GPX4 is not required for the thermogenesis function of brown adipose tissue in mice
Yifan Zhang
1
,
Cheng Li
1
,
Yu Liu
1
,
Wei Wang
1
,
Wanling You
1
,
Zhenyu Ju
1,*
,
Fudi Wang
2,3,*

,
Qian Hu
1,*
*Correspondence to:
Zhenyu Ju, Key Laboratory of Regenerative Medicine of Ministry of Education, Institute of Aging and Regenerative Medicine, Department of Developmental & Regenerative Medicine, College of Life Science and Technology, Jinan University, Guangzhou 510632, Guangdong, China.
E-mail: zhenyuju2016@jnu.edu.cn
Fudi Wang, The Second Affiliated Hospital, School of Public Health, State Key Laboratory of Experimental Hematology, Zhejiang University School of Medicine, Hangzhou 310058, Zhejiang, China. E-mail: fwang@zju.edu.cn
Qian Hu, Key Laboratory of Regenerative Medicine of Ministry of Education, Institute of Aging and Regenerative Medicine, Department of Developmental & Regenerative Medicine, College of Life Science and Technology, Jinan University, Guangzhou 510632, Guangdong, China. E-mail: huqian@jnu.edu.cn
Fudi Wang, The Second Affiliated Hospital, School of Public Health, State Key Laboratory of Experimental Hematology, Zhejiang University School of Medicine, Hangzhou 310058, Zhejiang, China. E-mail: fwang@zju.edu.cn
Qian Hu, Key Laboratory of Regenerative Medicine of Ministry of Education, Institute of Aging and Regenerative Medicine, Department of Developmental & Regenerative Medicine, College of Life Science and Technology, Jinan University, Guangzhou 510632, Guangdong, China. E-mail: huqian@jnu.edu.cn
Ferroptosis Oxid Stress. 2026;2:202616. 10.70401/fos.2026.0033
Received: April 14, 2026Accepted: June 18, 2026Published: June 18, 2026
Abstract
Aims: Brown adipose tissue (BAT) relies heavily on mitochondrial activity and reactive oxygen species homeostasis to regulate thermogenesis and metabolic balance. However, the specific role of glutathione peroxidase 4 (GPX4), a critical antioxidant enzyme and central regulator of ferroptosis, in BAT remains unclear. This study aims to investigate the necessity of GPX4 for the functional integrity and thermogenic capacity of BAT.
Methods: Initially, we employed pharmacological inhibition of GPX4 in vitro using differentiated brown adipocytes. To investigate its role in vivo, we generated a BAT-specific Gpx4 knockout mouse model. The physiological and metabolic impacts of GPX4 deficiency were evaluated across three different conditions: cold exposure, high-fat diet, and vitamin E-deficient diet. Comprehensive evaluations were conducted using metabolic, histological, ultrastructural, and transcriptomic (RNA-seq) analyses.
Results: In vitro, pharmacological inhibition of GPX4 induced ferroptosis in differentiated brown adipocytes, suggesting its potential regulatory role. Strikingly, in vivo histological, ultrastructural, and metabolic analyses indicated that the genetic deletion of GPX4 does not impair BAT morphology or thermogenic function under any of the tested conditions. Consistent with these physiological findings, RNA-seq revealed that GPX4 deficiency did not significantly alter the expression of genes associated with ferroptosis or thermogenic pathways.
Conclusion: Although pharmacological inhibition of GPX4 triggers ferroptosis in brown adipocytes in vitro, GPX4 is not essential for maintaining the morphological integrity and thermogenic capacity of BAT in vivo under the specific experimental conditions tested.
Keywords
Glutathione peroxidase 4, thermogenesis, brown adipose tissue
1. Introduction
Brown adipose tissue (BAT) efficiently mobilizes stored energy through non-shivering thermogenesis, playing a central regulatory role in mammalian thermoregulation, energy expenditure, and metabolic homeostasis[1-3]. Recent studies highlight the thermogenic pathway as a critical therapeutic target for obesity and metabolic disorders, including insulin resistance and dysregulated glucose/lipid metabolism[4-8]. Thermogenesis is accompanied by metabolic reprogramming. Cold exposure enhances mitochondrial reactive oxygen species (ROS) production in brown adipocytes, which governs thermogenesis and energy dissipation[9]. Concurrently, the abundance of polyunsaturated fatty acid-containing phosphatidylethanolamine, which is one of the crucial lipids that undergo peroxidation and result in ferroptosis[10,11], significantly increases during thermogenesis[12].
Ferroptosis is a type of regulated cell death caused by the accumulation of lipid peroxidation[13]. It has been highlighted in various physiological and pathological conditions, including tissue ischemia/reperfusion, neurodegeneration, inflammation, kidney injury, and cancer[14,15]. Selenoprotein glutathione peroxidase 4 (GPX4) serves as a master suppressor of ferroptosis. Through its glutathione-dependent catalytic activity, GPX4 specifically reduces lipid peroxides (e.g., lipid hydroperoxides) in cellular membranes to non-toxic lipid alcohols, thereby directly blocking lipid radical chain reactions and maintaining membrane lipid integrity to prevent ferroptosis[16]. GPX4 is indispensable for murine embryonic development and tissue/organ function. Genetic ablation of GPX4 induces embryonic lethality in mice[17]. Tissue specific GPX4 deficiency triggers extensive lipid peroxidation and ferroptosis in kidneys, liver, and hematopoietic systems, leading to organ dysfunction[18-20]. GPX4 also plays a pivotal role in white adipose tissue (WAT), with studies demonstrating that white adipocyte development is critically dependent on GPX4 expression. Intriguingly, GPX4 deletion in WAT causes metabolic dysregulation and systemic low-grade inflammation through mechanisms independent of ferroptosis[21]. Recent work revealed that ferroptosis signaling is involved in the expansion of adipose tissue and promotes adaptive thermogenesis in beige adipocytes in a high-fat diet (HFD) mouse model[22]. Furthermore, ferroptosis has been implicated in adipose tissue wasting during cancer cachexia[23]. However, whether GPX4 deletion results in dysfunction of BAT via ferroptosis, as has been suggested in WAT remains unexplored.
In this study, we demonstrated that pharmacological inhibition of GPX4 induces ferroptosis in brown adipocytes in vitro. To further investigate its physiological relevance, we generated BAT-specific Gpx4 knockout mice and systematically evaluated their metabolic phenotypes under normal diet feeding, HFD feeding, and vitamin E (VE)-depleted dietary challenges. Notably, GPX4 deficiency in BAT exhibited no significant effects on thermogenic capacity or blood glucose homeostasis. RNA sequencing (RNA-seq) analysis further revealed no substantial alterations in ferroptosis-related pathways or thermogenic/metabolic gene signatures upon GPX4 deletion. Collectively, these findings highlight that GPX4 is not required for BAT functionality under the specific conditions tested.
2. Materials and Methods
2.1 Mice
Ucp1-CreERT2 mice and Gpx4flox/flox mice were a kind gift from Professor Zhinan Yin and Professor Fudi Wang, respectively. Gpx4flox/flox mice were crossed with Ucp1-CreERT2 mice to generate the Gpx4flox/flox Ucp1-CreERT2 mice. For Gpx4 deletion, Gpx4flox/flox Ucp1-CreERT2 mice were intraperitoneally injected with 100 mg/kg tamoxifen (TMX) (Sigma, St. Louis, USA) for three consecutive days. Mice were fed a fixed formulation diet without VE (Beijing HFK BioScience Company, Beijing, China) for one month in the VE depletion experiments. Mice were fed an HFD (60% kcal% fat, Research Diets, New Brunswick, USA) for 16 weeks in the HFD experiments. Except that steady state analyses were performed in male mice, other conditions, including the cold exposure, HFD, and VE-deficient diet challenges were conducted in female mice. All experimental protocols were approved by the Animal Care and Ethics Committee of Jinan University.
Mice were individually housed in pre-chilled cages without bedding and placed in a cold chamber maintained at 4-6 °C. Throughout the experiment, animals were allowed free access to pre-cooled food and water. Core body temperature was monitored at specified time points using a portable digital thermometer and rectal thermocouple probe (HICHANCE, Beijing, China).
2.2 Glucose tolerance test (GTT)
Mice were fasted overnight before receiving an intraperitoneal injection of glucose (1 g/kg). Blood was collected from the tail vein at 0, 15, 30, 60, 90, and 120 min, and blood glucose concentrations were measured using an Accu-Check Aviva glucometer (Roche, Basel, Switzerland).
2.3 Insulin tolerance test (ITT)
Mice were fasted overnight before receiving an intraperitoneal injection of insulin (1 U/kg) (MedChemExpress, New Jersey, USA) by intraperitoneal injection. Blood was collected from the tail vein at 0, 15, 30, 60, 90, 120, and 150 min, and blood glucose concentrations were measured using an Accu-Check Aviva glucometer (Roche).
2.4 Malondialdehyde (MDA) content measurement
Tissue MDA content was quantified using a thiobarbituric acid reactive substances (TBARS) assay (Solarbio, Beijing, China). Tissue samples were homogenized in MDA extraction reagent and centrifuged to collect the supernatant. Total protein concentration in the supernatant was determined using the PierceTM bicinchoninic acid (BCA) Protein Assay Kit (Thermo Fisher Scientific, Waltham, USA). The supernatant was then mixed with MDA detection reagent, incubated at 100 °C for 60 min, and centrifuged. Absorbance of the resulting supernatant was measured at 450 nm, 532 nm, and 600 nm using a Synergy H1 microplate reader (BioTek, Vermont, USA). MDA concentration was calculated as follows.
2.5 Serum VE test
Serum VE levels were measured using the CheKine VE Content Assay Kit (CheKine, Wuhan, KTB2400). Briefly, 0.1 mL of mouse serum was mixed with 0.9 mL Extraction Buffer, vortexed for 5 min, and centrifuged at 5,000 g for 10 min at 25 °C. The supernatant was collected for analysis. For each sample, 100 µL of supernatant was added to a microplate well, followed by Reagent I (20 µL) and either Reagent II or Reagent III (20 µL). After mixing and incubating at 25 °C for 5 min, 60 µL of Reagent IV was added. Absorbance was measured at 530 nm, and the VE concentration was calculated from ΔA values using the kit’s standard curve regression equation.
