Recent developments in dark photocatalytic hydrogen production over smart catalysts
*Correspondence to:
Yong Ding, State Key Laboratory of Natural Product Chemistry, Key Laboratory of Advanced Catalysis of Gansu Province, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, Gansu, China.
E-mail: dingyong1@lzu.edu.cn
Smart Mater Devices. 2026;2:202609. 10.70401/smd.2026.0032
Received: February 26, 2026Accepted: April 14, 2026Published: April 22, 2026
This article belongs to the Special lssue Smart Porous Materials and Catalysis
This manuscript is made available in its unedited form to allow early access to the
reported findings. Further editing will be completed before final publication. As
such,
the content may include errors, and standard legal disclaimers are applicable.
Abstract
Widespread application of solar-driven hydrogen production is hampered by two major challenges: the safety risks associated with
Keywords
Dark photocatalytic hydrogen production, photoinduced charge separation, electron storage, persistent catalysis, semiconductor
1. Introduction
Under the global drive toward carbon neutrality and the growing urgency to address fossil energy depletion, hydrogen energy has emerged as a pivotal clean energy carrier, thanks to its high energy density and zero-emission characteristics[1-7]. Photocatalytic hydrogen evolution has long been regarded as a green pathway to link solar utilization and hydrogen production[8-15]. However, it still faces a critical bottleneck: the intermittency of solar energy. When light ceases, photogenerated electron-hole pair separation halts, and hydrogen production stops entirely. This light dependence not only affects hydrogen production output but also raises costs for subsequent storage and transportation, creating an urgent need for strategies that enable solar storage by day and hydrogen production at any time.
Against this backdrop, dark photocatalytic hydrogen evolution has emerged as a transformative solution, integrating solar capture, energy storage, and dark-state hydrogen production into a single system[16-29]. Unlike traditional photocatalytic hydrogen evolution, dark photocatalytic hydrogen evolution achieves temporal and spatial decoupling of solar energy and hydrogen production. Under light irradiation, semiconductors absorb photons to generate electrons, which are separated and stored via structural design
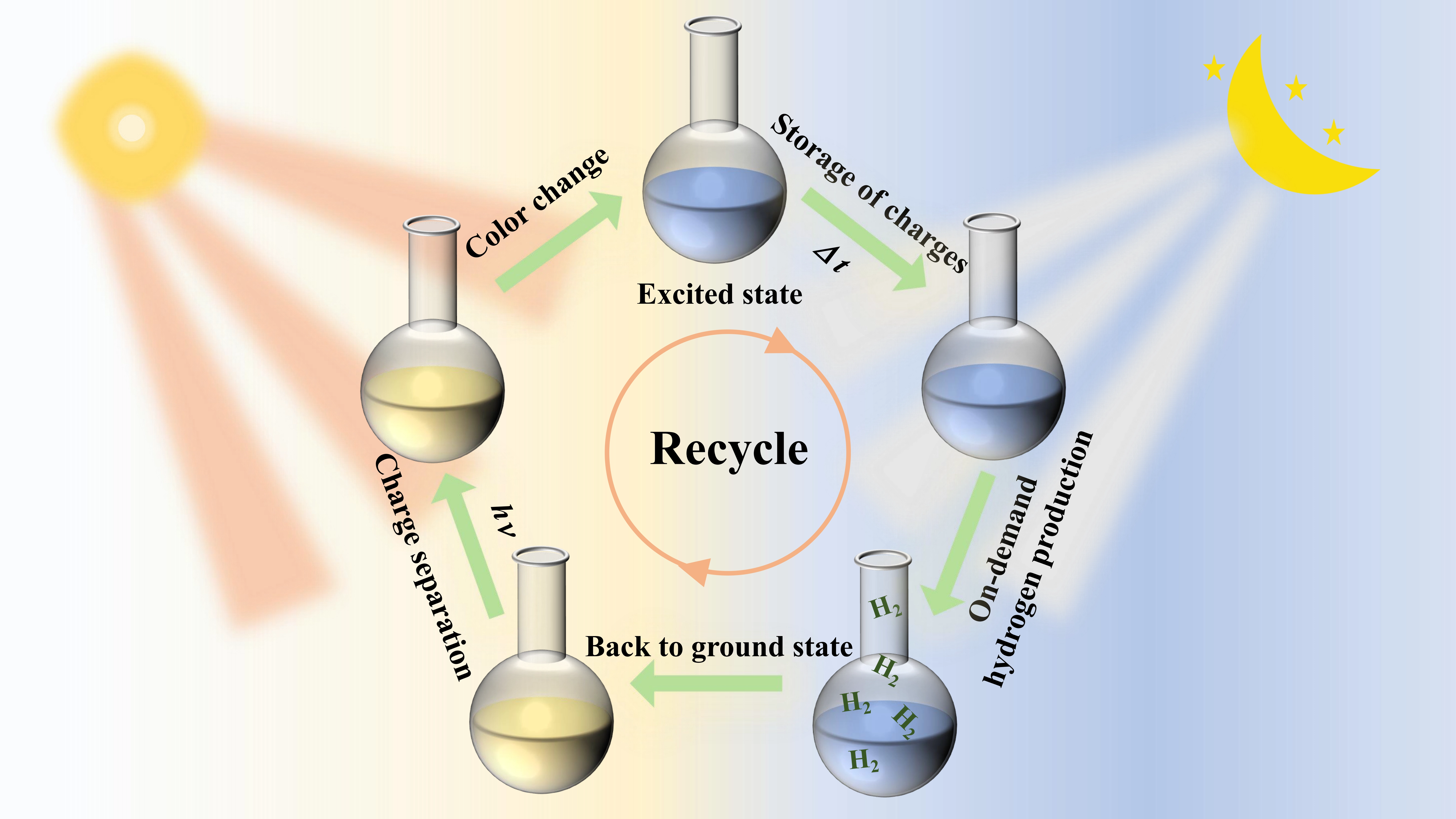
Scheme 1. The schematic diagram for dark photocatalytic hydrogen evolution mechanism.
In recent years, dark photocatalytic hydrogen evolution research has seen steady progress, with diverse material systems developed to support electron storage and dark hydrogen evolution (Figure 1), including carbon nitrides[16], polyoxometalates (POMs)[22],
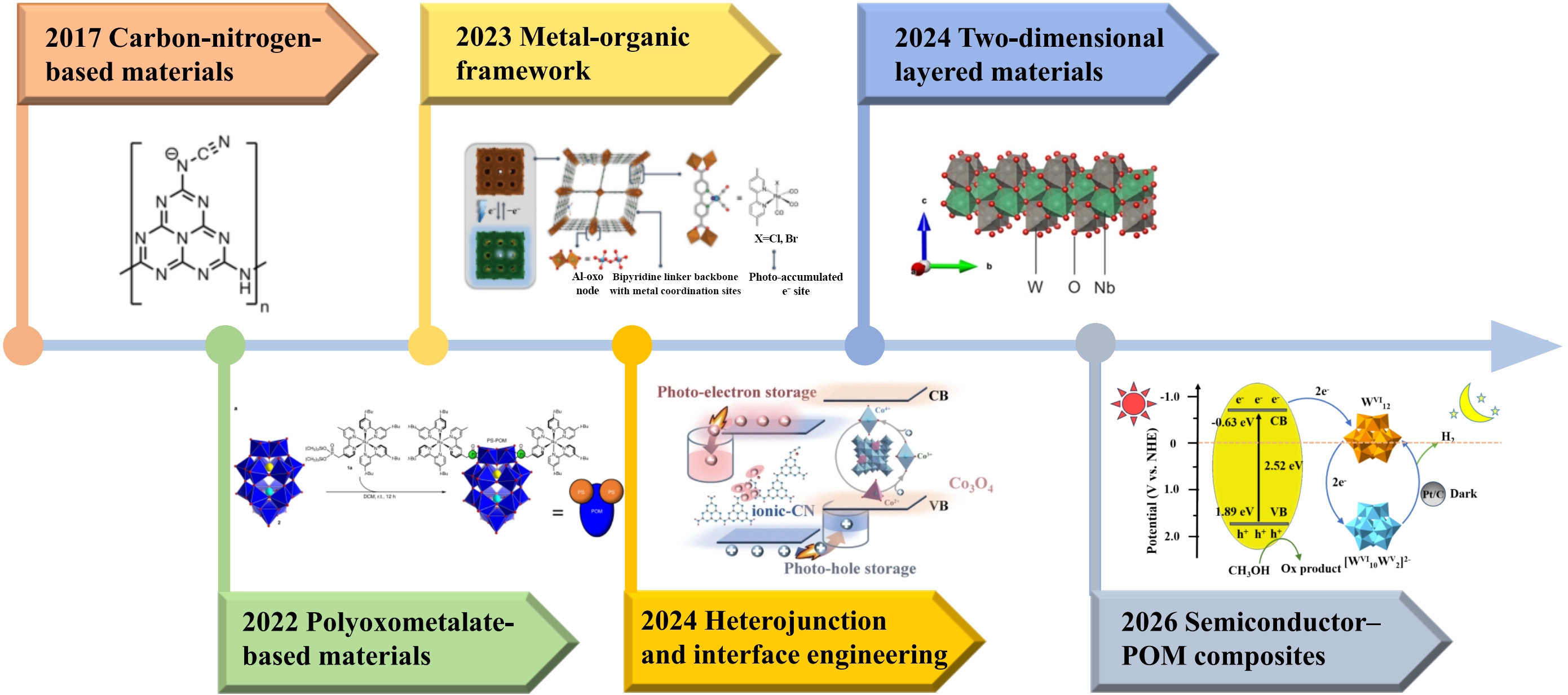
To synthesize these advances and guide future research, this review focuses on the core of dark photocatalytic hydrogen evolution, electron storage, by systematically summarizing progress in dark photocatalytic hydrogen evolution. It avoids a narrow focus on single materials or overly technical details, instead centering on the “structure-performance-mechanism” relationship across key material systems. The review first elaborates on dark photocatalytic hydrogen evolution’s basic principles and performance metrics, then classifies and discusses core material systems, analyzes factors influencing performance and corresponding regulation strategies, and finally outlines current challenges and future directions. By organizing these insights around electron storage, the review aims to provide a clear, holistic view of dark photocatalytic hydrogen evolution and inspire innovations to overcome its existing limitations.
