Regulating oxygen defects over CeO2 via rare earth oxide doping for Pt-catalyzed oxidative dehydrogenation of propane with carbon dioxide
*Correspondence to:
Zhong-Wen Liu, Key Laboratory of Syngas Conversion of Shaanxi Province, School of Chemistry & Chemical Engineering, Shaanxi Normal University, Xi’an 710119, Shaanxi, China.
E-mail: zwliu@snnu.edu.cn
Smart Mater Devices. 2026;2:202604. 10.70401/smd.2026.0029
Received: January 17, 2026Accepted: March 10, 2026Published: March 17, 2026
This article belongs to the Special lssue Smart Porous Materials and Catalysis
Abstract
The efficient activation of CO2 molecules is imperative for the development of a high-performance catalyst for the oxidative dehydrogenation of propane with carbon dioxide (CO2-ODP). To enhance the activation of CO2 over ceria, in this work, rare earth doped ceria (RE-CeO2, RE=La, Pr, Nd, and Sm) is comparatively studied as a support for PtSn for CO2-ODP. Compared with PtSn/CeO2, Ce1-xRExO2 solid solution is formed for the RE-doped PtSn/CeO2 catalysts, which increases the oxygen defect content, promotes the dispersion of Pt, and strengthens the ability of CO2 to supplement lattice oxygen in the catalyst, thereby enhancing the catalytic performance for CO2-ODP. Among the investigated catalysts, PtSn/ Nd-CeO2 shows the best CO2-ODP performance, with an initial propane conversion and propylene selectivity of 52.7% and 86.1%, respectively. Moreover, the Pt electron density and oxygen-defect content over PtSn/RE-CeO2 catalysts, which can be regulated to relatively large extents by doping ceria with different RE metals, are key factors in determining the activity of CO2-ODP. These findings regarding the CeO2-based binary RE solid solutions with adjustable oxygen defects and the derived impacts on supported Pt provide important references for advancing the design of oxygen-defect involved supported metal catalysts, including Pt-based catalysts, for the oxidative dehydrogenation of light alkanes with carbon dioxide.
Keywords
Propane, oxidative dehydrogenation, carbon dioxide, platinum, rare earth oxide, solid solution
1. Introduction
Propylene (C3H6), a crucial foundational feedstock in the petrochemical industry, occupies a pivotal position in modern chemical manufacturing[1]. It is extensively utilized in the production of a wide range of chemical products and materials, including cleaning agents and synthetic fibers/plastics[2-4]. For decades, the primary industrial sources of propylene have relied on steam cracking and catalytic cracking of light oils such as naphtha[5]. However, these conventional processes are associated with significant drawbacks, such as substantial capital investment, high energy consumption, and limited feedstock flexibility, etc. The introduction of oxygen into the reaction system presents a promising alternative. This approach not only mitigates the thermodynamic equilibrium constraints inherent in direct dehydrogenation but also alters the thermal characteristics of the process, offering enhanced energy efficiency and notable environmental benefits. Consequently, the oxidative dehydrogenation of light alkanes, including propane, using molecular oxygen (O2-ODP) has emerged as one of the research focuses in recent years. Nevertheless, the stronger oxidation ability of O2 often leads to undesirable deep oxidation of the light hydrocarbons, resulting in the formation of COx and reduced selectivity toward the target olefins[6,7]. A key challenge for O2-ODP, therefore, lies in achieving a reasonably high reaction rate while simultaneously maintaining economically high selectivity of light olefins such as propylene. In contrast, carbon dioxide (CO2), as an alternative to O2, is thermodynamically stable and kinetically inert, exhibiting significantly weaker oxidizing power. Employing CO2 as a mild oxidant for the oxidative dehydrogenation of propane (CO2-ODP) offers a compelling strategy. This method effectively circumvents the deep-oxidation issues observed in O2-based oxidative dehydrogenation processes, while still improving the thermodynamic equilibrium conversion compared to direct dehydrogenation[8]. Furthermore, it enables the utilization of CO2 resources, a major greenhouse gas, aligning with global efforts toward carbon emission reduction. As a result, CO2-ODP has attracted considerable attention and is regarded as a promising energy-efficient and environmentally benign green synthesis pathway for propylene.
At present, the reported CO2-ODP catalysts are mainly based on supported noble metals (e.g., Pt) and various metal oxides (e.g., CrOx, V2O5, Ga2O3, MoO3 and In2O3)[9-13]. Among these, supported Pt-based catalysts have attracted significant attention due to their high propylene yield, strong ability to activate CO2, and good stability[14]. In recent years, extensive research has been conducted to understand the influences and mechanisms of the structure and electronic properties of Pt, additives (such as Sn and Ce), and support materials (including SiO2 and CeO2)[15,16] on the catalytic performance of Pt in CO2-ODP, leading to substantial progress. A comprehensive review of the literature on CO2-ODP reveals that while the catalytic conversion of propane over Pt and its influencing factors have been thoroughly investigated, controversies persist regarding the activation mechanism of CO2 and the detailed reaction pathway. This is largely due to the complexity of the reaction system and catalyst structure, which in turn hinders the rational design of high-performance Pt-based catalysts. Considering the specific characteristics of the CO2-ODP reaction, the key to designing effective catalysts lies in the selective catalytic activation of C–H bonds in propane and the construction of dual-functional active sites capable of efficiently activating CO2. Our research group combined PtSn with CeO2 and employed a sequential impregnation method to prepare a PtSnCe/SiO2 catalyst[17]. This approach not only significantly enhanced the CO2-ODP performance of the Pt-based catalyst but also verified the crucial role of CeO2, with its abundant oxygen vacancies, in facilitating the catalytic activation of CO2. Furthermore, using hydrothermally synthesized CeO2 as a support, a PtSn/CeO2 catalyst was prepared via co-impregnation[18]. This study elucidated the role of Sn in modulating the Pt–CeO2 interface properties and the electron density of Pt, as well as its influence on the selective activation of both the C=O bond in CO2 and the C–H bond in propane. It was proposed that the Sn-modified Pt–O–Ce interface likely serves as the active center for catalyzing CO2-ODP over PtSn/CeO2. Building on this foundation, PtSn/CeO2 catalysts with different morphologies were designed[19]. This work revealed the shape-dependent influence of CeO2’s oxygen vacancy concentration on the structural and electronic properties of the catalysts. A higher concentration of oxygen vacancies was found to enhance Pt dispersion and electron density, thereby strengthening the selective cleavage of C–H bonds and significantly improving the initial propane conversion and propylene selectivity in the CO2-ODP reaction. However, the high-temperature and redox atmosphere inherent in the CO2-ODP reaction are major factors that can destabilize the CeO2 structure, leading to the degradation of its oxygen storage and release capacity. Introducing other metals into CeO2 to form solid solutions is an effective strategy for increasing both the oxygen vacancy content and the thermal stability of CeO2. For instance, in Fe-doped CeO2 catalysts for CO2-ODP[20], the incorporation of an optimal amount of Fe to form a Ce1-xFexO2 solid solution enhanced oxygen mobility and oxygen storage capacity (OSC), resulting in stable propane conversion and propylene selectivity for over 20 h. Similarly, under comparable CO2-ODP reaction conditions, studies on the oxidative dehydrogenation of ethylbenzene with CO2 have shown that forming a Ce1-xZrxO2 solid solution can significantly increase the oxygen defect concentration and OSC performance of CeO2, thereby promoting CO2 activation and conversion[21]. Nevertheless, achieving a homogeneous solid solution without phase separation is critical for ensuring the long-term stability of the OSC.
Based on the foregoing analyses and understandings, the introduction of rare earth (RE = La, Pr, Nd, Sm) metals into CeO2 to form a homogeneous solid solution of CeO2-based binary oxides represents a promising strategy to increase the oxygen vacancy concentration within CeO2. Consequently, the activation and conversion of CO2 are intrinsically enhanced over CeO2-based binary oxides. Diverging from prior research that predominantly focused on modifying the structure and electronic properties of the active Pt component via additives, the present work adopts a support-engineering perspective with enhanced oxygen vacancies for accelerating the redox cycle of the CO2-ODP reaction. To this end, a series of RE-doped CeO2 (RE-CeO2) supports were first synthesized via a facile co-precipitation method. Subsequently, PtSn/RE-CeO2 catalysts were prepared using a co-impregnation technique. The influence of different RE dopants on the catalyst’s structure, chemical properties, and performance was systematically investigated by correlating comprehensive characterization results with catalytic evaluation in the CO2-ODP reaction. This allowed for an in-depth analysis and discussion of the key factors governing catalyst activity, selectivity, and stability. The results demonstrate that the incorporation of RE elements and the subsequent formation of a Ce1-xRExO2 solid solution effectively increase the oxygen vacancy concentration in the catalyst system. This modification leads to multiple beneficial effects, such as improved dispersion of Pt nanoparticles, altered electron density of the active metal, and enhanced capability for replenishing lattice oxygen via CO2 activation. Collectively, these changes contribute to a significant enhancement in the catalytic performance for CO2-ODP. Therefore, the findings of this study provide crucial insights and important guidance for the rational design and development of high-performance catalysts for CO2-ODP.
