Forward genetic approaches using Caenorhabditis elegans for uncovering novel regulators of ferroptosis
Chong Yi Ng
1,2

,
Nicole L. Jenkins
1
,
Ashley I. Bush
1,2,*
,
Gawain McColl
1,2,*
*Correspondence to:
Ashley I. Bush, The Florey Institute of Neuroscience, Melbourne, VIC 3010, Australia.
E-mail: ashley.bush@florey.edu.au
Gawain McColl, The Florey Institute of Neuroscience, Melbourne, VIC 3010, Australia. E-mail: gawain.mccoll@florey.edu.au
Gawain McColl, The Florey Institute of Neuroscience, Melbourne, VIC 3010, Australia. E-mail: gawain.mccoll@florey.edu.au
Ferroptosis Oxid Stress. 2026;2:202610. 10.70401/fos.2026.0034
Received: March 24, 2026Accepted: June 22, 2026Published: June 23, 2026
Abstract
Ferroptosis, an iron-dependent form of regulated cell death characterised by lipid peroxidation, has emerged as a critical pathway in cancer, neurodegeneration, and ischaemia-reperfusion injury. While reverse genetic approaches have dominated ferroptosis research, forward genetic strategies offer unique advantages for discovering novel regulatory mechanisms without prior knowledge of pathway components. This perspective explores how classical forward genetic methodologies, including chemical mutagenesis screens, genetic modifier studies, and quantitative trait locus mapping, can be adapted to systematically identify ferroptosis regulators. We focus on employing the nematode model, Caenorhabditis elegans, and discuss the inherent advantages and disadvantages of this system. Technical considerations for designing phenotype-based screens are discussed, highlighting successful examples from related cell death pathways. Experimental frameworks for leveraging alternate model organisms to uncover conserved ferroptosis mechanisms are also explored. Forward genetics promises to reveal unexpected connections between ferroptosis and cellular processes, potentially identifying new therapeutic targets and biomarkers for ferroptosis-related diseases.
Keywords
Ferroptosis, forward genetics, Caenorhabditis elegans, mutagenesis, phenotype screening, cell death
1. Introduction
Ferroptosis represents an important conceptual advancement in our understanding of regulated cell death, complementing other established and well characterised mechanisms such as apoptosis and necrosis. Since its definition in 2012[1], ferroptosis has been implicated in diverse pathological conditions, from tumour suppression to neurodegeneration. The hallmarks of ferroptosis: iron dependence, lipid peroxidation, and sensitivity to radical-trapping antioxidants (RTAs), distinguish it from other forms of regulated cell death and suggest unique regulatory networks that determine this process.
The current understanding of ferroptosis regulation has emerged primarily through reverse genetic approaches, with investigation of the function of candidate genes based on prior knowledge of lipid biochemistry, antioxidant systems, and iron metabolism. Key discoveries include the role of glutathione peroxidase 4 (GPX4) as a central protective enzyme, the solute carrier family 7 member 11 (SLC7A11)-glutathione (GSH) axis in ferroptosis resistance, and Acyl-CoA synthetase long-chain family member 4 (ACSL4) function in incorporating polyunsaturated fatty acids (PUFAs) into membrane phospholipids. While these targeted approaches have been highly informative, they inherently favour discovery of pathways with established connections to iron or lipid metabolism.
Forward genetic screening is a powerful complementary approach that begins with a phenotype and systematically works backward to identify causal genes, potentially revealing unexpected regulators and novel pathway connections[2]. This approach operates without a priori assumptions of what cellular processes or signalling pathways will be altered and is therefore intrinsically unbiased relative to reverse genetics approaches. Consequently, this allows for the discovery of genes previously not associated with ferroptosis. Forward genetics has proven invaluable in other areas of cell death research, from the discovery of core apoptotic machinery in Caenorhabditis elegans (C. elegans)[3,4] to the identification of necroptosis components through chemical-genetic screens[5]. Some success has also been seen employing hypothesis-free screening approaches in ferroptosis research, such as the use of cDNA library expression in GPX4-deficient cells to identify ferroptosis suppressor protein 1 (FSP1) as a suppressor of ferroptosis[6], and the use of near genome-wide clustered regularly interspersed short palindromic repeats (CRISPR) activation screens to reveal the role of GCH1-BH4 pathway in limiting lipid peroxidation[7]. As the exploration of ferroptosis matures, forward genetic strategies present an opportunity to systematically explore the full regulatory landscape governing this critical cell death pathway.
1.1 Fundamental gaps in ferroptosis research
Fundamental gaps exist in ferroptosis research that forward genetics is uniquely positioned to address. Firstly, there is a lack of definitive ferroptosis markers as well as an incomplete understanding of the physiological functions of ferroptosis[8-10]. While investigations of developmental biology using C. elegans have centred on the role of apoptotic and, more recently, non-apoptotic cell death, the physiological role of ferroptosis remains under investigation[11]. By interrogating this cell death mechanism in an unbiased approach, novel genes and pathways that interact with ferroptosis may reveal insight into these fundamental questions and yield novel biomarkers.
In addition, there is a critical gap in clinical translation[8,9]. Targeting ferroptosis in the treatment of therapy-resistant tumours, neurodegeneration, cardiovascular diseases, and ischaemia-reperfusion injury has been a key research focus. However, current targeting of known regulators of ferroptosis is challenged by issues of compound solubility, bioavailability, and stability in vivo[12,13]. Targeting iron load with chelators to treat neurodegeneration has also proven complex, with negative results from clinical trials[14]. Furthermore, cellular variation in tumours and tissues means targeting known regulators may not work universally. There is a strong need for additional therapeutic targets, which forward genetics approaches may uncover.
Forward genetics offers an approach to identify novel targets without prior assumptions of mechanism, and without restriction to specific developmental, signalling, behavioural, or disease contexts. The use of forward genetics in model organisms also does not need to be restricted to any specific type of tissue or system. Unlike omics-based approaches, which are limited by the quality of their curated data (known genes, proteins, etc.), forward genetics can reveal entirely unexpected regulators. For example, unbiased approaches have identified novel targets in disease contexts that have led to potential new treatment strategies[15-17].
1.2 Non-canonical and emerging ferroptosis pathways
Beyond the canonical hallmarks of ferroptosis of iron regulation, GPX4-glutathione antioxidant pathway and PUFAs content in the cell membrane, recently discovered non-canonical pathways are shaping the complexity of ferroptosis regulation. Vesicular trafficking has recently been shown to be a novel mechanism for ferroptosis resistance by expelling iron-rich ferritin[18,19]. Mice obesity models have also revealed the effect of mitochondrial iron handling through Mitoferrin 1 and Mitoferrin 2 on ferroptosis[20], bringing in considerations of metabolism in ferroptosis regulation. Intercellular exchange adds another layer of complexity, where ferroptosis has been shown to propagate to neighbouring cells[21]. Factors directly impacting membrane integrity of cells could also serve as a new hallmark of ferroptosis[22]. Forward genetics has the potential to provide further evidence of the link between these non-canonical pathways and ferroptosis, and other iron and metabolic pathways not yet linked.
2. The Forward Genetics Toolkit for Ferroptosis Discovery
2.1 Chemical mutagenesis screens
Classical mutagenesis screens use chemical mutagens like ethyl methanesulfonate (EMS) or N-ethyl-N-nitrosourea to induce single DNA nucleotide changes[23]. Alternatively, irradiation can be used, for example via ultraviolet (UV) exposure alone or in combination with trimethylpsoralen (TMP) to generate small deletions, usually around 0.1-15 kb[24]. For ferroptosis research, these screens can be designed to identify both sensitising and protective mutations. Sensitising screens would involve treating mutagenised populations with sub-lethal ferroptosis inducers (such as RSL3, erastin, or FIN56[25]) and selecting for hypersensitive variants. Conversely, protective screens could apply lethal ferroptosis conditions and isolate resistant survivors.
The choice of ferroptosis inducer is crucial for screen design. Iron supplementation (such as ferric ammonium citrate (FAC) or ferrous sulphate) could reveal novel interactions between iron regulation and ferroptosis, such as ferritin turnover, iron compartmentalisation, or the recently discovered link between exosome export of iron loaded ferritin and ferroptosis[18]. RSL3 directly inhibits GPX4, making it useful for identifying downstream protective mechanisms but potentially missing upstream regulators of GPX4 expression or activity. Erastin blocks system xc- (SLC7A11), creating glutathione depletion that may reveal novel GSH-independent protective pathways. FIN56 depletes both GPX4 and CoQ10[25,26], potentially identifying regulators of CoQ10 biosynthesis or alternative antioxidant systems.
2.2 Genetic modifier screens
Modifier screens leverage known ferroptosis-related mutations as sensitised backgrounds for identifying genetic interactions. Starting with partial loss-of-function alleles of key ferroptosis genes (such as gpx4 hypomorphs or slc7a11 mutants), screens are then performed for second-site mutations that either enhance or suppress the ferroptosis phenotype. Enhancer mutations can identify genes operating in parallel protective pathways, while suppressor mutations could reveal negative regulators or alternative compensatory mechanisms. However, this also means that modifier screens are not unbiased, as they have a higher chance of identifying genes linked to the perturbed pathways.