2. The Basic Mechanism of Dark Light Catalysis
2.1 Separation and storage of photogenerated charges
The storage of photogenerated electrons is achieved primarily through intrinsic “electron traps” or integrated “electron reservoirs” within the material design. In carbon nitride-based materials, functionalization with cyanamide groups (e.g., in NCN-CNx or K+-poly (heptazine imide))[16] creates effective sites for electron localization. Upon irradiation in the presence of a sacrificial electron donor, long-lived radical species are formed, with lifetimes exceeding 12 hours (Figure 2a). Electron paramagnetic resonance (EPR) spectroscopy and computational studies indicate that the unpaired electron is localized on a single heptazine unit, stabilized by the cyanamide group and charge-balancing cations, endowing the system with a highly reductive character (Figure 2b). Furthermore, optimizing the crystallinity and cyanamide concentration through molten-salt synthesis can enhance this electron-storage capability. An optimal concentration balances efficient electron trapping against the detrimental formation of recombination centers.
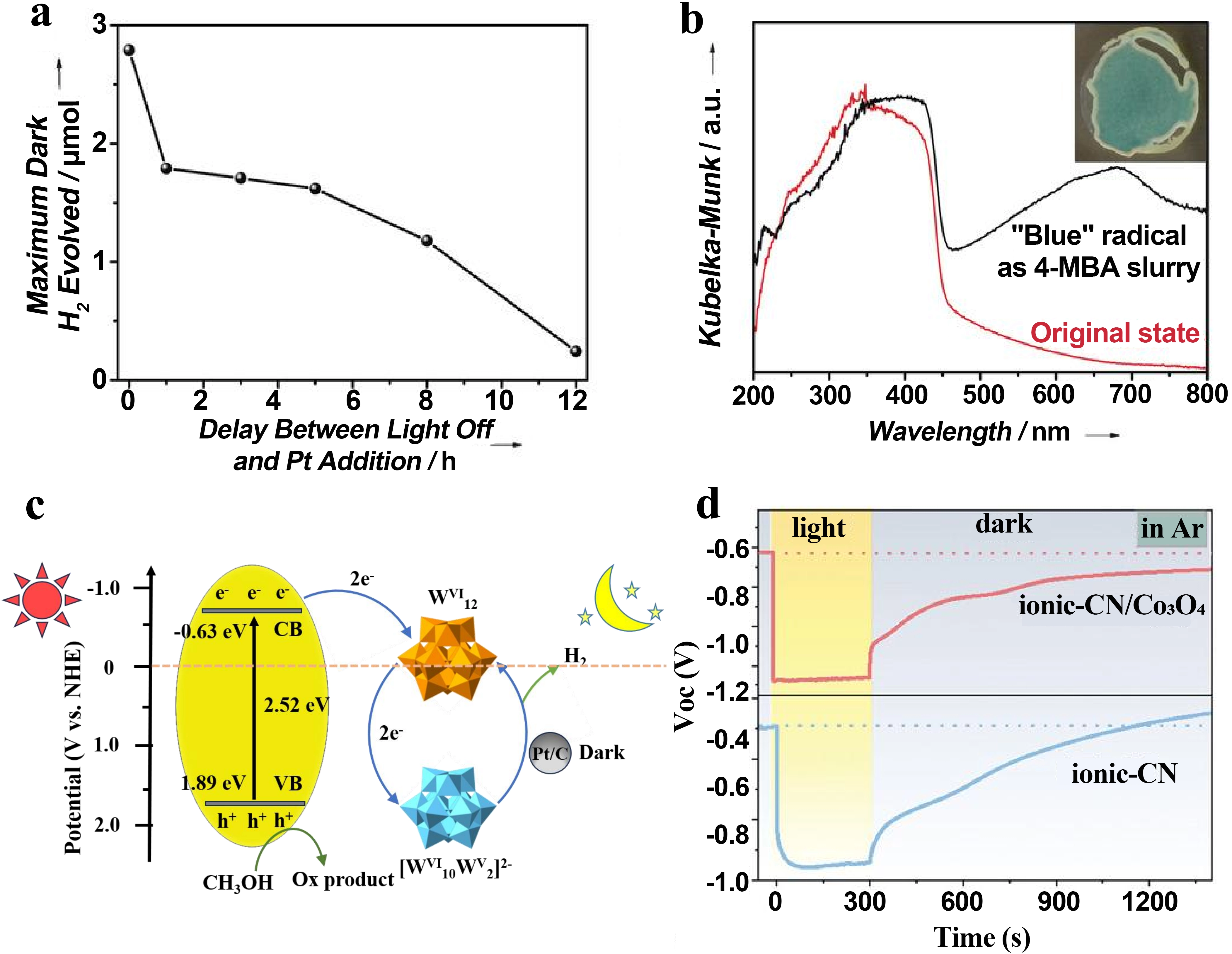
Figure 2. (a) Maximum dark hydrogen yield as a function of the delay time between light cessation and Pt colloid injection[16]; (b) UV-vis diffuse reflectance spectra of NCN-CN before and after irradiation[16]; (c) Semiconductor/POM composites. Republished with permission from[29]; (d) Open circuit voltage decay curves of ionic-CN and
POMs represent another major class of electron storage media due to their well-defined structures and remarkable capacity for reversible multi-electron redox reactions[29]. In composite systems, POMs such as ammonium metatungstate can accept electrons from an excited semiconductor like graphitic carbon nitride. Efficient electron transfer and storage, evidenced by the formation of heteropoly blue species (e.g., W5+), are facilitated by favorable band alignment and robust electrostatic self-assembly between oppositely charged components (Figure 2c). Beyond these, heterojunction engineering and interface design also promote charge separation and intermediate storage. For instance, bipolar charge storage junctions formed between ionic carbon nitride and metal oxides can drive directional electron transfer and temporary trapping at the interface (Figure 2d)[26].
Two-dimensional layered materials may stabilize electrons through polaron formation aided by ion intercalation (Figure 3a)[23]. The performance of electron storage is governed by multiple factors, including the intrinsic material properties such as crystallinity and defect concentration, the interfacial structure governing charge transfer, and external conditions like solution pH and light intensity. The regulatory effect of external experimental conditions on charge storage performance cannot be ignored. The pH value of the solution changes the accessibility of storage sites by affecting the surface charge state of materials. For the g-C3N4/(NH4)6H2W12O40 system, the protonation degree of the g-C3N4 surface is the highest at pH = 1.5, and the electrostatic interaction with W12 is the strongest (Figure 3b)[29]. The type of sacrificial agent determines the efficiency of hole quenching, thereby affecting the overall quantum efficiency of photoreduction. In the study by Amthor et al., when ascorbic acid (Asc) is used as the sacrificial agent, the quantum efficiency of photoreduction of PS-POM (Φ = 0.25%) is significantly higher than that when triethanolamine (TEOA) is used
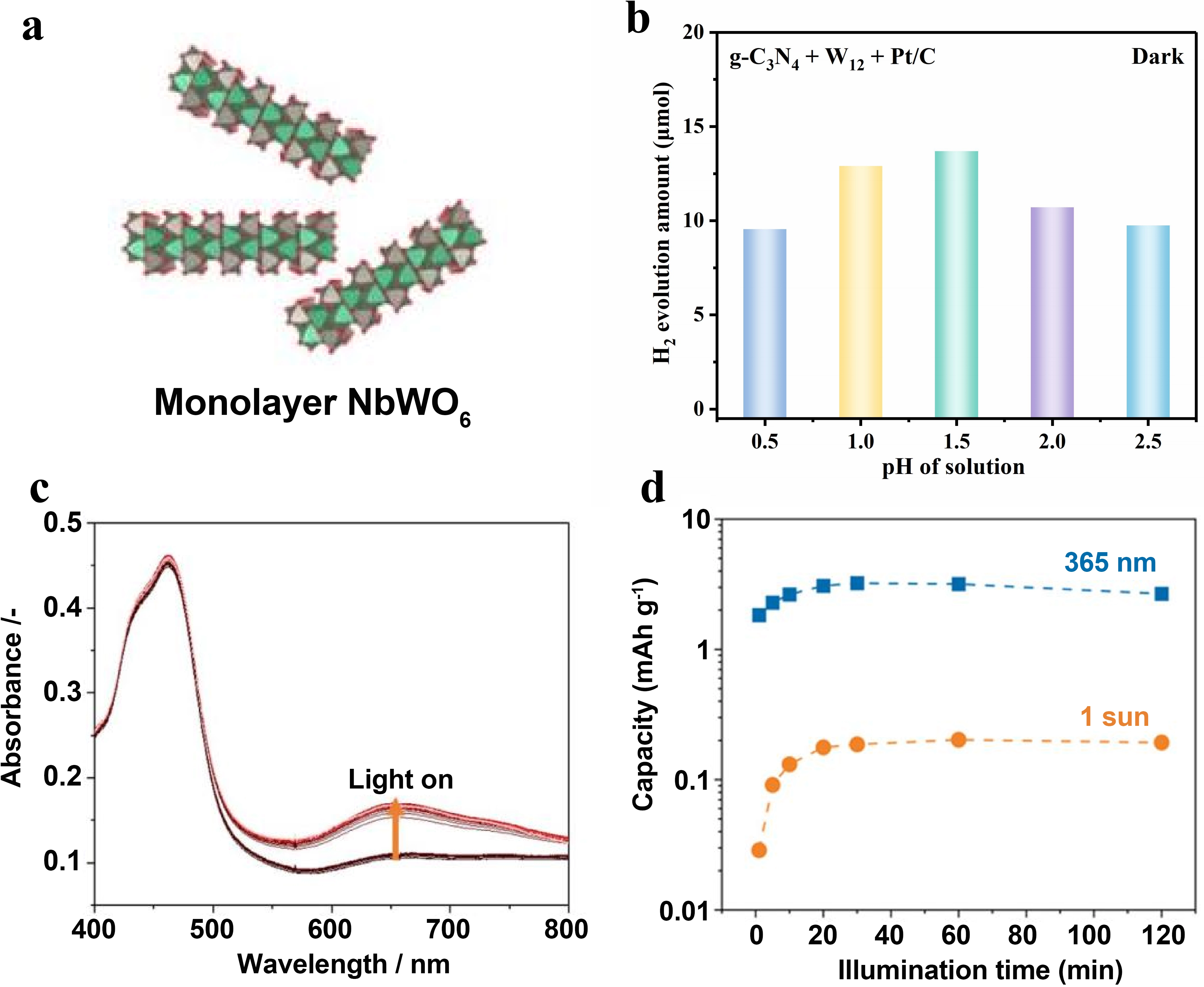
Figure 3. (a) Schematic illustration of 2D NbWO6 nanosheets[23]; (b) Dark H2 evolution activity under various pH conditions. Republished with permission from[29];
2.2 Charge release and catalytic reactions in the dark state
The ultimate goal of dark photocatalysis is the controlled and efficient release of stored electrons to drive proton reduction, achieving hydrogen production independent of illumination. This dark reaction is typically triggered by introducing a catalytic site for proton reduction, most commonly a hydrogen evolution co-catalyst[51-55].