2. Experimental Section
2.1 Catalyst preparation
2.1.1 Materials
All chemical reagents used in this study were obtained from Shanghai Macklin Biochemical Co., Ltd. and employed without further purification. These include the metal precursors for support synthesis, cerium nitrate hexahydrate (Ce(NO3)3·6H2O), lanthanum nitrate hexahydrate (La(NO3)3·6H2O), praseodymium nitrate hexahydrate (Pr(NO3)3·6H2O), neodymium nitrate hexahydrate (Nd(NO3)3·6H2O), samarium nitrate hexahydrate (Sm(NO3)3·6H2O), and ammonium bicarbonate (NH4HCO3). The active metal sources were chloroplatinic acid hexahydrate (H2PtCl6·6H2O) and tin(IV) chloride pentahydrate (SnCl4·5H2O).
2.1.2 Synthesis of RE-doped CeO2 supports
The RE-doped CeO2 supports (RE = La, Pr, Nd, Sm) were synthesized via a co-precipitation method[22,23], with the dopant content fixed at a nominal atomic ratio of RE/(RE + Ce) = 0.05. In a typical procedure, 12.8 mmol of Ce(NO3)3·6H2O and 0.64 mmol of the corresponding RE nitrate were dissolved in 200 mL of deionized water at 30 °C under continuous stirring. Subsequently, 200 mL of a 0.179 mol·L-1 NaHCO3 aqueous solution was rapidly added to the mixed metal solution. The resulting slurry was vigorously stirred for 0.5 h and then aged statically at 30 °C for 24 h. The precipitate was collected by filtration, thoroughly washed with deionized water to remove residual ions, and dried overnight at 80 °C. Finally, the obtained solid was calcined at 550 °C for 4 h under flowing air to yield the RE-CeO2 support. For comparison, pure CeO2 was synthesized following the same procedure in the absence of any dopant, while keeping all other conditions unchanged.
2.1.3 Synthesis of PtSn/RE-CeO2 catalysts
The PtSn/CeO2 and PtSn/RE-CeO2 catalysts were prepared via the incipient wetness impregnation method, with the nominal loading of both platinum and tin fixed at 1 wt% relative to the support. In a typical procedure, calculated volumes of aqueous H2PtCl6·6H2O and SnCl4·5H2O solutions were sequentially impregnated onto the CeO2 or RE-CeO2 support at room temperature. The mixture was then allowed to equilibrate for 6 h. Subsequently, the impregnated solids were dried in air at 80 °C for 8 h. Finally, the samples were calcined and reduced in a tubular furnace under a flowing H2 atmosphere (50 mL·min-1) at 500 °C for 4 h. The resulting catalysts are denoted as PtSn/CeO2, PtSn/La-CeO2, PtSn/Pr-CeO2, PtSn/Nd-CeO2, and PtSn/Sm-CeO2, respectively.
2.2 Characterizations
The crystalline structures of the synthesized materials were characterized by powder X-ray diffraction (XRD) on a Bruker D8 Advance diffractometer employing Cu Kα radiation (λ = 0.15406 nm). Data were collected in the 2θ range of 5°-85° with a scanning rate of 2°·min-1.
Raman spectra were acquired to probe the local structure and defects using a HORIBA LabRAM HR Evolution spectrometer. Measurements were performed with a 532 nm laser excitation source at a controlled output power of 2.5 mW to avoid thermal damage, utilizing a 50× long-focus objective lens. Spectra were recorded in the range of 200-1,800 cm-1 with an acquisition time of 10 s per scan.
Textural properties, including specific surface area, pore volume, and pore size distribution, were evaluated based on N2 adsorption-desorption isotherms measured at 77 K using a Micromeritics TriStar II 3020 analyzer. Prior to analysis, all samples were degassed under vacuum at 300 °C for 10 h to remove physisorbed species. The Brunauer–Emmett–Teller (BET) method was applied to calculate the specific surface area.
Morphological and structural analyses were conducted by high-resolution transmission electron microscopy (HRTEM) using an FEI Tecnai G2 F20 microscope operating at an accelerating voltage of 200 kV.
Surface elemental compositions and chemical states were investigated by X-ray photoelectron spectroscopy (XPS) on a KRATOS Analytical AXIS Ultra DLD spectrometer with monochromatic Al Kα X-rays (1,486.6 eV). All binding energies were referenced to the adventitious carbon C 1s peak set at 284.8 eV.
The platinum loading in the catalysts was quantitatively determined by inductively coupled plasma mass spectrometry (ICP-MS) on a Bruker Aurora M90 instrument. For analysis, approximately 0.01 g of each sample was digested in 2 mL of aqua regia at 100 °C, and the resulting solution was diluted with 1wt% HNO3 to a concentration suitable for measurement (50-200 ppm).
Thermogravimetric analysis was performed to assess coke deposition using a TA Instruments Q1000 system. Approximately 10 mg of sample was heated in an alumina crucible under a 30 mL·min-1 air flow from room temperature to 800 °C at a heating rate of 10 °C·min-1.
Hydrogen chemisorption experiments were carried out on a Micromeritics Autochem-II 2020 analyzer to estimate metallic Pt dispersion. Prior to measurement, a 0.1 g sample was reduced in a 10 vol% H2/Ar flow (30 mL·min-1) at 500 °C for 30 min, purged with Ar at the same temperature for 120 min, and then cooled to 50 °C under Ar. Pulses of pure H2 (0.5 mL per pulse) were subsequently introduced at 50 °C at 3 min intervals until saturation was achieved.
CO2 pulse experiments were conducted on a Micromeritics Autochem 2920 system to evaluate CO2 activation and conversion behavior. A sample (0.3 g) was pretreated in Ar at 500 °C for 60 min, reduced in a 10 vol% H2/Ar flow (30 mL·min-1) for 30 min, and purged again with Ar for 30 min. Pulses of CO2 (0.5 mL) in an Ar carrier gas (30 mL·min-1) were then injected into the catalyst bed. The effluent gases were monitored in real-time using a Hiden HPR QIC20 mass spectrometer, specifically tracking the signals for CO2 (m/z = 44) and CO (m/z = 28).
2.3 Catalytic experiments
The CO2-ODP catalytic performance was evaluated in a fixed-bed quartz reactor (inner diameter: 6 mm) at atmospheric pressure (0.1 MPa) and a reaction temperature of 550 °C. Prior to activity testing, 0.25 g of catalyst (40-60 mesh) was uniformly mixed with 0.75 g of quartz sand (of the same mesh size) and loaded into the reactor. The catalyst bed was then reduced in situ at 500 °C for 30 min under a flow of 50 vol.% H2/Ar (15 mL·min-1). Following reduction, the reactor was purged with He and heated to the target reaction temperature of 550 °C. A fully premixed reactant gas with a composition of C3H8/CO2/He/Ar/ = 4/4/16/1 (by volume) was then introduced at a total flow rate of 25 mL·min-1. Argon (Ar) served as an internal standard for accurate quantification. The effluent gas stream was analyzed using an online gas chromatography (GC) system equipped with two detectors: a Thermal Conductivity Detector and a Flame Ionization Detector. Gas separation was achieved using a combination of a TDX-01 packed column and an HP-PLOT Al2O3/KCl capillary column. Key catalytic performance parameters, including propane conversion, product selectivity, propylene yield, and carbon balance, were calculated based on the GC analysis data using Eq. (1) to Eq. (8) as follows.