Modifier screens are particularly powerful because they can detect subtle effects masked in wild-type backgrounds. They also provide functional context, i.e., enhancers likely operate in the same pathway or process as the primary mutation, while suppressors may reveal regulatory hierarchies or compensatory systems. The genetic architecture revealed by modifier screens can guide subsequent mechanistic studies and provide insights into evolutionary constraints.
2.3 Quantitative trait locus (QTL) mapping
Ferroptosis sensitivity likely exhibits quantitative variation in natural populations, making QTL mapping a viable approach for identifying genetic variants that modulate ferroptosis responses. By crossing genetically diverse strains and measuring ferroptosis-related phenotypes in segregating populations, the loci contributing to this variation can be mapped and identified. This approach has proven highly successful for identifying disease susceptibility genes[27,28] and could reveal ferroptosis regulators with more subtle effects than those detected in knockout (i.e., mutagenesis based) studies.
The choice of the specific phenotype measured for QTL mapping requires careful consideration. Traditional viability assays may lack the resolution needed to detect quantitative differences, but this will be dependent on the available phenotypic variability. Use of potentially more sensitive measurements, such as lipid peroxidation levels, GSH depletion kinetics, or expression of ferroptosis-responsive genes, could provide better quantitative traits, but will present a greater technical challenge. Advanced techniques like bulk segregant analysis combined with whole-genome sequencing (WGS) can accelerate QTL identification and fine-mapping[29-31].
3. Model Organism Considerations for Ferroptosis Genetics
3.1 C. elegans as a ferroptosis model
C. elegans offers exceptional advantages for forward genetic screens, including rapid generation time (~three days per generation depending on temperature), large brood sizes, and sophisticated and well-developed genetic tools. The conservation of core ferroptosis machinery, including GPX4 orthologues and iron homeostasis pathways, makes C. elegans a relevant model for ferroptosis research. However, compared to vertebrates, significant differences exist, including the absence of clear SLC7A11 orthologues and differences in fatty acid metabolism that may affect lipid peroxidation pathways. Importantly, ferroptosis-like cell death has been reported in C. elegans following treatment with ferroptosis inducers, suggesting conserved core mechanisms despite pathway differences. While it is still unknown if this form of cell death is truly conserved across species, analogous inducers, inhibitors, and biochemical changes consistent with ferroptosis imply similar lipid-peroxidation driven death in C. elegans[32].
The transparency of C. elegans also enables real-time monitoring of cell death progression and lipid peroxidation using fluorescent reporters, providing quantitative phenotypes ideal for genetic screens. Additionally, the compact genome and excellent RNAi libraries (for gene silencing) facilitate rapid candidate gene validation.
3.1.1 Key advantages of C. elegans
The key advantages of C. elegans in a genetics screen are its short life cycle and high fecundity, allowing for generation of large mutant libraries[33]. The use of a whole organism allows for studying different cell types and tissue-specific effects. Maintenance of C. elegans is also relatively low cost and not laborious. C. elegans has two sexes: hermaphrodites and males. Hermaphrodites can self-fertilise and facilitate rapid inbreeding to achieve homozygosity, while males enable mating for reintroduction of wild-type gene copies for rescue experiments, or combining gene variants for genetic complementation studies.
3.1.2 Limitations of C. elegans
Despite these advantages, the nematode lacks cardinal mammalian tissues, cell types, and systems such as haematopoietic tissues, a cardiovascular system, an adaptive immune system, and iron transport by transferrin. Thus, fewer disease models are possible in worms compared to mammals, e.g., modelling the complexity of the tumour microenvironment. While having advantages as an efficient discovery platform, subsequent validation in appropriate mammalian models is essential. This requires validation in human cell, organoid, and other pre-clinical models in the discovery process for therapeutically relevant ferroptosis mechanisms. Additionally, while all known ferroptosis pathways identified in mammalian culture have not yet been completely mapped out in C. elegans, the field-wide issue of a lack of biomarkers affects all model systems. Notably, transferrin receptor 1 is a biomarker for ferroptosis in mammalian cells, but is not expressed in C. elegans[34].
3.2 Ferroptosis pathways in C. elegans
C. elegans is a promising animal model to study ferroptosis and is gaining popularity[35]. Most of the key genes and pathways that have been identified in regulating ferroptosis are conserved in C. elegans. Studies using C. elegans have uncovered the involvement of ferroptosis in development, neurodegeneration, and ageing[32,36].
3.2.1 Ferroptosis in developmental and physiological contexts
In germline cells, dietary ingestion of certain PUFAs by C. elegans was able to trigger germ-cell ferroptosis and sterility[36]. Phosphatidylcholine level was also shown to impact nematode sterility through ferroptosis[37]. For somatic cells, C. elegans models of neurodegeneration, such as Parkinson’s disease and Alzheimer’s disease, have shown a link to iron dyshomeostasis and ferroptosis[38,39]. C. elegans has been broadly used for ageing studies due to the short lifespan (2-3 weeks). Over the adult lifetime, these animals accumulate intracellular iron and lose antioxidant GSH, resulting in late-life frailty[32,40]. Treatment with ferroptosis inhibitors or iron chelation mitigates age-related cell death and significantly and markedly increases lifespan[32].
3.2.2 Iron metabolism in C. elegans
The contribution of ferrous iron to ferroptosis is similar in C. elegans compared to mammalian cells, with the same iron-catalysed spontaneous peroxyl radical-mediated chain reaction (autoxidation). Many of the proteins involved in iron homeostasis are well conserved, with homologues of DMT1 (SMF-1, SMF-2, SMF-3), ferritin (FTN-1, FTN-2), and ferroportin (FPN-1.1, FPN-1.2) in C. elegans[41]. There are some differences in the iron metabolism pathway, such as a lack of a transferrin homologue, circulatory system, and erythropoiesis, obviously unnecessary in an animal 1 mm in length.
During biological ageing, intracellular iron accumulates in C. elegans, as in mammalian ageing tissue[42], with a failure of the capacity of the iron-storage protein ferritin to safely sequester iron[43]. This emergence of labile ferrous iron with age correlates with signalling effects that modulates ageing and becomes a lifespan hazard[44]. C. elegans that lack the iron storage protein, ferritin, have an elevated Fe2+:Fe3+ ratio and are short-lived compared to wild types[45].
The Fe2+-glutathione complex is considered the dominant form of iron in the cell labile iron pool and acts as one regulatory system to prevent abnormal redox cycling of iron[46]. At the same time, (phospho)lipid hydroperoxides can be terminated by glutathione peroxidases which depend on the reduction of glutathione. During normal ageing, the loss of glutathione in worms increases vulnerability to ferroptosis[32].
Beyond these classical pathways of iron uptake, storage and export, recent work has uncovered active iron-expulsion mechanisms that impact ferroptosis sensitivity. In human cell models, ovarian cancer cells exposed to erastin secrete iron loaded ferritin via CD63-mediated extracellular vesicles (EVs), reducing intracellular labile iron pool and lipid peroxidation, and conferring resistance to ferroptosis[18]. This process was shown to be dependent on iron overload conditions, where iron regulatory proteins increase CD63 expression[47]. On the other hand, general ferroptotic stress can also trigger prominin2-mediated EV export of ferritin[19]. Although both EV-mediated ferritin trafficking pathways have not been identified in C. elegans, homologues of the components such as CD63 (tsp-7) exist in this organism[48].
3.2.3 Glutathione and antioxidant systems
In mammals, glutathione synthesis is catalysed by γ-glutamylcysteine synthetase (γ-GCS) and glutathione synthetase from cysteine. For C. elegans, gcs-1 is the homologue of γ-GCS, while gss-1 is the homologue of glutathione synthetase[49]. In mammalian cells, GPX4 is a checkpoint enzyme that reduces lipid hydroperoxides to non-toxic lipid alcohols, protecting against ferroptosis. GPX4 is a selenoprotein which contains the amino acid residue selenocysteine in its catalytic centre and uses glutathione as a substrate.
In C. elegans, four primary glutathione peroxidase homologues have been identified so far, namely gpx-1, gpx-2, gpx-6, and gpx-7[50]. These four homologues share similar functions but are not selenoproteins; instead, they utilise cysteine residues to replace the selenocysteine in their active sites to clear phospholipid hydroperoxides. This renders the enzyme less efficient than the selenocysteine-containing homologues[51]. For the other major antioxidant system regulating ferroptosis, the FSP1/CoQ10 pathway[52], homologues of FSP1 and genes in this pathway have not yet been identified in worms.