In a charged system, these co-catalysts provide a pathway for the stored electrons to reduce protons. As demonstrated in the
The hydrogen evolution reactions (HER) pathway in the dark follows the fundamental steps of proton reduction, albeit with electrons supplied from the storage reservoirs rather than directly from a photoexcited state. Electrons migrate from storage sites
Different charge release mechanisms exhibit significant differences in applicable scenarios and efficiency. The noble metal
For practical application, the cyclic stability of the entire system is important. This involves the stability of both the electron storage material and the catalyst over repeated charging-discharging cycles. Promisingly, some systems, like the g-C3N4/W12 composite, show nearly constant dark activity over multiple cycles, indicating good stability of the POM storage unit. However, challenges remain, including the potential degradation of functional groups in carbon nitrides and the reliance on scarce noble metal co-catalysts for high performance. Developing efficient, stable, and earth-abundant alternatives to platinum and engineering robust interfaces between storage materials and co-catalysts are critical directions for future research aimed at enabling scalable implementation of dark photocatalytic hydrogen production.
3. Key Material System and Design Strategy
3.1 Mechanisms and structural design of typical dark photocatalytic systems
Figure 4 systematically compares the charge storage-release mechanisms of six representative material systems, highlighting both their common operating principles and structural specificities[16,17,22,23,26,29]. All systems operate via a core “light excitation-charge separation-storage-dark release-hydrogen evolution” process. Under illumination, semiconductors or light-absorbing units generate electron-hole pairs. Holes are rapidly quenched by sacrificial agents to prevent recombination, while electrons are stored at specific sites within the material. In the subsequent dark phase, these stored electrons are released in a controlled manner to drive proton reduction, thereby achieving the spatiotemporal decoupling of light absorption and HER.
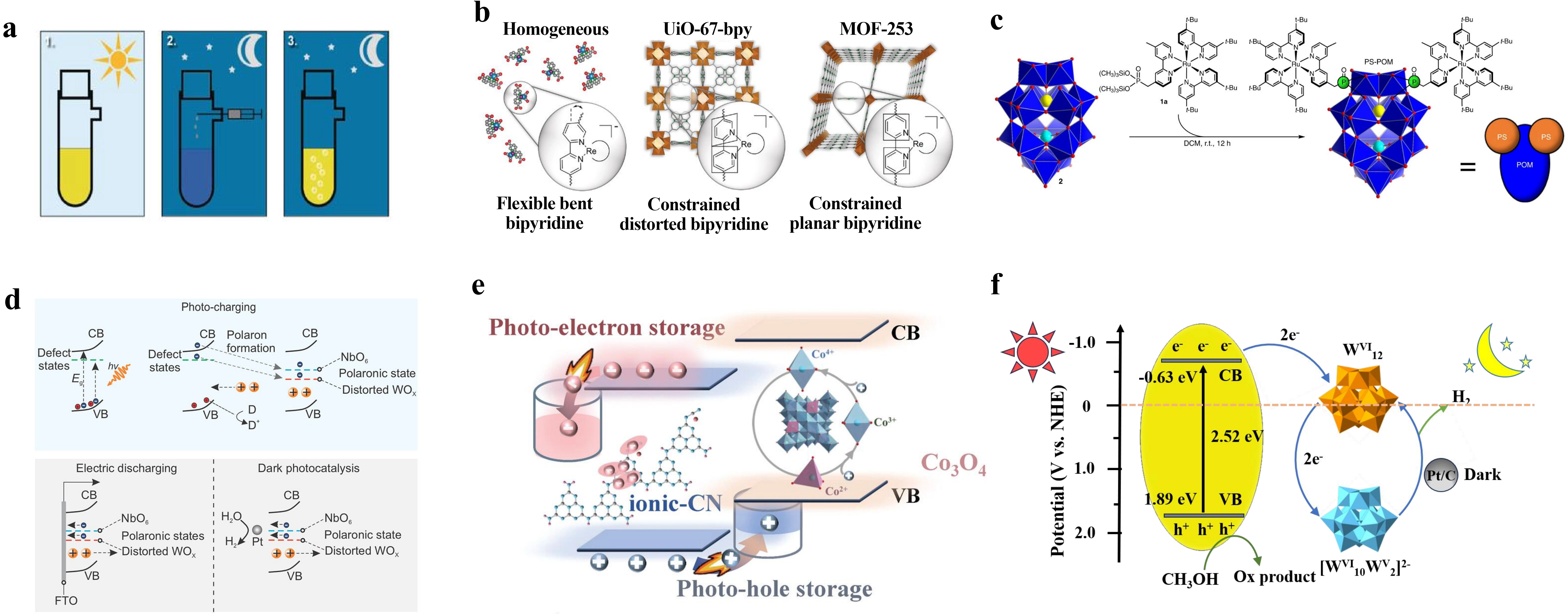
Figure 4. Schematic diagrams of six systems for dark photocatalytic hydrogen evolution. (a) Carbon-nitride-based materials[16]; (b) MOF-based materials[17]; (c) POM-based materials. Republished with permission from[22]; (d) 2D layered materials[23]; (e) Heterojunctions. Republished with permission from[26]; (f) Semiconductor/POM composites. Republished with permission from[29]. MOF: metal-organic framework; POM: polyoxometalate.
Despite these shared principles, distinct mechanistic differences arise from the unique structural design of each material system. Carbon-nitrogen-based materials (NCN-CNx) create electron localization sites within the heptazine unit network through the synergistic effect of cyanamide groups (-NCN) and K+ ions (Figure 4a)[16]. Photogenerated electrons are stored as long-lived radicals with a lifetime exceeding 12 hours. In the presence of a sacrificial electron donor, long-lived radicals with lifetimes exceeding the diurnal cycle are generated upon illumination. EPR spectroscopy[56-59] and density functional theory[60-65] calculations confirm that unpaired electrons are localized on single heptazine units, stabilized by the synergistic effect of cyanamide groups and
POM-based materials (PS-POM) integrate Ru-based photosensitizers with Dawson-type POMs (P2W17O6110-)[66-75] via
Heterojunction systems (ionic-CN/Co3O4) are constructed via electrostatic self-assembly to form a bipolar charge storage junction. With matched energy levels, ionic-CN functions as the electron storage component while Co3O4 serves as the hole storage layer, enabling efficient charge separation (Figure 4e)[26]. This design yields a high photo-charging rate of 0.86 A g-1, which is 6.61 times that of pristine ionic-CN. At an optimal Co3O4 loading of 10 wt.%, the system achieves a dark hydrogen evolution yield of 1.57 mmol g-1. Finally, semiconductor-POM composites (g-C3N4/W12) form a tight interface via electrostatic interactions between protonated -NH3+ groups on the g-C3N4 surface and anionic W12 clusters (Figure 4f)[29]. The favorable energy-level alignment ensures spontaneous electron transfer from g-C3N4 to W12, resulting in the formation of heteropoly blue species. This system exhibits a remarkable dark hydrogen evolution rate of 3,220 μmol g-1 h-1, and maintains a rate of 954 μmol g-1 h-1 under natural sunlight.
In essence, these mechanistic variations originate from the rational design of electron storage sites whether radicals, polarons, or heteropoly blues, and the nature of interfacial interactions, including covalent bonding, electrostatic forces, and confinement effects. These design choices fundamentally dictate the performance trade-offs of each system regarding storage lifetime, capacity, and release kinetics.
3.2 Dark hydrogen evolution activity of dark photocatalytic systems
Table 1 compares the dark hydrogen evolution activity of six typical dark photocatalytic material systems, intuitively presenting the differences in catalytic activity of each system[16,17,22,23,26,29]. In terms of activity data, the semiconductor-POM composite (g-C3N4/W12) exhibits the optimal dark hydrogen evolution rate of 3,220 μmol g-1 h-1, with an outdoor hydrogen evolution rate of 954 μmol g-1 h-1 under natural sunlight[29]. Its high activity originates from the efficient energy level matching and tight electrostatic interface between g-C3N4 and W12. The heterojunction system (ionic-CN/Co3O4) ranks second, with a dark hydrogen evolution rate of
Table 1. Summary of the catalysts for the dark photocatalytic hydrogen production.
| Catalyst | Sacrificial reagent | Catalyst amount (mg) | Dark reaction time (hour) | Dark H2 rate (µmol·g-1·h-1) | Stability |
| NCN-CNx | 4-MBA | 20 | 2.0 | 37.5 | over 15 cycles with gradual decay. |
| PS-POM | Sodium ascorbate | 0.96 | 0.22 | 284.04 | - |
| ReCl-253 | TEOA | 3 | 0.67 | 110 | 90% capacity retention over 10 cycles |
| ionic-CN/Co3O4 | MeOH | 10 | 0.2 | 1,350 | - |
| NbWO6 | MeOH | 95 | 2.5 | 2.86 | 76% capacity retention over 30 cycles |
| g-C3N4/W12 | MeOH | 34.5 | 0.08 | 3,220 | Stable in 5 cycles |
NCN: nitrogen–carbon–nitrogen; CN: carbon nitride; 4-MBA: 4-mercaptobenzoic acid; PS: polystyrene; POM: polyoxometalate; TEOA: triethanolamine.
Overall, the activity differences among various systems are closely related to the structure of electron storage sites, the mode of interfacial interaction, and the type of co-catalyst. High-activity systems mostly rely on efficient charge transfer and rapid electron release, which reflects the inherent trade-off relationships between “activity-lifetime-capacity” in the field of dark photocatalysis.