In these equations,
3. Results and Discussion
3.1 Characterization of the catalyst
The morphological and structural characteristics of the catalysts were examined using transmission electron microscopy (TEM), as shown in Figure 1a,b,c,d and Figure S1. The morphology of PtSn/CeO2 catalyst resembled a belt-like nanosheet, the size parameters of which are 20 to 60 nm in thickness, 5 to 10 μm in length, and 0.5-5 μm in width. After RE doping, the morphological structure of the catalyst remained well preserved, indicating that the introduction of RE elements into the CeO2 support essentially does not alter the architecture of the catalyst. To determine the elemental composition and spatial distribution, energy-dispersive X-ray spectroscopy mapping was performed for all of the catalysts. The results illustrated in Figure 1a,b,c,d confirm the successful loading of platinum (purple) and tin (yellow) species onto the CeO2 surface. Moreover, the RE dopants (orange), along with cerium (red) and oxygen (blue), were uniformly distributed throughout the PtSn/CeO2 catalyst. These results provide direct evidence that the doped RE cations were incorporated into the CeO2 lattice, likely forming a homogeneous single-phase solid solution. From the N2 adsorption–desorption isotherms and the derived pore-size distributions in Figure S2, all of the catalysts display type IV isotherms with H3-type hysteresis loops in P/P0 of 0.69-1.0 (Figure S2a), which could be assigned to mesopores following the IUPAC classification. Moreover, the hysteresis loops may have been induced by slit-shaped pores due to the aggregation of nanoparticles. As indicated in Figure S2b, all catalysts show a narrower pore-size distribution centered between 2.0 and 5.0 nm. The calculated BET surface area, pore volume, and average pore diameter based on the isotherms summarized in Table S1 could be applied to give quantitative comparisons between different samples. The PtSn/CeO2 catalyst exhibited a BET surface area of 49.0 m2·g-1, a pore volume of 0.105 cm3·g-1, and an average pore diameter of 10.0 nm. In comparison, RE-doped PtSn/CeO2 catalysts showed a clear increase in both BET surface area and pore volume. Among them, the PtSn/Nd-CeO2 catalyst possessed the highest values, with a BET surface area of 66.4 m2·g-1 and a pore volume of 0.137 cm3·g-1. Conversely, the average pore diameter of RE-doped catalysts was slightly reduced relative to the undoped counterpart, ranging from 9.5 to 9.1 nm, which was consistent with the refinement of the porous structure upon RE incorporation.
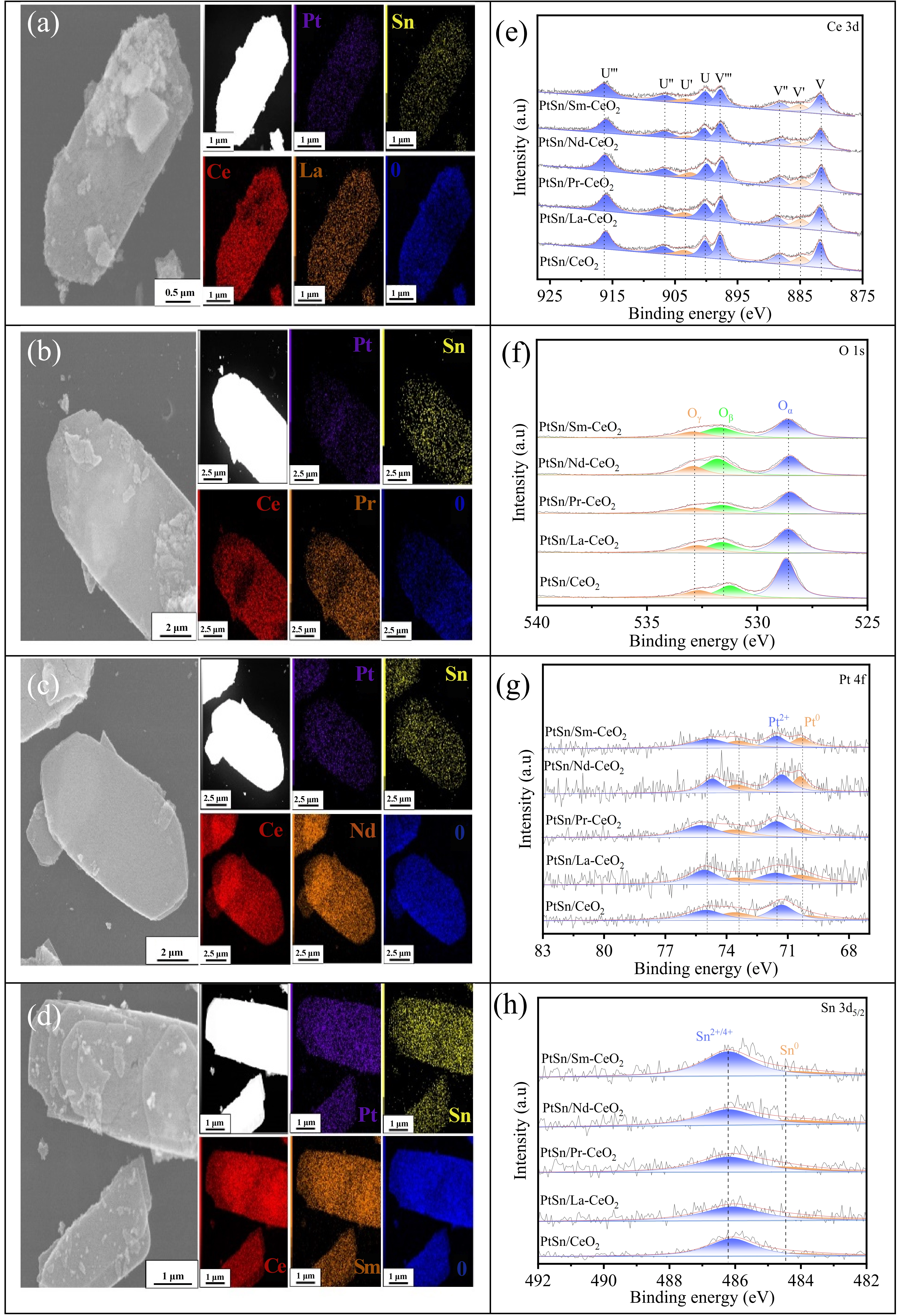
Figure 1. TEM images and STEM with EDS mapping(a-d), XPS of Ce 3d (e), O 1s (f), Pt 4f (g), and Sn 3d (h) over undoped or RE-doped PtSn/CeO2 catalysts. TEM: transmission electron microscopy; STEM: scanning transmission electron microscopy; EDS: energy-dispersive X-ray spectroscopy.
The structural properties of undoped and RE-doped PtSn/CeO2 catalysts were investigated using XRD, with the corresponding patterns presented in Figure S3a. The XRD pattern of the undoped PtSn/CeO2 catalyst displayed prominent diffraction at 2θ values of 28.54°, 33.12°, 47.54°, 56.34°, 59.13°, 69.34°, 76.59°, and 79.12°, corresponding to the (111), (200), (220), (311), (222), (400), (331), and (420) planes, respectively. All diffraction peaks were indexed to the cubic fluorite structure of CeO2 (Fm-3m space group, JCPDS card no. 34-0394, marked as ♦)[24]. Upon RE doping, the XRD patterns of all PtSn/RE-CeO2 catalysts retained only the characteristic peaks of the cubic CeO2 phase. However, a noticeable shift of the predominant (111) peak to a lower angle was observed, indicating a lattice expansion of the cubic phase (Figure S3b). This was reasonably attributed to the formation of a Ce1-xRExO2 solid solution as a result of the substitution of larger RE3+ cations (La3+: 0.110 nm, Pr3+: 0.113 nm, Nd3+: 0.113 nm, and Sm3+: 0.108 nm) for Ce4⁺ cations having a smaller radius of 0.097 nm[25,26]. Moreover, the formation of Ce1-xRExO2 solid solutions was also manifested from the lattice parameters calculated from XRD patterns, which were slightly increased from 0.541 nm for PtSn/CeO2 to 0.543, 0.542, 0.544, and 0.545 nm for La-, Pr-, Nd-, and Sm-doped samples, respectively. Considering the limitations of XRD as reflected by the slight variations in the characteristic diffractions, HRTEM tests of the representative PtSn/CeO2, PtSn/Nb-CeO2, and PtSn/Sm-CeO2 samples were conducted for the possible direct observation of the solid solutions, and the results are shown in Figure S4. In the TEM-FFT images, the distances from the center of the (111) diffraction spot to the spots corresponding to Nb- and Sm-doped samples were measured to be 3.147 nm-1 and 3.117 nm-1, respectively, which corresponded to lattice spacings of 0.3209 nm and 0.3177 nm. For the undoped sample, the (111) diffraction spot was located at a distance of 0.3227 nm-1 from the center, corresponding to a lattice spacing of 0.3099 nm. These results indicated that doping with Nb and Sm led to an expansion of the CeO2 lattice, consistent with the shift in diffraction peaks observed in the XRD patterns. Notably, no distinct XRD diffractions from either metallic Pt, platinum oxides (PtOx), or tin oxides (SnOx) were detected in any catalyst. This might be due to the high dispersion of these species and low loading or the amorphous nature of the active metal species, which cannot be detected by XRD.