2.6 Isolation of brown adipose stromal vascular fraction (SVF) and in vitro differentiation
After isolation, the BAT tissue was minced into fine fragments using scissors. The fragments were then digested in phosphate-buffered saline (PBS) solution containing 0.2% bovine serum albumin, 1 mM MgCl2, 1.2 mM CaCl2, and 1 mg/ml collagenase type I at 37 °C for 1 hour to obtain the SVF. The SVF was seeded in high-glucose Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S), followed by overnight incubation for 24 hours. The medium was subsequently replaced with fresh culture medium, and this refreshment was repeated every two days until cells reached contact inhibition.
For adipogenic differentiation, the culture medium was switched to an induction cocktail containing 20 nM/mL insulin, 1 μM dexamethasone, 1 μM rosiglitazone, 1 nM T3, 0.5 mM 3-isobutyl-1-methylxanthine, and 125 μM indomethacin. After two days, the medium was replaced with DMEM supplemented with 10% FBS, 1% P/S, 5 μg/mL insulin, and 2 μM rosiglitazone, which was refreshed every other day until the cells were harvested.
2.7 Western blot
The tissues were lysed using radioimmunoprecipitation assay (Beyotime, Shanghai, China) lysis buffer supplemented with protease and phosphatase inhibitor cocktail. After centrifugation, the supernatant was collected, and the protein concentration was measured using a Pierce BCA Protein Assay Kit (Thermo). Then, the proteins were run on standard 12% SDS-PAGE gels, transferred onto polyvinylidene fluoride membranes (Bio-Rad, California, USA), and blocked with TBST containing 5% milk for 1 h at room temperature (RT). The membranes were then incubated with primary antibodies overnight at 4 °C, washed with TBST, incubated with secondary antibodies for 1 h at RT, and detected on an Amersham Imager 600 System (GE, Boston, USA). Primary antibodies against GPX4 (Abcam, ab125066, Cambridge, UK) and β-actin (abclonal, Wuhan, AC026) and secondary antibodies against rabbit IgG (CST, 7074, Massachusetts, USA) and mouse IgG (CST, 7076, Massachusetts, USA) were used. The data were analyzed using ImageJ.
2.8 Cell viability assay
The SVF isolated from BAT was seeded in 24-well plates and differentiated into mature brown adipocytes using the protocol described above. Since rosiglitazone inhibits ACSL4, it was excluded from the culture medium during treatment with 2 μM RSL3 (Selleck, Houston, USA). After 24 hours, the medium was removed, and cells were washed once with PBS. Hoechst 33342 (Sangon Biotech, Shanghai, China) and propidium iodide (Sangon Biotech) were then used to stain total and dead cells, respectively, followed by incubation at 37 °C for 15 minutes. The cell images were captured by ImageXpress Micro Confocal (Molecular Devices, San Jose, CA, USA), and cell viability was measured with MetaXpress version 6.6.1.42 (Molecular Devices). Cell viability was assessed using a fluorescence microscope.
2.9 RNA-seq
Total RNA was extracted using Trizol (Takara, Tokyo, Japan). The mRNA was enriched and reverse transcribed into cDNA, followed by library construction through end repair, A-tailing, adapter ligation, size selection, amplification, and purification. The quality of the library was verified using Qubit quantification, quantitative polymerase chain reaction (qPCR), and Agilent 2100 bioanalyzer for size distribution analysis. The final library preparations were sequenced on an Illumina NovaSeq platform, which employs bridge amplification and reversible terminator chemistry for high-throughput sequencing (Novogene, Shenzhen, China). The sequencing data met standard quality-control criteria, with an average depth of 42.62 million reads per sample, a mapping rate of 91.79%, and 4 biological replicates in each group, ensuring the reliability of downstream analyses. Bioinformatics analysis identified differentially expressed genes and performed gene set enrichment analysis (GSEA) for functional enrichment.
2.10 Isolation of RNA and qPCR
Trizol (Takara, Tokyo, Japan) was used to collect RNA. RNA concentration was measured by nanodrop (Thermo Fisher Scientific). cDNA was synthesized using HiScript III RT SuperMix for qPCR (Vazyme, Nanjing, China). qPCR was performed using SYBR qPCR Master Mix (Vazyme, Nanjing, China), and a QuantStudio 6 Flex system (Applied Biosystems, California, USA). The amount of target mRNA was normalized to that of 18s mRNA. The gene expression quantities were determined according to the relative Ct.
GPX4 Primers (forward primer: GATGGAGCCCATTCCTGAACC; and reverse primer: CCCTGTACTTATCCAGGCAGA), UCP1 Primers (forward primer: TGGAAAGGGACGACCCCTAA; and reverse primer: CAGGAGTGTGGTGCAAAACC), AIFM2 Primers (forward primer: GCGACCTTCAAGGACAACTTCC; and reverse primer: GCCAGGATAAGATGTGAGAAGGG), DHODH Primers (forward primer: TGAGGAGCCTACAGGGAAAGAC; and reverse primer: ACGCTGGCAATGTCCTCCTTGT), GCH1 Primers (forward primer: AGCAAGTCCTTGGTCTCAGTAAAC; and reverse primer: ACCGCAATCTGTTTGGTGAGGC), SLC7A11 Primers (forward primer: TGGGTGGAACTGCTCGTAAT; and reverse primer: AGGATGTAGCGTCCAAATGC), 18s Primers (forward primer: ATTGGAGCTGGAATTACCGC; and reverse primer: CGGCTACCACATCCAAGGAA).
2.11 Histology
Following dissection, tissues were immediately fixed in 4% formaldehyde solution, paraffin-embedded, and sectioned. The sections were stained with hematoxylin and eosin (H&E) for routine histological examination under a light microscope.
2.12 Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining
TUNEL staining was performed on frozen sections according to the manufacturer’s protocol with minor modifications. Briefly, sections were permeabilized with 0.1% Triton X-100/0.1% sodium citrate, treated with proteinase K (20 μg/mL), and equilibrated in 1× Equilibrium Buffer. Samples were then incubated with TUNEL reaction mixture containing FITC-12-dUTP and recombinant TdT enzyme at 37 °C for 60 min in the dark. Negative control sections were processed in parallel without TdT enzyme. After washing, nuclei were counterstained with DAPI, and images were acquired by fluorescence microscopy.
2.13 Transmission electron microscopy sample preparation and observation
Tissue samples were cut into approximately 1 mm3 pieces and immediately fixed in electron microscopy fixative at 4 °C. After washing with 0.1 M phosphate buffer (pH 7.4), samples were post-fixed with 1% osmium tetroxide, dehydrated in a graded ethanol series, and acetone, and embedded in 812 resin. Ultrathin sections (70 nm) were cut, mounted on 200-mesh copper grids, stained with uranyl acetate and lead citrate, and examined using a transmission electron microscope (HITACHI HT7800).
2.14 Statistics
Sample sizes (specified in figure legends) included ≥ 3 biological replicates. No data exclusions impacting conclusions were performed. Age-, gender-, and genotype-matched animals were randomized into experimental groups. Statistical analyses (GraphPad Prism 8) are presented as means ± standard deviation. One-way or two-way analysis of variance was applied for single- or dual-factor comparisons, respectively, followed by Tukey’s or Bonferroni post-hoc tests for multiple comparisons. For comparisons between two groups, an unpaired Student’s t-test was also performed. Significance: ns (no significant), *p < 0.05, **p < 0.01, ***p < 0.001.