3.3 Photo-charging characterization of dark photocatalytic catalysts
The color change of the dark photocatalytic catalyst before and after the reaction is very obvious, which can be observed through the significant changes in the UV-vis diffuse reflectance spectrum[76-85] before and after the reaction. Figure 5a shows the photo-charging characterization features of carbon-nitrogen-based material NCN-CNx[16]. Irradiating NCN-CNx under oxygen-free conditions leads to the formation of characteristic blue radicals in the system, and its UV-vis diffuse reflectance spectrum changes significantly. The electron-localized blue radical state extends the material’s light absorption to 500-750 nm, thus enhancing solar energy utilization. Meanwhile, the EPR signal shows an obvious growth trend of radical signals with irradiation time, confirming the generation and accumulation of long-lived radicals. Figure 5b provides a comparative visual analysis of the ReCl-253 material in two distinct end states[17]. It shows the ReCl-253 after undergoing a full discharge and subsequent drying process, where the color reverts to its original orange appearance. This comparison provides clear and immediate visual proof of the reversibility of the photo-charging and discharging cycles, which confirms the reversible switching characteristics between the charging state and the discharge state endpoints. While Figure 5c exhibits the photochromic and absorption spectral evolution characteristics of two-dimensional layered material NbWO6 under 365 nm UV light irradiation[23]. With the irradiation time extending from 0 min to 60 min, the UV-vis absorption spectrum of NbWO6 suspension shows a broadened absorption enhancement in the range of 400~800 nm, among which the absorption intensity at 575 nm increases continuously with irradiation time and gradually stabilizes. This characteristic absorption is directly related to polarons formed by trapped photogenerated electrons, which intuitively reflects the photo-charging process of the material and the change of charge storage capacity, and is also the spectral embodiment of the photochromism of NbWO6 (changing from milky white to blue).
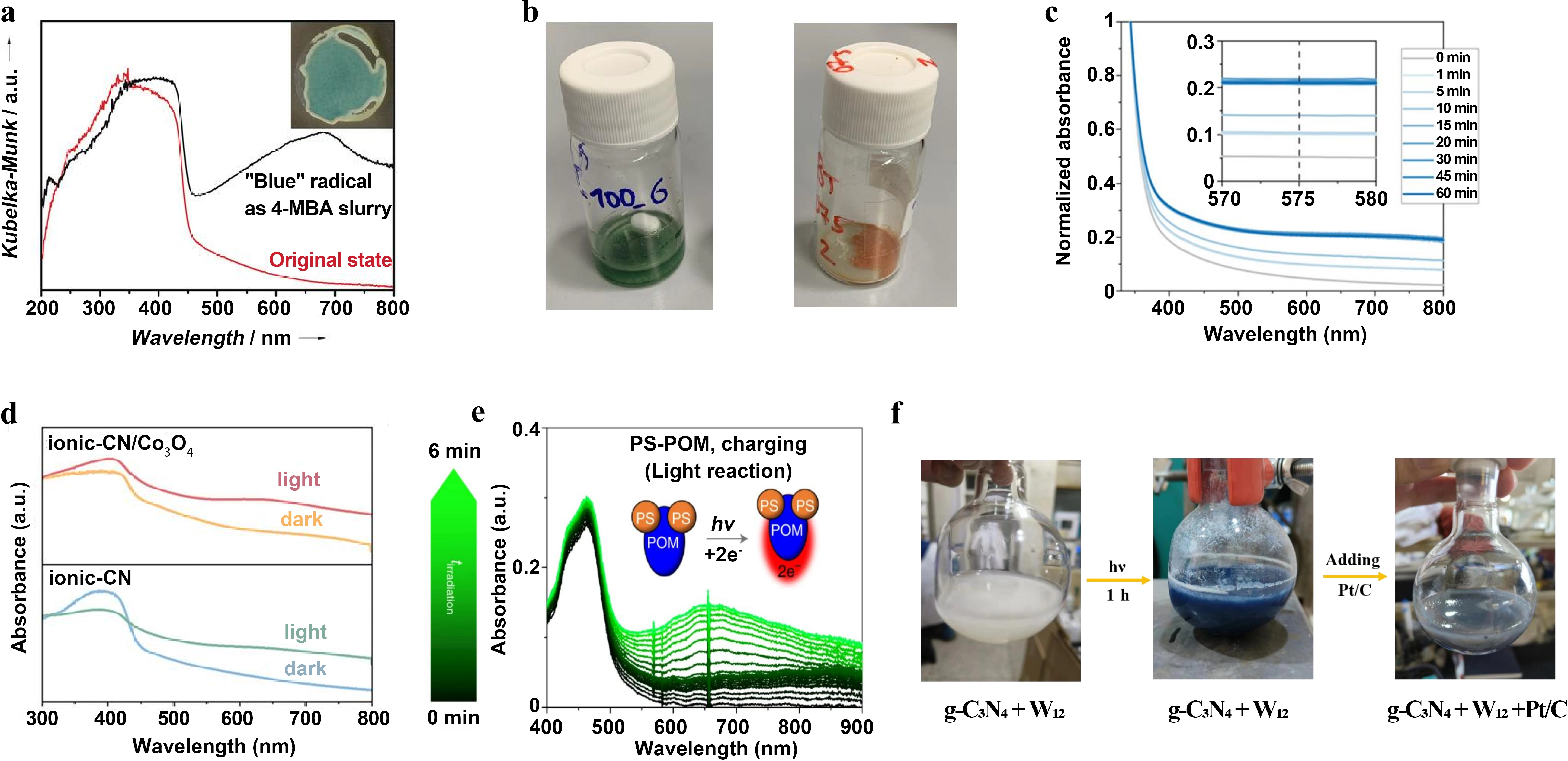
Figure 5. (a) UV-vis diffuse reflectance spectra of NCN-CN before and after irradiation[16]; (b) Visual comparison of the photo-charged and discharged states[17]; (c) Normalized operando UV-vis absorbance spectra of NbWO6 suspension under 365 nm UV illumination for different times. The inset shows the magnified region between 570 and
In addition, Figure 5d presents the UV-vis absorption spectrum comparison of the ionic-CN/Co3O4 bipolar charge storage junction before and after photo-charging[26]. After light irradiation, an obvious characteristic absorption band appears in the material in the range of 550~750 nm, and the color of the sample changes from initial yellow to blue. This phenomenon is directly related to the formation of long-lived photoinduced radical species by heptazine units in ionic-CN capturing photogenerated electrons, confirming the efficient photogenerated electron storage capacity of the system. Meanwhile, the rapid capture of photogenerated holes by Co3O4 further promotes charge separation and storage. In contrast, Figure 5e shows the photo-charging spectral characterization of the POM-based material PS-POM[22]. Under visible light irradiation and in the presence of a sacrificial electron donor, a broadened characteristic absorption band gradually appears and intensifies at 650 nm for PS-POM, which is a typical spectral signal of its
Finally, Figure 5f depicts the light absorption change of semiconductor-POMs composite g-C3N4/W12 during the photo-charging process and the dark-state triggering process[29]. Under light irradiation, the UV-vis absorption spectrum of g-C3N4/W12 system changes significantly. Due to the rapid transfer of photogenerated electrons from g-C3N4 to W12 and the reduction of W6+ to W5+, heteropoly blue species are formed, and the system suspension changes from pale yellow to deep blue. When Pt/C co-catalyst is added to the photo-charged system and placed in the dark state, the characteristic absorption of the system gradually disappears with the release of stored electrons and proton reduction for hydrogen evolution, which intuitively reflects the complete process of electron storage during photo-charging and electron release in the dark for this composite material.
3.4 Key photo-charging performance of various dark photocatalytic systems
Photo-charging performance is a core index for evaluating dark photocatalytic materials, which directly determines the electron storage capacity and subsequent dark hydrogen evolution efficiency of materials. Different material systems exhibit distinctive
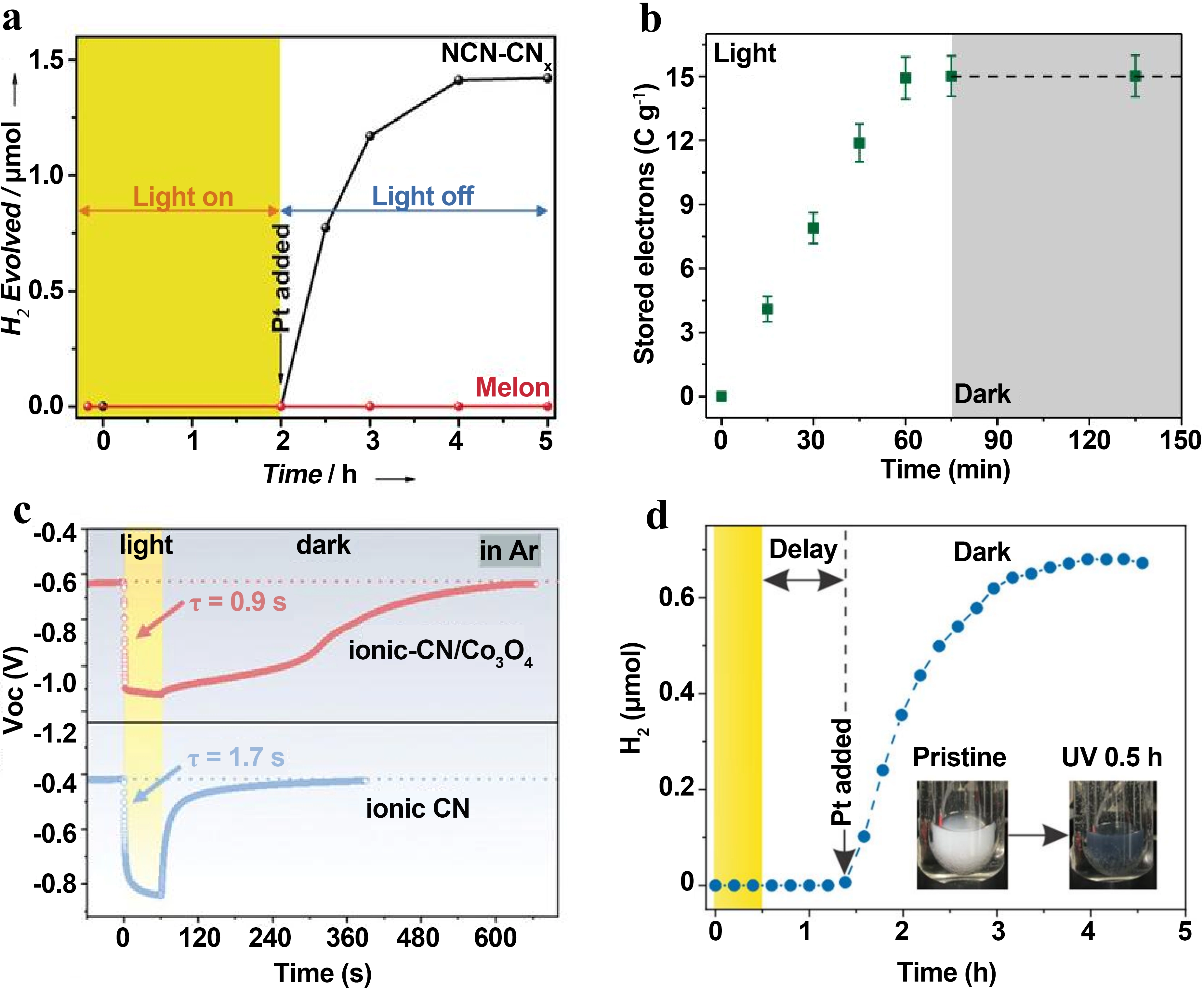
Figure 6. (a) Dark hydrogen evolution kinetic curves of NCN-CNx and Melon[16]; (b) Change in charge storage capacity of ReCl-253 during photo-charging[17]; (c) Open circuit voltage decay curves of ionic-CN and ionic-CN/Co3O4. Republished with permission from[26]; (d) Dark hydrogen evolution kinetic curve of NbWO6 after photo-charging[23].