To further probe the influence of RE doping on the lattice dynamics and defect structure, Raman spectroscopy was employed. As shown in Figure S5a, a principal peak at ~462 cm-¹ appeared for all of the catalysts. This was the characteristic F2g vibrational mode of the fluorite structure (Fm3m space group), i.e., the symmetric stretching vibration of Ce–O bonds. Moreover, a shoulder-like, broader but weaker band centered around 595 cm-1, enlarged in Figure S5b, was also observable as the defect-induced mode, which is commonly associated with the presence of oxygen vacancies within the lattice[27]. To quantitatively compare the relative concentration of these oxygen defects, the ratio for the intensity of the defect band to that of the principal F2g band (I595/I462) was calculated. The results in Figure S5b indicated that the promoted formation of oxygen vacancies by RE doping was obvious from the higher values of I595/I462 for the PtSn/RE-CeO2 catalysts relative to that of the undoped PtSn/CeO2 catalyst, i.e., in a decreasing order of PtSn/La-CeO2 (0.052) < PtSn/Pr-CeO2 (0.058) < PtSn/Sm-CeO2 (0.065) < PtSn/Nd-CeO2(0.074). Thus, the PtSn/Nd-CeO2 catalyst had the highest density of oxygen vacancies. It is well-established that lattice defects in CeO2-based oxides are intimately linked to crystallite size, where a reduction in particle dimensions typically promotes the formation of surface and sub-surface defects, thereby facilitating the generation of reactive oxygen species[28]. Consistent with this principle, the estimated crystallite sizes for the oxides within the PtSn/CeO2, PtSn/La-CeO2, PtSn/Pr-CeO2, PtSn/Sm-CeO2, and PtSn/Nd-CeO2 catalysts were calculated to be 17.8 nm, 15.2 nm, 14.5 nm, 13.9 nm, and 13.1 nm, respectively. This inverse correlation between crystallite size and the measured I595/I462 ratio strongly suggests that the RE doping strategy not only modified the electronic structure but also refined the oxide matrix. The resultant decrease in crystallite size provided a complementary pathway for augmenting the oxygen vacancy content, thereby enhancing the catalyst’s redox functionality.
To further investigate the influence of RE doping on the oxygen-vacancy content, XPS measurement was applied to characterize/analyze the chemical states of Ce and O species on the top surface of the reduced catalysts. As shown in Figure 1e, the Ce 3d XPS could be fitted to eight peaks from two pairs of spin orbits, in which V, V'', V''' and U, U'', U''' represented the characteristic peaks of Ce4+3d5/2 and Ce4+ 3d3/2, respectively, while V' and U' were the characteristic peaks of Ce3+ 3d5/2 and Ce3+3d3/2[29,30]. Based on the peak results, namely the changes in the peak areas of V' and U', it could be preliminarily determined that the content of Ce3+ on the catalyst surface was closely related to the type of dopant element. To conduct a quantitative comparison, the relative content of Ce3+ on different catalyst surfaces was calculated using the ratio of peak areas, that is, Ce3+/(Ce3+ + Ce4+), and the results are shown in Table S2. Obviously, RE doping increased the concentration of Ce3+ on the catalyst surface. Moreover, the concentration of Ce3+ on the catalyst surface was related to the type of doping element. Among them, the PtSn/Nd-CeO2 catalyst had the highest concentration of Ce3+. If part of Ce4+ in CeO2 was replaced by Ce3+, surface oxygen species were required for charge balance, which means a quantitative relationship was established between the Ce3+ content on the catalyst surface and the oxygen species content. For this purpose, the XPS spectrum of O 1s was analyzed (Figure 1f). According to the relevant literature reports of O 1s XPS, three oxygen species were identified on the surface of the catalyst, namely surface OH (Oγ, 529.0~529.4 eV), surface adsorbed oxygen (Oβ, 529.2~529.3 eV), and lattice oxygen (Oα, 532.1~533.6 eV)[31]. Compared with Oα, the catalytic oxidation activity of the high mobility Oβ was higher. The value of Oβ/(Oα + Oβ + Oγ) was often used to evaluate the concentration of surface oxygen vacancies (Ov). As shown in Table S2, the content of Oβ on the surface of each catalyst decreased in the following order of PtSn/Nd-CeO2 (44.6%) > PtSn/Sm-CeO2 (39.9%) > PtSn/Pr-CeO2 (36.7%) > PtSn/La-CeO2 (33.2%) > PtSn/CeO2 (24.1%), which was consistent with the variation pattern of Ce3+ concentration.
As shown in Table S3, the measured Pt contents by ICP-MS were 0.98 ± 0.02 wt.% for all of the catalysts, which were similar to those of the designed Pt loadings (1.0 wt.%). Based on the measured Pt contents and results of the H2 chemical adsorption, the calculated Pt dispersions for all of the catalysts are also provided in Table S3. Compared with the unmodified PtSn/CeO2, the RE-modification significantly increased the Pt dispersion, which increased in the order of PtSn/CeO2 (22.1%) < PtSn/La-CeO2 (24.6%) < PtSn/Pr-CeO2 (25.7%) < PtSn/Sm-CeO2 (28.5%) < PtSn/Nd-CeO2(30.4%). As for the reasons, on the one hand, the higher specific surface area of the RE-doped catalysts was favorable to disperse Pt species. On the other hand, the abundant defects on the surface of the Ce1-xRExO2 solid solution were helpful in anchoring Pt and improving its dispersion. Previous studies have demonstrated that surface oxygen vacancies play a pivotal role in determining the structural configuration of supported Pt species in heterogeneous catalysts[32]. The Pt 4f XPS spectra (Figure 1g) exhibited characteristic doublets of Pt 4f7/2 and Pt 4f5/2 centered at 72.1 eV and 75.4 eV, respectively, indicative of the coexistence of Pt0 (70.9 eV and 73.5 eV) and Pt2+ (71.7 eV and 75.5 eV) species[33]. Based on the peak areas of Pt0 and Pt2+, the relative content of Pt0 on the surface of different catalysts was calculated, and the results are shown in Table S4. Compared with the PtSn/CeO2 catalyst, the Pt0 content in the PtSn/RE-CeO2 catalyst was significantly increased. The content of Pt0 in different catalysts increased in the following order of PtSn/CeO2(30.5%) < PtSn/La-CeO2(34.8%) < PtSn/Pr-CeO2(36.7%) < PtSn/Sm-CeO2(37.8%) < PtSn/Nd-CeO2(38.9%), indicating that the electronic density of Pt gradually increased. Analysis of Sn 3d5/2 XPS spectra (Figure 1h) identified two predominant states of Sn0 (~484.8 eV) and Sn2+/Sn4+ (~486.4 eV)[34]. Quantitative evaluation yielded a decreased Sn0 concentration in the order of PtSn/Nd-CeO2 (17.2%) > PtSn/Sm-CeO2 (16.1%) > PtSn/Pr-CeO2 (14.9%) > PtSn/La-CeO2 (13.7%) > PtSn/CeO2 (11.3%). The same changing trend of Sn0 and electron-rich Pt0 species was beneficial for the formation of Pt-Sn alloy.