3.2.4 Lipid composition and ferroptosis sensitivity
The ratio of peroxidation-susceptible PUFAs to peroxidation-resistant monounsaturated fatty acids (MUFAs) also regulates ferroptosis sensitivity in C. elegans. Dietary supplementation of dihomo-gamma-linolenic acid (DGLA; 20:3n-6) triggers ferroptotic cell death in the germline[36]. However, not every type of PUFA is able to trigger ferroptosis, where supplementation of arachidonic acid, eicosapentaenoic acid, and docosahexaenoic acid did not trigger ferroptosis in C. elegans[36]. Conversely, MUFAs such as oleic acid confer protection from iron-overload-induced ferroptosis[53]. The PUFA to MUFA ratio can also be manipulated by altering ether lipid production, where ether lipid-deficient mutants result in a higher PUFA to MUFA ratio[54]. For example, ads-1 mutants grown on DGLA exhibited higher sterility that was rescued by the addition of ferroptosis inhibitors[36]. Research into metabolic plasticity, such as lipidome changes in C. elegans during adaptation to temperature shifts[55] could also serve as an emerging paradigm for modulating ferroptosis.
3.2.5 Ferroptosis inducers in C. elegans
For cell culture models of ferroptosis, chemical toxins like erastin and RSL3 are typically used to induce cell death. Erastin blocks cystine uptake by inhibiting system xc-, resulting in glutathione depletion, while RSL3 directly inhibits GPX4; either compound prevents the reduction of lipid peroxides, resulting in ferroptosis[1,56]. However, neither erastin nor RSL3 is suitable for inducing ferroptosis in C. elegans. Nematodes do not harbour a homologue of system xc- for erastin to act on[57]; whereas RSL3 binds to cysteine 66 in an allosteric site on mammalian GPX4[58], but there is neither evidence of this site being conserved in C. elegans nor evidence to suggest that RSL3 can inhibit nematode glutathione peroxidases, which lack selenocysteine.
To induce ferroptosis in C. elegans (Figure 1), one strategy utilises the thiol-oxidising agent diethyl maleate (DEM) to deplete glutathione[32]. Although glutathione is the most abundant intracellular thiol, off-target protein thiols may also be oxidised by DEM, and glutathione also protects against other forms of oxidative attack. The use of DEM as an inducer has also been studied in cell culture for non-cysteine depletion-induced ferroptosis[59]. To model iron overload-induced ferroptosis, medium is supplemented with FAC[43,53,60] or ferrous sulphate[61]. Dietary supplementation of lipids, in particular certain types of PUFA such as DGLA, promotes ferroptosis in C. elegans[36]. As ferroptosis is also thought to contribute to normal ageing in worms[32], ageing can also be used as a more gradual and naturalistic paradigm to investigate ferroptosis resistance.
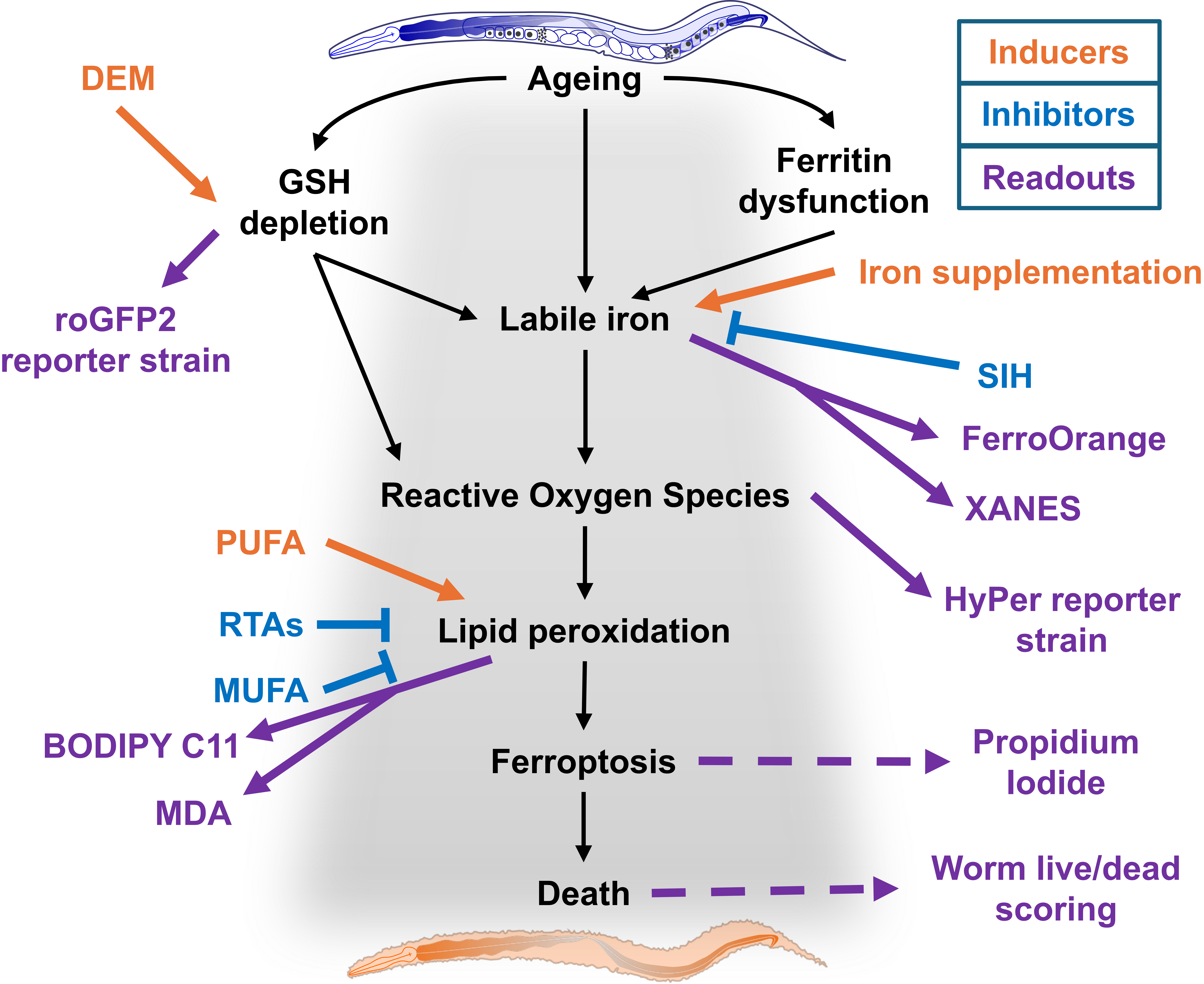{kind=link}
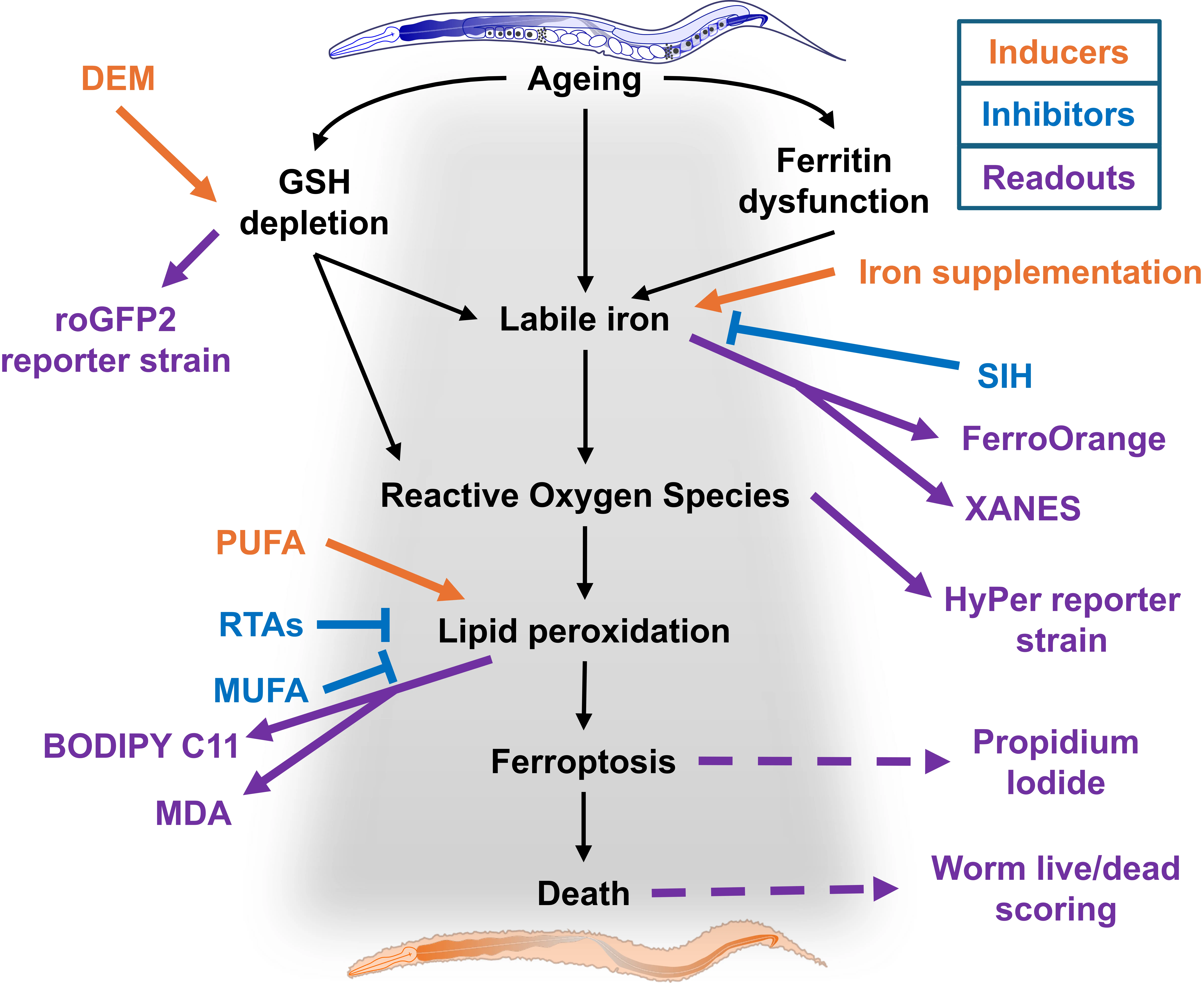
Figure 1. Summary of ferroptosis inducers, inhibitors and readouts in C. elegans. Inducers of ferroptosis in C. elegans include DEM to deplete GSH, iron supplementation with ferric ammonium citrate or ferrous sulphate, and dietary supplementation of certain PUFAs such as DGLA. Inhibitors can target labile iron with SIH or lipid peroxidation with RTAs such as ferrostatin-1 or liproxstatin-1. Dietary supplementation of MUFAs such as oleic acid can also reduce lipid peroxide levels. Readouts are available at multiple stages in the induction of ferroptosis. DEM: diethyl maleate; GSH: glutathione; PUFAs: polyunsaturated fatty acids; DGLA: dihomo-gamma-linolenic acid; SIH: salicylaldehyde isonicotinoyl hydrazone; RTAs: radical trapping antioxidants; MUFAs: monounsaturated fatty acids; XANES: X-ray absorption near edge structure; MDA: Malondialdehyde.