In comparison, MOF-based materials exhibit excellent charge storage stability by virtue of the unique spatial confinement effect[17]. Figure 6b shows the change of charge storage performance of ReCl-253 under 450 nm light irradiation. With TEOA as the sacrificial agent, its charge storage capacity increases nearly linearly with the extension of irradiation time and reaches saturation at about
Heterojunction engineering, on the other hand, achieves the dual improvement of photo-charging rate and charge storage stability by constructing a bipolar charge storage junction[26]. Figure 6c shows the comparison of open circuit voltage decay curves between ionic-CN and ionic-CN/Co3O4 bipolar charge storage junction. After 60 s of light irradiation, the ionic-CN/Co3O4 heterojunction has an open circuit voltage drop of about 0.50 V, and it takes 600 s to complete the potential recovery, while pure ionic-CN only has a voltage drop of 0.44 V and can quickly recover to the initial potential within 180 s. The performance difference between the two directly proves that the heterojunction can more stably confine photogenerated electrons in trapping sites through the directional separation and separate storage of electrons and holes, effectively suppressing non-radiative recombination pathways. Precisely based on this core optimization effect, ionic-CN/Co3O4 achieves a high photo-charging rate of 0.86 A g-1, which is 6.61 times higher than that of pure ionic-CN (0.13 A g-1), making it a highly advantageous efficient dark photocatalytic system under
Two-dimensional layered materials realize the effective storage and dark-state controllable release of photogenerated charges relying on the synergistic effect of ion intercalation and lattice distortion[23]. Figure 6d shows the dark hydrogen evolution kinetic characteristics of two-dimensional layered material NbWO6 after 0.5 h of 365 nm UV light irradiation. After adding platinum nanoparticle co-catalyst to the system in the dark state, the photogenerated electrons stored in the material are gradually released to drive the reduction of protons to generate hydrogen, with a final hydrogen production of about 0.68 µmol and a maximum turnover frequency of 0.13 h-1. At the same time, the system has good cyclic usability, which can complete multiple “light-charge-dark discharge” hydrogen evolution cycles with only a single addition of catalyst. In addition, its charge storage capacity shows significant light source dependence, with a storage capacity of 3.2 mA h g-1 under 365 nm UV light irradiation, about 18 times that under 1 sun simulated sunlight. This difference does not stem from a simple increase in light intensity, but from the efficient matching of light wavelength and the intrinsic absorption characteristics of the material, which also provides a key basis for the light source selection and performance optimization of such materials.
Overall, the above four types of core material systems have realized the optimized regulation of photo-charging performance from different perspectives: carbon-nitrogen-based materials construct efficient electron trapping sites by virtue of functional group modification[16], MOF-based materials achieve long-term stable charge storage through spatial confinement[17], heterojunction materials break through the charge recombination bottleneck to realize fast charging via bipolar storage[23], and two-dimensional layered materials realize controllable charge storage and release relying on ion intercalation[23]. These differentiated regulation strategies and performance characteristics provide valuable insights and new avenues for the subsequent design and development of high-efficiency dark photocatalytic catalysts.
3.5 Structural and interfacial characterization of dark photocatalytic catalysts
Accurate structural and interfacial characterization is a key tool to analyze the structure-performance relationship and reveal the charge transport mechanism. Figure 7 systematically demonstrates the structural design and interfacial regulation rules of different dark photocatalytic catalysts, and it intuitively reflects the regulatory effects of interface engineering and electrostatic self-assembly on material structure construction and charge transport performance. Among them, Figure 7a shows the crystal structure of
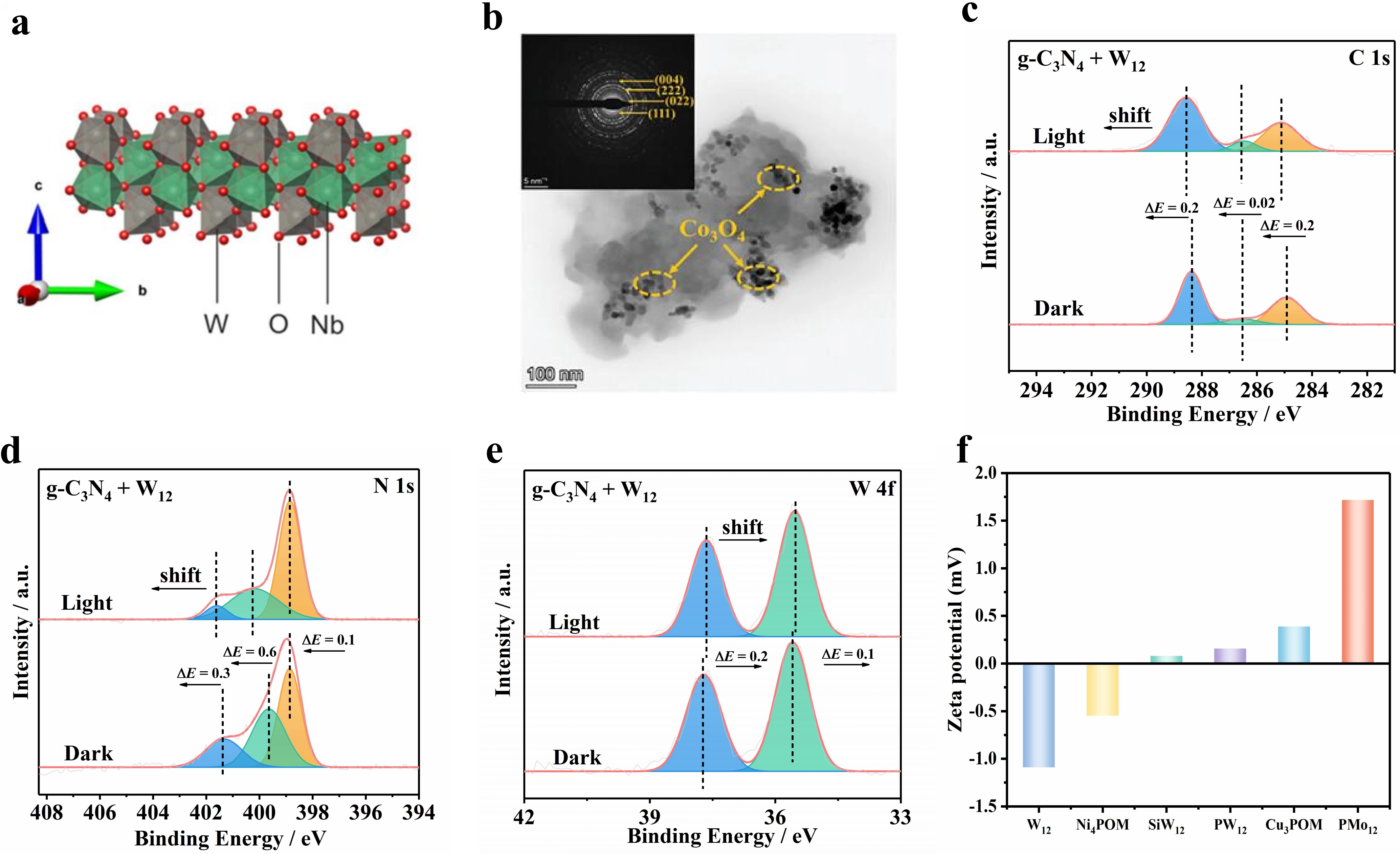
Figure 7. (a) Schematic illustration of the layered crystal structure of 2D NbWO6[23]; (b) TEM image of the ionic-CN/Co3O4 heterojunction. Inset: SAED pattern of the spinel
Furthermore, Figure 7c,d,e display the in-situ XPS spectra of g-C3N4/W12 composite under simulated reaction conditions, which are direct spectral evidence for light-induced interfacial charge transfer[29]. Under light irradiation, the binding energies of C 1s and N 1s orbitals of g-C3N4 shift to higher energy, indicating the decrease of electron cloud density due to the migration of photogenerated electrons. The binding energy of the W 4f orbital of W12 clusters shifts to lower energy, corresponding to the reduction of W6+ to W5+ by accepting photogenerated electrons from g-C3N4, thus forming stable heteropoly blue species. The reverse shift of binding energies of C, N and W directly confirms the directional and efficient trans-interfacial transfer of photogenerated electrons from
Generally speaking, Figure 7 clarifies the internal correlation of structural design-interfacial regulation-charge transport in dark photocatalytic catalysts through four core characterization methods. The 2D layered structure of NbWO6 provides intrinsic sites for charge storage[23]. The tight heterojunction interface of ionic-CN/Co3O4 lays a structural foundation for directional charge
3.6 Cycling stability of dark photocatalytic catalysts
Cycling stability and environmental tolerance are core indices for evaluating the practical potential of dark photocatalytic catalysts, which directly determine their long-term working capacity and practical application adaptability. Figure 8 systematically characterizes the cycling performance and environmental adaptability of typical dark photocatalytic catalysts from four aspects: charge-discharge cycle, light-dark hydrogen evolution cycle, composite system cycle and oxygen tolerance, fully reflecting the performance retention capacity of different material systems in cyclic use and actual working conditions. Among them, Figure 8a shows the charge-discharge cycle capacity retention curve of ReCl-253 in an acetonitrile system under an argon atmosphere[17]. After 10 consecutive photo-charging and dark-discharging cycles, the material retains about 90% of its initial charging capacity with an average capacity loss of only 1% per cycle. The slight attenuation is only caused by the gradual blocking of MOF pores by sacrificial agents and solvent molecules during the cycle, which directly confirms the good reversibility of ReCl-253 in the photo-charging and discharging process. While Figure 8b presents the cyclic hydrogen evolution stability curve of NbWO6[23]. After photo-charging with 365 nm UV light, the catalyst can complete multiple “light-charge-dark discharge” hydrogen evolution cycles with a single addition of co-catalyst, and the hydrogen evolution performance remains stable during the cycle. Meanwhile, as a solar battery anode, it still retains 76% of its initial capacity after 30 photo-charging and electro-discharging cycles, fully demonstrating the excellent structural stability and cyclic service performance of NbWO6. In addition, Figure 8c displays the cycling stability curve of the g-C3N4/W12 composite[29]. After multiple photo-charging and dark discharging cycles, the dark hydrogen evolution activity of the composite remains nearly constant without obvious attenuation, which confirms the high stability of the W12 electron storage unit in the composite system. The robust interface between g-C3N4 and W12 ensures efficient charge transport in the cyclic reaction, laying a foundation for the long-term operation of the composite system.