3. Results
3.1 Inhibition of GPX4 induces ferroptosis in brown adipocytes in vitro
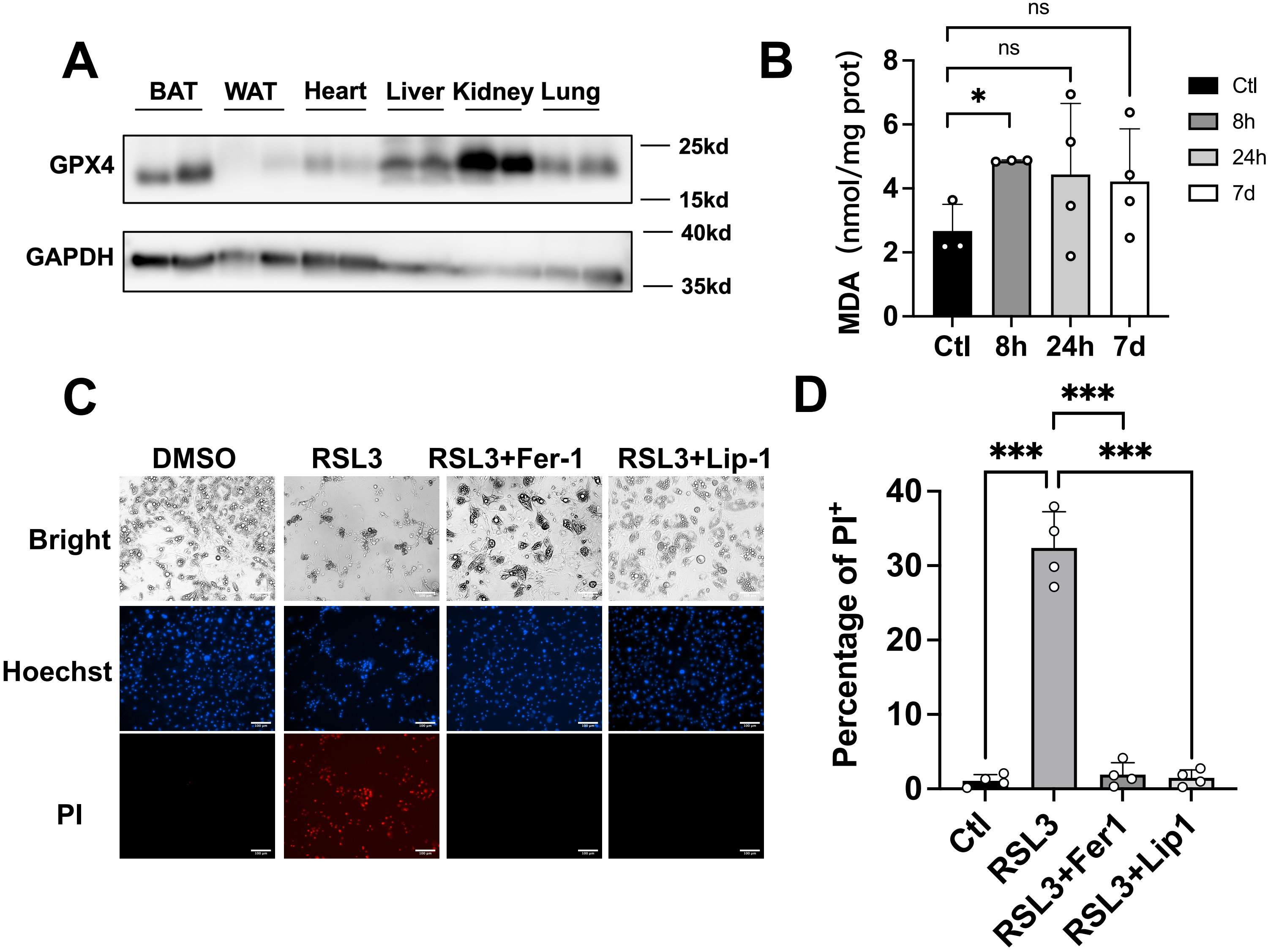
Given the critical role of GPX4 in tissue homeostasis, we conducted a comprehensive analysis of its protein expression across multiple organs. Western blot results revealed that GPX4 exhibits the highest expression in kidneys, with BAT showing significantly higher levels compared to subcutaneous inguinal white adipose tissue (iWAT) (Figure 1A). To investigate the level of lipid ROS under cold exposure, we measured MDA, a terminal product of lipid peroxidation, in BAT following cold exposure at 4 °C. This acute cold challenge triggered a substantial increase in MDA accumulation (Figure 1B), indicative of enhanced lipid peroxidation. To validate the functional significance of GPX4 in brown adipocytes, SVF cells isolated from wild-type mouse BAT were differentiated into mature brown adipocytes in vitro. Pharmacological inhibition of GPX4 with RSL3 for 24 hours triggered robust ferroptosis, which was effectively rescued by co-treatment with ferroptosis inhibitors ferrostatin-1 or liproxstatin-1 (Figure 1C,D).
{kind=link}
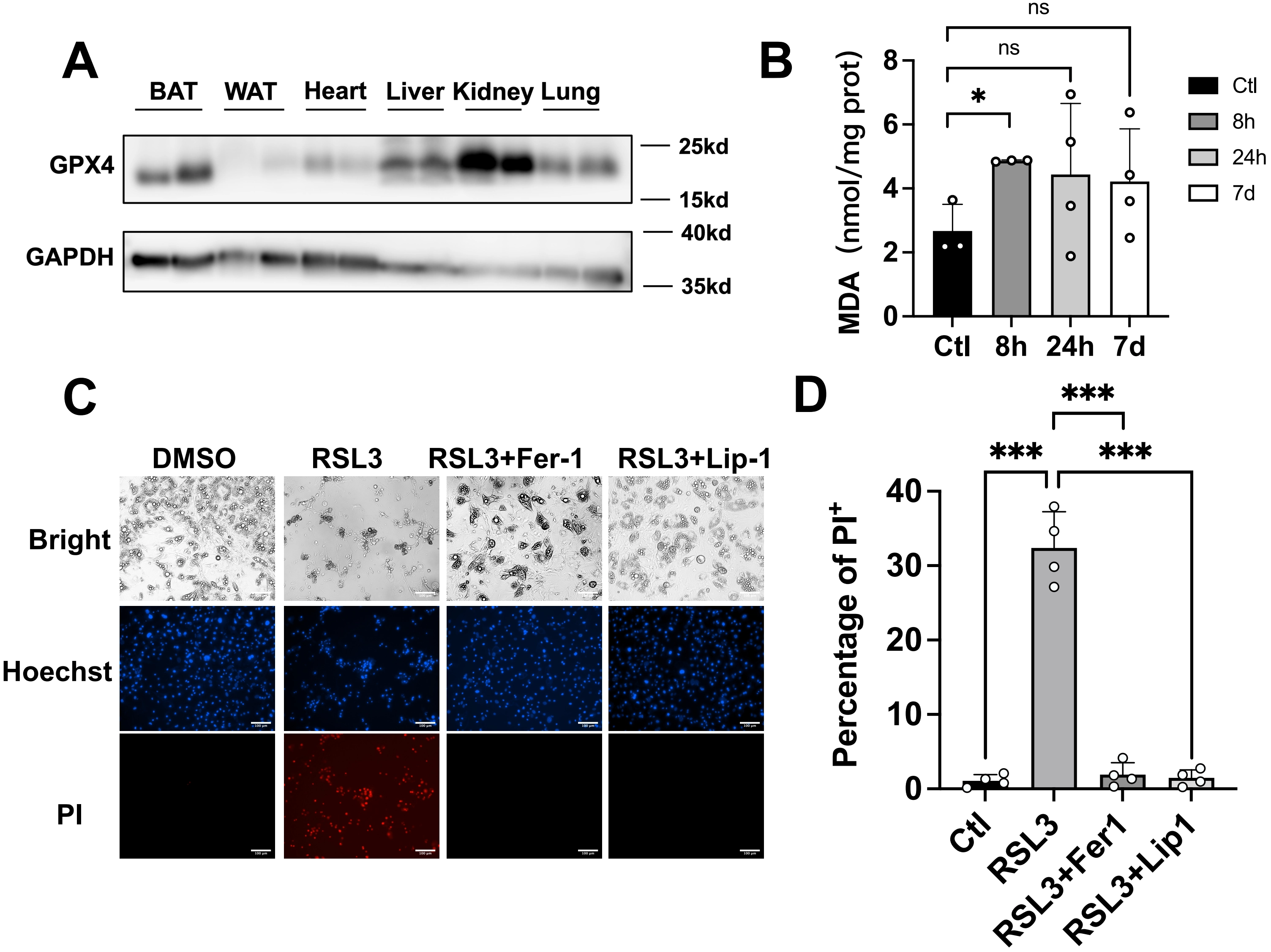
Figure 1. Inhibition of GPX4 induces ferroptosis in brown adipocytes in vitro. (A) Representative immunoblot showing GPX4 protein levels across murine tissues (n = 2); (B) MDA levels in BAT from mice exposed to cold for the indicated times were determined by TBARS assay (n = 3-4 mice per group); (C-D) The representative images of mature brown adipose cells treated with RSL3 (2 μM) and Ferrostatin-1 (2 μM), Liproxstatin-1 (1 μM) for 24 h following Hoechst and PI staining. Scale bar: 100 μm; (D) The statistical data of cell viability. The values were presented as mean ± SD; ns, no significance, *p < 0.05, ***p < 0.001. GPX4: glutathione peroxidase 4; MDA: malondialdehyde; BAT: brown adipose tissue; TBARS: thiobarbituric acid reactive substances; RSL3: RAS-selective lethal 3; SD: standard deviation.
3.2 Gpx4 knockout has no effect on BAT function in mice under cold exposure
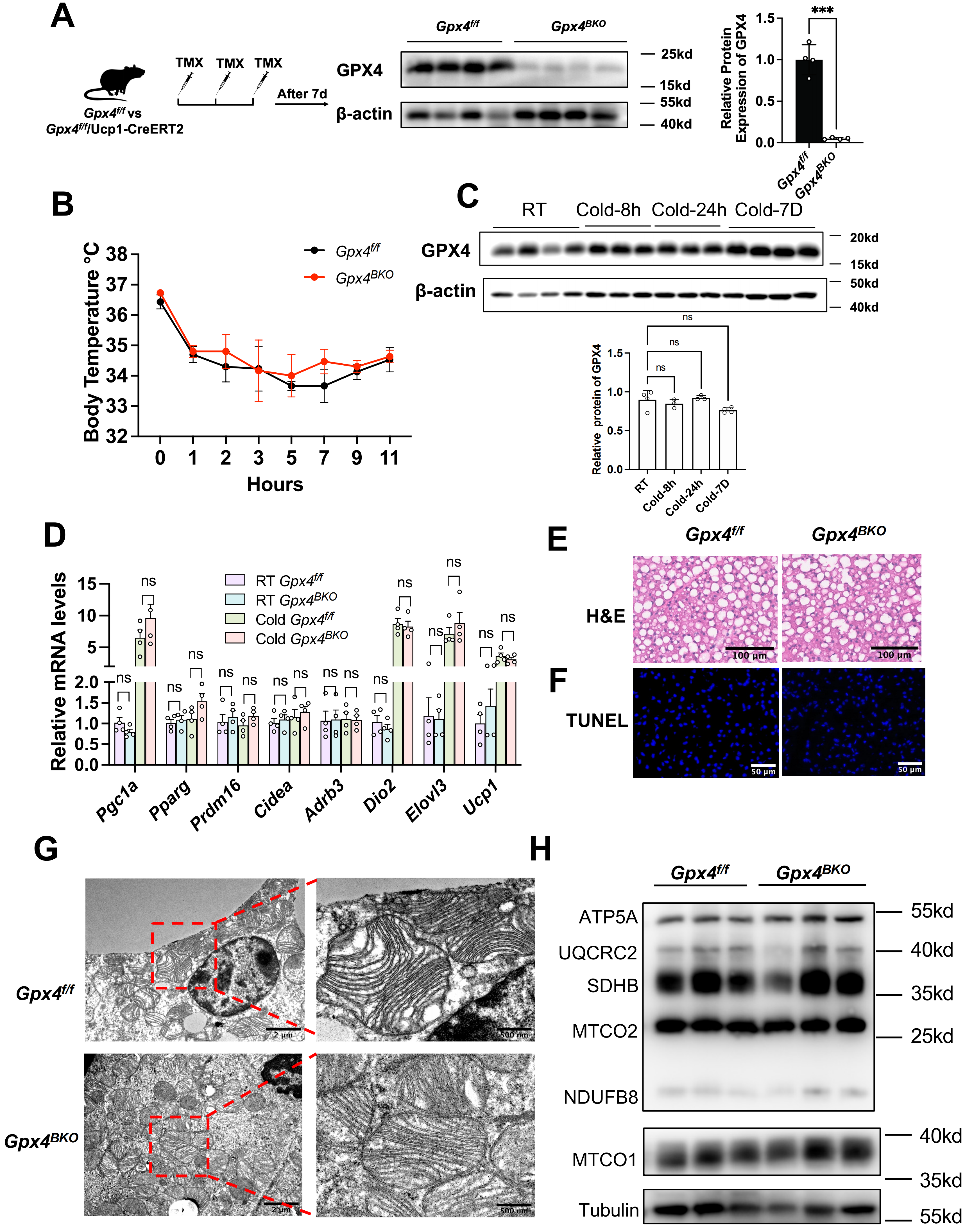
To delineate the in vivo functional relevance of GPX4 in BAT, we generated brown adipose-specific knockout mice by crossing Gpx4f/f (Gpx4flox/flox) with Ucp1-CreERT2 strains, obtaining Gpx4BKO (BAT-specific knockout) mice. Inducible Gpx4 ablation was achieved via sequential TMX administration (Figure 2A). Strikingly, Gpx4BKO mice exhibited comparable body weight, BAT mass index, and blood glucose levels relative to Gpx4f/f (Figure S1A,B,C). Notably, GPX4 deficiency did not alter the protein levels of ferroptosis markers ACSL4 and TFR1 in BAT tissues (Figure S1D,E).