Conversely, to inhibit ferroptosis in C. elegans (Figure 1), salicylaldehyde isonicotinoyl hydrazone can be used to chelate intracellular labile iron, while RTAs such as liproxstatin-1 inhibit lipid peroxidation[32,62]. Supplementation of the MUFA oleic acid has also been used as an inhibitor[53].
3.3 Drosophila melanogaster for tissue-specific studies
The vinegar fly, Drosophila melanogaster, offers an alternate or additional model system that provides specific advantages for studying ferroptosis in specific tissues and developmental contexts. The galactose-responsive transcription factor 4/upstream activating sequence system enables precise spatial and temporal control of gene expression[63], facilitating examination of tissue-specific ferroptosis regulators or investigation of how ferroptosis sensitivity changes during development. This is particularly relevant for neurodegeneration research, where ferroptosis may contribute to age-related pathology.
Drosophila also offers sophisticated behavioural assays that could serve as readouts for ferroptosis-related phenotypes in the nervous system. Locomotor defects, learning impairments, or shortened lifespan following iron exposure might reflect ferroptosis in neural tissues and provide quantitative traits for genetic mapping[64,65]. The availability of natural variation panels in Drosophila facilitates QTL mapping approaches for identifying naturally occurring variants that modulate ferroptosis sensitivity[66].
3.4 Zebrafish for vertebrate validation
Zebrafish, Danio rerio, offer a good balance of genetic tractability and vertebrate physiology for ferroptosis research[67]. The conservation of ferroptosis pathways in zebrafish, including clear orthologues of GPX4, SLC7A11, and ACSL4, makes this an ideal system for validating discoveries from invertebrate screens. Forward genetic approaches in zebrafish include chemical mutagenesis screens, and natural variation studies using wild-caught strains. Alternatively, CRISPR-based knockout offers forward genetics-like characteristics, where variants are curated rather than random, but is constrained by the library of targets.
The transparency of zebrafish embryos also enables in vivo monitoring of ferroptosis using fluorescent indicators for iron, reactive oxygen species, or cell death. This real-time visualisation capability is particularly valuable for studying ferroptosis during development, ischaemia-reperfusion injury models, or cancer progression. Additionally, zebrafish models of human diseases (including neurodegeneration, heart disease, and cancer) provide pathophysiologically relevant contexts for ferroptosis related research.
4. Technical Considerations for Forward Genetic Screens
4.1 Phenotype selection and quantification
The success of forward genetic screens depends critically on phenotype selection and measurement. Traditional viability assays may be too crude to detect subtle changes in ferroptosis sensitivity, while overly sensitive assays may yield too many hits for practical follow-up. Intermediate phenotypes, such as developmental delays, behavioural changes, or stress-response defects, might provide more tractable screening targets while still reflecting meaningful alterations in ferroptosis pathways.
Quantitative measurements of ferroptosis hallmarks may offer more precise phenotypes for genetic analysis. These include lipid peroxidation levels (measured by thiobarbituric acid reactive substances (TBARS) assays, C11-BODIPY fluorescence, or lipidomics)[68], glutathione depletion kinetics (monitored using ThiolTracker[69] or biochemical assays[70]), iron accumulation (detected by calcein-AM quenching[71] or cell permeable iron-sensitive fluorophores like FerroOrange[72]), or potentially mitochondrial morphological changes (using live-cell dyes like MitoTracker)[73].
4.2 Roadmap of classical mutagenesis screening approach
As previously discussed, forward genetic approaches do not require a priori assumptions about how the selected phenotype will be produced, making them intrinsically unbiased. This approach has been a successful strategy for other cell death pathways, such as for apoptosis, where unexpected pathways and regulatory connections have been revealed. However, it is important to acknowledge that there are still some intrinsic limitations of using forward genetics. For example, this approach is unable to elucidate genes whose mutation results in lethality or severe fertility defects.
4.2.1 Generation of mutant lines
To generate mutant lines, random mutagenesis can be performed using chemicals such as EMS, which typically causes single nucleotide changes[23]. Alternatively, irradiation can be used, for example via UV exposure alone or in combination with TMP to generate small deletions. CRISPR can also be used to induce DNA double-strand breaks, with guide RNA libraries targeting nearly the full genome[74], but it is not as cost- and time-efficient for large-scale genetic screening approaches[75].
Mutagenesis is carried out on a parental line (P0), just before the germline matures (L4 larval stage in C. elegans). The resulting F1 generation is then left to self-fertilise, followed by initial screening of the F2 generation (Figure 2)[76]. Phenotype screening is then carried out on tens of thousands of mutant progeny, as only one null mutation is typically expected for every 2,000 haploid copies of the genome[33]. Here, the choice of phenotype and strategy of screening must be carefully considered.