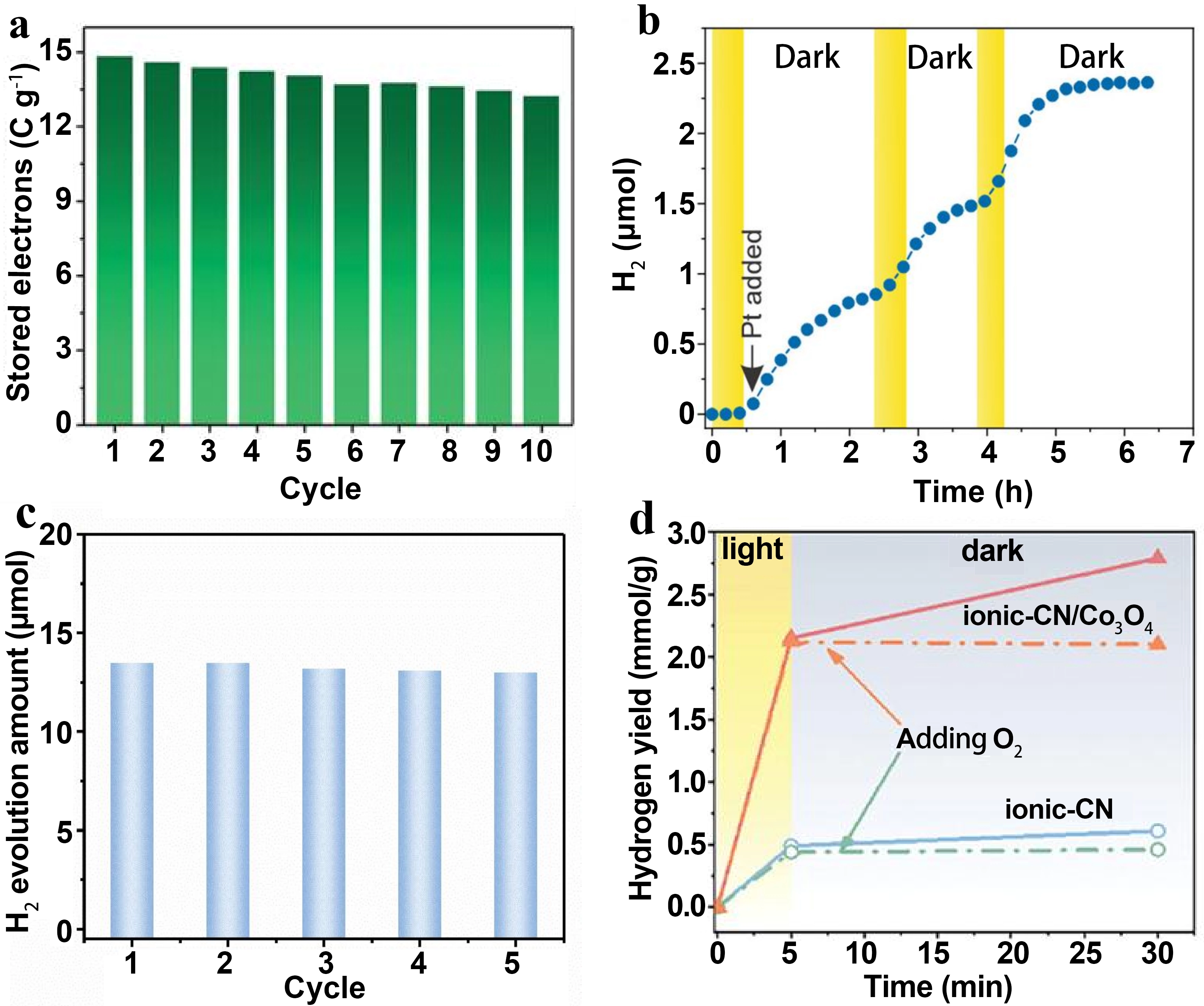
Figure 8. (a) Cyclic stability test of ReCl-253 under inert atmosphere[17]; (b) Hydrogen evolution stability of NbWO6 over multiple light-dark cycles[23]; (c) Cyclic stability tests of g-C3N4/W12. Republished with permission from[29]; (d) Effect of oxygen on dark H2 evolution over ionic-CN and the ionic-CN/Co3O4 junction. Republished with permission
Finally, Figure 8d compares the oxygen’s effect on the dark hydrogen evolution performance of ionic-CN and ionic-CN/Co3O4[26]. Oxygen quenching experiments show that after introducing oxygen as an electron scavenger, the dark hydrogen evolution rates of both catalysts drop sharply to near zero, in sharp contrast to the high efficiency hydrogen evolution under oxygen free conditions. This result directly proves that the dark hydrogen evolution completely depends on the photogenerated electrons stored during the photo-charging stage. Oxygen will rapidly consume the stored electrons and terminate the HER, clarifying that an oxygen-free environment is a key working condition requirement for dark photocatalytic hydrogen evolution.
In summary, the above three core material systems all exhibit good cycling stability. Materials with different structural designs achieve long term charge storage and cyclic hydrogen evolution through their own structural advantages. The significant inhibitory effect of oxygen on dark hydrogen evolution also provides a clear reference for the environmental conditions of the practical application of dark photocatalytic materials. These characterization results not only verify the long-term working capacity of typical dark photocatalytic catalysts, but also provide an important basis for the subsequent structural optimization and application scenario design of such catalysts.
4. Conclusion and Outlook
The inherent contradiction between charge storage capacity, storage lifetime, and release kinetics is a core scientific issue in dark photocatalytic hydrogen evolution. High-capacity charge storage often requires deep energy level traps, which reduce the electron release rate and result in slow dark hydrogen evolution kinetics; conversely, shallow traps can accelerate electron release but significantly decrease storage capacity and lifetime[16]. This contradiction stems from the correlation between electron trap depth and electron transfer barrier: the deeper the trap, the stronger the electron binding, the more stable the storage, but the higher the energy required for electrons to escape the trap and transfer to catalytic sites, resulting in slower kinetics. How to break this
In addition to the above fundamental bottleneck, the practical application of dark photocatalytic hydrogen evolution faces dual challenges of system integration and energy efficiency. In terms of system integration, most studies focus on dispersed systems of powder catalysts, lacking device design for practical applications, such as structural optimization and stability improvement of integrated devices for solar energy capture-charge storage-hydrogen evolution. In terms of energy efficiency, the overall
Against these challenges, dark photocatalytic hydrogen evolution, as an emerging solar-energy utilization strategy, successfully realizes the capture-storage-on-demand conversion of solar energy by simulating the spatiotemporal decoupling mechanism of natural photosynthesis, providing an effective way to overcome the intermittency of solar energy and the challenges of hydrogen storage and transportation. In this review, we systematically summarize the basic principles, charge storage mechanisms, key material systems, and design strategies of dark photocatalytic hydrogen evolution, clarifying the core progress and challenges in this field. Existing studies have developed various high-performance systems such as carbon-nitrogen-based materials, MOF-based materials, POM-based materials, two-dimensional layered materials, and heterojunction composites. Through design strategies such as functional modification, confinement effect, and interface engineering, significant improvements in charge storage capacity, lifetime, and hydrogen evolution efficiency have been achieved. Among them, the TiOx/CN/Pt composite has become the most efficient system currently, with a dark hydrogen evolution rate of 4,800 μmol g-1 h-1, MOF-253-Re shows long-term energy storage potential with an electron storage lifetime exceeding 4 weeks, and NbWO6 realizes the integration of solar battery and dark hydrogen evolution dual functions.
Future research should be advanced in a coordinated manner from the following dimensions: Firstly, in material design, innovative strategies such as multi-element doping and defect engineering must be employed to overcome the inherent trade-off between charge storage and release, thereby developing new material systems with broad-spectrum response, high storage capacity, and long cycle life. Secondly, in mechanistic investigation, advanced in-situ characterization techniques including in-situ EPR, in-situ XPS, and femtosecond transient absorption spectroscopy should be extensively applied to dynamically elucidate the microscopic mechanisms and evolution pathways during charge storage and release. In addition, in system integration, focused efforts are needed to construct integrated devices that combine solar energy capture, charge storage, and hydrogen evolution functions, with optimization of electrode structures and interfacial contacts to enhance operational stability and scalability. Finally, in sustainability, priority should be given to develop sacrificial-agent-free systems and non-noble-metal co-catalysts to effectively reduce overall costs and environmental impacts, thereby steering the technology toward practical and eco-friendly applications. With the deepening of material design and mechanism research, dark photocatalytic hydrogen evolution is expected to realize practical applications in distributed energy supply, hydrogen production and storage, providing important support for building a carbon-neutral energy system.
Acknowledgements
Doubao and DeepSeek were employed during manuscript preparation, solely for language polishing to improve the fluency of the text. The authors take full responsibility for the integrity, originality, and accuracy of the work.
Authors contribution
Ding Y: Conceptualization, supervision, writing-review & editing.
Dong X: Writing-original draft.
Zhou Y, Fang X: Writing-review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 22472071, 22075119 and 12404325), the Natural Science Foundation of Gansu Province (Grant No. 21JR7RA440), the Lanzhou Municipal-Level Science and Technology Reserve Project
Copyright
© The Author(s) 2026.