{kind=link}
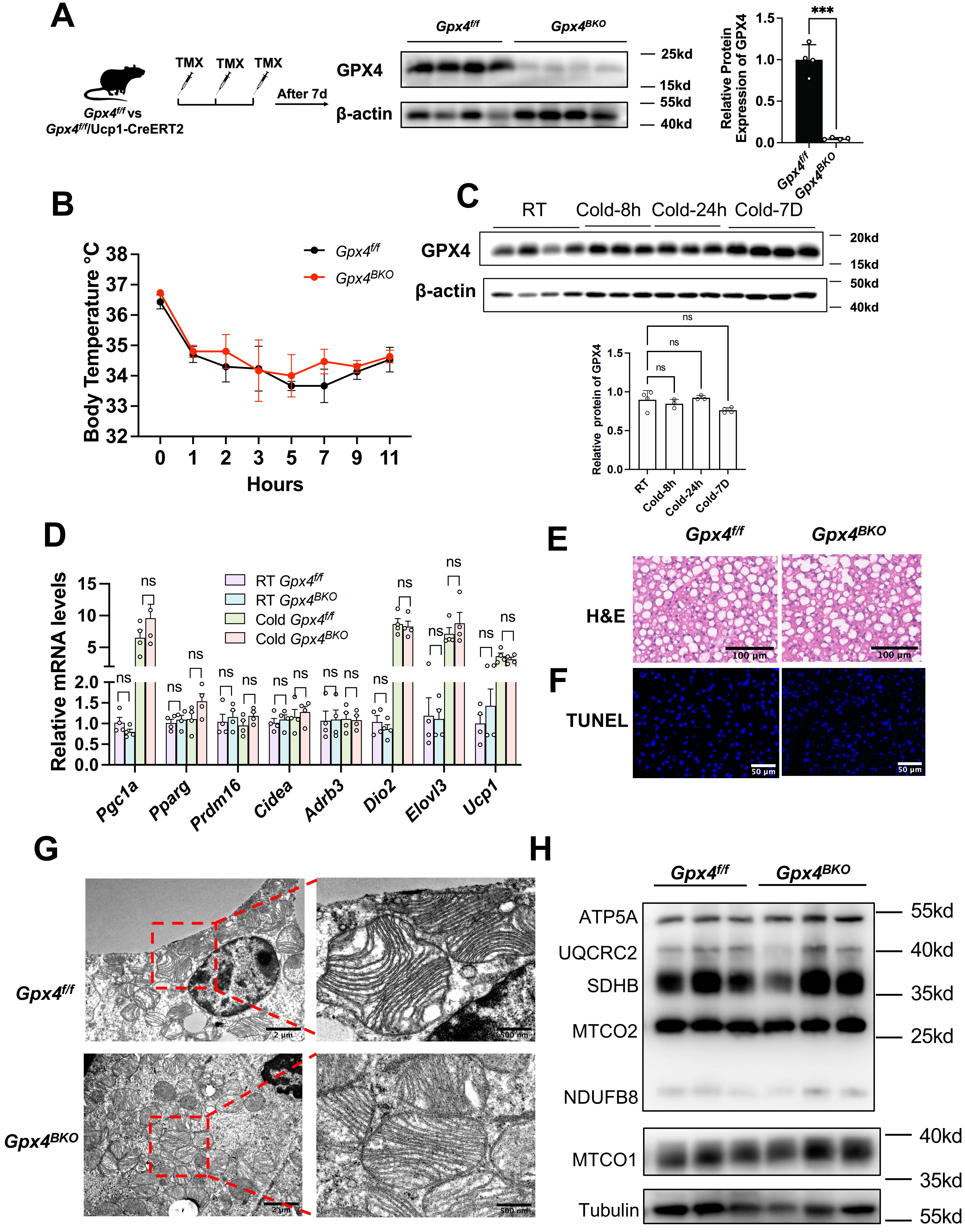
Figure 2. Gpx4 knockout has no effect BAT function in mice under cold exposure. (A) Left panel: The schematic of GPX4 knockout in mice. Male Gpx4f/f mice or Gpx4BKO mice (aged 8 weeks) were injected with TMX (100 mg/kg) three times continuous. After 7 days, the mice were sacrificed. Middle panel: Gpx4 deletion in the BAT of Gpx4BKO was verified by Western Blot. Right panel: The relative GPX4 levels were normalized to β-actin; (B) Rectal temperature measurement after cold exposure for 11 h (n = 4); (C) Up panel: Immunoblot analysis of GPX4 in BAT from Gpx4f/f and Gpx4BKO mice after cold exposure for different time. Down panel: Quantification of protein expression levels normalized to β-actin; (D) QPCR analysis of thermogenic gene expression in BAT from 8-week-old Gpx4f/f and Gpx4BKO mice exposed to 4 °C for 8 h, the mRNA levels were normalized to 18 s; (E) H&E staining of BAT in mice after cold stimulation. Scale bars, 100 μm; (F) TUNEL staining of BAT in mice after cold stimulation. Scale bars, 50 μm; (G) TEM analysis of BAT ultrastructure in mice after cold stimulation. Scale bars, 500 nm; (H) Immunoblot analysis of mitochondrial respiratory chain complexes in BAT from Gpx4f/f and Gpx4BKO mice after cold exposure. The values were presented as mean ± SD; ns, no significance, ***p < 0. 001. GPX4: glutathione peroxidase 4; BAT: brown adipose tissue; TMX: tamoxifen; QPCR: quantitative polymerase chain reaction; H&E: hematoxylin and eosin; TUNEL: Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling; TEM: transmission electron microscopy; SD: standard deviation.
Given the pivotal role of BAT in thermogenesis, we subjected mice to acute cold exposure. Notably, GPX4 ablation failed to impair cold-induced adaptive thermogenesis, as evidenced by unchanged core body temperature trajectories (Figure 2B). Moreover, cold exposure did not alter GPX4 protein expression levels in BAT (Figure 2C). To further evaluate the consequence of GPX4 deficiency under cold challenge, BAT was harvested after 8 h of exposure for biochemical and molecular analyses. QPCR analysis showed that GPX4 deficiency did not affect the expression of thermogenic genes (Figure 2D). H&E staining revealed no obvious alterations in BAT architecture following GPX4 deletion, and TUNEL staining showed no detectable cell death in the BAT of either Gpx4BKO or Gpx4f/f mice (Figure 2E,F). Transmission electron microscopy also showed no apparent abnormalities in mitochondrial ultrastructure in GPX4-deficient BAT (Figure 2G). Consistently, the protein levels of mitochondrial respiratory chain components, including MTCO1, NDUFB8, MTCO2, SDHB, UQCRC2, and ATP5A, remained unchanged (Figure 2H).
Interestingly, MDA levels were not significantly different between GPX4-deficient and control BAT, indicating that GPX4 loss did not exacerbate lipid peroxidation in this setting (Figure S1F). Consistently, the transcription levels of key lipid peroxidation regulators, including Aifm2 (FSP1 coding gene), Dhodh, Gch1, and Slc7a11, in BAT after 8 h of cold exposure showed no significant changes in their expression profiles (Figure S1G), arguing against compensatory transcriptional activation of canonical lipid peroxidation defense pathways.
Collectively, these findings indicate that GPX4 deficiency does not cause overt alterations in BAT morphology, mitochondrial ultrastructure, thermogenic gene expression, or lipid peroxidation under cold exposure.
3.3 BAT thermogenesis in HFD-induced obesity is unaffected by GPX4 ablation
Maintaining metabolic homeostasis through excess energy dissipation constitutes a fundamental function of BAT, with elevated ROS levels observed in obese individuals[24]. To investigate GPX4’s role in BAT metabolic regulation under diet-induced obesity, we subjected mice to HFD feeding for a longitudinal cohort following TMX-induced GPX4 ablation (Figure S2A,B). BAT-specific GPX4 deficiency did not alter body weight progression (Figure 3A) or the mass of BAT, iWAT, and liver (Figure 3B,C).
