived lactate promotes microglial activation in Parkinson’s disease via histone lactylation. npj Park Dis. 2025;11:3.[DOI]
-
168. Sui H, Sun Z, Liu C, Xi H. Ferritinophagy promotes microglia ferroptosis to aggravate neuroinflammation induced by cerebral ischemia-reperfusion injury via activation of the cGAS-STING signaling pathway. Neurochem Int. 2025;183:105920.[DOI]
-
171. Zhao X, Hu X, Wang W, Lu S. Macrophages dying from ferroptosis promote microglia-mediated inflammatory responses during spinal cord injury. Int Immunopharmacol. 2024;143:113281.[DOI]
-
172. Zeng F, Chen A, Chen W, Cheng S, Lin S, Mei R, et al. Knockout of TNF-α in microglia decreases ferroptosis and convert microglia phenotype after spinal cord injury. Heliyon. 2024;10(17):e36488.[DOI]
-
175. Lopez-Ortiz AO, Doceti M, Thomas J, Duffy A, Coburn M, Okojie AK, et al. Transcriptional regulation of microglial metabolic and activation states byP2RY12. Glia. 2025;73(12):2464-2482.[DOI]
-
176. Kim SU, Park YH, Min JS, Sun HN, Han YH, Hua JM, et al. Peroxiredoxin I is a ROS/p38 MAPK-dependent inducible antioxidant that regulates NF-κB-mediated iNOS induction and microglial activation. J Neuroimmunol. 2013;259(1-2):26-36.[DOI]
-
179. Zhong S, Xu M, Wang Q, Wang S, Li X, Guo Y, et al. Thioredoxin protects against diabetic hearing loss by regulating TOMM22 mediated mitochondrial autophagy in hair cells and inhibiting microglial M1 polarization. Sci Rep. 2026;16:14332.[DOI]
-
181. Mahoney-Sánchez L, Lucas-Clarke H, Penverne A, Evans JR, D'Sa K, Strohbuecker S, et al. The SNCA A53T mutation sensitizes human neurons and microglia to ferroptosis. bioRxiv [Preprint]. 2025.[DOI]
-
185. Elyaman W, Stern LJ, Jiang N, Dressman D, Bradley P, Klatzmann D, et al. Exploring the role of T cells in Alzheimer’s and other neurodegenerative diseases: Emerging therapeutic insights from the T Cells in the Brain symposium. Alzheimers Dement. 2025;21(2):e14548.[DOI]
-
186. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. CD8+ T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. 2022;40(4):365-378.e6.[DOI]
-
188. Kłopotowska M, Baranowska I, Hajduk S, Jurga A, Leśniowska N, Łaźniewski M, et al. GPX4 is a key ferroptosis regulator orchestrating T cells and CAR-T-cells sensitivity to ferroptosis. Cancer Immunol Immunother. 2025;74(9):280.[DOI]
-
190. Liu J, Dong R, Yuan B, Xie Y, Feng Z, Zhou S, et al. Immune cells dying from ferroptosis: Mechanisms and therapeutic opportunities. Cell Death Dis. 2025;16:878.[DOI]
-
197. Harada S, Hashimoto D, Saito Y, Miyajima T, Li W, Senjo H, et al. Ferroptosis inhibition generates TCF-1 + CAR-T cells with enhanced persistence and cytotoxicity. Blood. 2023;142:97.[DOI]
-
200. Jo Y, Lee B, Joo M, Hong C. Nrf2 expression is upregulated in tumor infiltrating T cells and induces T cell anergy. J Immunol. 2016;196(1_Supplement):143.15.[DOI]
-
202. Jo Y, Shim JA, Jeong JW, Kim H, Lee SM, Jeong J, et al. Targeting ROS-sensing Nrf2 potentiates anti-tumor immunity of intratumoral CD8+ T and CAR-T cells. Mol Ther. 2024;32(11):3879-3894.[DOI]
-
207. Moskowitz N, Schook W, Puszkin S. Interaction of brain synaptic vesicles induced by endogenous Ca2+-dependent phospholipase A2. Science. 1982;216(4543):305-307.[DOI]
-
210. Dalton TP, Dieter MZ, Yang Y, Shertzer HG, Nebert DW. Knockout of the mouse glutamate cysteine ligase catalytic subunit (gclc) gene: Embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem Biophys Res Commun. 2000;279(2):324-329.[DOI]
-
211. Kim SW, Kim Y, Kim SE, An JY. Ferroptosis-related genes in neurodevelopment and central nervous system. Biology. 2021;10(1):35.[DOI]
-
213. Tschuck J, Padmanabhan Nair V, Galhoz A, Zaratiegui C, Tai HM, Ciceri G, et al. Suppression of ferroptosis by vitamin A or radical-trapping antioxidants is essential for neuronal development. Nat Commun. 2024;15:7611.[DOI]
-
217. Tu C, Tan CW, Monkman J, Almeida AC, Antonio-Carreon G, Omer N, et al. Spatial multi-omics characterization of neuroblastoma reveals ferroptosis-associated metabolic features in high-risk tumors. Genome Med. 2026;18(1):35.[DOI]
-
219. Singh S, Mohapatra I, Barik D, Zheng H, Kim S, Sharma M, et al. Harnessing ferroptosis to transform glioblastoma therapy and surmount treatment resistance. Cell Death Discov. 2025;11:448.[DOI]
-
220. Mashayekhi S, Majedi H, Dehpour AR, Dehghan S, Jafarian M, Hadjighassem M, et al. Ferroptosis as a therapeutic target in glioblastoma: Mechanisms and emerging strategies. Mol Ther Nucleic Acids. 2025;36(3):102649.[DOI]
-
221. Koeken I, Walravens M, Fernández-Acosta R, Van Hoyweghen R, Vintea I, Kong Y, et al. Dual lipid modulation overcomes ferroptosis resistance in high-risk neuroblastoma. Cell Death Differ. 2026;33(5):903-913.[DOI]
-
225. Gohil K, Oommen S, Quach HT, Vasu VT, Aung HH, Schock B, et al. Mice lacking α-tocopherol transfer protein gene have severe α-tocopherol deficiency in multiple regions of the central nervous system. Brain Res. 2008;1201:167-176.[DOI]
-
226. Woo MS, Therriault J, Ali Hosseini S, Wang YT, Macedo AC, Rahmouni N, et al. Glia inflammation and cell death pathways drive disease progression in preclinical and early AD. EMBO Mol Med. 2025;17(11):3064-3079.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Winneberger J, Raich L, Potthast T, Woo MS. Non-neuronal ferroptosis in the central nervous system. Ferroptosis Oxid Stress. 2026;2:202609. https://doi.org/10.70401/fos.2026.0030
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Oligodendrocyte Lineages
- 3. Brain Endothelial Cells
- 4. Astrocytes
- 5. Microglia
- 6. CNS Infiltrating T Cells
- 7. Physiological Role of Ferroptosis
- 8. Conclusion
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Winneberger J, Raich L, Potthast T, Woo MS. Non-neuronal ferroptosis in the central nervous system. Ferroptosis Oxid Stress. 2026;2:202609. https://doi.org/10.70401/fos.2026.0030
copy
Share Link
copy