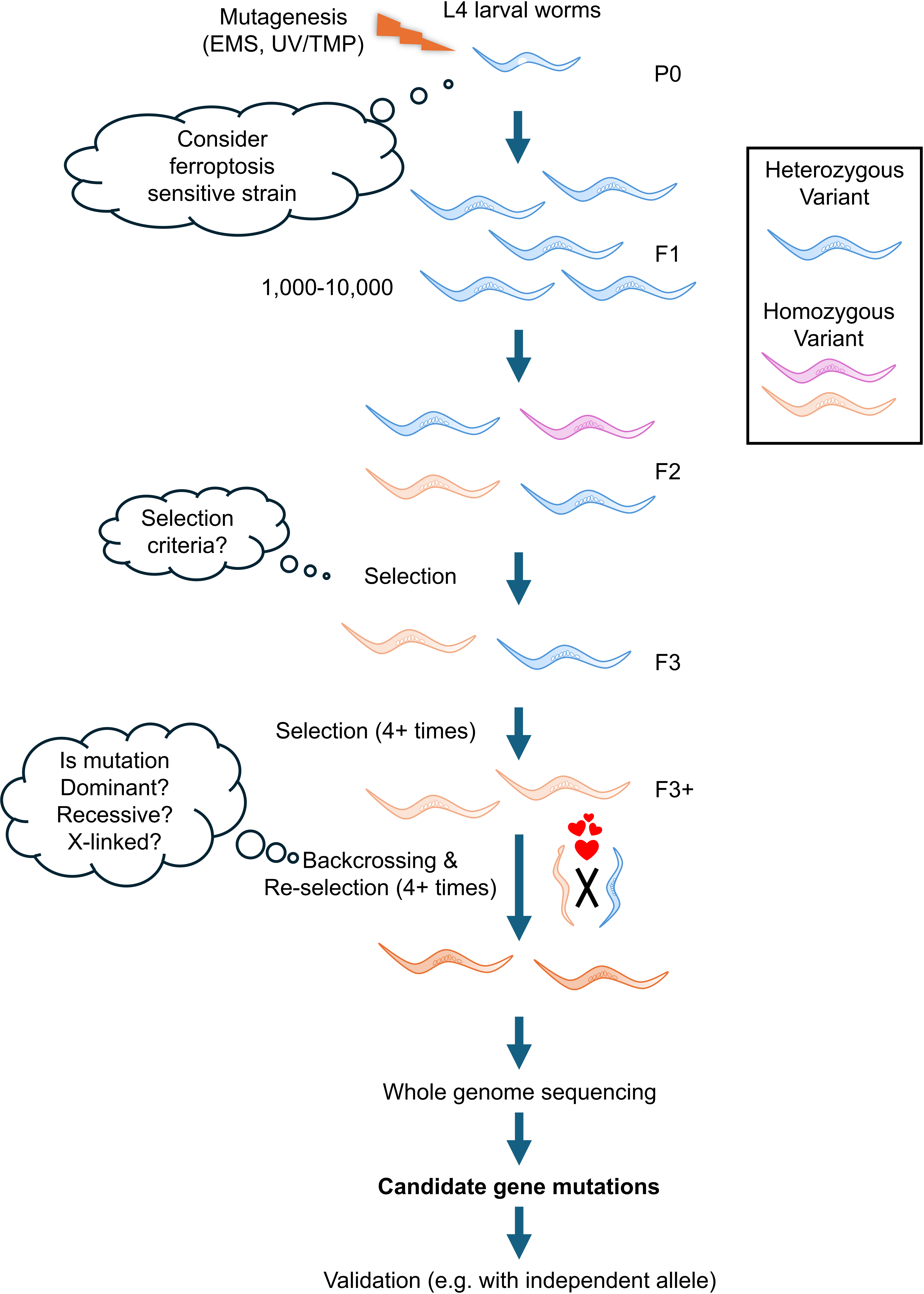{kind=link}
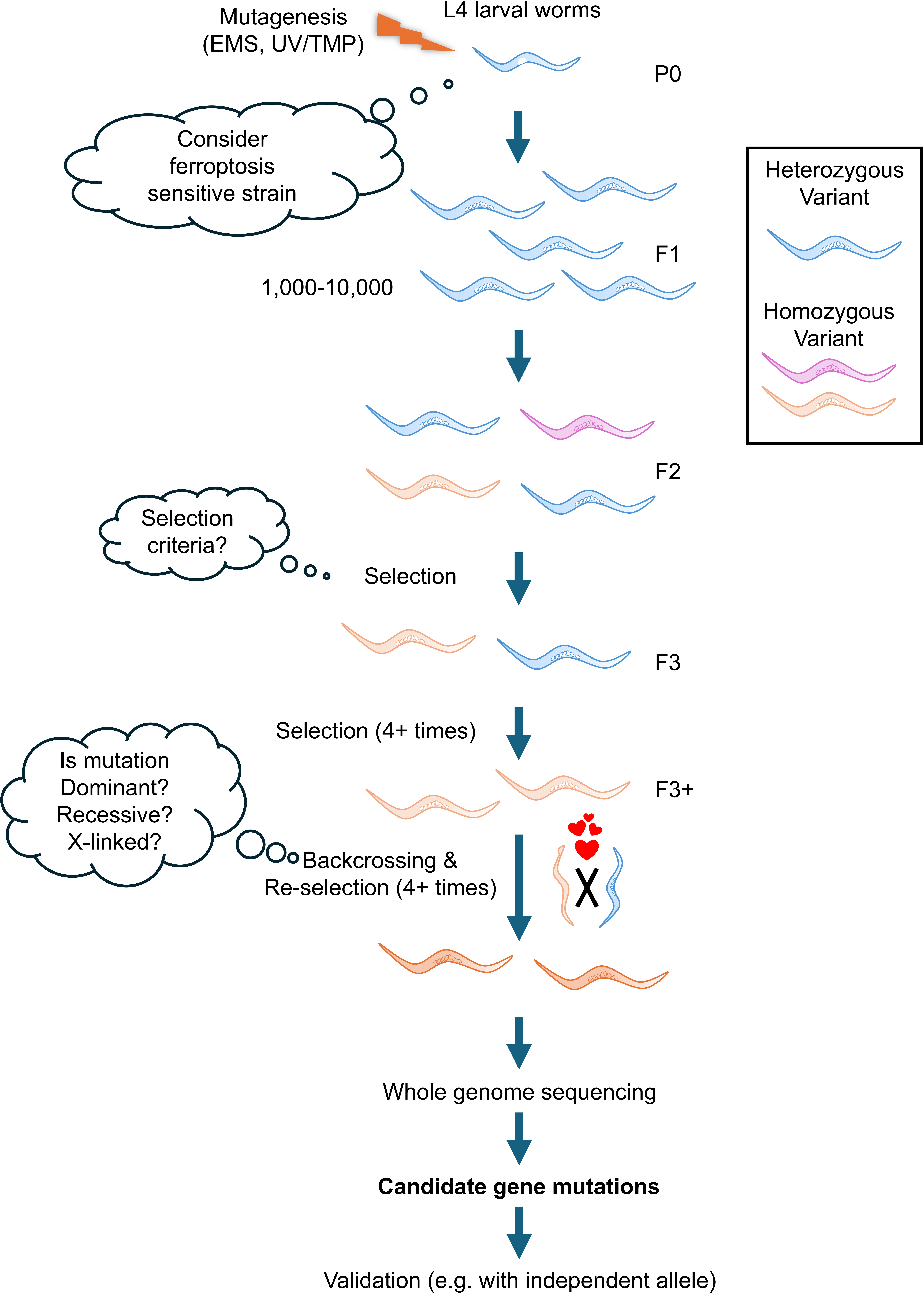
Figure 2. Roadmap of performing a forward genetics screen in C. elegans. The main steps involve mutagenesis, screening and rounds of selection, followed by backcrossing and selection again. Finally, the resulting strains can be sent for whole genome sequencing. Candidate mutations would then need to be validated. EMS: ethyl methanesulfonate; UV: ultraviolet; TMP: trimethylpsoralen.
4.2.2 Screening for mutants in ferroptosis pathways
The potentially easiest method to screen for mutants in ferroptosis pathways would be to select for resistance to a ferroptosis inducer, thereby selecting for variants that provide survival to an otherwise lethal treatment. Alternative approaches could include measurement of lipid peroxidation levels, with individuals measuring either high or low being isolated. This allows for screening for mutations conferring increased sensitivity or resistance to ferroptosis by selecting for strains having higher or lower lipid peroxide levels respectively. Another consideration is the use of ferroptosis-sensitive strains of C. elegans to create a larger difference in phenotype for ease of selection. However, it is worth noting that this is likely to bias a screen toward uncovering mutations in genes that can rescue the perturbed pathway of choice.
Derived clonal lines (F3 generation lines) are then screened for variants with the desired phenotype (Figure 2). Typically, repeated testing is used to confirm the phenotype of resistance and to reduce the number for subsequent analyses. At the same time, selection against variants with severe defects in development, viability, and fertility is done to remove any confounding effects. Once variants are identified, this is followed by iterative rounds of backcrossing, which is the process of crossing to the parental line and then selection to remove any unrelated mutations. This is also a chance to identify if the mutation is dominant or recessive, and if the mutation is present on the sex chromosomes or autosomes. Finally, the isolated strains showing a robust phenotype can be sent for WGS to identify the causal mutations. WGS provides reliable detection of single nucleotide variants, balancing high accuracy without the need for high costs[76].
4.2.3 Identifying variants of interest and hit validation
Once a candidate mutation is identified, validation can be done either through reverting the change in phenotype with insertion of a wild-type transgene or testing of mutant strains with independent alleles of the candidate gene. For C. elegans, there is an extensive library of mutants available through the Caenorhabditis Genetics Center and National BioResource Project[77], enabling rapid validation of candidate genes. Alternatively, investigation of phenotypes can be undertaken using RNAi knockdown of the candidate gene, which is performed by feeding worms with bacteria expressing dsRNA targeting the gene[78]. RNAi is a simple and effective method for determining loss-of-function phenotypes of genes, with the added advantage of being able to separate developmental effects from adult effects of gene knockdown. Two near full-genome RNAi libraries are commercially available for use, allowing for ease of gene knockdown for virtually every gene in C. elegans[79,80].
4.2.4 Investigation of genetic pathways
Using known mutants and RNAi, genes associated with or in the same pathway as candidate genes can be investigated. The specificity of the mutation in affecting ferroptosis compared to other cell death mechanisms can also be investigated, using inducers or inhibitors of other forms of programmed cell death[81]. A wealth of genetic and chemical assays are available to investigate apoptosis in C. elegans[82]. For example, strains harbouring mutations in candidate genes can be treated with cisplatin to induce intrinsic apoptosis. Survival of these strains can then be compared to wild-type worms to deduce if the candidate genes are also involved in intrinsic apoptosis. Similar approaches can then be carried out for extrinsic apoptosis, pyroptosis, necroptosis, and so on.