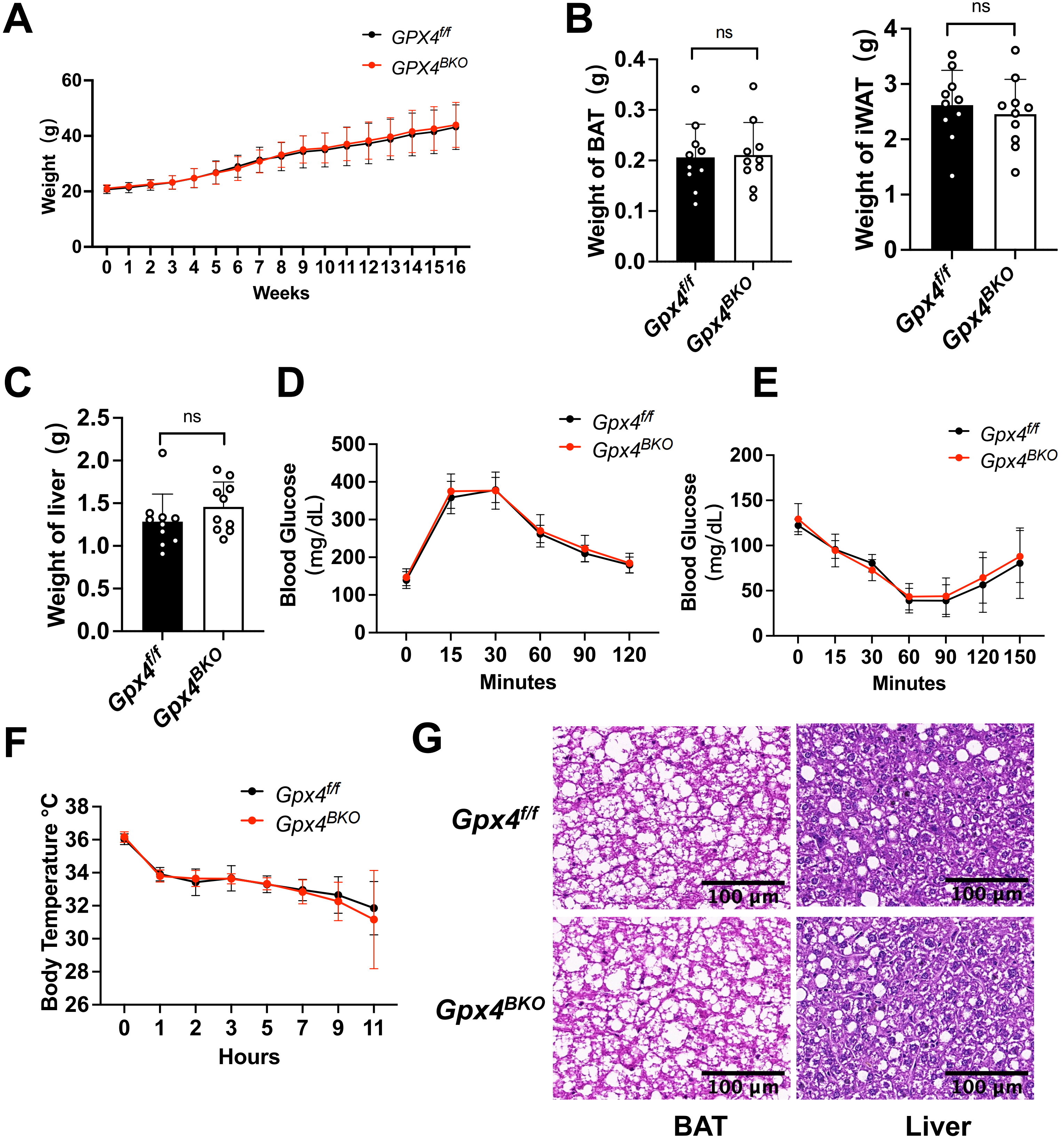{kind=link}
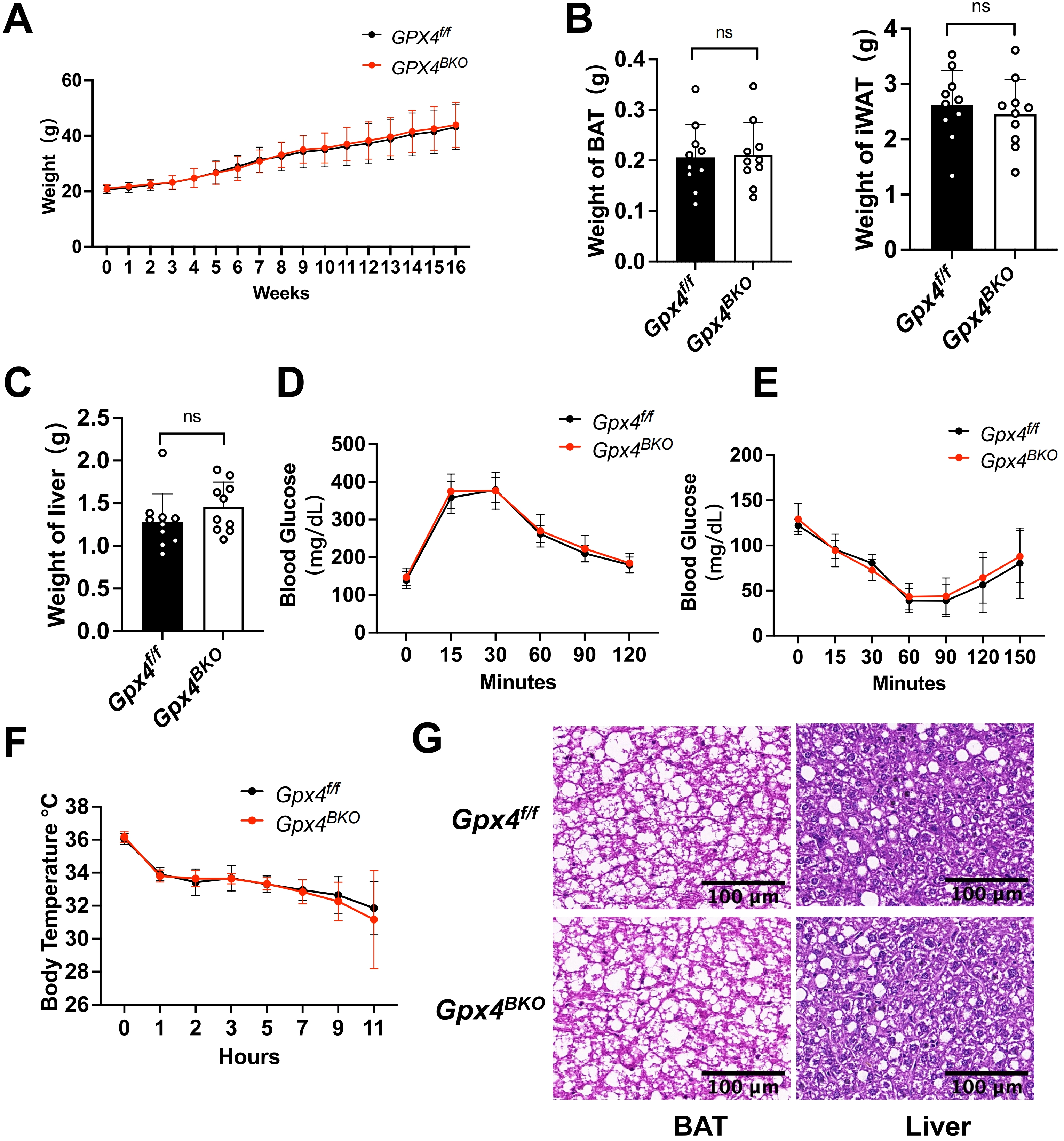
Figure 3. HFD-induced obesity unaffected by GPX4 ablation in BAT thermogenesis. Gpx4f/f and Gpx4BKO mice (aged 8 weeks) were treated with an HFD for 16 weeks. (A) Body weight was recorded each week (n = 10); (B-C) iWAT weight, BAT mass and liver weight of Gpx4BKO mice compared with Gpx4f/f mice (n = 10); (D-E) (D) Intraperitoneal glucose tolerance tests and (E) Insulin tolerance tests of high-fat diet (n = 10); (F) Rectal temperature measurement after cold exposure for 11 h (n = 10); (G) H&E-stained BAT and liver samples. Scale bars, 100 μm. The values were presented as mean ± SD, ns, no significance. HFD: high-fat diet; GPX4: glutathione peroxidase 4; BAT: brown adipose tissue; iWAT: inguinal white adipose tissue; H&E: hematoxylin and eosin; SD: standard deviation.
A glucose challenge test and ITT revealed comparable glucose tolerance (Figure 3D) and insulin sensitivity (Figure 3E) between Gpx4BKO and Gpx4f/f mice. Furthermore, GPX4 ablation failed to impair cold-induced thermoregulation in obese mice, as evidenced by unchanged core body temperature during cold exposure (Figure 3F). Histological analysis demonstrated no significant differences in adipocyte diameter within BAT or hepatic lipid droplet morphology (Figure 3G). Furthermore, western blot results demonstrated that GPX4 deficiency did not affect the protein levels of ferroptosis markers ACSL4 and TFR1 in mouse BAT tissues under HFD conditions (Figure S2C,D). Collectively, these data indicate that under HFD-induced obesity conditions, GPX4 is not required for BAT function.
3.4 Combined GPX4 and VE deficiency fails to compromise BAT thermogenic function in mice
Given the potential compensatory role of dietary VE in mitigating lipid peroxidation under GPX4 inhibition[20,25-27], we established a dual intervention model combining TMX induced GPX4 ablation (Figure S3A) with VE-deficient diet feeding. Measurement of serum VE confirmed that mice receiving the VE-deficient diet had significantly lower VE levels than mice fed a normal diet. (Figure S3B). However, the results revealed that 4 weeks VE deprivation in the diet did not alter the body weight, the mass of BAT, iWAT, and liver (Figure 4A,B,C,D) of the GPX4 deficient mice. Furthermore, neither GTT nor ITT (Figure 4E,F) was impacted by VE deprivation.