3.2 Catalytic performance
The results of CO2-ODP over undoped and RE-doped PtSn/CeO2 catalysts are summarized in Figure 2. As shown in Figure 2a, the initial propane conversion at a time on stream (TOS) of 5 min varied significantly, in the increasing order of PtSn/CeO2 (34.3%) < PtSn/La-CeO2 (41.1%) < PtSn/Pr-CeO2 (45.4%) < PtSn/Sm-CeO2 (48.0%) < PtSn/Nd-CeO2 (52.7%). In the case of the CO2 conversion (Figure 2b), it was clearly lower than that of the propane conversion, although the same changing pattern was still there, i.e., PtSn/CeO2 (12.3 %) < PtSn/La-CeO2 (20.9%) < PtSn/Pr-CeO2 (23.5%) < PtSn/Sm-CeO2 (31.2 %) < PtSn/Nd-CeO2 (26.4 %). Moreover, RE doping led to a modest but consistently improved propylene selectivity (Figure 2c), from 82.1% for PtSn/CeO2 to approximately 86.3% for the PtSn/RE-CeO2 catalysts. These results indicate that the CO2-ODP reaction did not occur stoichiometrically, and the non-oxidative dehydrogenation of propane (PDH) for producing propylene simultaneously happened as a side reaction, the extent of which strongly depended on the specific catalyst. To give an estimate, the selectivity of by-products was examined. As indicated from Figure 2d, the summed selectivity of hydrocarbons (CH4, C2H4, and C2H6) originated from cracking reactions was the highest over the undoped PtSn/CeO2 catalyst (9.3%), while it decreased over the RE-doped catalysts (6.8 to 4.2%), the lowest of which was observed over PtSn/Nd-CeO2 (4.2%). Furthermore, the selectivity of CO formed via side reactions (primarily from the dry reforming of propane (DRP)) rather than CO2-ODP is 11.3% for PtSn/CeO2, while it decreased to 6.2-7.8% for the RE-doped counterparts (Figure 2e). Thus, the enhanced propylene selectivity upon RE introduction was principally attributed to the suppression of side reactions, including cracking and DRP, and the extent of CO2-ODP relative to PDH was the highest over PtSn/Nd-CeO2 catalyst. Consequently, the initial propylene yield followed the decreasing order of PtSn/Nd-CeO2 (45.3%) > PtSn/Sm-CeO2 (41.3%) > PtSn/Pr-CeO2 (38.6%) > PtSn/La-CeO2 (33.7%) > PtSn/CeO2 (27.6%). Considering the clearly varied Pt dispersions over different catalysts (Table S3), the turnover frequency for the formation of propene (TOFpropylene) was calculated using the CO2-ODP results at a TOS of 5 min. As shown in Figure S6a, TOFpropylene decreased in the order of PtSn/Pr-CeO2 (0.359 s-1) ≈ PtSn/Nd-CeO2 (0.356 s-1) > PtSn/Sm-CeO2 (0.347 s-1) > PtSn/La-CeO2 (0.325 s-1) > PtSn/CeO2 (0.293 s-1). Considering the slight differences in TOFpropylene for all of the catalysts and the errors in measuring Pt dispersions, the intrinsic rate for the formation of propylene was not clearly different during CO2-ODP over all of the catalysts. In contrast, more significant variations of TOFCO2 were manifested from Figure S6b, with a decreasing order of PtSn/Nd-CeO2 (0.243 s-1) > PtSn/Sm-CeO2 (0.222 s-1) > PtSn/Pr-CeO2 (0.219 s-1) > PtSn/La-CeO2 (0.201 s-1) > PtSn/CeO2 (0.130 s-1). These results revealed that the modification of PtSn/CeO2 with RE clearly improved the Pt dispersion, and the activity of CO2-ODP was enhanced with the promoted CO2 activation by oxygen vacancies. This was consistent with the highest activity of PtSn/Nd-CeO2 for the formation of propylene along with the simultaneous conversion of CO2. In the case of catalyst stability, it was assessed by calculating the relative deactivation rate (RD), defined as RD = (X0-Xf)/X0, where X0 and Xf represented the propane conversions at 5 min and 200 min TOS, respectively. As illustrated in Figure 2f, all RE-doped catalysts exhibited significantly lower RD values than that of undoped PtSn/CeO2, confirming that RE incorporation enhanced catalytic stability. Among the investigated catalysts, PtSn/Nd-CeO2 demonstrated the highest stability, as revealed from the smallest RD value (48.2%). To estimate its long-term stability, CO2-ODP over the PtSn/Nd-CeO2 catalyst was operated for a TOS of 1000 min, and the results of the propane conversion and propylene selectivity are shown in Figure S7. The catalyst underwent a fast deactivation in the first 400 min, where the propane conversion changed from 52.7% to 10.7%, while the propylene selectivity remained at 85.4%. The propane conversion dropped slowly to 5.1% with extended TOS from 400 to 1000 min, while the propylene selectivity remained at 90.2%. The C3H8 conversion gradually decreased during the relatively longer catalytic test, which may have been due to the gradual accumulation of coke on the catalyst. The overall performance of the optimal PtSn/Nd-CeO2 catalyst was benchmarked against prominent catalysts reported in the literature for CO2-ODP. A comparison of catalyst names, reaction conditions, and the calculated propylene space-time yield (STY) is provided in Table S5. Remarkably, a propylene STY of 0.82 kg(C3H6)·kg(cat)-1·h-1 over PtSn/Nd-CeO2 was higher than those of the most reported Pt-based catalysts, provided comparable reaction conditions were taken into account. Furthermore, the CO2 conversion over PtSn/Nd-CeO2 remained consistently high throughout the stability test (31.6 to 15.7%), a merit that was important for the simultaneous conversion of CO2, a greenhouse gas, to valuable CO during propylene production.
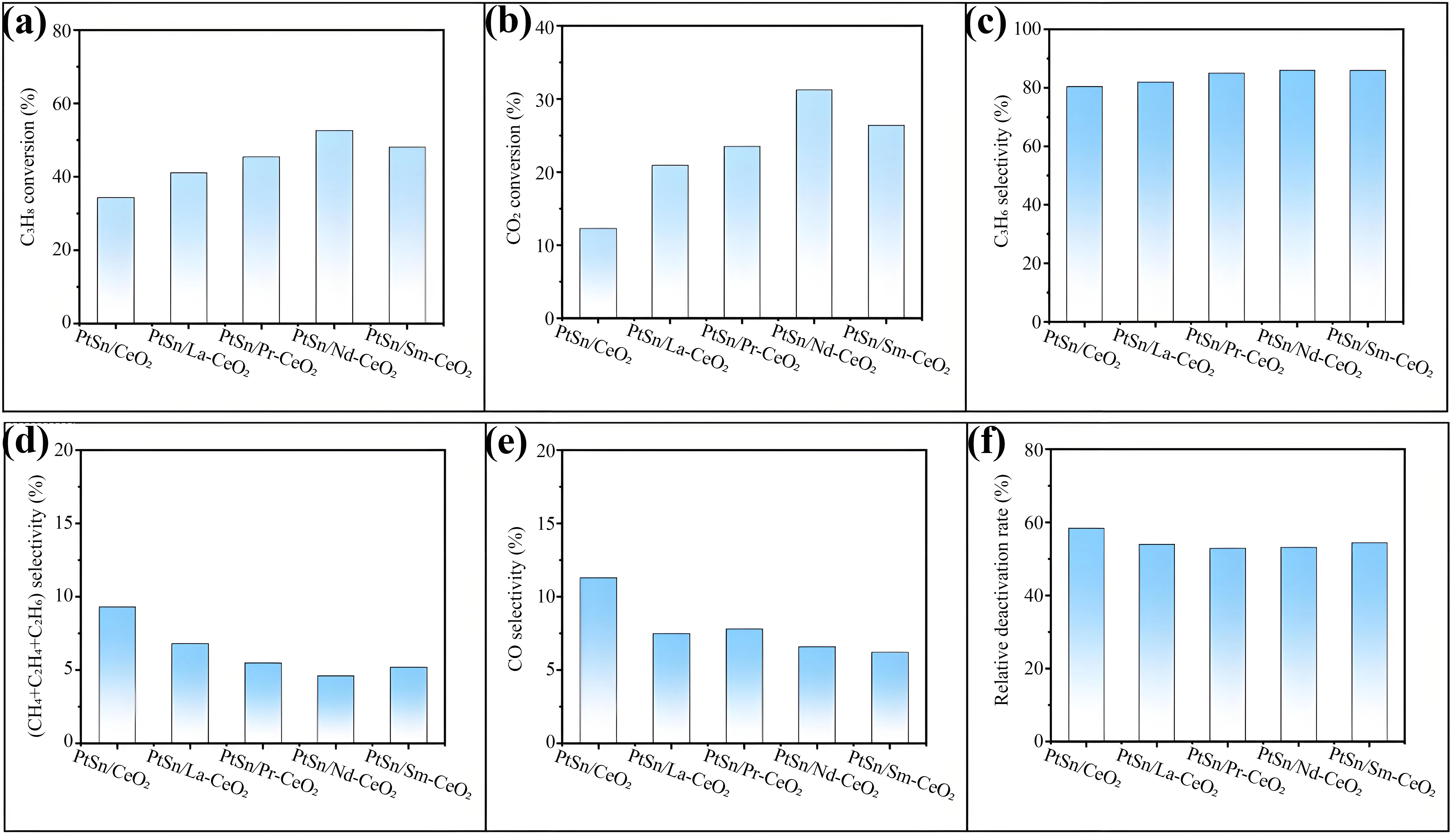
Figure 2. The initial C3H8 conversion (a), CO2 conversion (b), C3H6 selectivity (c), CH4+C2H4+C2H6 selectivity (d), selectivity of CO produced from C3H8 (e) at a time on stream of 5 min, and the relative deactivation rate (f) over undoped or RE-doped PtSn/CeO2 catalysts for CO2-ODP.