It is also possible that the candidate genes identified are involved in generalised stress response pathways. We recommend checking targets such as insulin-like signalling, oxidative stress resistance, and the skn-1 pathway (homologue of the mammalian Nrf2), all with well characterised effects on C. elegans stress resistance and lifespan. Given enough candidate mutant strains, typical workflows should yield multiple new genes associated with ferroptosis (Figure 2).
4.3 Screen design and controls
Effective screen design requires careful attention to controls and statistical power. Positive controls should include known ferroptosis regulators (e.g., ads-1 mutants, ferritin knockouts[36,45]) to validate screen conditions and detection methods. Negative controls using non-mutagenised populations help establish background variation and false-positive rates. The choice of mutagenesis density must balance genome coverage with practical screening constraints. Too few mutations may miss relevant genes, while excessive mutagenesis can yield non-specific or overwhelming toxic effects.
Statistical considerations include multiple testing corrections for large-scale screens, appropriate sample sizes for detecting expected effect sizes, and strategies for handling batch effects across screening plates or time periods. Clear criteria for hit selection can improve reproducibility and reduce bias in candidate gene identification.
4.4 Complementation testing and validation
Rigorous validation of screen hits requires complementation testing to confirm that phenotypes result from single-gene mutations rather than genetic background effects or multiple mutations. For organisms with efficient transformation systems, transgenic rescue experiments provide definitive evidence for gene identification. In cases where transgenics or genome editing is challenging, genetic complementation using deficiency lines or independent alleles can establish gene identity.
Following expert advice on identifying ferroptosis regulating mechanisms[81], mechanistic validation should extend beyond complementation to examine how identified genes regulate ferroptosis. This includes determining whether mutations affect known ferroptosis markers (perturbed iron regulation, GPX4 activity, glutathione levels, lipid peroxidation), investigating genetic interactions with characterised ferroptosis genes, and testing sensitivity to different ferroptosis inducers to understand pathway relationships. Once the mechanism has been characterised, perturbation of the pathway should be tested on human homologues in appropriate clinical or disease models, such as cancer cells.
5. Measures of Ferroptosis in C. elegans
Unlike other types of cell death, there is no single definitive readout for ferroptosis[81]. Markers of ferroptosis are still being determined, such as hyperoxidised PRDX3 in chronic liver diseases[83]. The C. elegans homologue prdx-3 may potentially serve as a ferroptosis marker in the same manner. Currently, usually several readouts of ferroptosis pathways in addition to evidence of cell death are required to confirm ferroptosis. In C. elegans, cell-specific death can be observed following induction of ferroptosis and occurs prior to organismal death[32]. One advantage of C. elegans is that the worms are optically transparent, hence vital dyes and genetic in vivo fluorescent sensors can be applied to visualise cell death and other hallmarks of cell stress (Figure 1).
5.1 Cell death detection
Propidium iodide is a fluorescent dye that binds to DNA but can only permeate dead cells. This vital dye can therefore be used to assay dead or dying cells within intact nematodes and determine cell-specific or tissue localisation[32]. However, this dye cannot distinguish between types of cell death and requires additional supporting evidence, such as lipid peroxidation and rescue by ferroptosis inhibitors.
5.2 Lipid peroxidation assays
Assays for lipid peroxidation are particularly valuable, as ferroptosis is fundamentally driven by oxidised PUFAs. Staining can be carried out with BODIPY-C11, which is a lipid-containing dye that changes in fluorescence wavelength when oxidised (note that ratiometric analysis is essential for this approach)[40,53,84]. Another potential method of identifying lipid peroxidation within individual worms is with Liperfluo, a specialised fluorescent probe used to detect and image lipid peroxides in living cells[85]. However, further optimisation is required before it can be used as a tool to study ferroptosis in C. elegans. While a ratio of oxidised to non-oxidised states of BODIPY-C11 can be measured, Liperfluo lacks a means of ratiometric comparison and can be biased by variations in uptake for in vivo use.
To avoid bias from the uptake of fluorescent probes by the nematodes, additional measurement of malondialdehyde (MDA) can also be employed. MDA is a key end-product of PUFA peroxidation and can be measured ex vivo as a readout of lipid peroxidation[84]. The downside of MDA measurement is the lack of spatial information of lipid peroxidation in situ, since tissue lysis is typically required.
5.3 Transgenic redox sensors
Transgenic strains to study oxidative load have also been developed, expressing HyPer, a YFP-based hydrogen peroxide sensor, and another line expressing roGFP2, a fluorescent sensor for GSSG/2GSH ratio[86]. While these provide useful information through imaging, to our knowledge, these strains are yet to be applied to study candidate genes for ferroptosis.
5.4 Iron measurements
Iron measurements have been reported using synchrotron-based X-ray absorption near edge structure imaging methods to spatially resolve altered iron coordination (the shift towards Fe2+), but the technique’s specialised instrumentation restricts its availability[32,45]. A potentially more accessible approach may be to use live imaging via FerroOrange dye, which selectively binds to labile Fe2+ in vivo and fluoresces at emission wavelength of 572 nm[60]. Imaging is then performed on stained live worms or quantification performed on a microplate reader. Lastly, it is possible to track ferritin expression levels and localisation using C. elegans strains with green fluorescent protein tagged ftn-1 and ftn-2[87], which can reveal perturbations in iron homeostasis.
6. Lessons from Forward Genetics of Cell Death Pathways
6.1 Apoptosis: From morphology to mechanism
The discovery of apoptotic machinery through forward genetics in C. elegans provides a compelling roadmap for ferroptosis research. Starting with the initial observation that specific cells die during early development, Horvitz and colleagues used genetic screens to identify cell death defective (ced) genes that disrupted normal developmental cell death[3]. This unbiased approach revealed the core apoptotic machinery: CED-3 (caspase), CED-4 (Apaf-1), and CED-9 (Bcl-2), whose conservation across species established the fundamental principles of apoptotic regulation[88,89].
The success of apoptosis forward genetics demonstrates several key principles applicable to ferroptosis research. First, starting with clear, observable phenotypes enables systematic genetic dissection even without prior mechanistic knowledge. Second, the conservation of core pathway components across species validates the use of simple model organisms for discovering human disease-relevant mechanisms. Finally, forward genetics can reveal unexpected pathway connections, e.g., the relationship between apoptosis and cell cycle control, DNA damage responses, and developmental patterning emerged from genetic interaction studies rather than biochemical approaches.
6.2 Necroptosis: Chemical-genetic approaches
Necroptosis research illustrates how chemical-genetic screens can complement traditional forward genetics for cell death pathway discovery. The identification of necrostatin-1 as a necroptosis inhibitor enabled chemical-genetic screens that revealed RIPK1 kinase as the specific cellular target and led to the discovery of the RIPK1-RIPK3-MLKL pathway[90]. This approach combined small molecule screening with genetic validation, providing both tool compounds and mechanistic insights.
The necroptosis investigations highlight the value of chemical diversity in forward genetic approaches. Different necroptosis inducers, TNF-α, TRAIL, and TLR ligands, engage distinct upstream pathways while converging on the core RIPK1/RIPK3 machinery. Similarly, ferroptosis research may benefit from using diverse inducers (system xc- inhibitors, GPX4 inhibitors, iron overload) to capture the full regulatory network governing ferroptotic cell death.
7. Future Directions and Emerging Technologies
7.1 CRISPR-based forward screens
CRISPR technology is revolutionising forward genetics by enabling targeted mutagenesis at unprecedented scale and precision. Genome-wide CRISPR knockout screens can systematically interrogate every gene for ferroptosis-related phenotypes. CRISPR activation and interference screens offer complementary, genome-wide technologies used for loss- and gain-of-function studies. The ability to multiplex CRISPR targeting allows for combination screens that test genetic interactions directly.
Base editing and prime editing technologies extend CRISPR’s utility for forward genetics by enabling precise nucleotide changes (e.g., causing loss or gain of function) beyond knockouts. This is particularly valuable for studying essential genes where null mutations are lethal, but hypomorphic (partial loss of function) alleles might reveal ferroptosis-related functions. The ability to generate allelic series through base editing can allow detailed structure-function analysis within forward genetic frameworks.
7.2 Leveraging natural variation
Natural populations harbour extensive genetic variation that could provide insights into ferroptosis regulation. Genome-wide association studies (GWAS) in humans have identified genetic variants associated with iron homeostasis disorders, neurodegenerative diseases, and cancer susceptibility, phenotypes potentially linked to ferroptosis sensitivity. Forward genetic approaches can complement GWAS by testing whether identified variants affect ferroptosis responses in a controlled experimental system.