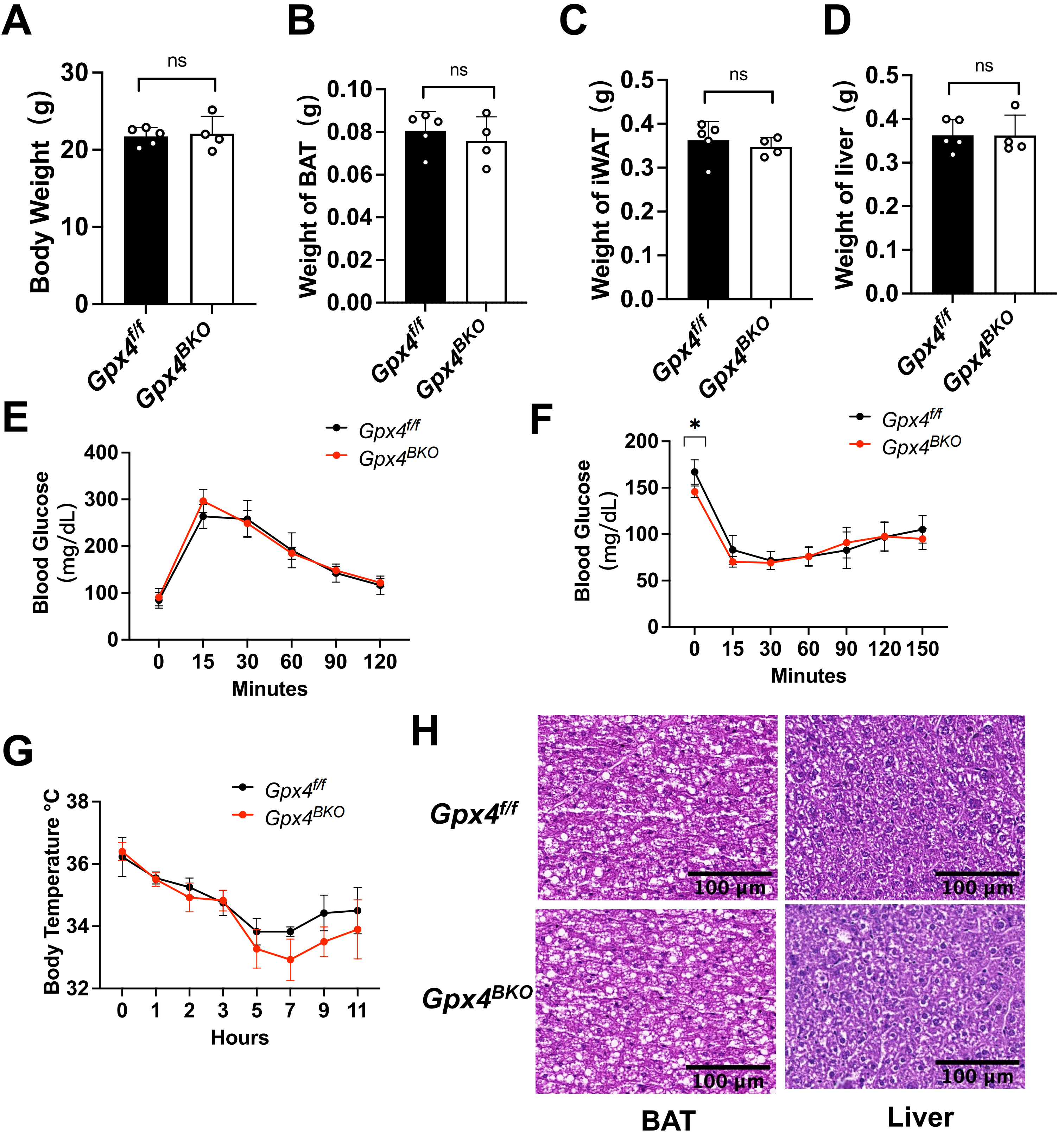{kind=link}
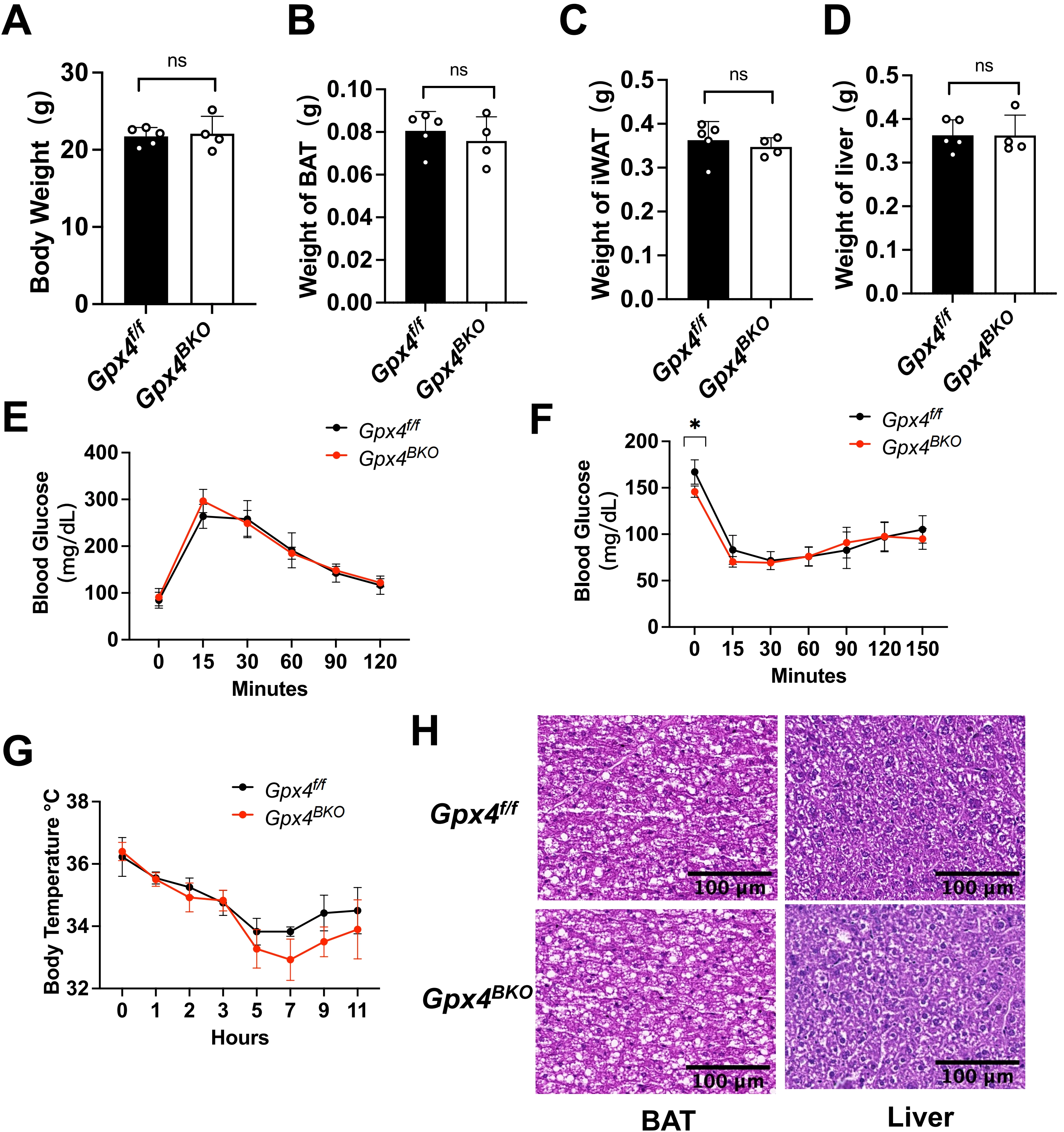
Figure 4. Combined GPX4 and vitamin E deficiency fails to compromise BAT thermogenic function in mice. Gpx4f/f and Gpx4BKO mice (aged 8 weeks) were treated with a vitamin E depletion diet for 4 weeks. (A-D) Body weight, iWAT weight, BAT mass and liver weight of Gpx4BKO mice compared with Gpx4f/f mice (n = 4-5); (E-F) (E) Intraperitoneal glucose tolerance tests and (F) Insulin tolerance tests (n = 4-5); (G) Rectal temperature measurement after cold exposure for 11 h (n = 4-5); (H) H&E-stained BAT and liver samples. Scale bars, 100 μm. The values were presented as mean ± SD, ns, no significance. GPX4: glutathione peroxidase 4; BAT: brown adipose tissue; iWAT: inguinal white adipose tissue; H&E: hematoxylin and eosin; SD: standard deviation.
Notably, Gpx4BKO mice maintained normal thermoregulatory capacity during cold challenge (Figure 4G). Histological quantification confirmed preserved BAT adipocyte diameter and hepatic architecture between Gpx4f/f mice and Gpx4BKO mice under a VE deprived diet (Figure 4H). Notably, even upon VE deprivation in the diet, GPX4 ablation still did not alter the levels of the ferroptosis indicators ACSL4 and TFR1 (Figure S3C,D). These findings demonstrate that under the combined conditions of GPX4 ablation and dietary VE deprivation, neither endogenous GPX4 nor dietary VE appears essential for maintaining the basal BAT functionality assessed in this study.