3.3 Key factors affecting catalytic activity and C3H6 selectivity
The characterization and catalytic evaluation results collectively demonstrated the RE doping significantly modulated the structure, chemical properties, and catalytic activity and selectivity of the PtSn/CeO2 catalyst for CO2-ODP. As shown in Figure 2, the PtSn/Nd-CeO2 catalyst exhibited the highest initial propane conversion (52.7%) and propylene yield (45.3%). In stark contrast, the pure CeO2 and Nd-CeO2 supports showed negligible activity (conversion < 2%), unequivocally identifying the PtSn species as the essential active component for this reaction (Figure S8). A positive linear correlation was observed between Pt dispersion and initial propane conversion across the catalyst series: higher Pt dispersion corresponded to improved catalytic activity (Figure 3a). Combined with the characterization data, this indicated that RE doping enhanced the specific surface area and surface oxygen vacancy concentration of the support, which in turn promoted Pt dispersion, a key factor contributing to the improved activity. The improved Pt dispersion by oxygen deficiency may be related to electronic and geometric interactions between Pt and RE-CeO2 carriers, similar to reported references[35,36]. Notably, while Pt dispersion increased from 22.1% for PtSn/CeO2 to 30.4% for PtSn/Nd-CeO2, the observed propane conversion (52.7%) significantly exceeded the value predicted solely from the increased active-site density (estimated at about 47.2%). This discrepancy suggested that factors beyond Pt dispersion played a crucial role in enhancing catalytic performance. According to the literature[18], CO2-ODP over PtSn/CeO2 likely followed a redox mechanism, where not only the number of active sites but also the rate of the redox cycle critically determines the overall activity. To validate this hypothesis, an additional PDH experiment was performed over the PtSn/Nd-CeO2 catalyst, and comparative results are given in Figure S9. Throughout the TOS of 200 min, the C3H8 conversion for CO2-ODP was significantly higher than that for PDH, indicating the positive role of CO2 in converting propane. If the CO2-ODP reaction followed the Mars–van-Krevelen (MvK) redox mechanism, oxygen defects over the catalysts participate in activating CO2 molecules. Thus, CO2 pulse experiments were conducted at the reaction temperature (550 °C) over pre-reduced catalysts. As shown in Figure 3b and Figure S10, CO2 was consumed with the simultaneous release of CO upon contact with all reduced catalysts, confirming the replenishment of oxygen vacancies over the reduced catalysts through activating CO2. The total amount of CO2 consumed during the consecutive pulses increased in the order of PtSn/CeO2 (70.3 μmol·g-1) < PtSn/La-CeO2 (158.2 μmol·g-1) < PtSn/Pr-CeO2 (178.4 μmol·g-1) < PtSn/Sm-CeO2 (194.2 μmol·g-1) < PtSn/Nd-CeO2(225.7 μmol·g-1), which aligned with the trend in oxygen vacancy concentration quantified by Ce3+/(Ce3+ + Ce4+) ratio, Oβ/(Oα + Oβ + Oγ) ratio, and Raman I595/I462 intensity ratio (Figure 3c). With these understandings, an MvK redox mechanism of CO2-ODP catalyzed by Nd-doped PtSn/CeO2 was proposed in Figure S11. The redox cycle starts with the propane dehydrogenation to propylene with the concurrent catalyst reduction, and the replenishment of oxygen vacancies over the reduced catalyst via the reduction of CO2 to CO completes the redox cycle. A strong linear correlation was observed between CO2 consumption and initial propane conversion (Figure 3d), indicating that the catalyst’s ability to activate CO2 and facilitate lattice oxygen replenishment was a key determinant of its activity. In terms of product selectivity, RE doping led to a slight but consistent increase in propylene selectivity. Analysis of by-product distribution revealed that RE incorporation effectively suppressed side reforming reactions. As supported by XPS results (Table S4), the formation of a defect-rich Ce1-xRExO2 solid solution promoted the formation of a PtSn alloy and increased the electron density of Pt. This electronic modification enhanced the catalyst’s ability to selectively activate C–H bonds in propane while maintaining relative inertness toward C–C bond cleavage, thereby improving propylene selectivity.
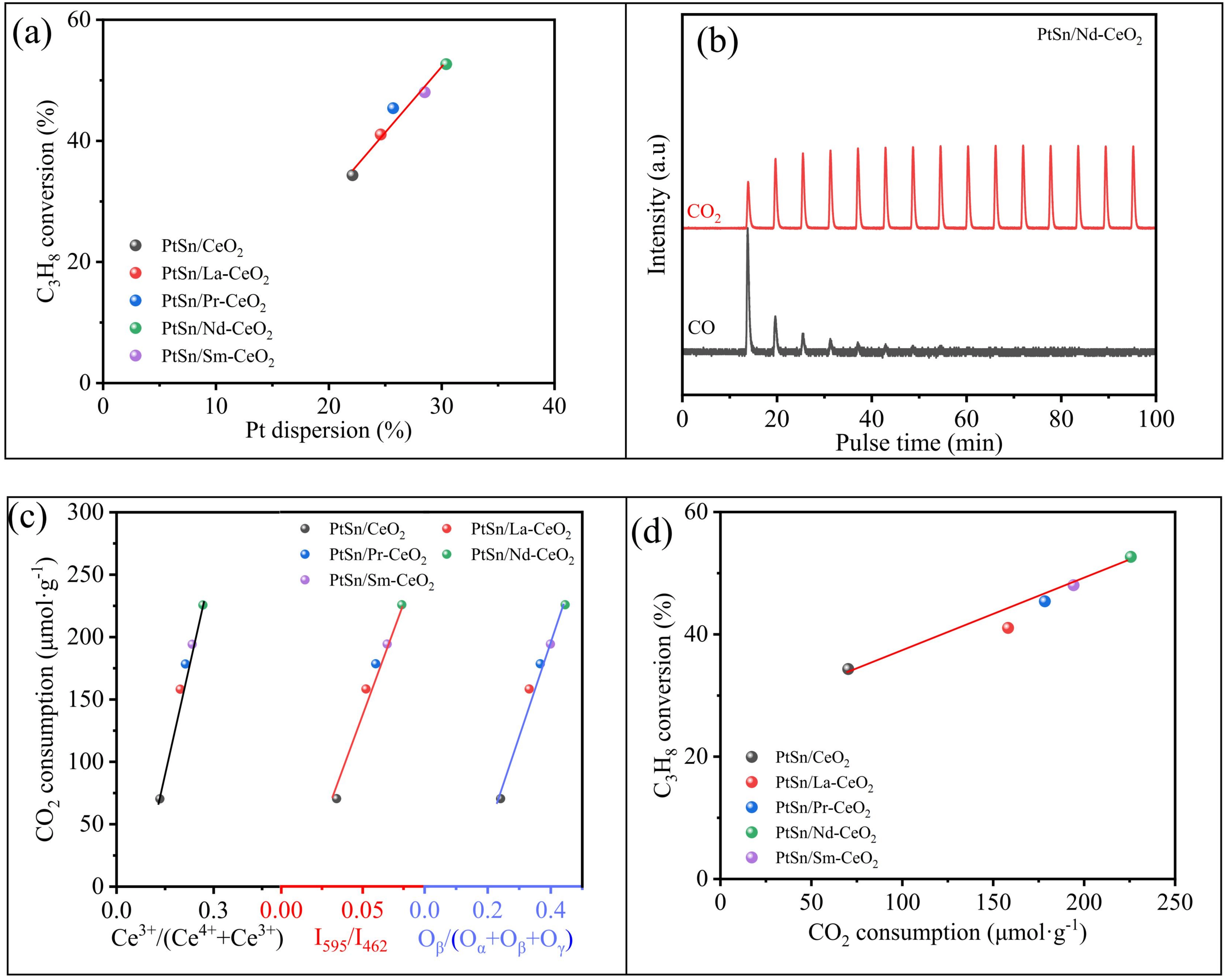
Figure 3. Correlation between the initial C3H8 conversion and Pt dispersion over undoped or RE-doped PtSn/CeO2 catalysts (a), representative transient MS responses of the consecutive CO2 pulses over the pre-reduced PtSn/Nd-CeO2 (b), the correlation of the Ce3+/(Ce3++Ce4+), I595/I462 and Oβ/(Oα+Oβ+Oγ) ratio with the cumulative amount of CO2 consumed during pulse experiments (c), and the correlation of the cumulative amount of CO2 consumed with the initial C3H8 conversion of CO2-ODP (d). CO2-ODP: oxidative dehydrogenation of propane with carbon dioxide.