References
-
1. Zhang DS, Wang L, Zhang X, Li XB, Xu H, Tung CH, et al. Emerging inorganic–organic hybrid photocatalysts for solar-driven overall water splitting: Progress and perspectives. Chem Soc Rev. 2025;54(21):9978-10005.[DOI]
-
3. Yasin G, Tabish M, Ajmal S, Zhuo Q, Mushtaq MA, Saad A, et al. Single atom horizons for shaping the future of catalysis and sustainability: The next frontiers in energy conversion and storage. Prog Mater Sci. 2026;155:101525.[DOI]
-
4. Lee WH, Lee CW, Cha GD, Lee BH, Jeong JH, Park H, et al. Floatable photocatalytic hydrogel nanocomposites for large-scale solar hydrogen production. Nat Nanotechnol. 2023;18(7):754-762.[DOI]
-
5. Choi J, Jung W, Gonzalez-Carrero S, Durrant JR, Cha H, Park T. Understanding charge carrier dynamics in organic photocatalysts for hydrogen evolution. Energy Environ Sci. 2024;17(21):7999-8018.[DOI]
-
6. Liu H, Cheng M, Liu Y, Wang J, Zhang G, Li L, et al. Single atoms meet metal–organic frameworks: Collaborative efforts for efficient photocatalysis. Energy Environ Sci. 2022;15(9):3722-3749.[DOI]
-
7. Navalón S, Dhakshinamoorthy A, Álvaro M, Ferrer B, García H. Metal–organic frameworks as photocatalysts for solar-driven overall water splitting. Chem Rev. 2023;123(1):445-490.[DOI]
-
8. Melchiorre P. Introduction: Photochemical catalytic processes. Chem Rev. 2022;122(2):1483-1484.[DOI]
-
9. Nishiyama H, Yamada T, Nakabayashi M, Maehara Y, Yamaguchi M, Kuromiya Y, et al. Photocatalytic solar hydrogen production from water on a 100-m2 scale. Nature. 2021;598(7880):304-307.[DOI]
-
10. Pavliuk MV, Wrede S, Liu A, Brnovic A, Wang S, Axelsson M, et al. Preparation, characterization, evaluation and mechanistic study of organic polymer nano-photocatalysts for solar fuel production. Chem Soc Rev. 2022;51(16):6909-6935.[DOI]
-
11. Mai H, Le TC, Chen D, Winkler DA, Caruso RA. Machine learning for electrocatalyst and photocatalyst design and discovery. Chem Rev. 2022;122(16):13478-13515.[DOI]
-
12. Jin HG, Zhao PC, Qian Y, Xiao JD, Chao ZS, Jiang HL. Metal–organic frameworks for organic transformations by photocatalysis and photothermal catalysis. Chem Soc Rev. 2024;53(18):9378-9418.[DOI]
-
13. Zhao M, Li W, Yang M, Zhao Z, Ye R, Mao X, et al. Long-range enhancements of micropollutant adsorption on metal-promoted photocatalysts. Nat Catal. 2024;7(8):912-920.[DOI]
-
14. Boettcher SW. Introduction to green hydrogen. Chem Rev. 2024;124(23):13095-13098.[DOI]
-
15. Odenweller A, Ueckerdt F. The green hydrogen ambition and implementation gap. Nat Energy. 2025;10(1):110-123.[DOI]
-
16. Lau VW, Klose D, Kasap H, Podjaski F, Pignié MC, Reisner E, et al. Dark photocatalysis: Storage of solar energy in carbon nitride for time-delayed hydrogen generation. Angew Chem Int Ed. 2017;56(2):510-514.[DOI]
-
17. Stanley PM, Sixt F, Warnan J. Decoupled solar energy storage and dark photocatalysis in a 3D metal–organic framework. Adv Mater. 2023;35:2207280.[DOI]
-
18. Pan Y, Wang J, Chen S, Yang W, Ding C, Waseem A, et al. Linker engineering in metal–organic frameworks for dark photocatalysis. Chem Sci. 2022;13(22):6696-6703.[DOI]
-
19. Zeng S, Zhang L, Wang W, Shao D, Hao H. Hydrogen evolution based on the electrons/protons stored on amorphous TiO2. Phys Chem Chem Phys. 2017;19(43):29053-29056.[DOI]
-
20. Luo L, Wang S, Zhang L, Xiao X, Wu B, Jaroniec M, et al. Unveiling enhanced dark photocatalysis: Electron storage-enabled hydrogen production in polymeric carbon nitride. Appl Catal B Environ. 2024;343:123475.[DOI]
-
21. Schlomberg H, Kröger J, Savasci G, Terban MW, Bette S, Moudrakovski I, et al. Structural insights into poly(heptazine imides): A light-storing carbon nitride material for dark photocatalysis. Chem Mater. 2019;31(18):7478-7486.[DOI]
-
22. Amthor S, Knoll S, Heiland M, Zedler L, Li C, Nauroozi D, et al. A photosensitizer–polyoxometalate dyad that enables the decoupling of light and dark reactions for delayed on-demand solar hydrogen production. Nat Chem. 2022;14(3):321-327.[DOI]
-
23. Wang Y, Chan YT, Oshima T, Duppel V, Bette S, Küster K, et al. Decoupling of light and dark reactions in a 2D niobium tungstate for light-induced charge storage and on-demand hydrogen evolution. J Am Chem Soc. 2024;146(37):25467-25476.[DOI]
-
24. Ruan Q, Xi X, Yan B, Kong L, Jiang C, Tang J, et al. Stored photoelectrons in a faradaic junction for decoupled solar hydrogen production in the dark. Chem. 2023;9(7):1850-1864.[DOI]
-
25. Li H, Dhar S, Kolora JA, Devulapalli VSD, Borguet E. All trapped electrons in metal oxides are equivalent in dark photocatalysis. ACS Catal. 2025;15(23):19768-19776.[DOI]
-
26. Yan B, Ruan Q, Wang S, Kong L, Zhang P, Wang H, et al. Expediting photo-charging of semiconductors through a bipolar charge storage junction for responsive dark photocatalysis. Adv Funct Mater. 2024;34(49):2408895.[DOI]
-
27. Wang Y, Yang M, Wang Y, Cheng J. Cyanamide-functionalized carbon nitride with ion modification for enhanced “dark” photocatalysis. Adv Funct Mater. 2025;35(31):2503353.[DOI]
-
28. Podjaski F, Lotsch BV. Optoelectronics meets optoionics: Light storing carbon nitrides and beyond. Adv Energy Mater. 2021;11(4):2003049.[DOI]
-
29. Dong X, Fang X, Li B, Feng Y, Liu Y, She L, et al. Solar energy storage in polyoxometalate for on-demand hydrogen transportation and evolution. Adv Mater. 2026;38(9):e19875.[DOI]
-
30. Aleena PA, Singh G, Vidyasagar D, Kumar P, Ahmed MI, Bahadur R, et al. Recent advancements in metal-free nitrides based single-atom catalysts: Nanoarchitectonics and applications. Prog Mater Sci. 2025;153:101490.[DOI]
-
31. Lu T, Xu N, Zhou B, Guo L, Wen X, Lou S, et al. Photo-electroactive p-n heterojunction catalyst with dual Co sites for high-performance light-enhanced zinc–air batteries. eScience. 2026;6(1):100450.[DOI]
-
32. Fang X, Li B, Huang J, Hu C, Yang X, Feng P, et al. Sodium/potassium poly(heptazine imide) with an electron sink effect for hydrogen peroxide photosynthesis. Energy Environ Sci. 2025;18(12):6202-6213.[DOI]
-
33. Feng C, Luo J, Chen C, Zuo S, Ren Y, Wu ZP, et al. Cooperative tungsten centers in polymeric carbon nitride for efficient overall photosynthesis of hydrogen peroxide. Energy Environ Sci. 2024;17(4):1520-1530.[DOI]
-
34. Rocha GFR, da Silva MA, Rogolino A, Diab GA, Noleto LF, Antonietti M, et al. Carbon nitride based materials: More than just a support for single-atom catalysis. Chem Soc Rev. 2023;52(15):4878-4932.[DOI]
-
35. Ding G, Li C, Chen L, Liao G. Porphyrin-based metal–organic frameworks for photo(electro)catalytic CO2 reduction. Energy Environ Sci. 2024;17(15):5311-5335.[DOI]
-
36. Chen J, Sun Z, Ma S, Wang B, Xu E, Wang Z, et al. Elastically bendable metal–organic framework crystals for efficient flexocatalytic hydrogen evolution from water. J Am Chem Soc. 2026;148(4):4217-4229.[DOI]
-
37. Hu S, Gao ML, Huang J, Wang H, Wang Q, Yang W, et al. Introducing hydrogen-bonding microenvironment in close proximity to single-atom sites for boosting photocatalytic hydrogen production. J Am Chem Soc. 2024;146(29):20391-20400.[DOI]
-
38. Liu Z, Su Y, Wang H, Ban T, Wang L, Lu S, et al. Domain-trained language model for inverse design and synthesis of high-performance hydrogen storage MOFs. Angew Chem Int Ed. 2026;65(2):e13366.[DOI]
-
39. Lei H, Zhang J, Wu Z, Ye Y, Li Z, Zhao Y, et al. Crystal facet-dependent photocatalytic hydrogen evolution from ultra-stable Cu-Zr/Hf heterobimetallic metal–organic frameworks. Angew Chem Int Ed. 2025;64(37):e202509572.[DOI]
-
40. Ruan W, Feng Y, Gao Y, Yonesato K, Lan L, Guo Z, et al. Homoleptic and heteroleptic polyoxotungstate–organic cages for efficient photocatalytic hydrogen evolution. Angew Chem Int Ed. 2025;64(37):e202508797.[DOI]
-
41. Tang W, Zhang L, Qiu T, Tan H, Wang Y, Liu W, et al. Efficient conversion of biomass to formic acid coupled with low energy consumption hydrogen production from water electrolysis. Angew Chem Int Ed. 2023;62(30):e202305843.[DOI]
-
42. Lu L, Luo J, Montag M, Diskin-Posner Y, Milstein D. Polyoxymethylene upcycling into methanol and methyl groups catalyzed by a manganese pincer complex. J Am Chem Soc. 2024;146(31):22017-22026.[DOI]
-
43. Shimoyama Y, Ogiwara N, Weng Z, Uchida S. Oxygen evolution reaction driven by charge transfer from a Cr complex to co-containing polyoxometalate in a porous ionic crystal. J Am Chem Soc. 2022;144(7):2980-2986.[DOI]
-
44. Gong X, Liu ZH, Xu Q, Wang L, Guo Q, Zhang J, et al. Single-molecule manipulation of copper nanoclusters for modulating nonlinear optics. Polyoxometalates. 2025;4(1):9140072.[DOI]
-
45. Liu WD, Zhu HT, Zhang X, Su F, Sang XJ, Zhang XL, et al. Room temperature self-assembly and nonenzymatic electrochemical sensing performance of transition metal-embedded wheel-shaped tungstophosphates. Polyoxometalates. 2025;4(1):9140073.[DOI]
-