3.5 GPX4 deficiency does not affect the transcriptomic profile in BAT
To gain further insights into GPX4 function on BAT from global gene expression analysis, we performed RNA-seq on BAT samples from Gpx4f/f and Gpx4BKO mice under RT and cold-stimulated (Cold) conditions. Firstly, the genetic ablation of Gpx4 was confirmed (Figure S4A). Analysis of all the expressed genes by a principal component analysis plot showed that samples derived from cold-stimulated and RT groups fell into distinct groups, suggesting variability between groups (Figure 5A). However, Gpx4BKO and Gpx4f/f groups are clustered close, indicating no statistically significant differences between the groups. Differential gene expression analysis identified 1,562 significantly altered genes (|Log2FC| ≥ 1, adjusted p < 0.05) between cold-stimulated and RT conditions in Gpx4f/f mice, while 1,478 significantly altered genes (|Log2FC| ≥ 1, adjusted p < 0.05) between cold-stimulated and RT conditions were observed in Gpx4BKO mice. Remarkably, comparison between Gpx4BKO and Gpx4f/f groups revealed only 7 differentially expressed genes under cold stimulation and 3 differentially expressed genes under RT conditions (Figure 5B).
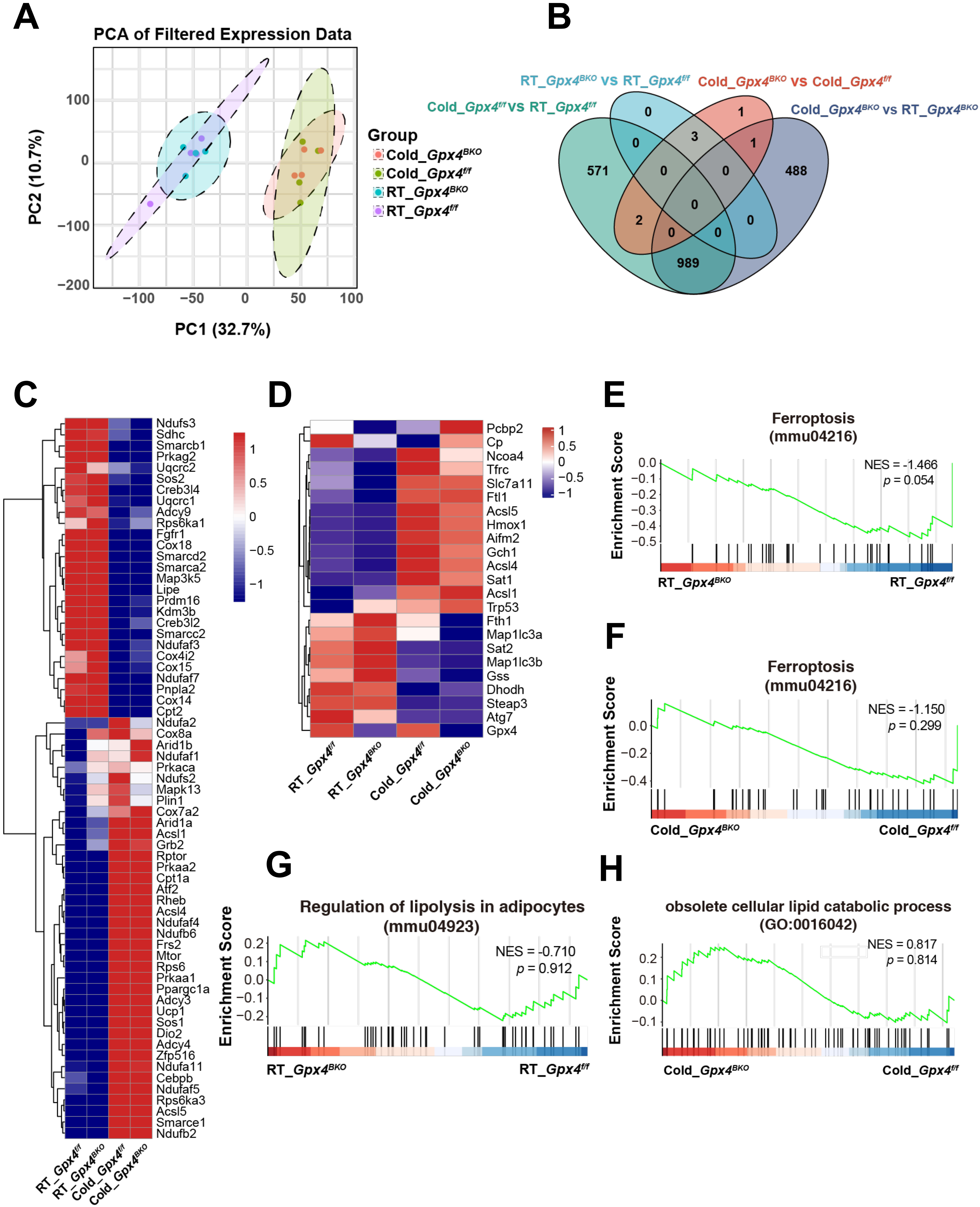{kind=link}
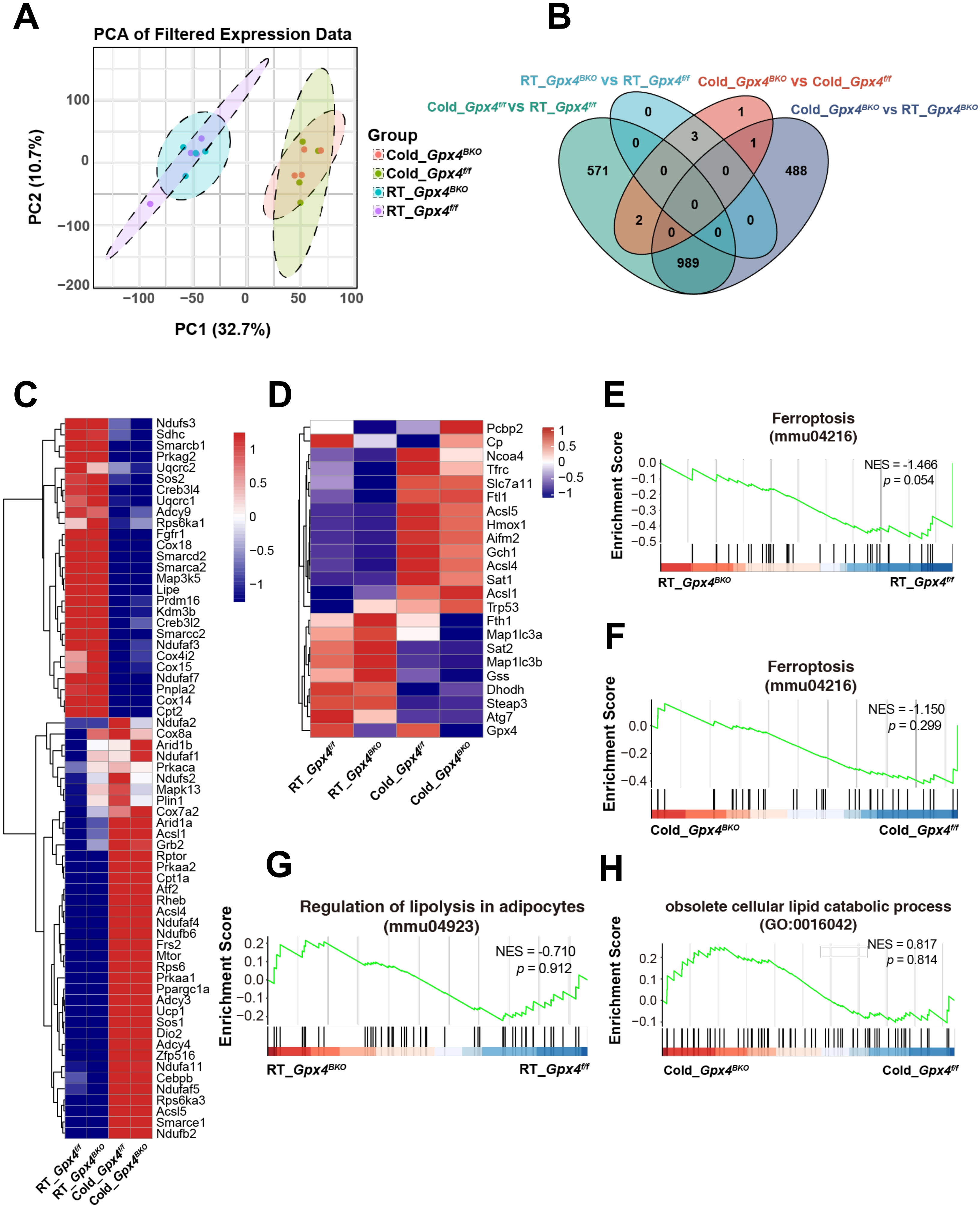
Figure 5. GPX4 deficiency does not affect the transcriptomic profile in BAT. (A) PCA plot based on all detected genes in RNA sequencing of BAT; (B) Venn diagram showing Differential Genes in BAT; (C) Heatmap of BAT thermogenic genes from the RNA-seq data; (D) Heatmap of BAT ferroptosis genes from the RNA-seq data; (E-F) GSEA of ferroptosis-related genes in BAT from the Gpx4f/f and Gpx4BKO mice BAT in room temperature or cold; (G-H) GSEA of lipolysis-related genes in BAT from the Gpx4f/f and Gpx4BKO mice BAT in room temperature or cold. GPX4: glutathione peroxidase 4; BAT: brown adipose tissue; PCA: principal component analysis; RNA-seq: RNA sequencing; GSEA: gene set enrichment analysis.
Further analysis of RNA-seq showed that the expression of thermogenesis-related genes, including Ucp1, Ppargc1a, Cebpb and Dio2, were significantly upregulated after cold-stimulation. However, the absence of Gpx4 did not affect the expression of thermogenic related genes (Figure 5C). Subsequent analysis of GPX4 deficiency on ferroptosis-associated gene networks revealed that GPX4 ablation did not significantly alter ferroptosis-related gene expression under either RT conditions or cold challenge. However, cold exposure markedly upregulated iron metabolism regulators (Tfrc, Ftl1) and Acsl4, and concomitant upregulation of ferroptosis-suppressive genes (e.g., Slc7a11, Aifm2, Gch1) was also observed (Figure 5D). qPCR analysis corroborated these findings, demonstrating an upregulation of the expression of Slc7a11 Gch1 and Aifm2 in BAT following acute (8 h), sustained (24 h), and prolonged (7 d) cold exposure (Figure S4B). Moreover, no significant genotype-dependent differences were observed in pathways related to oxidative phosphorylation, fatty acid oxidation, NRF2 signaling, or oxidative stress-associated metabolic regulation (Figure S4C,D,E,F,G).