3.4 Catalyst deactivation
The results in Figure 2f showed that as the reaction time increased, all the catalysts became deactivated, and the degree of deactivation was closely related to the type of dopant element. Generally speaking, catalyst deactivation of CO2-ODP is mainly related to the evolution of coke deposition and Pt species structure. As shown in Figure S12, the carbon balance over all catalysts remained above 95.6% throughout the entire CO2-ODP reaction, indicating a small amount of coke deposited over the catalysts. Raman analysis was conducted on the PtSn/CeO2 and PtSn/Nd-CeO2 catalysts after the reaction, and the results are shown in Figure S13a. After the reaction, the catalysts showed obvious signal values at the Raman shifts of 1345 cm-1 and 1600 cm-1, which could be attributed to the graphite carbon (D bond), representing the disordered structure, and the graphite carbon (G bond), representing the first-order scattering E2g mode[37,38]. To quantitatively characterize the degree of graphitization of the carbon deposits on the PtSn/CeO2 and PtSn/Nd-CeO2 catalysts, the ratio of D peak intensity (ID) to G peak intensity (IG) was calculated. The ID/IG of the PtSn/Nd-CeO2 catalyst was 0.74, which was much lower than the 0.92 of the PtSn/CeO2 catalyst. This indicates that the introduction of Nd significantly reduced the formation of graphitized carbon. According to relevant literature reports, the degree of graphitization of the carbon deposits produced by the CO2-RP reaction is significantly higher than that of the CO2-ODP reaction. Therefore, this may be due to the introduction of Nd into CeO2, which inhibited the occurrence of CO2-RP and other side reactions, thereby reducing the formation of graphitized carbon. To quantitatively analyze the carbon content in the catalysts, TG analysis was performed on the PtSn/CeO2 and PtSn/Nd-CeO2 catalysts after the reaction, as shown in Figure S13b. The catalysts showed two weight loss stages in the temperature range of 100-200 ℃ and 200-500 ℃, corresponding to the desorption of water on the catalyst surface and the combustion of carbon deposits[39]. The carbon content distribution of the PtSn/CeO2 and PtSn/Nd-CeO2 catalysts was 1.82% and 2.11%, respectively. The carbon generation rate was calculated and correlated with the relative deactivation rate, as shown in Figure S13c. The carbon generation rate of the PtSn/Nd-CeO2 catalyst was much higher than that of the PtSn/CeO2 catalysts. At the same time, the carbon generation rate was inversely proportional to the relative deactivation rate, indicating that carbon deposition may not be the main cause of catalyst deactivation. Therefore, XPS characterization was performed on the PtSn/Nd-CeO2 catalyst after the reaction. As shown in Figure S13d, compared with the fresh catalyst, the binding energy of Pt on the reacted catalyst shifted to a higher binding energy. The Pt0 species at the binding energies of 71.3 eV and 74.3 eV basically disappeared, which may be due to the segregation of Pt species from the PtSn alloy. The XRD spectrum of the spent PtSn/Nd-CeO2 catalyst shows a clear diffraction of platinum species at 2θ values of 39.56°, further confirming this conclusion (Figure S13e). To further verify the causes of the catalyst deactivation, reaction-regeneration experiments were conducted by using optimal PtSn/Nd-CeO2 catalyst. As shown in Figure S14a, the propane conversion was fully restored, while the propylene selectivity was slightly decreased after the first regeneration. However, as the number of regeneration cycles increases further, the propane conversion at the initial and end of the test showed a slight decrease. Combining the XRD results (Figure S14b) of the spent catalyst after reaction-regeneration tests, the change of the Pt species as the key factor of the catalyst deactivation was further confirmed. Thus, the structural evolution of Pt species on the catalyst after the reaction may be a key factor affecting the catalyst deactivation.
4. Conclusion
From the perspectives of enhancing the activation of the C=O bond in CO2 molecules, selectively activating the C-H bond in propane molecules, and the synergy between the two, a catalyst design concept was proposed to utilize different RE dopants to modulate the oxygen vacancy concentration and Pt electronic density of the PtSn/CeO2 catalyst. A systematic study was conducted on the effects of different RE dopants on the catalyst structure and the performance of CO2-ODP. The following conclusions were obtained: the cubic fluorite structure solid solution Ce1-xRExO2 was formed by doping different RE into CeO2 using the co-precipitation method. The dopants have a significant influence on the textural properties of the PtSn/CeO2 catalyst. The surface Pt dispersion and oxygen vacancy content decreased in the order of PtSn/Nd-CeO2 > PtSn/Sm-CeO2 > PtSn/Pr-CeO2 > PtSn/La-CeO2 > PtSn/CeO2. Different RE dopants significantly affected the performance of PtSn/CeO2 in catalyzing CO2-ODP. RE doping significantly increased the initial propane and CO2 conversion of the catalyst, but had a slight inhibitory effect on the selectivity of propylene. The propylene yield decreased in the order of PtSn/Nd-CeO2 > PtSn/Sm-CeO2 > PtSn/Pr-CeO2 > PtSn/La-CeO2 > PtSn/CeO2. The results of structural performance correlation analysis indicate that the oxygen vacancy content and Pt dispersion are the key factors affecting the activity of the RE-doped PtSn/CeO2 catalyst in catalyzing CO2-ODP, and the evolution of the Pt species structure is the key factor leading to catalyst deactivation. Therefore, based on the regularity of the RE modulation of the surface oxygen vacancy concentration and Pt dispersion of PtSn/ CeO2 catalysts, it is expected that better-performing Pt-based CO2-ODP catalysts are obtained.
Supplementary materials
The supplementary material for this article is available at: Supplementary materials.
Authors contribution
Peng C: Writing-original draft, data curation, formal analysis, methodology, investigation.
Yan M: Methodology, investigation.
Liu ZW: Writing-review & editing, data curation, formal analysis, supervision, funding acquisition.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
The data and materials supporting the findings of this study are available from the corresponding author upon reasonable request.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 22338010).
Copyright
© The Author(s) 2026.