Model organism populations offer advantages for natural variation studies, including controlled genetic backgrounds, standardised environments, and the ability to perform functional studies. The C. elegans Natural Diversity Resource[91], Drosophila Genetic Reference Panel[66], and zebrafish diversity panels[92] provide genetically characterised populations ideal for QTL mapping of ferroptosis-related traits. These resources enable the systematic identification of naturally occurring variants that modulate ferroptosis sensitivity under various environmental conditions.
7.3 Next-generation approaches and emerging technologies
Technological advances mean classical genetic screening is even more cost effective for elucidating potential new pathways regulating ferroptosis. Compared to early studies undertaken to identify apoptosis genes, it is now much faster and more cost-effective to perform WGS, with modern methods giving higher sequencing depth for more accurate analysis[2]. Emerging approaches collectively termed ‘next-generation forward genetic screens’ combine classical mutagenesis with modern genomic technologies to accelerate gene discovery.
8. Practical Considerations and Challenges
8.1 Gaps in current C. elegans ferroptosis models
While C. elegans is a promising model for ferroptosis research, some gaps still exist. All known ferroptosis pathways identified in mammalian culture have yet to be completely mapped out in C. elegans. Additionally, while iron distribution and lipid peroxidation have been imaged, single-cell resolution imaging of cells in worms undergoing ferroptosis is an area that warrants further development.
Compared to cell culture, the exploration of the genetic pathways that control ferroptosis in whole organisms, particularly during ageing, is less developed, partly due to the lack of suitable tools. There is a strong need for non-toxin models of inducible ferroptosis for spatial and temporal induction. Development of such new targeted approaches suitable for in vivo induction will significantly improve our understanding of this cell death mechanism. These tools could then be translatable to cell culture and other model systems.
8.2 Limitations of forward genetics
It is important to acknowledge that forward genetics has inherent limitations. The approach is less suited to elucidate genes whose removal results in lethality or fertility defects, as these mutations cannot be recovered in viable, fertile strains. However, strategies like synthetic lethal screens or conditional alleles can be employed to investigate essential genes[93,94]. Additionally, genetic changes may not be able to influence some ferroptosis processes involving lipid metabolism and plasma membrane dynamics if these processes are essential for cell viability.
Despite these limitations, forward genetics remains a powerful and complementary approach to reverse genetics and hypothesis-driven research. The relatively unbiased nature of the method means that unexpected regulators and pathway connections are likely to be discovered.
8.3 Future strategies for enhanced discovery
Once more forward genetic screens have been performed for ferroptosis studies, future work can look at more specific contexts. While ferroptosis can be modelled in the standard wild-type C. elegans strain (e.g., N2 Bristol), additional strategies using mutant strains sensitive to ferroptosis can be considered. Potentially, this can reveal additional pathways that are not involved during normal physiology. For example, C. elegans mutants lacking key stress response factors (e.g., DAF-16) may be more susceptible to ferroptosis. A strain with deletion of the four phospholipid hydroperoxide glutathione peroxidases is also available[50], which may be susceptible to ferroptosis. Additional future work includes the use of refined strategies to look at embryonic lethal genes or screen for gene mutations that have tissue-specific effects on ferroptosis[95].
9. Conclusions and Recommendations
Forward genetic approaches offer unique advantages for ferroptosis research that complement the insights gained from reverse genetic studies. By starting with a phenotype rather than candidate genes, forward genetics can reveal unexpected regulators, pathway connections, and evolutionary relationships that might otherwise remain hidden. The success of forward genetics in other cell death pathways provides a roadmap for systematic genetic dissection of ferroptosis regulation.
The integration of classical forward genetics with modern technologies such as CRISPR screening, single-cell analysis, and natural variation studies promises to accelerate ferroptosis gene discovery. These approaches can reveal not only individual regulators but also the genetic architecture underlying ferroptosis sensitivity variation. Understanding this architecture is crucial for predicting individual susceptibility to ferroptosis-related diseases and developing personalised therapeutic approaches.
Finally, forward genetics can address fundamental questions about ferroptosis evolution and conservation. Why has this iron-dependent cell death pathway been maintained across diverse species? What selective pressures shaped ferroptosis regulation? How do ferroptosis mechanisms vary among different cell types and developmental stages? Answering these questions may provide crucial insights into when and how ferroptosis contributes to human disease.
Novel genes identified from forward genetic screens can direct development of novel biotechnologies targeting ferroptosis in higher organisms and guide strategies to alter the rate of ageing and maximise late-life fitness. The field of ferroptosis research stands at an inflection point where forward genetic approaches can provide transformative insights into this critical cell death pathway. By embracing the power of phenotype-driven discovery, researchers can uncover the full complexity of ferroptosis regulation and identify new targets for therapeutic intervention. The lessons learned from classical genetics, careful phenotype selection, rigorous controls, and systematic genetic analysis, remain as relevant today as they were in the pioneering studies of development and behaviour that established the foundations of modern genetics.
Authors contribution
Ng CY: Conceptualisation, writing-original draft, writing-review & editing.
Jenkins NL, Bush AI: Writing-review & editing.
McColl G: Supervision, visualisation, writing-review & editing.
Conflicts of interest
Ashley I. Bush is an Editor of Ferroptosis and Oxidative Stress. He is also a shareholder in Alterity Therapeutics Ltd and Cogstate Ltd, and has a profit-share arrangement with Collaborative Medicinal Development Pty Ltd. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by an ARC Discovery project (Grant No. DP230102244), the Victorian Government’s Operational Infrastructure Support Program (This program does not have a grant number), Medical Research Future Fund Dementia, Ageing and Aged Care (Grant No. MRFF2007656 to Ashley I. Bush), a National Health & Medical Research Council Leadership 3 fellowship (Grant No. GNT2041278 to Ashley I. Bush), and the University of Melbourne through a Melbourne Research Scholarship to Chong Yi Ng (This program does not have a grant number).
Copyright
© The Author(s) 2026.
References
-
2. Ali Khan A, Raess M, De Angelis MH. Moving forward with forward genetics: A summary of the INFRAFRONTIER Forward Genetics Panel Discussion. F1000Research. 2021;10:456.[DOI]
-
3. Hengartner MO, Horvitz HR. Programmed cell death in Caenorhabditis elegans. Curr Opin Genet Dev. 1994;4(4):581-586.[DOI]
-
5. Liao D, Sun L, Liu W, He S, Wang X, Lei X. Necrosulfonamide inhibits necroptosis by selectively targeting the mixed lineage kinase domain-like protein. MedChemCommun. 2014;5(3):333-337.[DOI]
-
8. Linkermann A. Key questions in ferroptosis. Ferroptosis Oxid Stress. 2025;1:202503.[DOI]
-
9. Levkina A. The coming decade in ferroptosis research: Five riddles. Ferroptosis Oxid Stress. 2026;2:202516.[DOI]
-
10. Feinsod H, Stockwell BR. Fundamental mechanism of ferroptosis: Three unanswered questions. Ferroptosis Oxid Stress. 2026;2:202512.[DOI]
-
11. Horowitz LB, Shaham S. Apoptotic and nonapoptotic cell death in Caenorhabditis elegans development. Annu Rev Genet. 2024;58:113-134.[DOI]
-
12. Yan HF, Zou T, Tuo QZ, Xu S, Li H, Ali Belaidi A, et al. Ferroptosis: Mechanisms and links with diseases. Sig Transduct Target Ther. 2021;6:49.[DOI]
-
13. Singh M, Arora HL, Naik R, Joshi S, Sonawane K, Sharma NK, et al. Ferroptosis in cancer: Mechanism and therapeutic potential. Int J Mol Sci. 2025;26(8):3852.[DOI]
-
14. Alves F, Bush AI, Ayton S. Revisiting the role of iron in ferroptosis. Ferroptosis Oxid Stress. 2026;2:202507.[DOI]
-
15. Sheardown E, Mech AM, Petrazzini MEM, Leggieri A, Gidziela A, Hosseinian S, et al. Translational relevance of forward genetic screens in animal models for the study of psychiatric disease. Neurosci Biobehav Rev. 2022;135:104559.[DOI]
-
17. Widrick JJ, Kawahara G, Alexander MS, Beggs AH, Kunkel LM. Discovery of novel therapeutics for muscular dystrophies using zebrafish phenotypic screens. J Neuromuscul Dis. 2019;6(3):271-287.[DOI]
-
18. Battaglia AM, Sacco A, Giorgio E, Petriaggi L, Elzanowska J, Cruz AR, et al. Expulsion of iron-rich ferritin via CD63-mediated exosome drives ferroptosis resistance in ovarian cancer cells. Front Cell Dev Biol. 2025;13:1532097.[DOI]
-
20. Bucarey JL, Casas M, Espinosa A. Mitochondrial iron handling and lipid peroxidation as drivers of ferroptosis. Int J Mol Sci. 2026;27(5):2232.[DOI]
-
21. Roeck BF, Nasudivar SL, Vorndran MRH, Schueller L, Yapici FI, Rübsam M, et al. Ferroptosis spreads to neighboring cells via plasma membrane contacts. Nat Commun. 2025;16:2951.[DOI]
-
22. Hirata Y, Mishima E. Membrane dynamics and cation handling in ferroptosis. Physiology. 2024;39(2):73-87.[DOI]
-
23. Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77(1):71-94.[DOI]
-
24. Yandell MD, Edgar LG, Wood WB. Trimethylpsoralen induces small deletion mutations in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1994;91(4):1381-1385.[DOI]
-
26. Ferrada L, Barahona MJ, Salazar K, Godoy AS, Vera M, Nualart F. Pharmacological targets for the induction of ferroptosis: Focus on Neuroblastoma and Glioblastoma. Front Oncol. 2022;12:858480.[DOI]
-
27. Devoto M, Falchi M. Genetic mapping of quantitative trait loci for disease-related phenotypes. In: Rifkin SA, editor. Quantitative trait loci (QTL): Methods and protocols. Totowa: Humana Press; 2012. p. 281-311.[DOI]
-
31. Sharma SK, McLean K, Colgan RJ, Rees D, Young S, Sønderkær M, et al. Combining conventional QTL analysis and whole-exome capture-based bulk-segregant analysis provides new genetic insights into tuber sprout elongation and dormancy release in a diploid potato population. Heredity. 2021;127(3):253-265.