46. Lei S, Chen B, Wang L, Li J. Study on the regulatory mechanisms of mitochondrial biosynthesis by polyoxometalates. Polyoxometalates. 2025;4(1):9140074.[DOI]
-
47. Liu Z, Hao Y, Zhang J, He Y, Chen W. Polyoxovanadate-modified SnO2 electron transport layer for perovskite photodetectors. Polyoxometalates. 2025;4(1):9140076.[DOI]
-
48. Shang Q, Shen K, Zhao D, Wang X, Wei Y, Wang H, et al. Rare-earth-induced peroxo-phosphotungstates: Enhanced proton conductivity in corresponding membranes. Polyoxometalates. 2025;4(1):9140077.[DOI]
-
49. Hu B, Du Q, Yang Y, Li D, Fu J, Liu B, et al. Facile construction of polyoxometalate-modified polyaryletherketone-based hybrid membranes with enhanced proton conductivity. Polyoxometalates. 2025;4(1):9140079.[DOI]
-
50. Cui LP, Zhang S, Zhao Y, Ge XY, Yang L, Li K, et al. Tunable multi-electron redox polyoxometalates for decoupled water splitting driven by sunlight. Nat Commun. 2025;16:3674.[DOI]
-
51. Firth CR, Jeanguenat C, Lutz-Bueno V, Boureau V, Sivula K. A halted photodeposition technique controls co-catalyst loading and morphology on organic semiconductor nanoparticles for solar H2 Production. Adv Energy Mater. 2025;15(38):2403372.[DOI]
-
52. Yang M, Oldham LI, Daboczi M, Baghdadi Y, Cui J, Benetti D, et al. Advancing hematite photoanodes for photoelectrochemical water splitting: The impact of g-C3N4 supported Ni-CoP on photogenerated hole dynamics. Adv Energy Mater. 2024;14(29):2401298.[DOI]
-
53. Liu R, Chen Y, Yu H, Položij M, Guo Y, Sum TC, et al. Linkage-engineered donor–acceptor covalent organic frameworks for optimal photosynthesis of hydrogen peroxide from water and air. Nat Catal. 2024;7(2):195-206.[DOI]
-
54. Hu H, Sun X, Ma Y, Li H, Zhang W, Fan H, et al. Anchoring redox mediator on COFs for efficient solar to hydrogen conversion. Adv Mater. 2025;37(45):e10193.[DOI]
-
55. Feng J, Mak CH, Jia G, Han B, Shen HH, Santoso SP, et al. Unlocking interfacial interactions of in situ grown multidimensional bismuth-based perovskite heterostructures for photocatalytic hydrogen evolution. Adv Energy Mater. 2024;14(43):2470187.[DOI]
-
56. Allen JP, Szczuka C, Jónsson E, Eichel RA, Granwehr J, Grey CP. Transition metal coordination to degradation products in battery electrolytes revealed by NMR and EPR spectroscopy. Energy Environ Sci. 2025;18(23):10147-10163.[DOI]
-
57. Attar F, Yin H, Schumann SL, Langley J, Cox N, Zeng Z, et al. Advanced electron paramagnetic resonance in chemical energy conversion: Current status and future potential. Energy Environ Sci. 2024;17(10):3307-3328.[DOI]
-
58. Kulikov I, Panjwani NA, Vereshchagin AA, Spallek D, Lukianov DA, Alekseeva EV, et al. Spins at work: Probing charging and discharging of organic radical batteries by electron paramagnetic resonance spectroscopy. Energy Environ Sci. 2022;15(8):3275-3290.[DOI]
-
59. Miao Q, Nitsche C, Orton H, Overhand M, Otting G, Ubbink M. Paramagnetic chemical probes for studying biological macromolecules. Chem Rev. 2022;122(10):9571-9642.[DOI]
-
60. Ong WJ, Tan LL, Ng YH, Yong ST, Chai SP. Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: Are we a step closer to achieving sustainability? Chem Rev. 2016;116(12):7159-7329.[DOI]
-
61. Yuan A, Liu H, Wang H, He T, Zhang H, Hu D, et al. Lattice oxygen mechanism triggered via stable Ni–O–W for solar-boosted water electrolysis. eScience. 2026.[DOI]
-
62. Yang Q, Fedorova EA, Cao DB, Saraçi E, Kondratenko VA, Kreyenschulte CR, et al. Understanding Mn-modulated restructuring of
Fe-based catalysts for controlling selectivity in CO2 hydrogenation to olefins. Nat Catal. 2025;8(6):595-606.[DOI] -
63. Zhao T, Wang J, Wei Y, Zhuang Z, Dou Y, Yang J, et al. From lab-scale to industrialization: Atomically M–N–C catalysts for the oxygen reduction reaction. Energy Environ Sci. 2025;18(8):3462-3501.[DOI]
-
64. Barth S, Zengel D, Goncalves TJ, Plessow PN, Studt F, Grunwaldt JD, et al. Tailoring the active sites in Cu-SSZ-13 as a catalyst for the selective catalytic reduction of NH3 to minimize HCHO and HCN emissions. Nat Catal. 2025;8(8):804-821.[DOI]
-
65. Qiu C, Sellers C, Wu ZY, Cullen DA, Stavitski E, Tayal A, et al. Low-iridium stabilized ruthenium oxide anode catalyst for durable proton-exchange membrane water electrolysis. Nat Nanotechnol. 2025;20(12):1787-1795.[DOI]
-
66. Amao Y. Photo/biohybrid catalytic system for application in semiartificial photosynthesis of CO2 to chemicals. Chem Rev. 2026;126(2):1635-1685.[DOI]
-
67. Zhang T, Qu X, Shao J, Dong X. Organic photosensitizers: From molecular design to phototheranostics. Chem Soc Rev. 2025;54(18):8406-8433.[DOI]
-
68. Guo J, Xia Q, Tang WY, Li Z, Wu X, Liu LJ, et al. Visible light-mediated intermolecular crossed [2+2] cycloadditions using a MOF-supported copper triplet photosensitizer. Nat Catal. 2024;7(3):307-320.[DOI]
-
69. Chen LX, Yano J. Deciphering photoinduced catalytic reaction mechanisms in natural and artificial photosynthetic systems on multiple temporal and spatial scales using X-ray probes. Chem Rev. 2024;124(9):5421-5469.[DOI]
-
70. Ma XL, Ma LH, Guo S, Zhang ZM, Lu TB. Identifying the key photosensitizing factors over metal–organic frameworks for selective control of 1O2 and O2∙- Generation. Angew Chem Int Ed. 2025;64(17):e202423157.[DOI]
-
71. Sánchez-Naya R, Beuerle F. A BODIPY-containing covalent organic framework as a highly porous photosensitizer for environmental remediation and pollutants adsorption. Angew Chem Int Ed. 2025;64(13):e202423676.[DOI]
-
72. Xu SY, Wang P, Guo S, Yao S, Wang C, Lu TB, et al. Modulating electron-transfer via covalent assembly polyoxometalate with photosensitizer for efficient H2 evolution and tandem hydrogenation. Angew Chem Int Ed. 2026;65(5):e23472.[DOI]
-
73. Yin Y, Feng S, Xu X, Liu Y, Li Y, Gao L, et al. Multivariate tuning of photosensitization in mixed-linker metal–organic frameworks for efficient CO2 reduction. J Am Chem Soc. 2025;147(19):16481-16493.[DOI]
-
74. Wu J, Guo F, Yi C, Yang R, Lei X, Xia Z. Photosensitized gold-catalyzed cross-couplings of aryl bromides. J Am Chem Soc. 2025;147(7):5839-5850.[DOI]
-
75. Zheng HL, Zhao JQ, Ma XF, Li JW, Cheng YJ, Chen EX, et al. Electrostatic confinement of photosensitizers within molybdenum–sulfur cluster-based supramolecular matrices for efficient dual-photocatalysis: Hydrogen evolution and trifluoromethylation. J Am Chem Soc. 2026;148(1):652-664.[DOI]
-
76. Zhang D, Wang R, Wang X, Gogotsi Y. In situ monitoring redox processes in energy storage using UV–Vis spectroscopy. Nat Energy. 2023;8(6):567-576.[DOI]
-
77. Zhang D, Gogotsi Y. UV–vis spectroscopy for monitoring oxidation state changes during electrochemical energy storage. Nat Energy. 2023;8(6):565-566.[DOI]
-
78. Antonov L, Nedeltcheva D. Resolution of overlapping UV–Vis absorption bands and quantitative analysis. Chem Soc Rev. 2000;29(3):217-227.[DOI]
-
79. Schoonheydt RA. UV-VIS-NIR spectroscopy and microscopy of heterogeneous catalysts. Chem Soc Rev. 2010;39(12):5051.[DOI]
-
80. Jezkova T, Wiens JJ. Rates of change in climatic niches in plant and animal populations are much slower than projected climate change. Proc R Soc B. 2016;283(1843):20162104.[DOI]
-
81. Soorkia S, Jouvet C, Grégoire G. UV photoinduced dynamics of conformer-resolved aromatic peptides. Chem Rev. 2020;120(7):3296-3327.[DOI]
-
82. Suo ZY, Xiao GB, Su Z, Dong R, Mu X, Gao X, et al. Organic emitters with near-unity photoluminescence to reinforce buried interface of perovskite solar cells and modules. Energy Environ Sci. 2024;17(14):5115-5123.[DOI]
-
83. van der Zee LJ, Hofman J, van Gaalen JM, Slootweg JC. Mechanistic studies on single-electron transfer in frustrated Lewis pairs and its application to main-group chemistry. Chem Soc Rev. 2024;53(10):4862-4876.[DOI]
-
84. Vavra J, Ramona GPL, Dattila F, Kormányos A, Priamushko T, Albertini PP, et al. Solution-based Cu+ transient species mediate the reconstruction of copper electrocatalysts for CO2 reduction. Nat Catal. 2024;7(1):89-97.[DOI]
-
85. Alem A, Poormehrabi P, Lins J, Pachernegg-Mair L, Bandl C, Ruiz V, et al. Monitoring chemical processes in redox flow batteries employing in situ and in operando analyses. Energy Environ Sci. 2025;18(15):7373-7401.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Dong X, Zhou Y, Fang X, Ding Y. Recent developments in dark photocatalytic hydrogen production over smart catalysts. Smart Mater Devices. 2026;2:202609. https://doi.org/10.70401/smd.2026.0032
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. The Basic Mechanism of Dark Light Catalysis
- 3. Key Material System and Design Strategy
- 4. Conclusion and Outlook
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Dong X, Zhou Y, Fang X, Ding Y. Recent developments in dark photocatalytic hydrogen production over smart catalysts. Smart Mater Devices. 2026;2:202609. https://doi.org/10.70401/smd.2026.0032
copy
Share Link
copy