References
-
2. Wang H, Xiang Z, Zhang X, Su Z, Chen R, Chen T. Rare-earth-modified magnesium aluminate spinel-supported PtSn nanoparticles for propane dehydrogenation. ACS Catal. 2025;15(17):14682-14692.[DOI]
-
4. Ji J, Fan G, Zheng L, Li F. Confinement of atomically dispersed Ptδ+ sites in zinc-incorporated Silicalite-1 zeolite for enhanced propane dehydrogenation. ACS Catal. 2025;15(7):5858-5875.[DOI]
-
5. Monai M, Gambino M, Wannakao S, Weckhuysen BM. Propane to olefins tandem catalysis: A selective route towards light olefins production. Chem Soc Rev. 2021;50(20):11503-11529.[DOI]
-
6. Jiang X, Sharma L, Fung V, Park SJ, Jones CW, Sumpter BG, et al. Oxidative dehydrogenation of propane to propylene with soft oxidants via heterogeneous catalysis. ACS Catal. 2021;11(4):2182-2234.[DOI]
-
7. Nowicka E, Reece C, Althahban SM, Mohammed KMH, Kondrat SA, Morgan DJ, et al. Elucidating the role of CO2 in the soft oxidative dehydrogenation of propane over ceria-based catalysts. ACS Catal. 2018;8(4):3454-3468.[DOI]
-
8. Fan L, Wang S, Li W, Zhang L, Huang K. Influence of Ce doping on the activity of Ce-Cr/S-1 catalysts for the oxidative dehydrogenation of propane with CO2. Eur J Inorg Chem. 2025;28(9):e202400790.[DOI]
-
9. Dou J, Funderburg J, Yang K, Liu J, Chacko D, Zhang K, et al. CexZr1-xO2-supported CrOx catalysts for CO2-assisted oxidative dehydrogenation of propane─probing the active sites and strategies for enhanced stability. ACS Catal. 2023;13(1):213-223.[DOI]
-
10. Xing F, Nakaya Y, Yasumura S, Shimizu KI, Furukawa S. Ternary platinum–cobalt–indium nanoalloy on ceria as a highly efficient catalyst for the oxidative dehydrogenation of propane using CO2. Nat Catal. 2022;5(1):55-65.[DOI]
-
11. Liu Q, Li J, Zhao Z, Gao M, Kong L, Liu J, et al. Synthesis, characterization, and catalytic performances of potassium-modified molybdenum-incorporated KIT-6 mesoporous silica catalysts for the selective oxidation of propane to acrolein. J Catal. 2016;344:38-52.[DOI]
-
12. Wang L, Zhang HB, Hu R, Ge HQ, Song YH, Yang GQ, et al. Bridging the structural gap of supported vanadium oxides for oxidative dehydrogenation of propane with carbon dioxide. EES Catal. 2024;2(5):1126-1138.[DOI]
-
13. Xu B, Zheng B, Hua W, Yue Y, Gao Z. Support effect in dehydrogenation of propane in the presence of CO2 over supported gallium oxide catalysts. J Catal. 2006;239(2):470-477.[DOI]
-
15. Zhai P, Xie Z, Huang E, Aireddy DR, Yu H, Cullen DA, et al. CO2-mediated oxidative dehydrogenation of propane enabled by Pt-based bimetallic catalysts. Chem. 2023;9(11):3268-3285.[DOI]
-
16. Xing F, Ma J, Shimizu KI, Furukawa S. High-entropy intermetallics on ceria as efficient catalysts for the oxidative dehydrogenation of propane using CO2. Nat Commun. 2022;13:5065.[DOI]
-
17. Wang L, Yang GQ, Ren X, Liu ZW. CeO2-promoted PtSn/SiO2 as a high-performance catalyst for the oxidative dehydrogenation of propane with carbon dioxide. Nanomaterials. 2022;12(3):417.[DOI]
-
18. Yang GQ, Ren X, Kondratenko VA, Zhang HB, Kondratenko EV, Liu ZW. Promotional nature of Sn on Pt/CeO2 for the oxidative dehydrogenation of propane with carbon dioxide. Nano Res. 2023;16(5):6237-6250.[DOI]
-
19. Peng C, Yang G, Ge H, Liu Z. Morphology engineered PtSn/CeO2 catalysts for oxidative dehydrogenation of propane with carbon dioxide. J Rare Earths. 2025.[DOI]
-
20. Wang H, Tsilomelekis G. Catalytic performance and stability of Fe-doped CeO2 in propane oxidative dehydrogenation using carbon dioxide as an oxidant. Catal Sci Technol. 2020;10(13):4362-4372.[DOI]
-
21. Wang H, Yang GQ, Song YH, Liu ZT, Liu ZW. Defect-rich Ce1-xZrxO2 solid solutions for oxidative dehydrogenation of ethylbenzene with CO2. Catal Today. 2019;324:39-48.[DOI]
-
23. Rushton MJD, Chroneos A. Impact of uniaxial strain and doping on oxygen diffusion in CeO2. Sci Rep. 2014;4:6068.[DOI]
-
24. Li H, Meng F, Gong J, Fan Z, Qin R. Structural, morphological and optical properties of shuttle-like CeO2 synthesized by a facile hydrothermal method. J Alloys Compd. 2017;722:489-498.[DOI]
-
25. Rangaswamy A, Sudarsanam P, Reddy BM. Rare earth metal doped CeO2-based catalytic materials for diesel soot oxidation at lower temperatures. J Rare Earths. 2015;33(11):1162-1169.[DOI]
-
26. Xu B, Yang H, Zhang Q, Yuan S, Xie A, Zhang M, et al. Design and Synthesis of Sm, Y, La and Nd-doped CeO2 with a broom-like hierarchical structure: A photocatalyst with enhanced oxidation performance. ChemCatChem. 2020;12(9):2638-2646.[DOI]
-
27. Lee J, Ryou Y, Chan X, Kim TJ, Kim DH. How Pt interacts with CeO2 under the reducing and oxidizing environments at elevated temperature: The origin of improved thermal stability of Pt/CeO2 compared to CeO2. J Phys Chem C. 2016;120(45):25870-25879.[DOI]
-
29. Jiang F, Wang S, Liu B, Liu J, Wang L, Xiao Y, et al. Insights into the influence of CeO2 crystal facet on CO2 hydrogenation to methanol over Pd/CeO2 catalysts. ACS Catal. 2020;10(19):11493-11509.[DOI]
-
30. Peng R, Sun X, Li S, Chen L, Fu M, Wu J, et al. Shape effect of Pt/CeO2 catalysts on the catalytic oxidation of toluene. Chem Eng J. 2016;306:1234-1246.[DOI]
-
31. Huang H, Dai Q, Wang X. Morphology effect of Ru/CeO2 catalysts for the catalytic combustion of chlorobenzene. Appl Catal B Environ. 2014;158:96-105.[DOI]
-
32. Lee J, Ryou Y, Kim J, Chan X, Kim TJ, Kim DH. Influence of the defect concentration of ceria on the Pt dispersion and the CO oxidation activity of Pt/CeO2. J Phys Chem C. 2018;122(9):4972-4983.[DOI]
-
33. Gao Y, Wang W, Chang S, Huang W. Morphology effect of CeO2 support in the preparation, metal–support interaction, and catalytic performance of Pt/CeO2 catalysts. ChemCatChem. 2013;5(12):3610-3620.[DOI]
-
34. Motagamwala AH, Almallahi R, Wortman J, Igenegbai VO, Linic S. Stable and selective catalysts for propane dehydrogenation operating at thermodynamic limit. Science. 2021;373(6551):217-222.[DOI]
-
35. Pan YX, Sun ZQ, Cong HP, Men YL, Xin S, Song J, et al. Photocatalytic CO2 reduction highly enhanced by oxygen vacancies on Pt-nanoparticle-dispersed gallium oxide. Nano Res. 2016;9(6):1689-1700.[DOI]
-
36. Tovt A, Bagolini L, Dvořák F, Tran ND, Vorokhta M, Beranová K, et al. Ultimate dispersion of metallic and ionic platinum on ceria. J Mater Chem A. 2019;7(21):13019-13028.[DOI]
-
37. Nie R, Shi J, Du W, Ning W, Hou Z, Xiao FS. A sandwich N-doped graphene/Co3O4 hybrid: An efficient catalyst for selective oxidation of olefins and alcohols. J Mater Chem A. 2013;1(32):9037.[DOI]
-
38. Fan X, Liu D, Sun X, Yu X, Li D, Yang Y, et al. Mn-doping induced changes in Pt dispersion and PtxMny alloying extent on Pt/Mn-DMSN catalyst with enhanced propane dehydrogenation stability. J Catal. 2020;389:450-460.[DOI]
-
39. Wang J, Song YH, Yuan EH, Liu ZT, Liu ZW. Elucidating the support-size effect on the catalytic stability of CrOx/Silicalite-1 for oxidative dehydrogenation of propane with CO2. Catal Lett. 2023;153(3):790-804.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Peng C, Yan M, Liu ZW. Regulating oxygen defects over CeO2 via rare earth oxide doping for Pt-catalyzed oxidative dehydrogenation of propane with carbon dioxide. Smart Mater Devices. 2026;2:202604. https://doi.org/10.70401/smd.2026.0029
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Peng C, Yan M, Liu ZW. Regulating oxygen defects over CeO2 via rare earth oxide doping for Pt-catalyzed oxidative dehydrogenation of propane with carbon dioxide. Smart Mater Devices. 2026;2:202604. https://doi.org/10.70401/smd.2026.0029
copy
Share Link
copy