-
32. Jenkins NL, James SA, Salim A, Sumardy F, Speed TP, Conrad M, et al. Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging Caenorhabditis elegans. eLife. 2020;9:e56580.[DOI]
-
34. Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 2020;30(10):3411-3423.[DOI]
-
35. Martins AC, Virgolini MB, Tinkov AA, Skalny AV, Tirumala RP, Farina M, et al. Iron overload and neurodegenerative diseases: What can we learn from Caenorhabditis elegans? Toxicol Res Appl. 2022;6:23978473221091852.[DOI]
-
38. Majerníková N, Caiado MJ, Seinstra RI, Couzijn S, Goya ME, Salinas CS, et al. Ferroptosis inhibition protects against α-synuclein-related neuronal cell death. Cell Death Dis. 2026;17:78.[DOI]
-
39. Peng W, Chung KB, Lawrence BP, O’Banion MK, Dirksen RT, Wojtovich AP, et al. DMT1 knockout abolishes ferroptosis induced mitochondrial dysfunction in C. elegans amyloid β proteotoxicity. Free Radic Biol Med. 2024;224:785-796.[DOI]
-
40. Pensotti R, Sciandrone B, Bovio F, Schröter L, Maglioni S, Thewes L, et al. Redox homeostasis in ferroptosis and aging: A causal role for fard-1 and dhs-25 in Caenorhabditis elegans. Redox Biol. 2025;88:103912.[DOI]
-
41. Anderson CP, Leibold EA. Mechanisms of iron metabolism in Caenorhabditis elegans. Front Pharmacol. 2014;5:113.[DOI]
-
42. Zeidan RS, Han SM, Leeuwenburgh C, Xiao R. Iron homeostasis and organismal aging. Ageing Res Rev. 2021;72:101510.[DOI]
-
43. James SA, Roberts BR, Hare DJ, de Jonge MD, Birchall IE, Jenkins NL, et al. Direct in vivo imaging of ferrous iron dyshomeostasis in ageing Caenorhabditis elegans. Chem Sci. 2015;6(5):2952-2962.[DOI]
-
44. Xu J, Marzetti E, Seo AY, Kim JS, Prolla TA, Leeuwenburgh C. The emerging role of iron dyshomeostasis in the mitochondrial decay of aging. Mech Ageing Dev. 2010;131(7-8):487-493.[DOI]
-
49. Ferguson GD, Bridge WJ. The glutathione system and the related thiol network in Caenorhabditis elegans. Redox Biol. 2019;24:101171.[DOI]
-
55. VanHook AM. Compensating for the heat. Sci Signal. 2015;8(379):eaac6707.[DOI]
-
59. Ali Belaidi A, Masaldan S, Southon A, Kalinowski P, Acevedo K, Appukuttan AT, et al. Apolipoprotein E potently inhibits ferroptosis by blocking ferritinophagy. Mol Psychiatry. 2024;29(2):211-220.[DOI]
-
60. Gremme A, Gerisch E, Wieland D, Hillebrand J, Drews F, Pirritano M, et al. Consequences of iron exposure and glutathione depletion on redox balance, lipidome, and neurotransmission in C. elegans. Redox Biol. 2026;90:104023.[DOI]
-
62. Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25(6):424-442.[DOI]
-
65. Gomes KK, Dos Santos AB, Dos Anjos JS, Leandro LP, Mariano MT, Pinheiro FL, et al. Increased iron levels and oxidative stress mediate age-related impairments in male and female Drosophila melanogaster. Oxid Med Cell Longev. 2023;2023:7222462.[DOI]
-
66. MacKay TFC, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, et al. The Drosophila melanogaster genetic reference panel. Nature. 2012;482(7384):173-178.[DOI]
-
67. Cao Z, Liu G, Zhang R, Zhang H, Wang A, Zheng M, et al. Validation of ferroptosis in zebrafish as a reliable model for phenotypic studies. Sci Rep. 2026;16:14357.[DOI]
-
68. Stockwell BR, Jiang X. The chemistry and biology of ferroptosis. Cell Chem Biol. 2020;27(4):365-375.[DOI]
-
71. Wang L, Ouyang S, Li B, Wu H, Wang F. GSK-3β manipulates ferroptosis sensitivity by dominating iron homeostasis. Cell Death Discov. 2021;7:334.[DOI]
-
72. Yu F, Zhang Q, Liu H, Liu J, Yang S, Luo X, et al. Dynamic O-GlcNAcylation coordinates ferritinophagy and mitophagy to activate ferroptosis. Cell Discov. 2022;8:40.[DOI]
-
73. Zhang Q, Qu H, Chen Y, Luo X, Chen C, Xiao B, et al. Atorvastatin induces mitochondria-dependent ferroptosis via the modulation of Nrf2-xCT/GPx4 axis. Front Cell Dev Biol. 2022;10:806081.[DOI]
-
75. Kim HM, Hong Y, Chen J. A decade of CRISPR-Cas gnome editing in C. elegans. Int J Mol Sci. 2022;23(24):15863.[DOI]
-
78. Conte D, MacNeil LT, Walhout AJM, Mello CC. RNA interference in Caenorhabditis elegans. Curr Protoc Mol Biol. 2015;109(1):26.[DOI]
-
80. Rual JF, Hill DE, Vidal M. ORFeome projects: Gateway between genomics and omics. Curr Opin Chem Biol. 2004;8(1):20-25.[DOI]
-
81. Mishima E, Nakamura T, Doll S, Proneth B, Fedorova M, Pratt DA, et al. Recommendations for robust and reproducible research on ferroptosis. Nat Rev Mol Cell Biol. 2025;26(8):615-630.[DOI]
-
85. Wang Y, Ye Y, Ji J, Wang JS, Tang L, Zhang L, et al. Effect of cadmium and fumonisin B1 co-exposure on mitochondrial dysfunction and ferroptosis pathway in Caenorhabditis elegans. J Hazard Mater. 2025;483:136504.[DOI]
-
88. Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44(6):817-829.[DOI]
-
89. Malin JZ, Shaham S. Cell death in C. elegans development. Curr Top Dev Biol. 2015;114:1-42.[DOI]
-
90. Pan P, Cai Z, Zhuang C, Chen X, Chai Y. Methodology of drug screening and target identification for new necroptosis inhibitors. J Pharm Anal. 2019;9(2):71-76.[DOI]
-
93. Tang S, Gökbağ B, Fan K, Shao S, Huo Y, Wu X, et al. Synthetic lethal gene pairs: Experimental approaches and predictive models. Front Genet. 2022;13:961611.[DOI]
-
94. Cronan JE. Approaches to study proteins encoded by essential genes. Proteins. 2026;94(2):463-476.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Ng CY, Jenkins NL, Bush AI, McColl G. Forward genetic approaches using Caenorhabditis elegans for uncovering novel regulators of ferroptosis. Ferroptosis Oxid Stress. 2026;2:202610. https://doi.org/10.70401/fos.2026.0034
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. The Forward Genetics Toolkit for Ferroptosis Discovery
- 3. Model Organism Considerations for Ferroptosis Genetics
- 4. Technical Considerations for Forward Genetic Screens
- 5. Measures of Ferroptosis in C. elegans
- 6. Lessons from Forward Genetics of Cell Death Pathways
- 7. Future Directions and Emerging Technologies
- 8. Practical Considerations and Challenges
- 9. Conclusions and Recommendations
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Ng CY, Jenkins NL, Bush AI, McColl G. Forward genetic approaches using Caenorhabditis elegans for uncovering novel regulators of ferroptosis. Ferroptosis Oxid Stress. 2026;2:202610. https://doi.org/10.70401/fos.2026.0034
copy
Share Link
copy