GSEA further confirmed that there was no significant association between BAT-specific GPX4 deficiency and canonical ferroptosis pathways (Figure 5E,F). In addition, the loss of GPX4 had no significant effect on the lipolysis pathway in BAT (Figure 5G,H). These findings demonstrate that GPX4 ablation induces minimal alteration in differential gene expression profiles within BAT tissue.
4. Discussion
Ferroptosis has emerged as a research hotspot in recent years, with its key inhibitory protein GPX4 demonstrating critical roles in multiple tissues including the kidney[18], liver[19], and WAT[21] (affecting adipocyte differentiation). Our study focuses on GPX4’s functional significance in BAT, a metabolic thermogenic organ recognized as a therapeutic target for obesity and metabolic syndromes. While GPX4 inhibition induces ferroptosis in brown adipocytes in vitro, our in vivo data suggest that BAT’s metabolic and thermogenic functions are not overtly dependent on GPX4 under the specific challenges tested (cold exposure, HFD, and VE-deficient diet).
A notable finding of this study is the discrepancy between the in vitro and in vivo phenotypes. While cultured brown adipocytes were sensitive to GPX4 loss, BAT in vivo remained structurally and functionally preserved. This difference likely reflects the substantial gap between simplified cell culture conditions and the physiological BAT microenvironment. In vitro assays impose acute oxidative stress and limited antioxidant compensation, primarily revealing cell-autonomous vulnerability. In contrast, in vivo BAT is protected by systemic antioxidant supply, including dietary factors, as well as by vascularization, sympathetic innervation, endocrine regulation, extracellular matrix context, cellular heterogeneity, and compensatory lipid- and ROS-buffering pathways. In addition, standard cell culture is typically performed under supraphysiological oxygen conditions compared with native tissue, which may further influence oxidative stress, mitochondrial activity, and lipid peroxidation. Therefore, the in vitro findings are best interpreted as evidence of cell-intrinsic ferroptotic susceptibility rather than a full recapitulation of the in vivo phenotype.
Our findings further support the idea that adipose tissue possesses strong intrinsic resistance to ferroptosis. In contrast to the marked ferroptosis sensitivity reported in other organs, adipose depots appear relatively resilient to GPX4 loss. Together with previous observations that ferroptosis-related perturbations do not readily trigger overt adipocyte death, our results suggest that BAT is equipped with potent anti-ferroptosis defense systems. One possible mechanism underlying this resistance may involve FSP1, an alternative ferroptosis suppressor pathway independent of GPX4[28,29]. Previous studies have suggested that BAT expresses relatively high levels of FSP1, and our data further showed that FSP1 was upregulated in BAT after cold exposure (Figure S4B), which is consistent with a potential protective role[30,31]. Future studies incorporating FSP1 localization analysis, pharmacological inhibition, and genetic loss-of-function models will be necessary to determine whether FSP1 is functionally required for BAT protection in vivo. Overall, while our study indicates that GPX4 is dispensable for BAT function under the conditions tested, the molecular basis of BAT ferroptosis resistance remains an important question for further investigation.
5. Limitations
Several limitations should be acknowledged. Firstly, only acute cold exposure was examined. Whether GPX4 contributes to chronic thermogenic remodeling or long-term cold adaptation remains unknown. Secondly, potential sex-specific effects should be considered[32]. In this study, except that steady state analyses were performed in male mice (Figure 2A,B and Figure S1A,B,C), other conditions including the cold exposure, HFD, and VE-deficient diet challenges were conducted in female mice. Although no overt BAT dysfunction or cell death was observed in either setting, future side-by-side studies in both sexes will help clarify the interplay among sex, lipid peroxidation, and BAT function. Thirdly, TBARS provides only an indirect and non-specific estimate of MDA and lipid peroxidation[33]. More specific approaches, such as liquid chromatography–mass spectrometry or antibody-based detection of MDA, 4-HNE adducts, or peroxidized lipids, will be helpful to further validate and refine these measurements. Finally, assessment of direct mitochondrial functional assays, such as Seahorse oxygen consumption rate analysis, mitochondrial membrane potential measurement, and ATP production assay, remains an important goal for future studies.
6. Conclusion
In summary, under the specific conditions tested (cold exposure, HFD-induced obesity, and VE deficiency), GPX4 is not required for BAT thermogenesis in mice. BAT-specific Gpx4 deletion cause overt impairment of cold-induced thermoregulation, glucose homeostasis, or detectable ferroptosis-related gene expression as assessed by the methods used in this study. These findings reveal that BAT may possess a degree of resilience to ferroptosis that is independent of GPX4, at least under the tested challenges, distinguishing it from other highly sensitive organs. Understanding the basis of this resilience could inform future research into metabolic disease therapies.
Supplementary materials
The supplementary material for this article is available at: Supplementary materials.
Acknowledgements
We are grateful to Prof. Zhinan Yin (Jinan University, Guangzhou, China) for generously providing the Ucp1-CreERT2 mouse strains essential for this study. The authors acknowledge the use of DeepSeek-V4 for language editing and polishing of the manuscript. No AI tool was used for data analysis, figure generation, or scientific interpretation. The authors take full responsibility for the integrity, originality, and accuracy of the work.
Authors contribution
Hu Q: Conceptualization, writing-original draft, supervision, data curation, writing-review & editing.
Ju Z, Wang F: Conceptualization, supervision, data curation, writing-review & editing.
Zhang Y: Methodology, investigation, writing-original draft, data curation, writing-review & editing.
Wang W, Li C, You W: Methodology, investigation, data curation, writing-review & editing.
Liu Y: Formal analysis, data curation, visualization, writing-review & editing.
Conflicts of interest
Fudi Wang is an Editorial Board Member of Ferroptosis and Oxidative Stress. The other authors declare no conflicts of interest.
Ethical approval
All animal procedures were carried out in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of Experimental Animal Center of Jinan University. The study protocol was reviewed and approved by the Animal Care and Ethics Committee of Jinan University (Approval No. 20240731-12).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
The data and materials supporting the findings of this study are available from the supplementary materials and corresponding authors upon reasonable request.
Funding
This work was supported by the Guangzhou Science and Technology Program (Grant No. 2024A04J4046), the Innovation Team Project of Universities in Guangdong Province (Grant No. 2023KCXTD004), and the National Key R&D Program of China (Grant No. 2021YFA1100103).
Copyright
© The Author(s) 2026.
References
-
2. Morrison SF, Madden CJ, Tupone D. Central neural regulation of brown adipose tissue thermogenesis and energy expenditure. Cell Metab. 2014;19(5):741-756.[DOI]
-
6. Broeders EPM, Nascimento EBM, Havekes B, Brans B, Roumans KHM, Tailleux A, et al. The bile acid chenodeoxycholic acid increases human brown adipose tissue activity. Cell Metab. 2015;22(3):418-426.[DOI]
-
7. Xiao L, De Jesus DF, Ju CW, Wei JB, Hu J, DiStefano-Forti A, et al. m6A mRNA methylation in brown fat regulates systemic insulin sensitivity via an inter-organ prostaglandin signaling axis independent of UCP1. Cell Metab. 2024;36(10):2207-2227.[DOI]
-
11. Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. 2022;82(12):2215-2227.[DOI]
-
12. Johnson JM, Peterlin AD, Balderas E, Sustarsic EG, Maschek JA, Lang MJ, et al. Mitochondrial phosphatidylethanolamine modulates UCP1 to promote brown adipose thermogenesis. Sci Adv. 2023;9(8):eade7864.[DOI]
-
13. Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25(6):424-442.[DOI]
-
14. Berndt C, Alborzinia H, Amen VS, Ayton S, Barayeu U, Bartelt A, et al. Ferroptosis in health and disease. Redox Biol. 2024;75:103211.[DOI]
-
15. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185(14):2401-2421.[DOI]
-
16. Jiang X, Stockwell BR, Conrad M. Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266-282.[DOI]
-
20. Hu Q, Zhang Y, Lou H, Ou Z, Liu J, Duan W, et al. GPX4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis. 2021;12(7):706.[DOI]
-
25. Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016;9:22-31.[DOI]
-
27. Luo S, Zeng Y, Chen B, Yan J, Ma F, Zhuang G, et al. Vitamin E and GPX4 cooperatively protect Treg cells from ferroptosis and alleviate intestinal inflammatory damage in necrotizing enterocolitis. Redox Biol. 2024;75:103303.[DOI]
-
30. Nguyen HP, Yi D, Lin F, Viscarra JA, Tabuchi C, Ngo K, et al. Aifm2, a NADH oxidase, supports robust glycolysis and is required for cold- and diet-induced thermogenesis. Mol Cell. 2020;77(3):600-617.[DOI]
-
33. Ito F, Sono Y, Ito T. Measurement and clinical significance of lipid peroxidation as a biomarker of oxidative stress: Oxidative stress in diabetes, atherosclerosis, and chronic inflammation. Antioxidants. 2019;8(3):72.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Zhang Y, Li C, Liu Y, Wang W, You W, Ju Z, et al. GPX4 is not required for the thermogenesis function of brown adipose tissue in mice. Ferroptosis Oxid Stress. 2026;2:202616. https://doi.org/10.70401/fos.2026.0033
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Materials and Methods
- 3. Results
- 4. Discussion
- 5. Limitations
- 6. Conclusion
- Supplementary materials
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Zhang Y, Li C, Liu Y, Wang W, You W, Ju Z, et al. GPX4 is not required for the thermogenesis function of brown adipose tissue in mice. Ferroptosis Oxid Stress. 2026;2:202616. https://doi.org/10.70401/fos.2026.0033
copy
Share Link
copy